Integrative Analysis of the lncRNA and mRNA Transcriptome Revealed Genes and Pathways Potentially Involved in the Anther Abortion of Cotton (Gossypium hirsutum L.)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Library Construction and RNA-Sequencing (RNA-Seq)
2.3. Transcript Assembly, Alignment, and Identification of Genes
2.4. Differential mRNA and lncRNA Expression Analyses
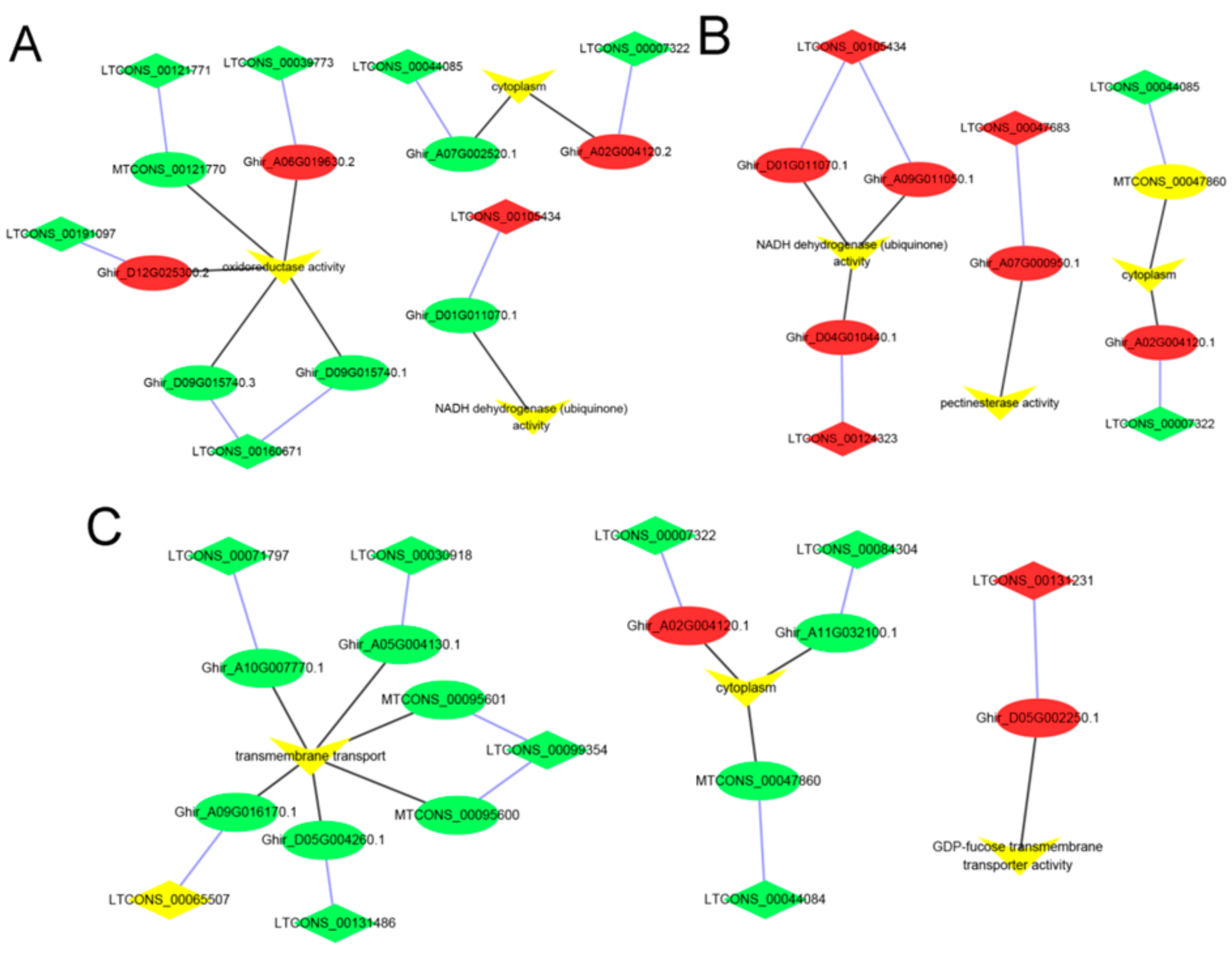
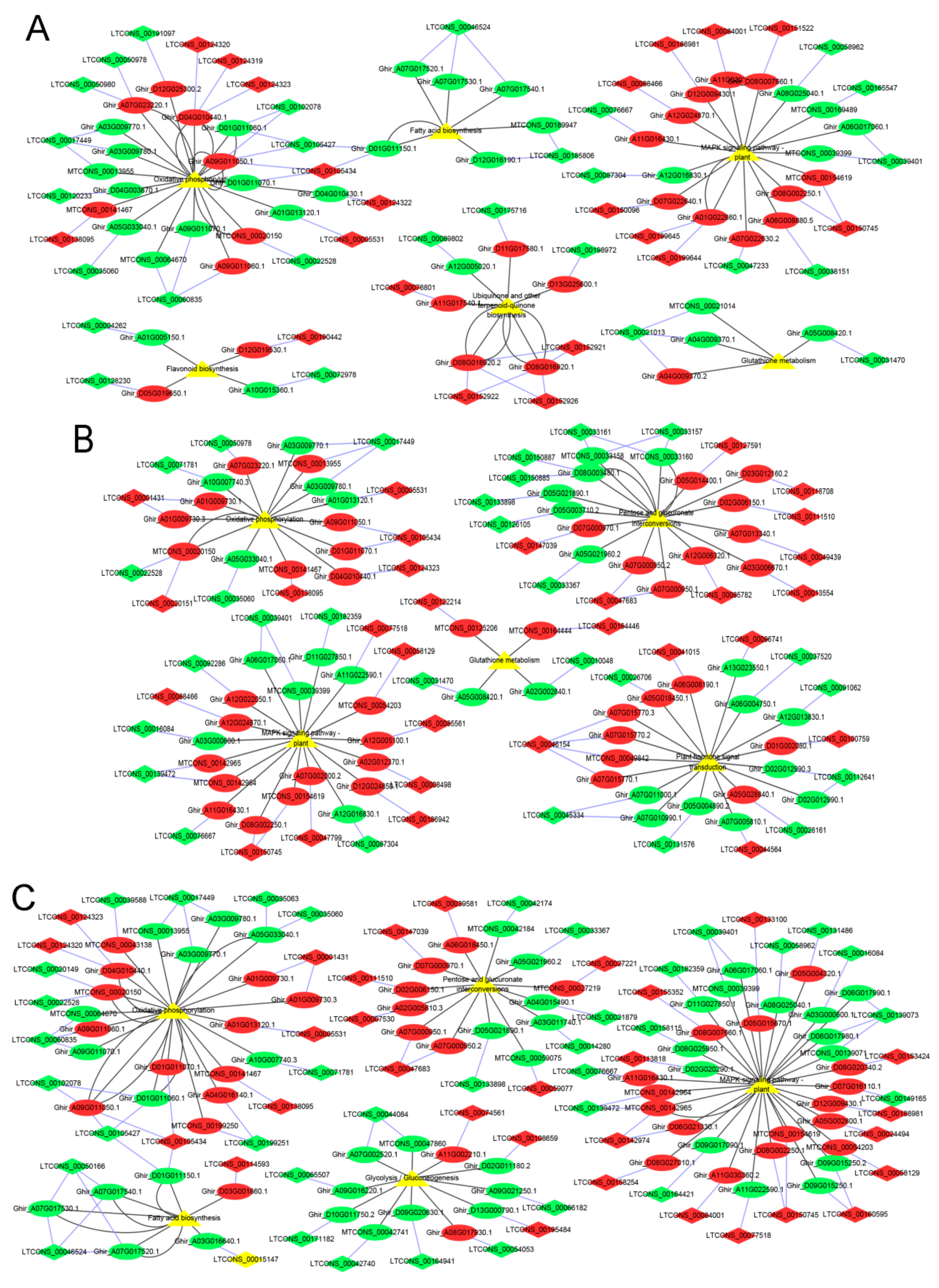
2.5. lncRNA–mRNA Pathway Network Construction
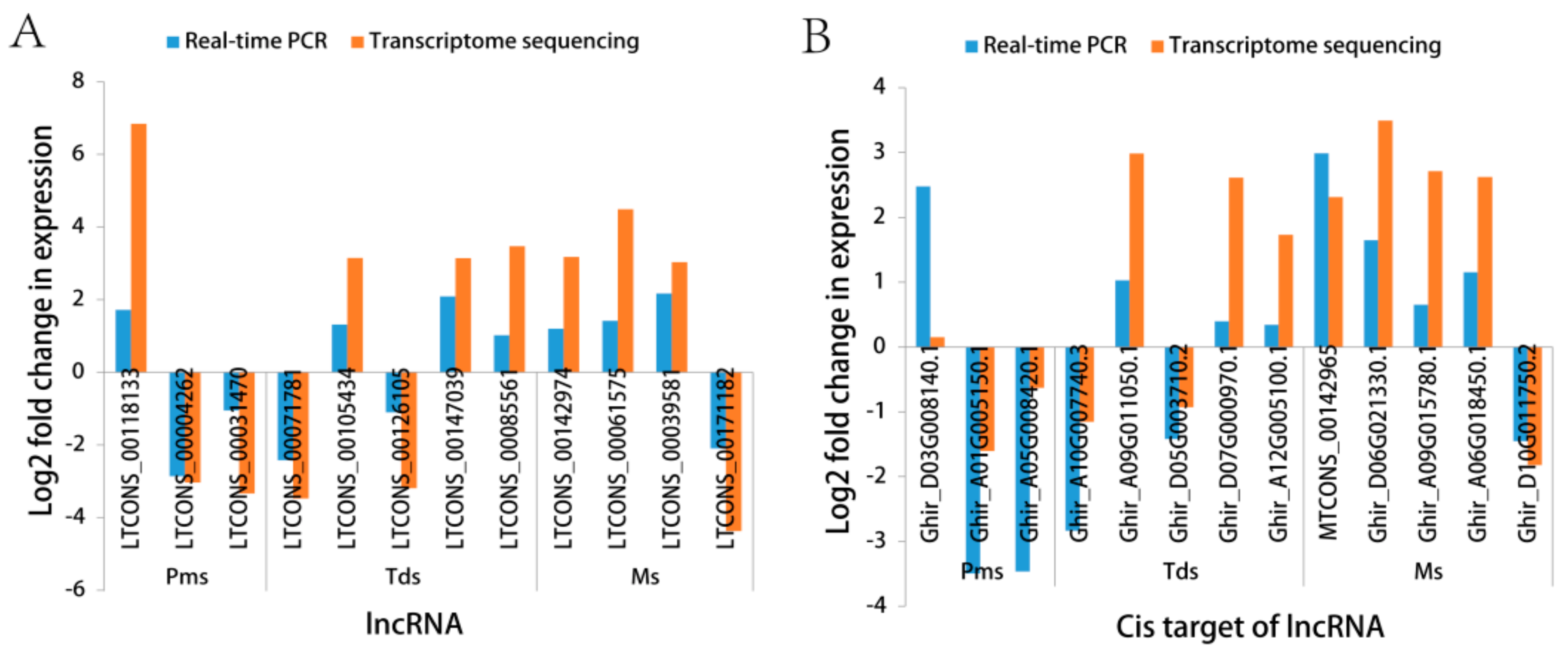
2.6. Gene Expression Confirmed by Real-Time Quantification PCR (RT-qPCR)
3. Results
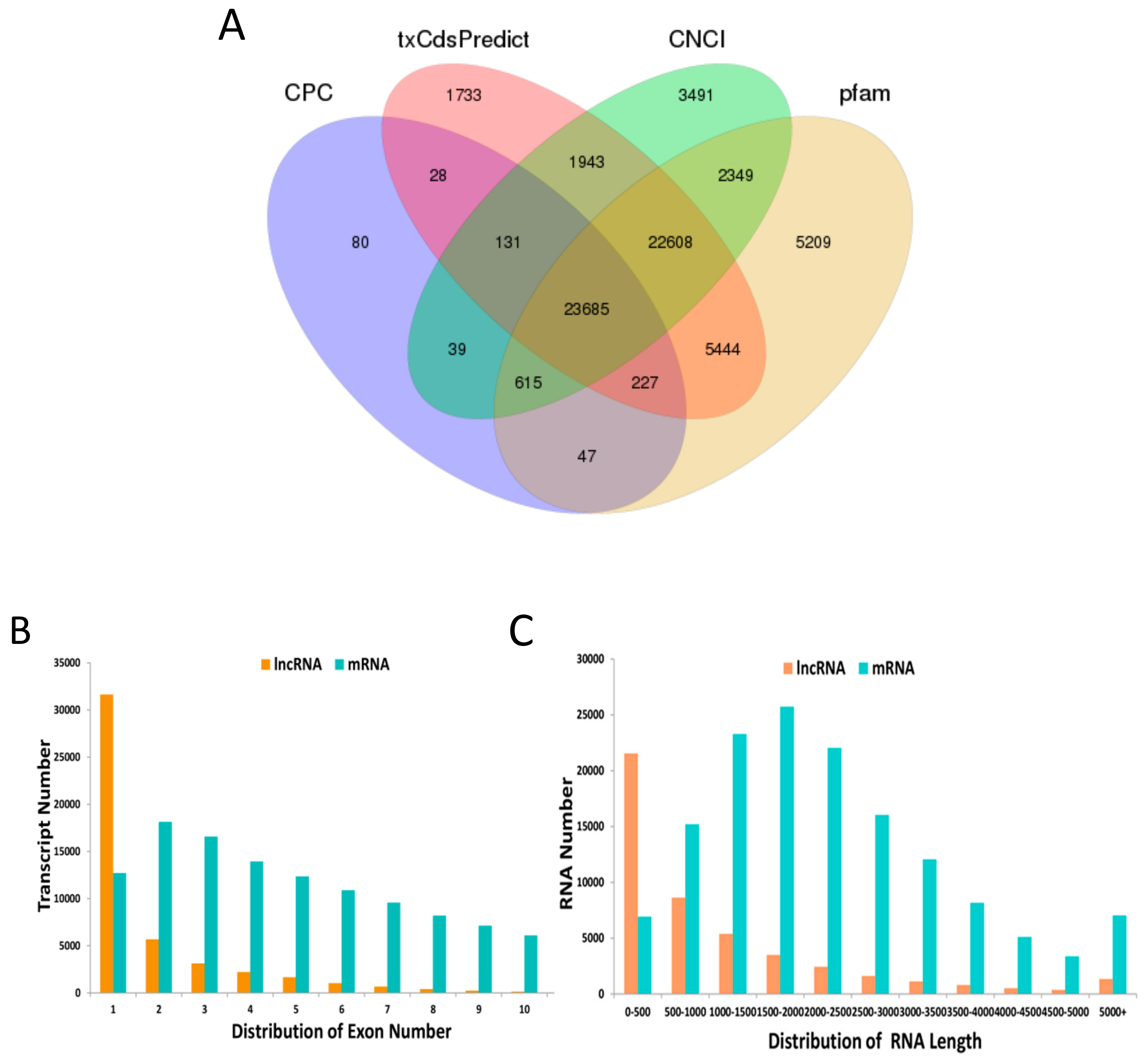
3.1. Identification and Characterization of lncRNA and mRNA
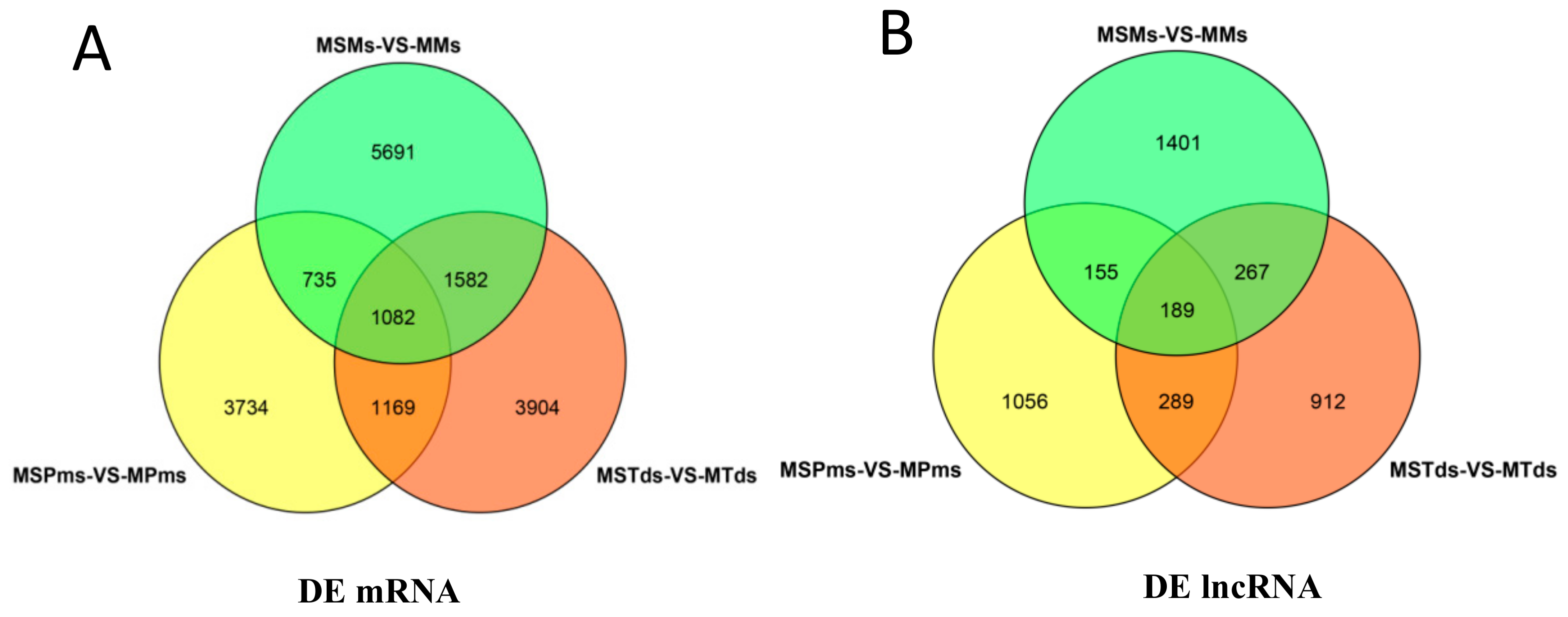
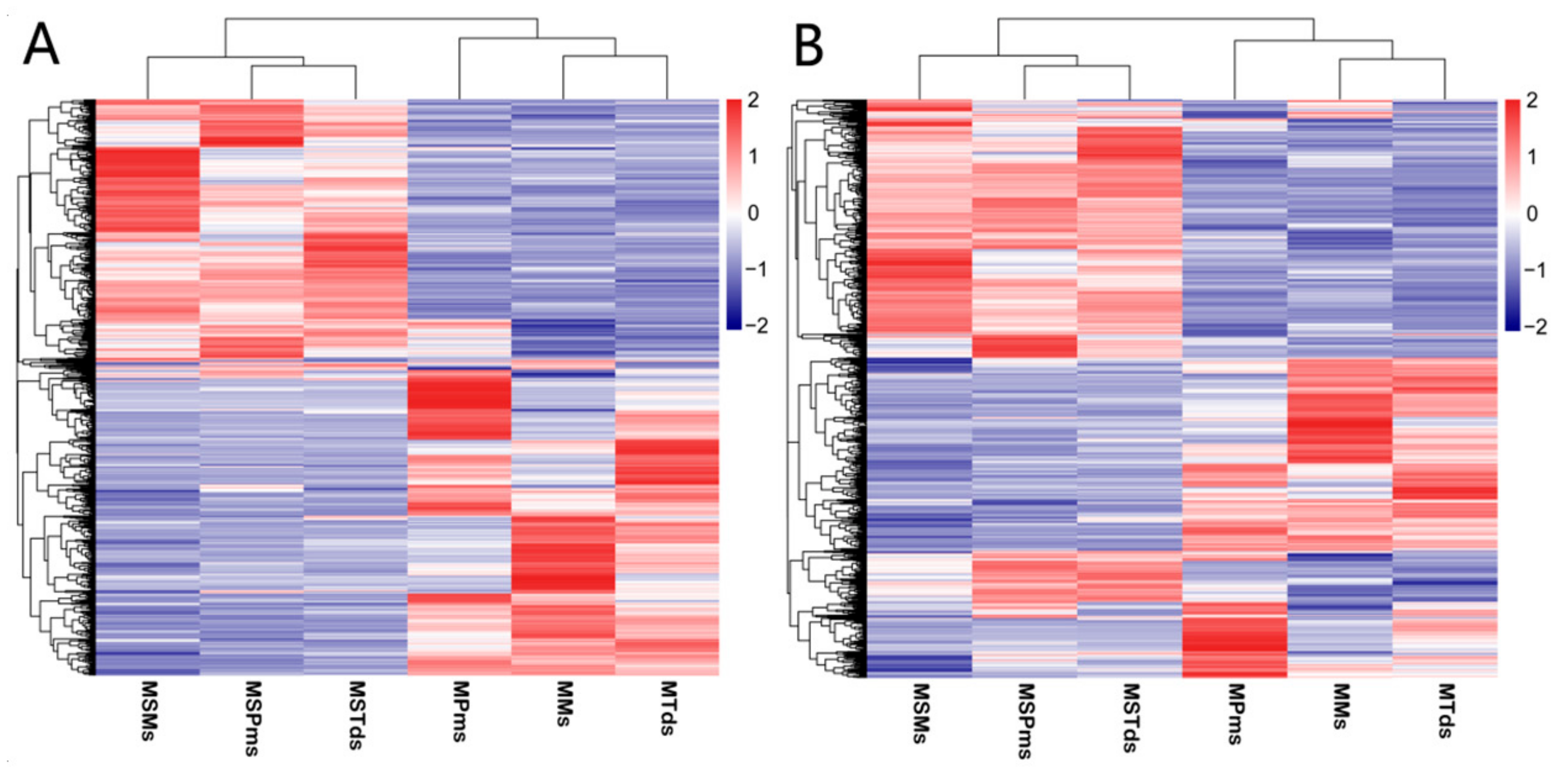
3.2. Differentially Expressed (DE) mRNAs and lncRNAs
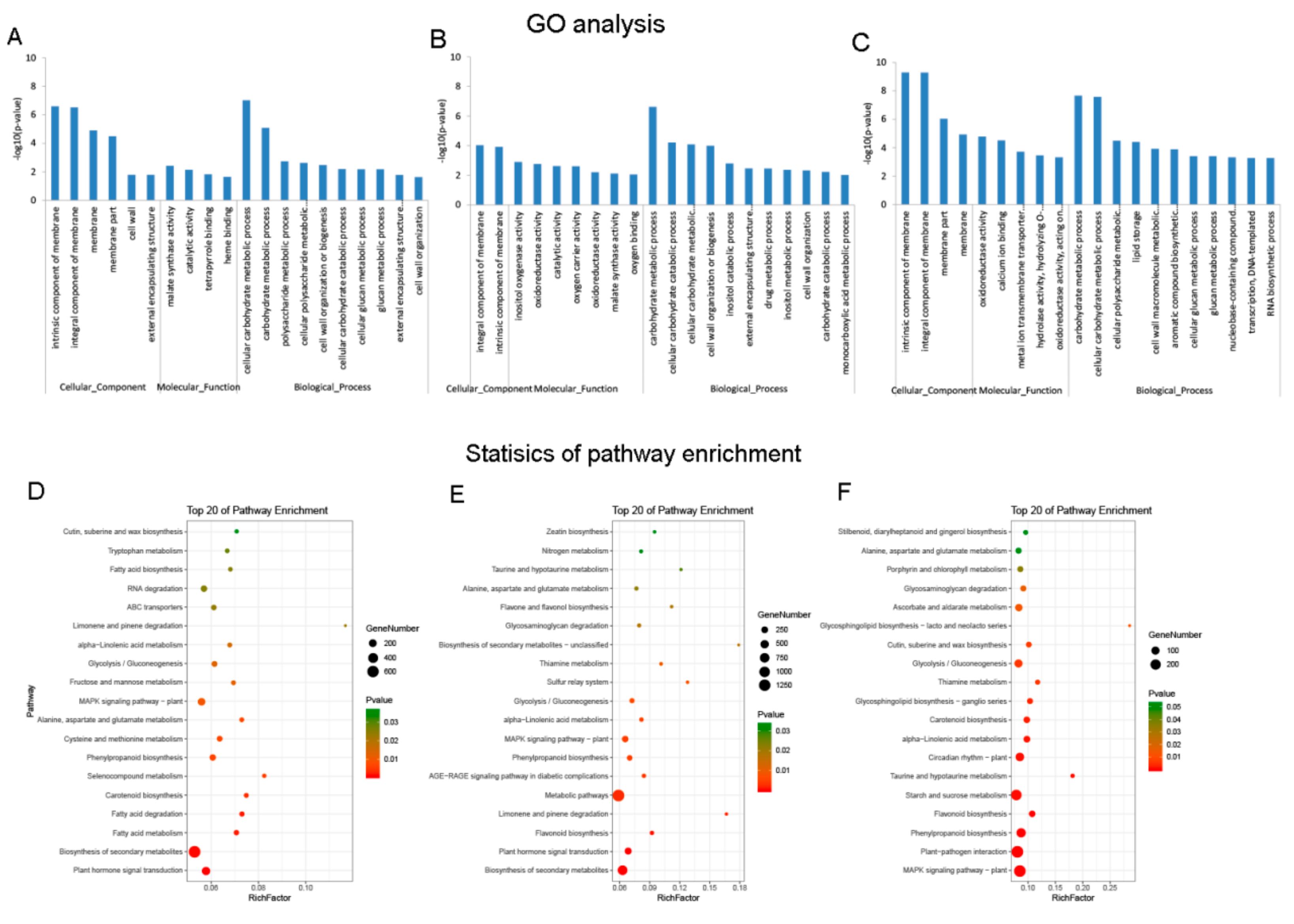
3.3. DE mRNAs Enrichment Analyses
3.4. Identification and Enrichment Analyses of Cis-Target Genes of lncRNAs
3.5. Validation of RNA-Sequencing (RNA-Seq) by Real-Time Quantitative PCR (RT-qPCR)
4. Discussion
4.1. Transcriptome mRNA in the Anther Development of Cotton
4.2. lncRNA in the Anther Development of Cotton and Predicted Functions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Derrien, T.; Guigo, R.; Johnson, R. The Long Non-Coding RNAs: A New (P)layer in the “Dark Matter”. Front. Genet. 2011, 2, 107. [Google Scholar] [CrossRef]
- Ma, X.; Shao, C.; Jin, Y.; Wang, H.; Meng, Y. Long non-coding RNAs: A novel endogenous source for the generation of Dicer-like 1-dependent small RNAs in Arabidopsis thaliana. Rna Biol. 2014, 11, 373–390. [Google Scholar] [CrossRef]
- Murakami, K. Non-coding RNAs and hypertension-unveiling unexpected mechanisms of hypertension by the dark matter of the genome. Curr. Hypertens. Rev. 2015, 11, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant. Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zou, J.; Meng, J.; Wang, J. Integrative analysis of genome-wide lncRNA and mRNA expression in newly synthesized Brassica hexaploids. Ecol. Evol. 2018, 8, 6034–6052. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Mandal, S.S. Long noncoding RNAs: Emerging stars in gene regulation, epigenetics and human disease. Chemmedchem. 2014, 9, 1932–1956. [Google Scholar] [CrossRef]
- Shafiq, S.; Li, J.; Sun, Q. Functions of plants long non-coding RNAs. BBA Gene Regul. Mech. 2016, 1859, 155–162. [Google Scholar] [CrossRef]
- Lu, Z.; Xia, X.; Jiang, B.; Ma, K.; Zhu, L.; Wang, L.; Jin, B. Identification and characterization of novel lncRNAs in Arabidopsis thaliana. Biochem Biophys Res. Commun. 2017, 488, 348–354. [Google Scholar] [CrossRef]
- Zhang, Y.; Liao, J.; Li, Z.; Yu, Y.; Zhang, J.; Li, Q.; Qu, L.; Shu, W.; Chen, Y. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, W.; Gao, L.; Zhao, L. Genome-wide profiling of long non-coding RNAs from tomato and a comparison with mRNAs associated with the regulation of fruit ripening. BMC Plant. Biol. 2018, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hu, J.; Gao, C.; Chen, G.; Wang, B.; Lin, C.; Song, L.; Ding, Y.; Zhou, G. Genome-wide analysis of long non-coding RNAs unveils the regulatory roles in the heat tolerance of Chinese cabbage (Brassica rapa ssp.chinensis). Sci. Rep. 2019, 9, 5002. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Wang, Y.; Yao, Y.; Song, N.; Hu, Z.; Qin, D.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant. Biol. 2011, 11, 61. [Google Scholar] [CrossRef]
- Seo, J.S.; Sun, H.X.; Park, B.S.; Huang, C.H.; Yeh, S.D.; Jung, C.; Chua, N.H. ELF18-INDUCED LONG-NONCODING RNA Associates with Mediator to Enhance Expression of Innate Immune Response Genes in Arabidopsis. Plant. Cell 2017, 29, 1024–1038. [Google Scholar] [CrossRef]
- Cui, J.; Luan, Y.; Jiang, N.; Bao, H.; Meng, J. Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance to Phytophthora infestans by co-expressing glutaredoxin. Plant. J. 2017, 89, 577–589. [Google Scholar] [CrossRef]
- Chen, L.; Shi, S.; Jiang, N.; Khanzada, H.; Wassan, G.M.; Zhu, C.; Peng, X.; Xu, J.; Chen, Y.; Yu, Q.; et al. Genome-wide analysis of long non-coding RNAs affecting roots development at an early stage in the rice response to cadmium stress. BMC Genom. 2018, 19, 460. [Google Scholar] [CrossRef]
- Burleigh, S.H.; Harrison, M.J. A novel gene whose expression in Medicago truncatula roots is suppressed in response to colonization by vesicular-arbuscular mycorrhizal (VAM) fungi and to phosphate nutrition. Plant. Mol. Biol. 1997, 34, 199–208. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Y.; Dong, J.; Sun, Y.; Lim, B.L.; Liu, D.; Lu, Z.J. Systematic characterization of novel lncRNAs responding to phosphate starvation in Arabidopsis thaliana. BMC Genom. 2016, 17, 655. [Google Scholar] [CrossRef]
- Chen, M.; Wang, C.; Bao, H.; Chen, H.; Wang, Y. Genome-wide identification and characterization of novel lncRNAs in Populus under nitrogen deficiency. Mol. Genet. Genom. 2016, 291, 1663–1680. [Google Scholar] [CrossRef]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, Y.G. Long non-coding RNAs play an important role in regulating photoperiod- and temperature-sensitive male sterility in rice. Sci. China Life Sci. 2017, 60, 443–444. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Liu, Z. Global identification and analysis of long non-coding RNAs in diploid strawberry Fragaria vesca during flower and fruit development. BMC Genom. 2015, 16, 815. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Liu, S.R.; Zhang, X.Y.; Ma, Y.J.; Hu, C.G.; Zhang, J.Z. Genome-wide screening and characterization of long non-coding RNAs involved in flowering development of trifoliate orange (Poncirus trifoliata L. Raf.). Sci Rep. 2017, 7, 43226. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 3, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Li, F.G. The Gossypium raimondii Genome, a Huge Leap Forward in Cotton Genomics. J. Integr. Plant. Biol. 2013, 55, 570–571. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, M.; Li, N.; Wang, H.; Qiu, P.; Pei, L.; Xu, Z.; Wang, T.; Gao, E.; Liu, J.; et al. Long noncoding RNAs involve in resistance to Verticillium dahliae, a fungal disease in cotton. Plant. Biotechnol. J. 2018, 16, 1172–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Chen, X.; Mu, M.; Wang, J.; Wang, X.; Wang, D.; Yin, Z.; Fan, W.; Wang, S.; Guo, L. Genome-Wide Analysis of Long Noncoding RNAs and Their Responses to Drought Stress in Cotton (Gossypium hirsutum L.). PLoS ONE 2016, 11, e0156723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant. Biol. 2018, 18, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Yang, Z.; Zhao, G.; Zhang, X.; Wang, L.; Zheng, L.; Zhuo, F.; Yin, H.; Ge, X.; et al. Target of Rapamycin (TOR) Regulates the Expression of lncRNAs in Response to Abiotic Stresses in Cotton. Front. Genet. 2018, 9, 690. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.; Wang, Q.; Lu, C.; Yang, W.; Zhang, Y.; Cheng, H.; Feng, X.; Prosper, M.A.; Song, G. Transcriptome analysis reveals long noncoding RNAs involved in fiber development in cotton (Gossypium arboreum). Sci. China Life Sci. 2016, 59, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Wang, M.; Ding, Y.; Zhu, S.; Zhao, G.; Tu, L.; Zhang, X. Transcriptomic repertoires depict the initiation of lint and fuzz fibres in cotton (Gossypium hirsutum L.). Plant. Biotechnol J. 2018, 16, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, 279–285. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.; Liu, X.; Zhao, S.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Wang, H.V.; Chekanova, J.A. Long Noncoding RNAs in Plants. Adv. Exp. Med. Biol. 2017, 1008, 133–154. [Google Scholar]
- Carlin, D.E.; Demchak, B.; Pratt, D.; Sage, E.; Ideker, T. Network propagation in the cytoscape cyberinfrastructure. PLoS Comput. Biol. 2017, 13, e1005598. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Shu-Lan, Y.; Li-Fen, X.; Hui-Zhu, M.; Ching San, P.; Wei-Cai, Y.; Lixi, J.; Venkatesan, S.; De, Y. Tapetum determinant1 is required for cell specialization in the Arabidopsis anther. Plant. Cell 2003, 15, 2792–2804. [Google Scholar]
- Catherine, A.; Eugenia, R.; Valerie, H.; Erik, B.; Sacco, D.V. The Arabidopsis thaliana SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASES1 and 2 control male sporogenesis. Plant. Cell 2005, 17, 3337–3349. [Google Scholar]
- Fu, Z.; Yu, J.; Cheng, X.; Zong, X.; Xu, J.; Chen, M.; Li, Z.; Zhang, D.; Liang, W. The Rice Basic Helix-Loop-Helix Transcription Factor TDR INTERACTING PROTEIN2 Is a Central Switch in Early Anther Development. Plant. Cell 2014, 26, 1512–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.L.; Li, X.B. The GhACS1 gene encodes an acyl-CoA synthetase which is essential for normal microsporogenesis in early anther development of cotton. Plant. J. 2009, 57, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Li, X.B.; Xu, D.; Wang, X.L.; Huang, G.Q.; Luo, J.; Li, D.D.; Zhang, Z.T.; Xu, W.L. Three cotton genes preferentially expressed in flower tissues encode actin-depolymerizing factors which are involved in F-actin dynamics in cells. J. Exp. Bot. 2010, 61, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jiang, J.; Du, M.L.; Li, L.; Wang, X.L.; Li, X.B. A cotton gene encoding MYB-like transcription factor is specifically expressed in pollen and is involved in regulation of late anther/pollen development. Plant. Cell Physiol. 2013, 54, 893–906. [Google Scholar] [CrossRef]
- Oliver, S.N.; Dennis, E.S.; Dolferus, R. ABA regulates apoplastic sugar transport and is a potential signal for cold-induced pollen sterility in rice. Plant. Cell Physiol. 2007, 48, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Song, Y.; Guo, J.; Wang, J.; Zhang, L.; Niu, N.; Ma, S.; Zhang, G.; Zhao, H. Physiological and metabolome changes during anther development in wheat (Triticum aestivum L.). Plant. Physiol Biochem. 2018, 132, 18–32. [Google Scholar]
- Datta, R.; Chamusco, K.C.; Chourey, P.S. Starch biosynthesis during pollen maturation is associated with altered patterns of gene expression in maize. Plant. Physiol. 2002, 130, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.S.; Douglas, C.J. Sporopollenin monomer biosynthesis in arabidopsis. J. Plant. Biol. 2013, 56, 1–6. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Li, Y.; Ma, Y.; Zhao, Y.; Wang, C.; Chi, H.; Chen, M.; Ding, Y.; Guo, X.; et al. Proteomic analysis reveals that sugar and fatty acid metabolisms play a central role in sterility of the male-sterile line 1355A of cotton. J. Biol Chem. 2019, 294, 7057–7067. [Google Scholar] [CrossRef]
- Ma, Y.; Min, L.; Wang, M.; Wang, C.; Zhao, Y.; Li, Y.; Fang, Q.; Wu, Y.; Xie, S.; Ding, Y.; et al. Disrupted Genome Methylation in Response to High Temperature Has Distinct Affects on Microspore Abortion and Anther Indehiscence. Plant. Cell 2018, 30, 1387–1403. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Song, M.; Fan, S.; Yu, S. Transcriptomic analysis of differentially expressed genes during anther development in genetic male sterile and wild type cotton by digital gene-expression profiling. BMC Genom. 2013, 14, 97. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.J.; Chen, J.; Liu, J.B.; Xia, M.X.; Wang, W.; Shen, F.F. Transcriptome Analysis of Early Anther Development of Cotton Revealed Male Sterility Genes for Major Metabolic Pathways. J. Plant. Growth Regul. 2014, 34, 223–232. [Google Scholar] [CrossRef]
- Bagnall, D.J. Control of Flowering in Arabidopsis thaliana by Light, Vernalisation and Gibberellins. Funct. Plant. Biol. 1992, 19, 401–409. [Google Scholar] [CrossRef]
- Thornsberry, J.M.; Goodman, M.M.; Doebley, J.; Kresovich, S.; Nielsen, D.; Buckler, E.S.T. Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 2001, 28, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Tian, M.; Liu, F.; Wang, C.; Zhang, Y. Hormonal and morphological changes during seed development of Cypripedium japonicum. Protoplasma 2017, 254, 2315–2322. [Google Scholar] [CrossRef]
- Chen, S.X.; Zhao, F.; Huang, X.J. MAPK signaling pathway and erectile dysfunction. Zhonghua Nan Ke Xue Natl. J. Androl. 2018, 24, 442–446. [Google Scholar]
- Feng, X.; Li, F.; Wang, F.; Zhang, G.; Pang, J.; Ren, C.; Zhang, T.; Yang, H.; Wang, Z.; Zhang, Y. Genome-wide differential expression profiling of mRNAs and lncRNAs associated with prolificacy in Hu sheep. Biosci Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.H.; Chu, E.T.; Spektor, R.; Soloway, P.D. Long non-coding RNA regulation of reproduction and development. Mol. Reprod Dev. 2015, 82, 932–956. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, L.; Cui, J.; Che, S.; Song, Y. The mRNA and lncRNA landscape of the non-pregnant endometrium during the oestrus cycle in dairy goat. Anim. Prod. Sci. 2019. [Google Scholar] [CrossRef]
- Liang, W.C.; Ren, J.L.; Wong, C.W.; Chan, S.O.; Waye, M.M.; Fu, W.M.; Zhang, J.F. LncRNA-NEF antagonized epithelial to mesenchymal transition and cancer metastasis via cis-regulating FOXA2 and inactivating Wnt/beta-catenin signaling. Oncogene 2018, 37, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Li, Z.; Ma, M.; Wang, Z.; Han, P.; Abdalla, B.A.; Nie, Q.; Zhang, X. LncRNA-Six1 Encodes a Micropeptide to Activate Six1 in Cis and Is Involved in Cell Proliferation and Muscle Growth. Front. Physiol. 2017, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Bin, Z.; Gayatri, A.; Mao, Y.S.; Zsolt, L.; Gourab, B.; Xiaokun, X.; Booth, C.J.; Jie, W.; Chaolin, Z. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar]
- Lu, Z.; Li, Y.; Che, Y.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Li, N.; Sun, N.; He, J. The TGFβ-induced lncRNA TBILA promotes non-small cell lung cancer progression in vitro and in vivo via cis-regulating HGAL and activating S100A7/JAB1 signaling. Cancer Lett. 2018, 432, 156–168. [Google Scholar] [CrossRef]
- Carmona, S.; Lin, B.; Chou, T.; Arroyo, K.; Sun, S. LncRNA Jpx induces Xist expression in mice using both trans and cis mechanisms. PLoS Genet. 2018, 14, e1007378. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Lin, F.; He, G.; Terzaghi, W.; Zhu, D.; Deng, X.W. Arabidopsis noncoding RNA mediates control of photomorphogenesis by red light. Proc. Natl. Acad. Sci. USA 2014, 111, 10359–10364. [Google Scholar] [CrossRef] [Green Version]
- Csorba, T.; Questa, J.I.; Sun, Q.; Dean, C. Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [Green Version]
- He, Y. Chromatin regulation of flowering. Trends Plant. Sci. 2012, 17, 556–562. [Google Scholar] [CrossRef]
- Xiangyang, H.; Xiangxiang, K.; Chuntao, W.; Lan, M.; Jinjie, Z.; Jingjing, W.; Xiaoming, Z.; Loake, G.J.; Ticao, Z.; Jinling, H. Proteasome-mediated degradation of FRIGIDA modulates flowering time in Arabidopsis during vernalization. Plant. Cell 2014, 26, 4763–4781. [Google Scholar]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, D.; Zhang, D.; Yin, D.; Zhao, Y.; Ji, C.; Zhao, X.; Li, X.; He, Q.; Chen, R.; et al. A novel antisense long noncoding RNA, TWISTED LEAF, maintains leaf blade flattening by regulating its associated sense R2R3-MYB gene in rice. New Phytol. 2018, 218, 774–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Luo, X.; Sun, F.; Hu, J.; Zha, X.; Su, W.; Yang, J. Overexpressing lncRNA LAIR increases grain yield and regulates neighbouring gene cluster expression in rice. Nat. Commun. 2018, 9, 3516. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fu, D.; Zhu, B.; Luo, Y.; Zhu, H. CRISPR/Cas9-mediated mutagenesis of lncRNA1459 alters tomato fruit ripening. Plant. J. 2018, 94, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Bao, S.; Zhou, X.; Liu, J.; Zhuang, Y. The key genes and pathways related to male sterility of eggplant revealed by comparative transcriptome analysis. BMC Plant. Biol. 2018, 18, 209. [Google Scholar] [CrossRef]
- Nie, Z.; Zhao, T.; Yang, S.; Gai, J. Development of a cytoplasmic male-sterile line NJCMS4A for hybrid soybean production. Plant. Breed. 2017, 136, 516–525. [Google Scholar] [CrossRef]
- Wang, K.; Gao, F.; Ji, Y.; Liu, Y.; Dan, Z.; Yang, P.; Zhu, Y.; Li, S. ORFH79 impairs mitochondrial function via interaction with a subunit of electron transport chain complex III in Honglian cytoplasmic male sterile rice. New Phytol. 2013, 198, 408–418. [Google Scholar] [CrossRef]
- Liu, J.; Liang, L.; Jiang, Y.; Chen, J. Changes in Metabolisms of Antioxidant and Cell Wall in Three Pummelo Cultivars during Postharvest Storage. Biomolecules 2019, 9, 319. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wei, H.; Song, M.; Pang, C.; Liu, J.; Wang, L.; Zhang, J.; Fan, S.; Yu, S. Transcriptome profiling analysis reveals that flavonoid and ascorbate-glutathione cycle are important during anther development in Upland cotton. PLoS ONE 2012, 7, e49244. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Data | Clean Data | GC (%) | Q30 (%) | Clean Reads Rate (%) | novel_lncRNA.isoforms | novel_mRNA.isoforms | known_mRNA.isoforms |
|---|---|---|---|---|---|---|---|---|
| MsPms1 | 96339898 | 84112026 | 44.33 | 97.65 | 87.308 | 26669 | 22642 | 77144 |
| MsPms2 | 96339060 | 84728466 | 44.43 | 97.84 | 87.948 | 26079 | 22080 | 74534 |
| MsPms3 | 96340692 | 84822750 | 44.52 | 97.81 | 88.045 | 26723 | 22570 | 76486 |
| MsTds1 | 66418638 | 60016542 | 44.22 | 98.33 | 90.361 | 25569 | 22130 | 74937 |
| MsTds2 | 96341884 | 84562392 | 43.57 | 97.68 | 87.773 | 27125 | 23083 | 78797 |
| MsTds3 | 96340878 | 84830410 | 44 | 97.77 | 88.052 | 27515 | 22581 | 76691 |
| MsMs1 | 70226770 | 62112146 | 45.21 | 98.35 | 88.445 | 24684 | 21119 | 72145 |
| MsMs2 | 96340984 | 84269862 | 44.5 | 97.81 | 87.47 | 27175 | 22017 | 75181 |
| MsMs3 | 75363530 | 68729552 | 44.43 | 98.45 | 91.197 | 26502 | 22011 | 75483 |
| MPms1 | 78357630 | 71140510 | 43.98 | 98.42 | 90.79 | 26798 | 22389 | 74464 |
| MPms2 | 93592588 | 83300884 | 43.81 | 97.97 | 89.004 | 27279 | 22445 | 74560 |
| MPms3 | 94122576 | 81326894 | 44.24 | 97.83 | 86.405 | 27314 | 22397 | 74869 |
| MTds1 | 93071774 | 84922682 | 43.79 | 98.38 | 91.244 | 27246 | 22535 | 74405 |
| MTds2 | 79502370 | 72818484 | 43.85 | 98.23 | 91.593 | 26317 | 22163 | 73329 |
| MTds3 | 71210292 | 64700084 | 43.83 | 98.33 | 90.858 | 26046 | 21985 | 72553 |
| MMs1 | 96340778 | 84992800 | 44.36 | 98.03 | 88.221 | 26255 | 22543 | 75378 |
| MMs2 | 96340324 | 84734142 | 43.65 | 97.83 | 87.953 | 26319 | 22745 | 75915 |
| MMs3 | 74569750 | 67998900 | 43.6 | 98.23 | 91.188 | 25285 | 22181 | 73375 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Qin, T.; Dong, N.; Wei, C.; Zhang, Y.; Sun, R.; Dong, T.; Chen, Q.; Zhou, R.; Wang, Q. Integrative Analysis of the lncRNA and mRNA Transcriptome Revealed Genes and Pathways Potentially Involved in the Anther Abortion of Cotton (Gossypium hirsutum L.). Genes 2019, 10, 947. https://doi.org/10.3390/genes10120947
Li Y, Qin T, Dong N, Wei C, Zhang Y, Sun R, Dong T, Chen Q, Zhou R, Wang Q. Integrative Analysis of the lncRNA and mRNA Transcriptome Revealed Genes and Pathways Potentially Involved in the Anther Abortion of Cotton (Gossypium hirsutum L.). Genes. 2019; 10(12):947. https://doi.org/10.3390/genes10120947
Chicago/Turabian StyleLi, Yuqing, Tengfei Qin, Na Dong, Chunyan Wei, Yaxin Zhang, Runrun Sun, Tao Dong, Quanjia Chen, Ruiyang Zhou, and Qinglian Wang. 2019. "Integrative Analysis of the lncRNA and mRNA Transcriptome Revealed Genes and Pathways Potentially Involved in the Anther Abortion of Cotton (Gossypium hirsutum L.)" Genes 10, no. 12: 947. https://doi.org/10.3390/genes10120947