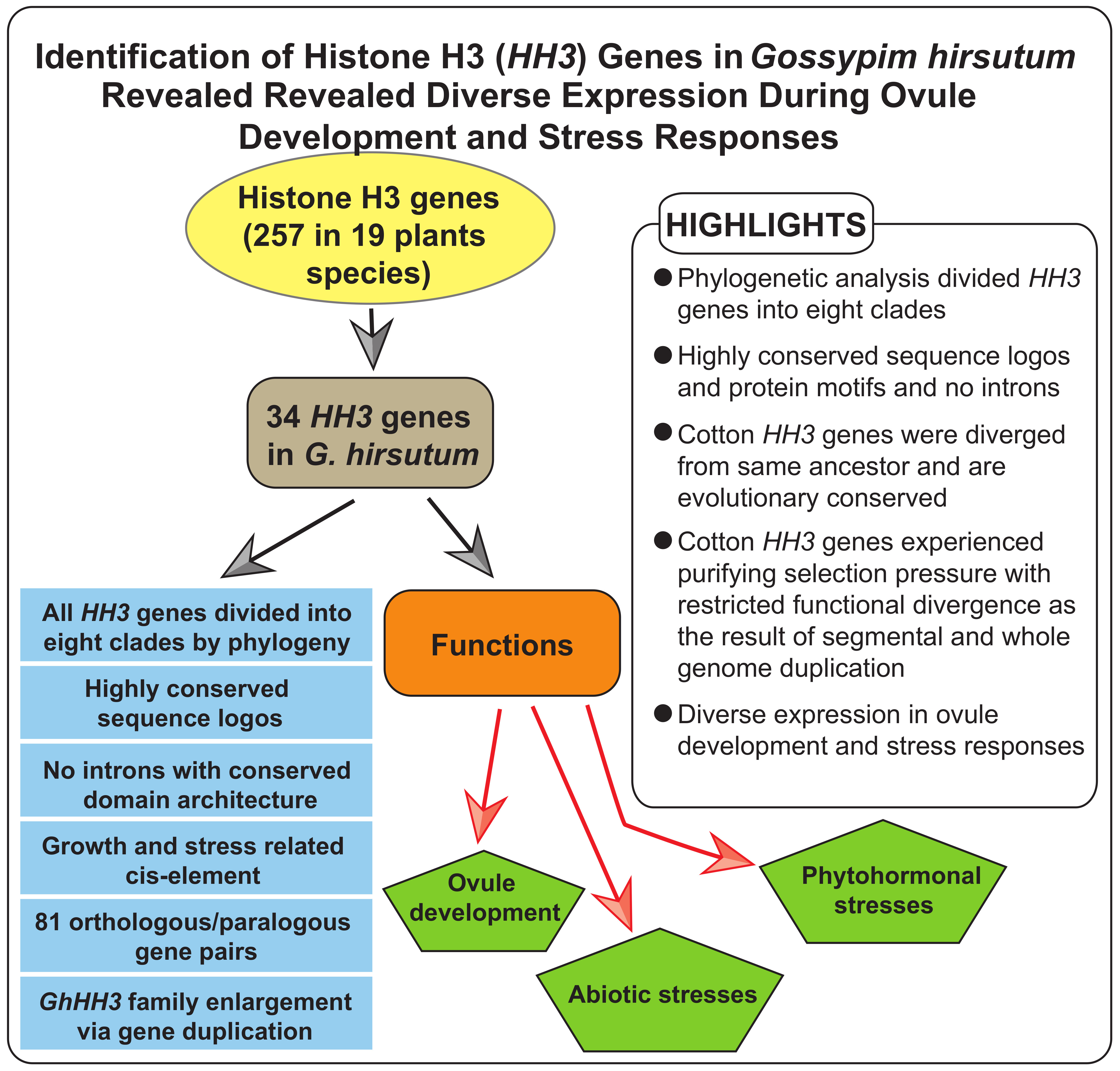
Identification of Histone H3 (HH3) Genes in Gossypium hirsutum Revealed Diverse Expression During Ovule Development and Stress Responses

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sequence Identification
2.2. Phylogenetic and Conserved Sequence Analysis
2.3. Gene Structure, Domain Architecture, and Cis-Elements Analysis
2.4. Chromosomal Localizations, Collinearity, and Ka/Ks Ratio Analysis
2.5. Plant Material, Treatment, and qRT-PCR Analysis
3. Results
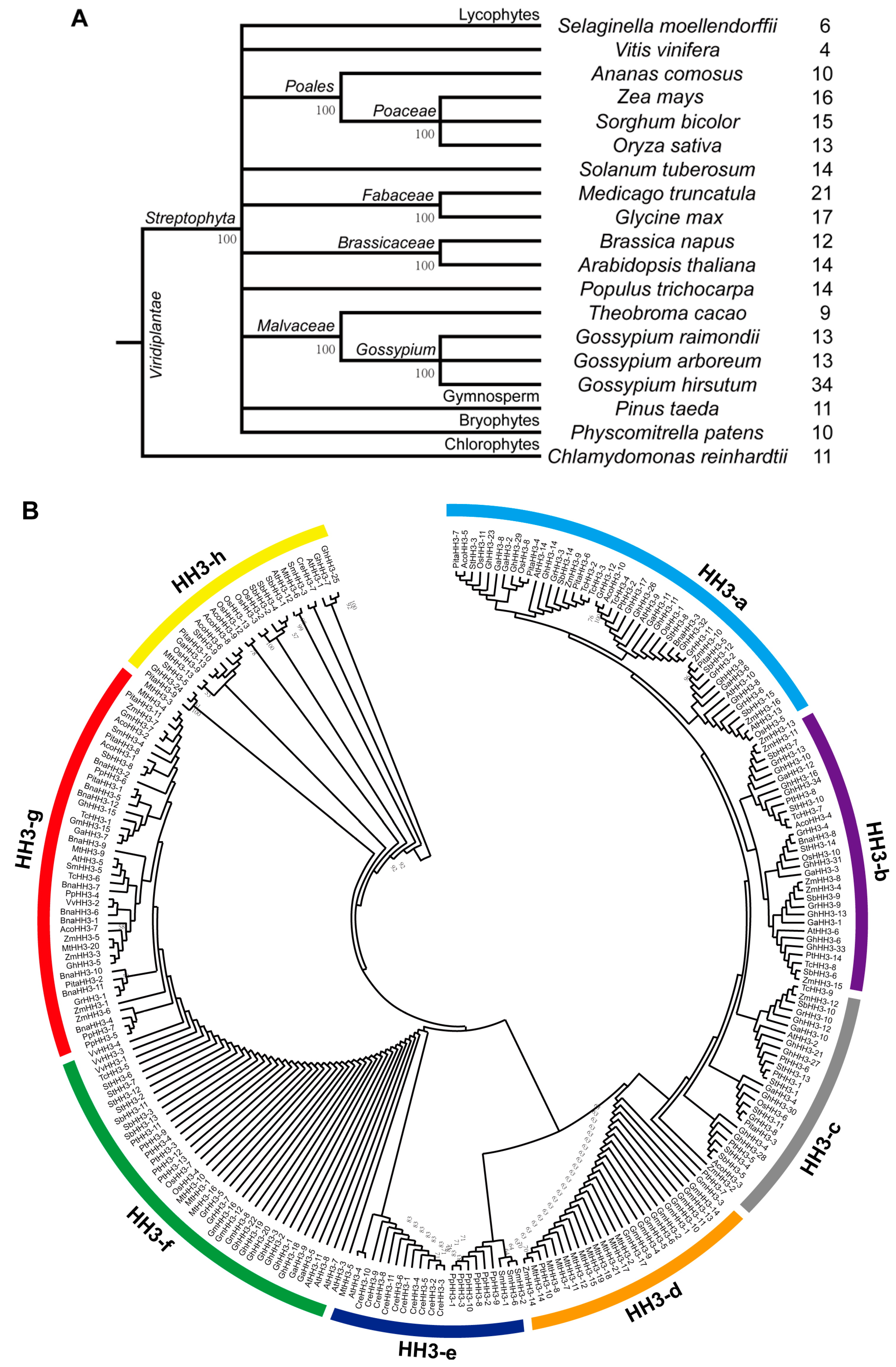
3.1. Identification of HH3 Family Members
3.2. Phylogenetic Analysis of HH3 Genes
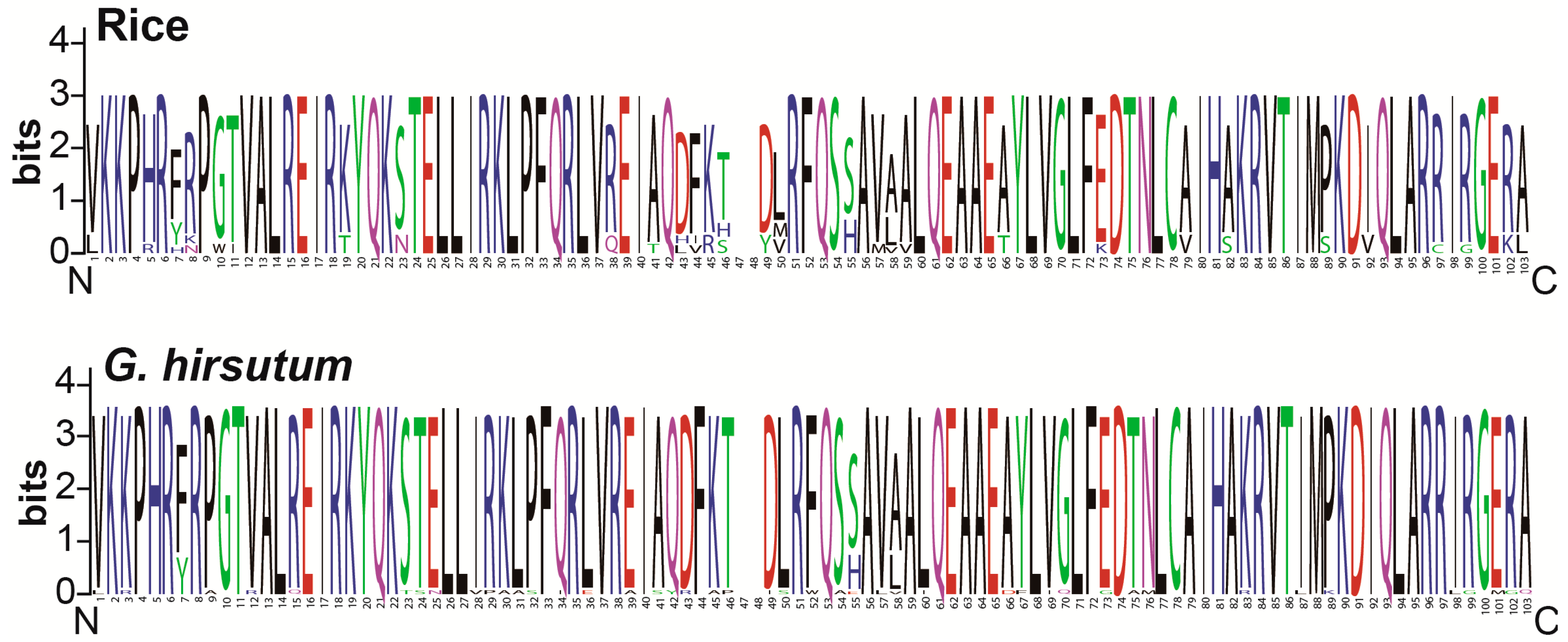
3.3. Conserved Amino Acid Residues Analysis
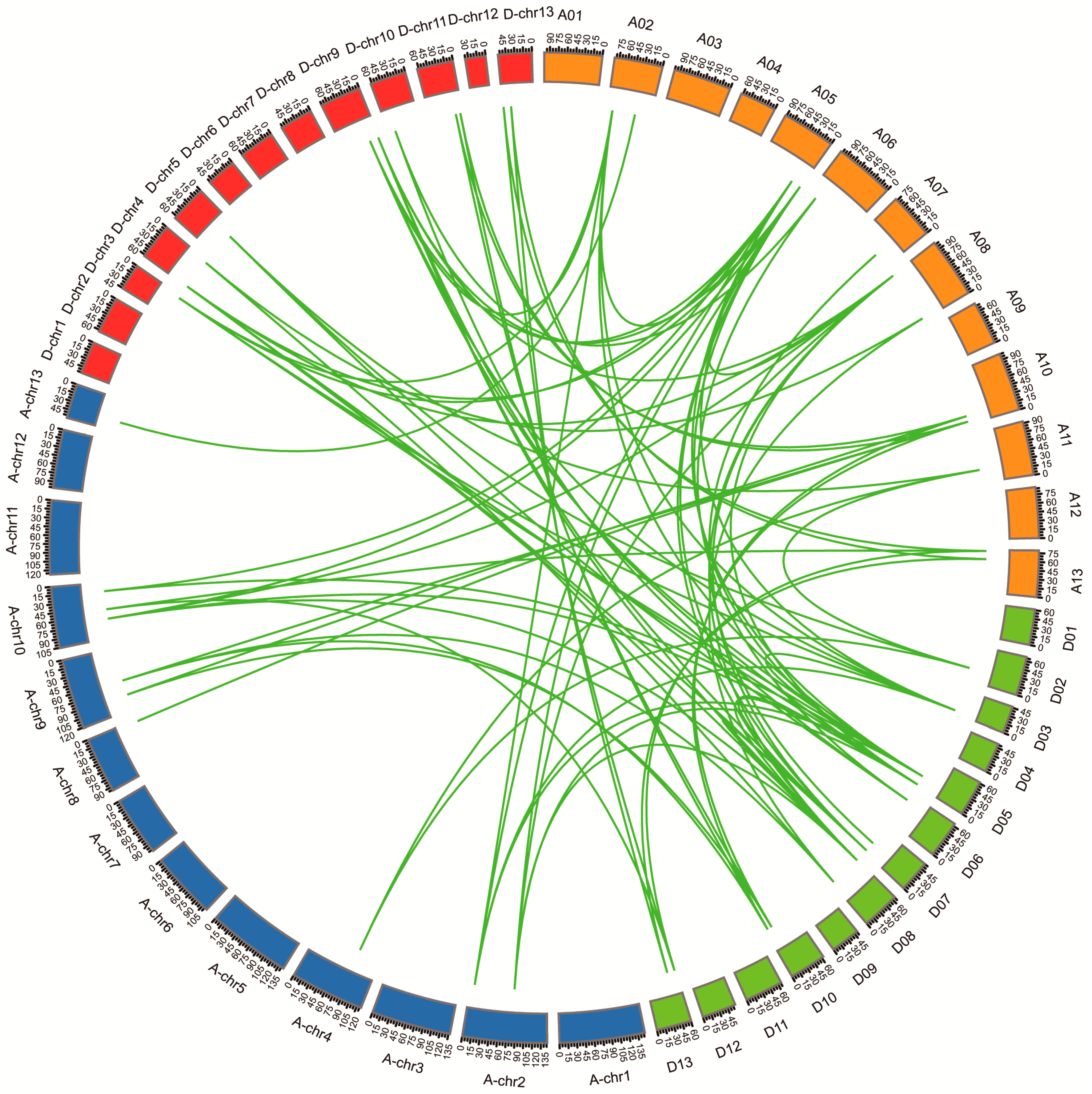
3.4. Chromosomal Locations, Duplication, and Collinearity Analysis of GhHH3 Members
3.5. Promoter Cis-Element, Gene Structure, and Domain Architecture of the GhHH3 Gene Family
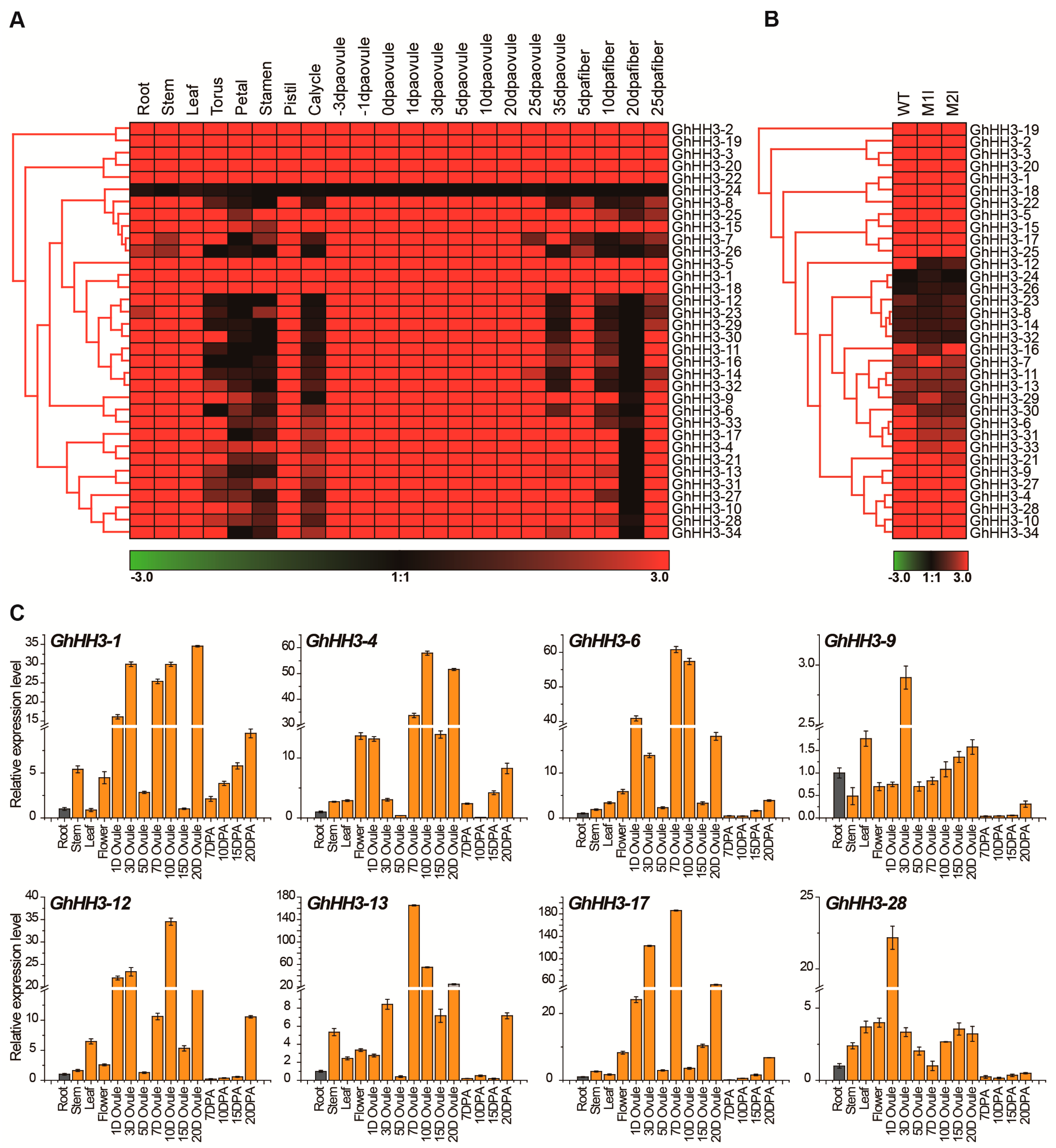
3.6. Expression Profiles of GhHH3 Members in Various Tissues and Developmental Stages
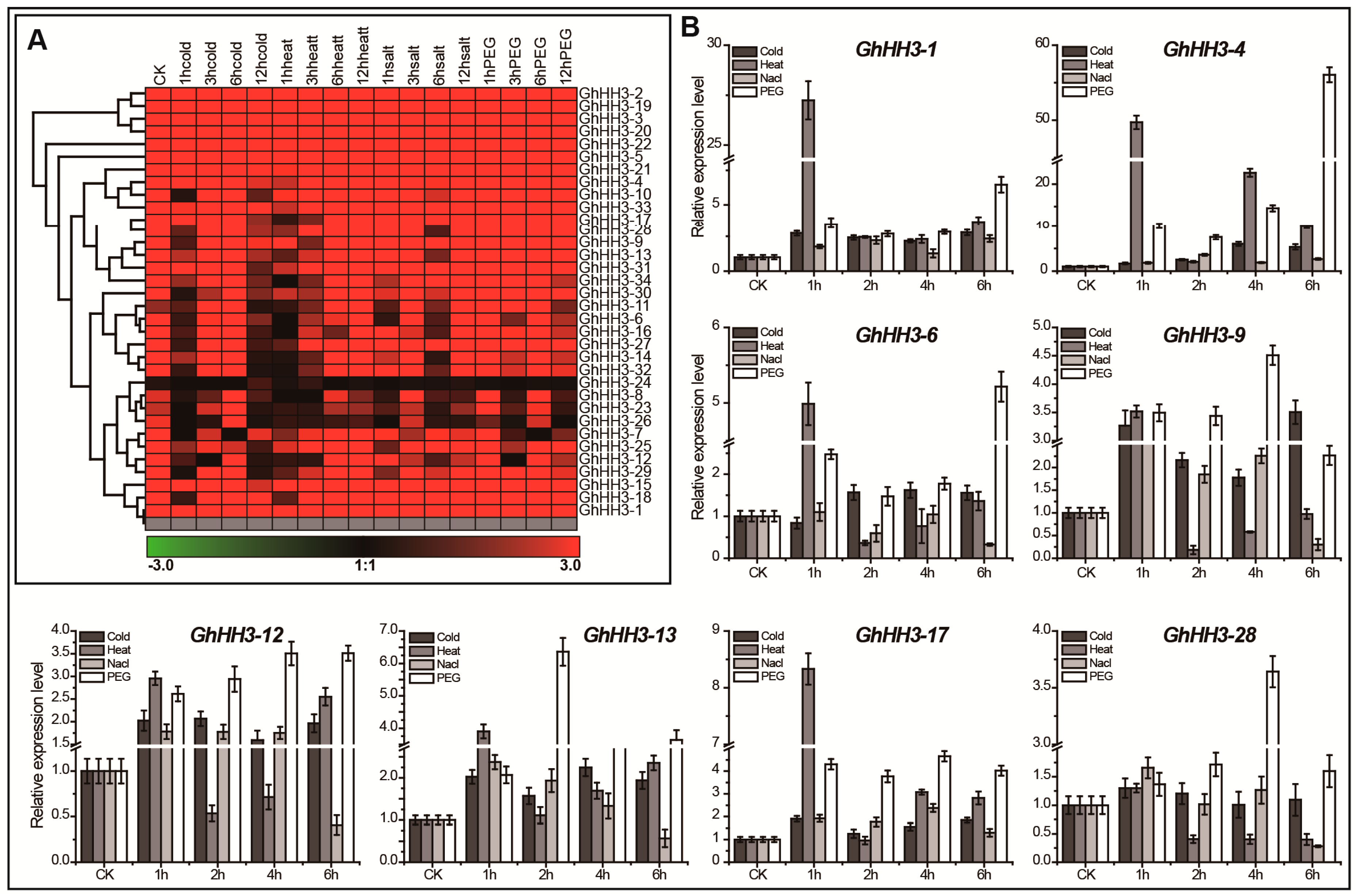
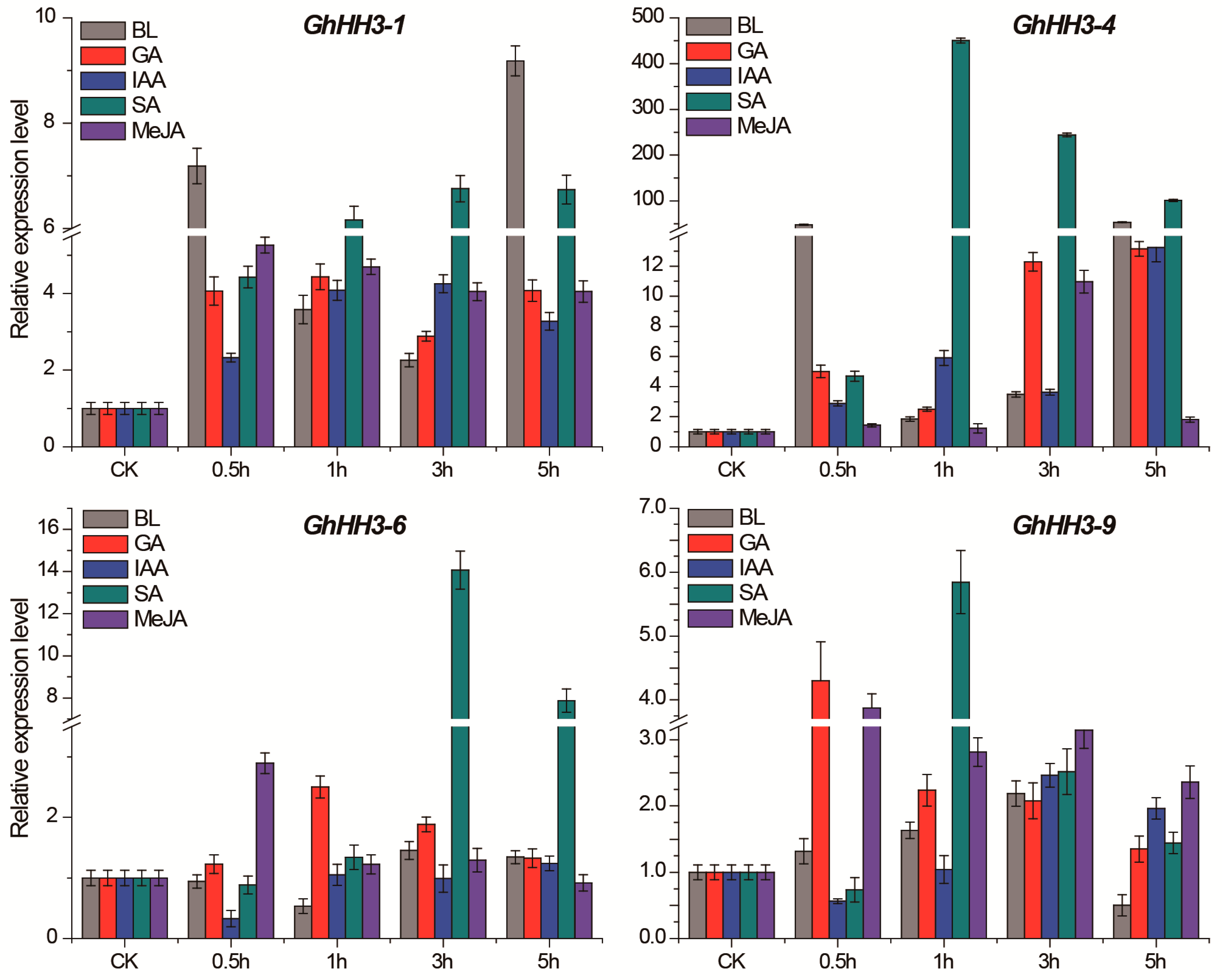
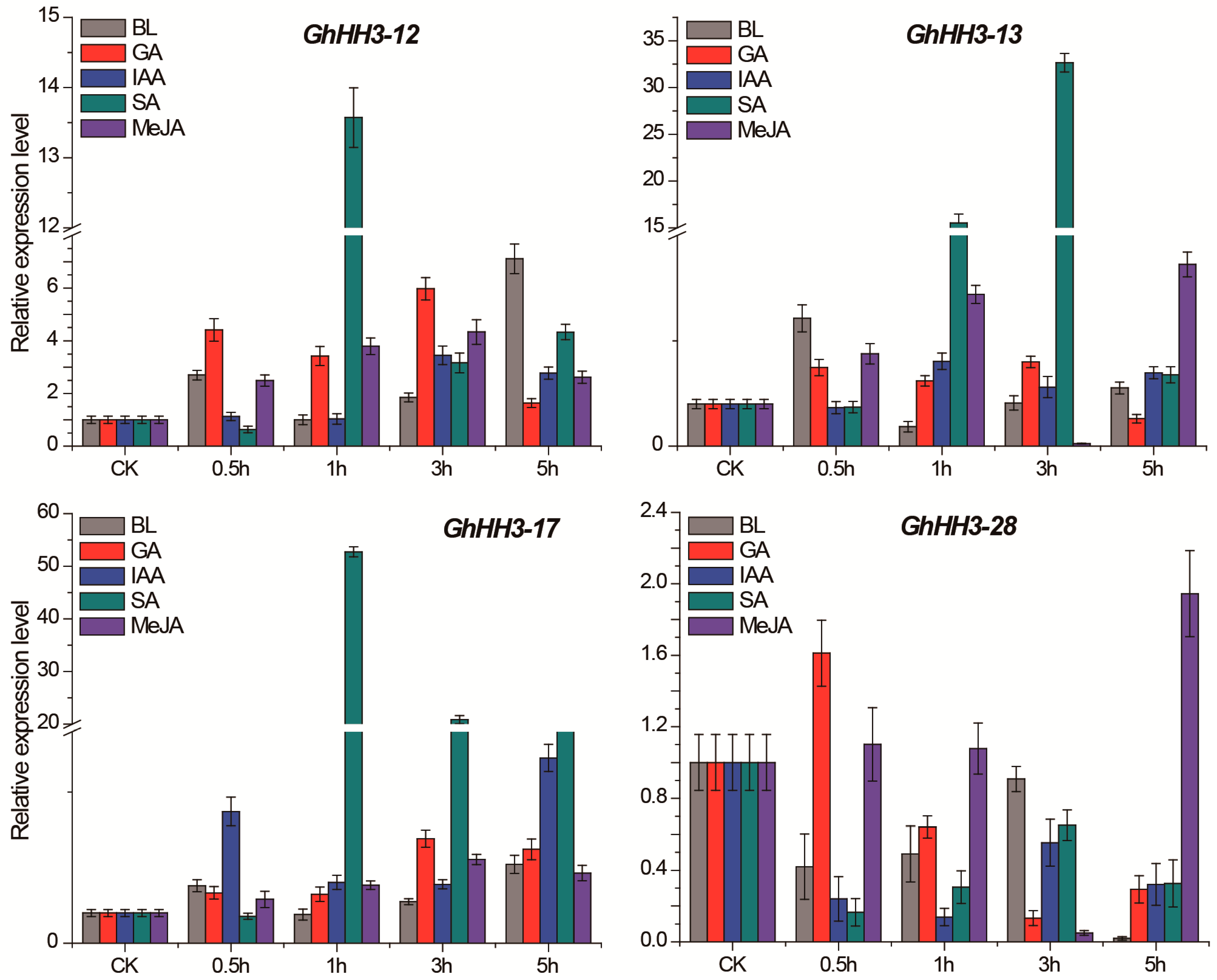
3.7. Responses of GhHH3 Members Under Various Abiotic Stresses and Phytohormonal Treatments
4. Discussion
4.1. Cotton HH3s Have Been Highly Conserved During Evolution
4.2. Cotton HH3 Gene Enlargement During Evolutionary Processes
4.3. Functional Diversifications of GhHH3 Family Members
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Okada, T.; Endo, M.; Singh, M.B.; Bhalla, P.L. Analysis of the histone H3 gene family in Arabidopsis and identification of the male-gamete-specific variant AtMGH3. Plant J. 2005, 44, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lai, Y. Identification and expression analysis of rice histone genes. Plant Physiol. Biochem. PPB 2015, 86, 55–65. [Google Scholar] [CrossRef]
- Shu, H.; Nakamura, M.; Siretskiy, A.; Borghi, L.; Moraes, I.; Wildhaber, T.; Gruissem, W.; Hennig, L. Arabidopsis replacement histone variant H3.3 occupies promoters of regulated genes. Genome Biol. 2014, 15, R62. [Google Scholar] [CrossRef]
- Stroud, H.; Otero, S.; Desvoyes, B.; Ramirez-Parra, E.; Jacobsen, S.E.; Gutierrez, C. Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2012, 109, 5370–5375. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, H.; Holec, S.; Alden, K.; Clarke, N.D.; Jacques, P.E.; Berger, F. Dynamic deposition of histone variant H3.3 accompanies developmental remodeling of the Arabidopsis transcriptome. PLoS Genet. 2012, 8, e1002658. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Histone variants--ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef]
- Deal, R.B.; Henikoff, S. Histone variants and modifications in plant gene regulation. Curr. Opin. Plant Biol. 2011, 14, 116–122. [Google Scholar] [CrossRef]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [Green Version]
- Elsaesser, S.J.; Goldberg, A.D.; Allis, C.D. New functions for an old variant: No substitute for histone H3.3. Curr. Opin. Genet. Dev. 2010, 20, 110–117. [Google Scholar] [CrossRef]
- Talbert, P.B.; Ahmad, K.; Almouzni, G.; Ausio, J.; Berger, F.; Bhalla, P.L.; Bonner, W.M.; Cande, W.Z.; Chadwick, B.P.; Chan, S.W.; et al. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 2012, 5, 7. [Google Scholar] [CrossRef]
- Postberg, J.; Forcob, S.; Chang, W.J.; Lipps, H.J. The evolutionary history of histone H3 suggests a deep eukaryotic root of chromatin modifying mechanisms. BMC Evol. Biol. 2010, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Deal, R.B.; Henikoff, J.G.; Henikoff, S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 2010, 328, 1161–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Zang, C.; Wei, G.; Cui, K.; Peng, W.; Zhao, K.; Felsenfeld, G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009, 41, 941–945. [Google Scholar] [CrossRef]
- Ahmad, K.; Henikoff, S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 2002, 9, 1191–1200. [Google Scholar] [CrossRef]
- Wong, L.H.; McGhie, J.D.; Sim, M.; Anderson, M.A.; Ahn, S.; Hannan, R.D.; George, A.J.; Morgan, K.A.; Mann, J.R.; Choo, K.H. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010, 20, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; et al. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Mahadevaiah, S.K.; Celeste, A.; Romanienko, P.J.; Camerini-Otero, R.D.; Bonner, W.M.; Manova, K.; Burgoyne, P.; Nussenzweig, A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell 2003, 4, 497–508. [Google Scholar] [CrossRef]
- Du, X.M.; Huang, G.; He, S.P.; Yang, Z.E.; Sun, G.F.; Ma, X.F.; Li, N.; Zhang, X.Y.; Sun, J.L.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Wang, M.; Wang, P.; Lin, M.; Ye, Z.; Li, G.; Tu, L.; Shen, C.; Li, J.; Yang, Q.; Zhang, X. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nat. Plants 2018, 4, 90–97. [Google Scholar] [CrossRef]
- Zhang, T.Z.; Hu, Y.; Jiang, W.K.; Fang, L.; Guan, X.Y.; Chen, J.D.; Zhang, J.B.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Li, F.G.; Fan, G.Y.; Wang, K.B.; Sun, F.M.; Yuan, Y.L.; Song, G.L.; Li, Q.; Ma, Z.Y.; Lu, C.R.; Zou, C.S.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.C.; Llewellyn, D.; Showmaker, K.C.; Shu, S.Q.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Li, J.; Yu, D.; Qanmber, G.; Lu, L.; Wang, L.; Zheng, L.; Liu, Z.; Wu, H.; Liu, X.; Chen, Q.; et al. GhKLCR1, a kinesin light chain-related gene, induces drought-stress sensitivity in Arabidopsis. Sci. China Life Sci. 2019, 62, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.; Misra, P.; Alok, A.; Kaur, N.; Sharma, S.; Lakhwani, D.; Asif, M.H.; Tiwari, S.; Trivedi, P.K. Genome-wide identification and expression analysis of homeodomain leucine zipper subfamily IV (HDZ IV) gene family from Musa accuminata. Front Plant Sci 2016, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.E.; Gong, Q.; Qin, W.Q.; Yang, Z.R.; Cheng, Y.; Lu, L.L.; Ge, X.Y.; Zhang, C.J.; Wu, Z.X.; Li, F.G. Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 2017, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Yang, Z.R.; Zhang, C.J.; Yang, X.J.; Liu, K.; Wu, Z.X.; Zhang, X.Y.; Zheng, W.; Xun, Q.Q.; Liu, C.L.; Lu, L.L.; et al. PAG1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol. 2014, 203, 437–448. [Google Scholar] [CrossRef]
- Wendel, J.F.; Cronn, R.C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 2003, 78, 139–186. [Google Scholar] [Green Version]
- Hu, Y.; Liu, D.; Zhong, X.; Zhang, C.; Zhang, Q.; Zhou, D.X. CHD3 protein recognizes and regulates methylated histone H3 lysines 4 and 27 over a subset of targets in the rice genome. Proc. Natl. Acad. Sci. USA 2012, 109, 5773–5778. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Chen, X.; Zhong, X.; Zhao, Y.; Liu, X.; Zhou, S.; Cheng, S.; Zhou, D.X. Jumonji C domain protein JMJ705-mediated removal of histone H3 lysine 27 trimethylation is involved in defense-related gene activation in rice. Plant Cell 2013, 25, 4725–4736. [Google Scholar] [CrossRef]
- Li, C.; Huang, L.; Xu, C.; Zhao, Y.; Zhou, D.X. Altered levels of histone deacetylase OsHDT1 affect differential gene expression patterns in hybrid rice. PLoS ONE 2011, 6, e21789. [Google Scholar] [CrossRef]
- Qin, F.J.; Sun, Q.W.; Huang, L.M.; Chen, X.S.; Zhou, D.X. Rice SUVH histone methyltransferase genes display specific functions in chromatin modification and retrotransposon repression. Mol. Plant 2010, 3, 773–782. [Google Scholar] [CrossRef]
- Fankhauser, C.; Chory, J. Light control of plant development. Annu. Rev. Cell Dev. Biol. 1997, 13, 203–229. [Google Scholar] [CrossRef]
- Diaz-Martin, J.; Almoguera, C.N.; Prieto-Dapena, P.; Espinosa, J.M.; Jordano, J. Functional interaction between two transcription factors involved in the developmental regulation of a small heat stress protein gene promoter. Plant Physiol. 2005, 139, 1483–1494. [Google Scholar] [CrossRef]
- Narusaka, Y.; Nakashima, K.; Shinwari, Z.K.; Sakuma, Y.; Furihata, T.; Abe, H.; Narusaka, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Interaction between two cis-acting elements, ABRE and DRE, in ABA-dependent expression of Arabidopsis rd29A gene in response to dehydration and high-salinity stresses. Plant J. 2003, 34, 137–148. [Google Scholar] [CrossRef]
- Song, C.P.; Agarwal, M.; Ohta, M.; Guo, Y.; Halfter, U.; Wang, P.C.; Zhu, J.K. Role of an Arabidopsis AP2/EREBP-type transcriptional repressor in abscisic acid and drought stress responses. Plant Cell 2005, 17, 2384–2396. [Google Scholar] [CrossRef]
- Wen, F.; Zhu, H.; Li, P.; Jiang, M.; Mao, W.Q.; Ong, C.; Chu, Z.Q. Genome-wide evolutionary characterization and expression analyses of WRKY family genes in Brachypodium distachyon. DNA Res. 2014, 21, 327–339. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Prince, V.E.; Pickett, F.B. Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 2002, 3, 827–837. [Google Scholar] [CrossRef]
- Vandepoele, K.; Simillion, C.; Van de Peer, Y. Evidence that rice and other cereals are ancient aneuploids. Plant Cell 2003, 15, 2192–2202. [Google Scholar] [CrossRef]
- Roy, S.W.; Gilbert, W. The evolution of spliceosomal introns: Patterns, puzzles and progress. Nat. Rev. Genet. 2006, 7, 211–221. [Google Scholar]
- Tuttle, J.R.; Nah, G.; Duke, M.V.; Alexander, D.C.; Guan, X.; Song, Q.; Chen, Z.J.; Scheffler, B.E.; Haigler, C.H. Metabolomic and transcriptomic insights into how cotton fiber transitions to secondary wall synthesis, represses lignification, and prolongs elongation. BMC Genom. 2015, 16, 477. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Liu, F.; Chen, X.S.; Ma, X.J.; Zeng, H.Q.; Yang, Z.M. Transcriptome profiling of early developing cotton fiber by deep-sequencing reveals significantly differential expression of genes in a fuzzless/lintless mutant. Genomics 2010, 96, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D. RNA-seq analysis of short fiber mutants Ligon-lintless-1 (Li 1 ) and -2 (Li 2 ) revealed important role of aquaporins in cotton (Gossypium hirsutum L.) fiber elongation. BMC Plant Biol. 2015, 15, 65. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Q.; Zhang, B. Evaluation and selection of reliable reference genes for gene expression under abiotic stress in cotton (Gossypium hirsutum L.). Gene 2013, 530, 44–50. [Google Scholar] [CrossRef]
- Du, Z.; Li, H.; Wei, Q.; Zhao, X.; Wang, C.; Zhu, Q.; Yi, X.; Xu, W.; Liu, X.S.; Jin, W.; et al. Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol. Plant 2013, 6, 1463–1472. [Google Scholar] [CrossRef]
- Zong, W.; Zhong, X.; You, J.; Xiong, L. Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol. Biol. 2013, 81, 175–188. [Google Scholar] [CrossRef]
- Malone, B.M.; Tan, F.; Bridges, S.M.; Peng, Z.H. Comparison of four chip-seq analytical algorithms using rice endosperm H3K27 trimethylation profiling data. PLoS ONE 2011, 6, e25260. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; He, K.; Ma, Y.; Su, N.; He, H.; Stolc, V.; Tongprasit, W.; Jin, W.; Jiang, J.; et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell 2008, 20, 259–276. [Google Scholar] [CrossRef]
- Yokoo, T.; Saito, H.; Yoshitake, Y.; Xu, Q.; Asami, T.; Tsukiyama, T.; Teraishi, M.; Okumoto, Y.; Tanisaka, T. Se14, encoding a JmjC domain-containing protein, plays key roles in long-day suppression of rice flowering through the demethylation of H3K4me3 of RFT1. PLoS ONE 2014, 9, e96064. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, X.; Wang, Q.; Zhang, F.; Lou, Z.; Zhang, Q.; Zhou, D.X. Structural basis of a histone H3 lysine 4 demethylase required for stem elongation in rice. PLoS Genet. 2013, 9, e1003239. [Google Scholar] [CrossRef]
- Ding, B.; Bellizzi Mdel, R.; Ning, Y.; Meyers, B.C.; Wang, G.L. HDT701, a histone H4 deacetylase, negatively regulates plant innate immunity by modulating histone H4 acetylation of defense-related genes in rice. Plant Cell 2012, 24, 3783–3794. [Google Scholar] [CrossRef]
- Sun, C.; Fang, J.; Zhao, T.; Xu, B.; Zhang, F.; Liu, L.; Tang, J.; Zhang, G.; Deng, X.; Chen, F.; et al. The histone methyltransferase SDG724 mediates H3K36me2/3 deposition at MADS50 and RFT1 and promotes flowering in rice. Plant Cell 2012, 24, 3235–3247. [Google Scholar] [CrossRef]
- Maestrini, P.; Cavallini, A.; Rizzo, M.; Giordani, T.; Bernardi, R.; Durante, M.; Natali, L. Isolation and expression analysis of low temperature-induced genes in white poplar (Populus alba). J. Plant Physiol. 2009, 166, 1544–1556. [Google Scholar] [CrossRef]
- Singh, K.B.; Foley, R.C.; Onate-Sanchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef]
- Roy, S.W.; Penny, D. A very high fraction of unique intron positions in the intron-rich diatom Thalassiosira pseudonana indicates widespread intron gain. Mol. Biol. Evol. 2007, 24, 1447–1457. [Google Scholar] [CrossRef]
- Roy, S.W.; Gilbert, W. Complex early genes. Proc. Natl. Acad. Sci. USA 2005, 102, 1986–1991. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Maekawa, M.; Saito, A.; Higo, H.; Higo, K. Evolutionary relationship of plant catalase genes inferred from exon-intron structures: Isozyme divergence after the separation of monocots and dicots. Theor. Appl. Genet. 1998, 97, 9–19. [Google Scholar] [CrossRef]
- Ramsey, J.; Schemske, D.W. Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 1998, 29, 467–501. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Chapman, B.A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA 2004, 101, 9903–9908. [Google Scholar] [CrossRef] [Green Version]
- Woodhouse, M.R.; Schnable, J.C.; Pedersen, B.S.; Lyons, E.; Lisch, D.; Subramaniam, S.; Freeling, M. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol. 2010, 8, e1000409. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, R.T.; Pires, J.C.; Iniguez-Luy, F.; Leon, E.; Osborn, T.C. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 2007, 19, 3403–3417. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, H.Z.; Wang, J.; Sun, R.F.; Wu, J.; Liu, S.Y.; Bai, Y.Q.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1359. [Google Scholar] [CrossRef]
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossa, K.; Diouf, D.; Cisse, N. Genome-wide investigation of Hsf genes in sesame reveals their segmental duplication expansion and their active role in drought stress response. Front. Plant Sci. 2016, 7, 1522. [Google Scholar] [CrossRef]
- Jia, J.T.; Zhao, P.C.; Cheng, L.Q.; Yuan, G.X.; Yang, W.G.; Liu, S.; Chen, S.Y.; Qi, D.M.; Liu, G.S.; Li, X.X. MADS-box family genes in sheepgrass and their involvement in abiotic stress responses. BMC Plant Biol. 2018, 18, 42. [Google Scholar] [CrossRef]
- Qanmber, G.; Yu, D.; Li, J.; Wang, L.; Ma, S.; Lu, L.; Yang, Z.; Li, F. Genome-wide identification and expression analysis of Gossypium RING-H2 finger E3 ligase genes revealed their roles in fiber development, and phytohormone and abiotic stress responses. J. Cotton Res. 2018, 1, 1. [Google Scholar] [CrossRef]
- Yang, Z.E.; Gong, Q.; Wang, L.L.; Jin, Y.Y.; Xi, J.P.; Li, Z.; Qin, W.Q.; Yang, Z.R.; Lu, L.L.; Chen, Q.J.; et al. Genome-wide study of YABBY genes in upland cotton and their expression patterns under different stresses. Front Genet 2018, 9, 33. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Yang, Z.E.; Chen, E.Y.; Zhang, C.J.; Zhang, X.Y.; Li, F.G. Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genom. 2018, 19, 348. [Google Scholar] [CrossRef]
- Yin, G.J.; Xu, H.L.; Xiao, S.Y.; Qin, Y.J.; Li, Y.X.; Yan, Y.M.; Hu, Y.K. The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013, 13, 148. [Google Scholar] [CrossRef]
- Ren, Z.; Yu, D.; Yang, Z.; Li, C.; Qanmber, G.; Li, Y.; Li, J.; Liu, Z.; Lu, L.; Wang, L.; et al. Genome-wide identification of the MIKC-type MADS-box gene family in Gossypium hirsutum L. unravels their roles in flowering. Front. Plant Sci. 2017, 8, 384. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qanmber, G.; Ali, F.; Lu, L.; Mo, H.; Ma, S.; Wang, Z.; Yang, Z. Identification of Histone H3 (HH3) Genes in Gossypium hirsutum Revealed Diverse Expression During Ovule Development and Stress Responses. Genes 2019, 10, 355. https://doi.org/10.3390/genes10050355
Qanmber G, Ali F, Lu L, Mo H, Ma S, Wang Z, Yang Z. Identification of Histone H3 (HH3) Genes in Gossypium hirsutum Revealed Diverse Expression During Ovule Development and Stress Responses. Genes. 2019; 10(5):355. https://doi.org/10.3390/genes10050355
Chicago/Turabian StyleQanmber, Ghulam, Faiza Ali, Lili Lu, Huijuan Mo, Shuya Ma, Zhi Wang, and Zuoren Yang. 2019. "Identification of Histone H3 (HH3) Genes in Gossypium hirsutum Revealed Diverse Expression During Ovule Development and Stress Responses" Genes 10, no. 5: 355. https://doi.org/10.3390/genes10050355