
Insights into the Dynamic Succession of Microbial Community and Related Factors of Vanillin Content Change Based by High-Throughput Sequencing and Daqu Quality Drivers

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
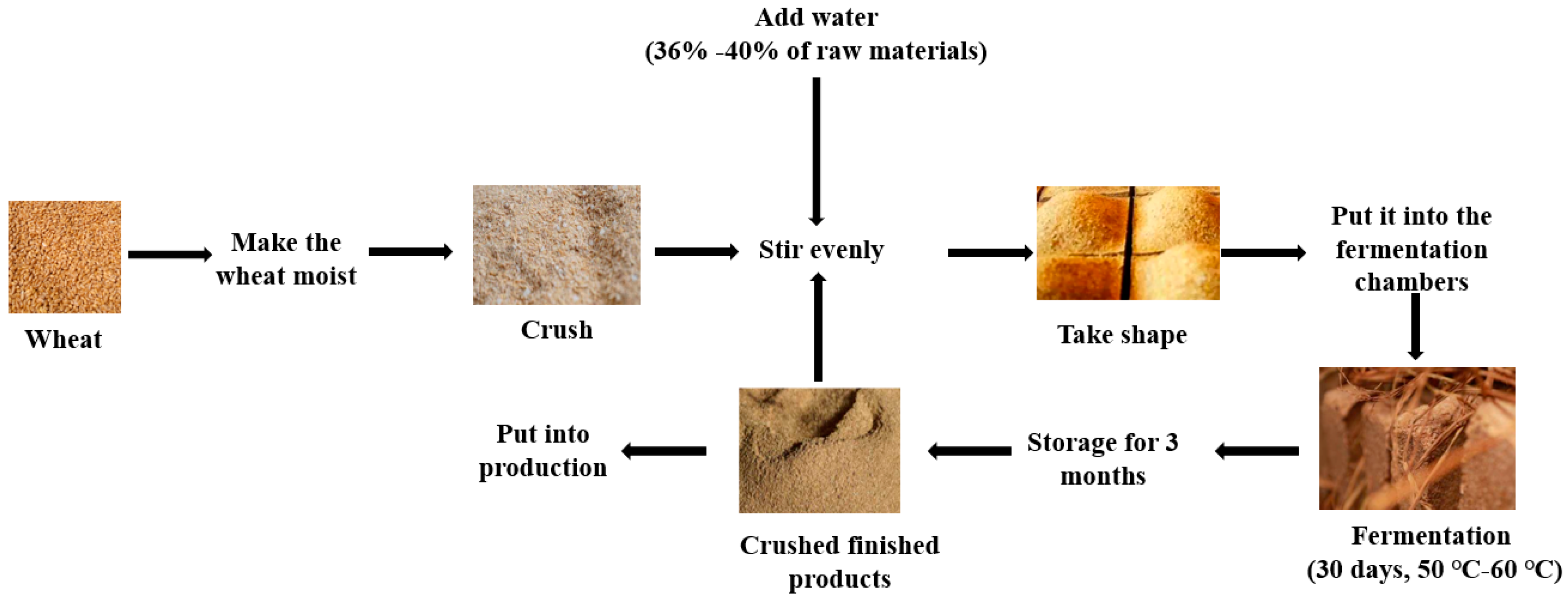
2.1. Sample Collection
2.2. Determination of Vanillin Content
2.3. Analysis of Physicochemical Properties
2.4. DNA Extraction, Amplification, and Sequencing
2.5. Processing of Raw Sequence Data
2.6. Data Analysis
3. Results
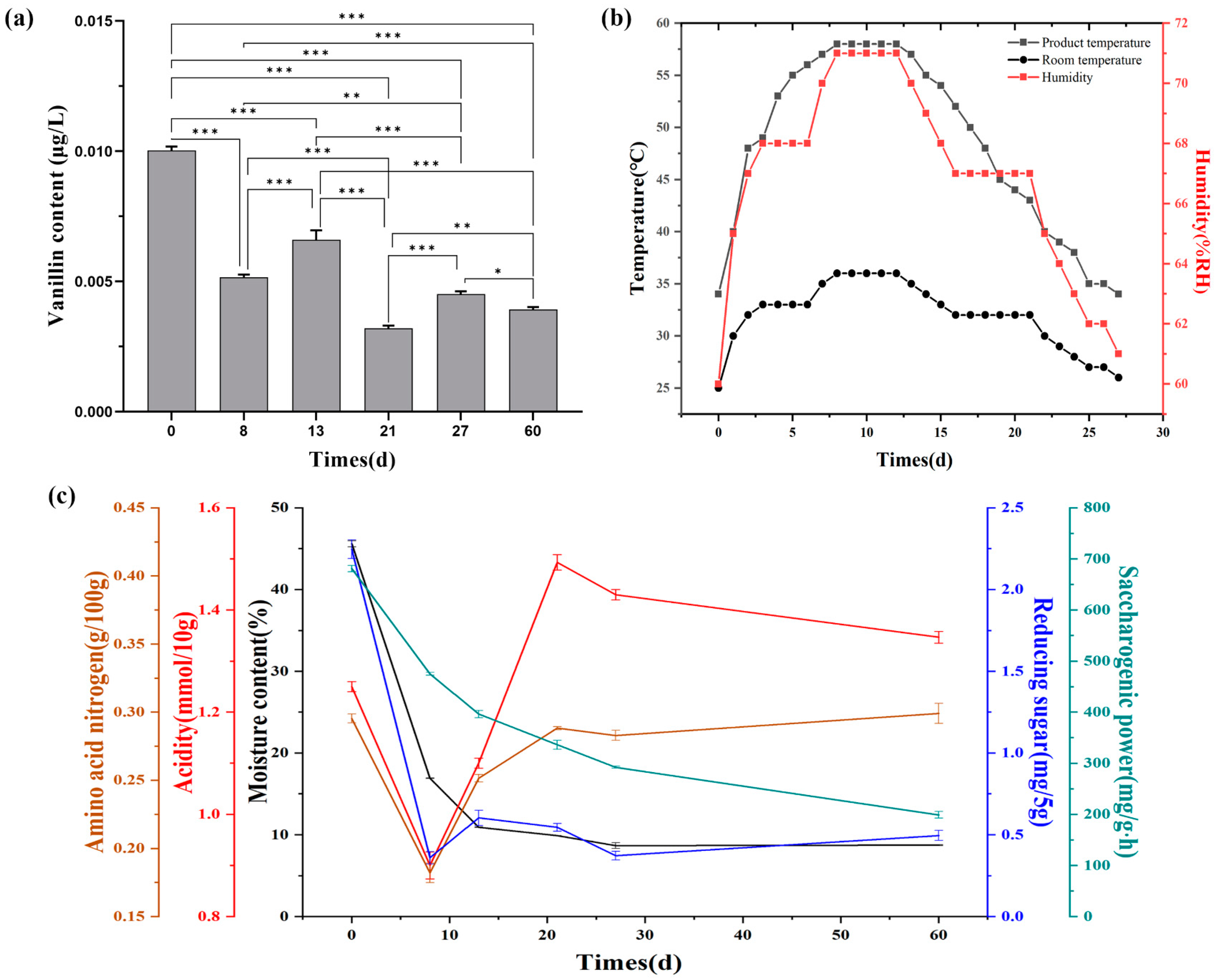
3.1. Vanillin Content and Physicochemical Properties during the Fermentation of Daqu
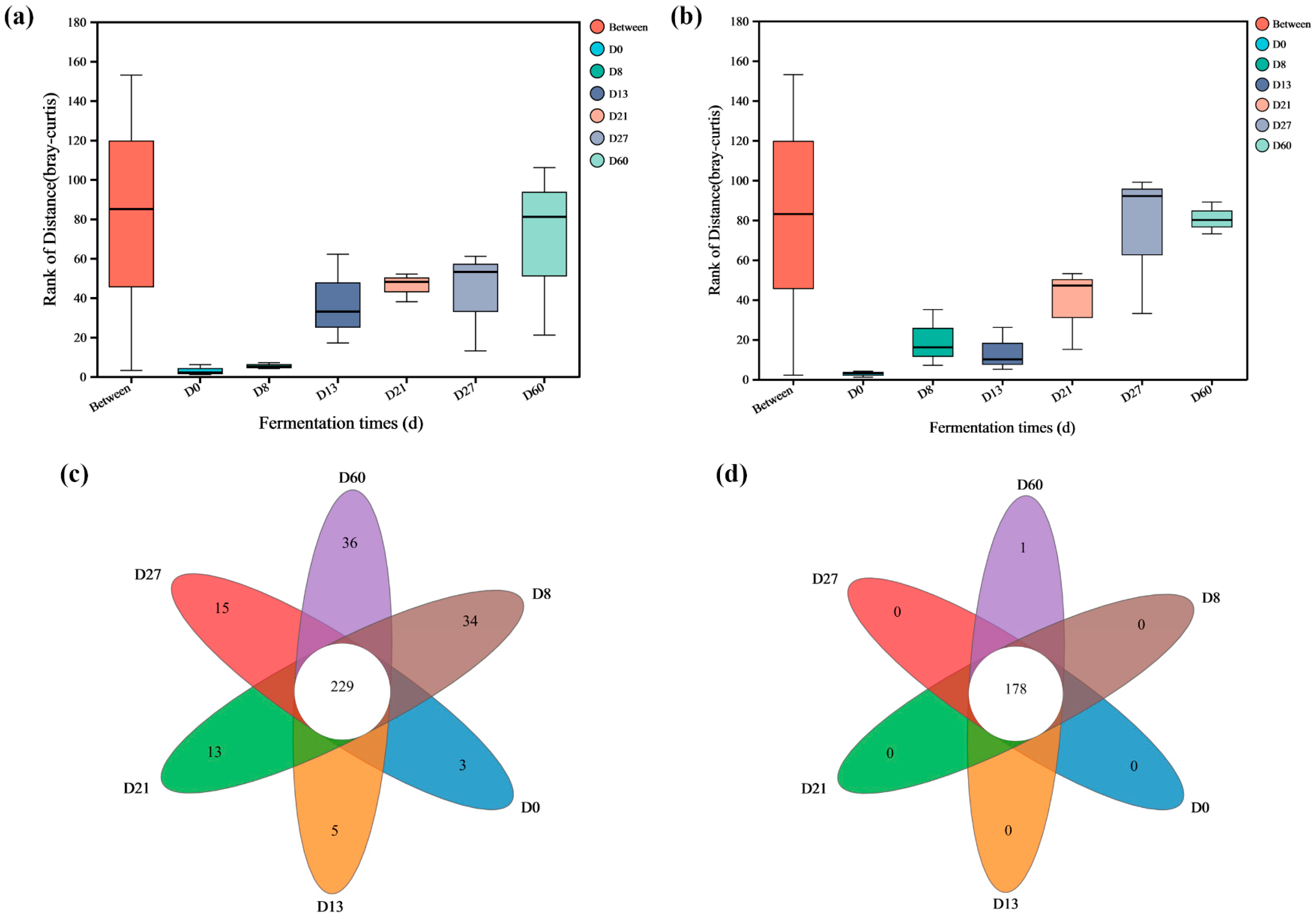
3.2. The Richness and Diversity of Microorganisms during the Fermentation Process of Daqu
3.3. Influence of Microbial Diversity and Change on Vanillin Content
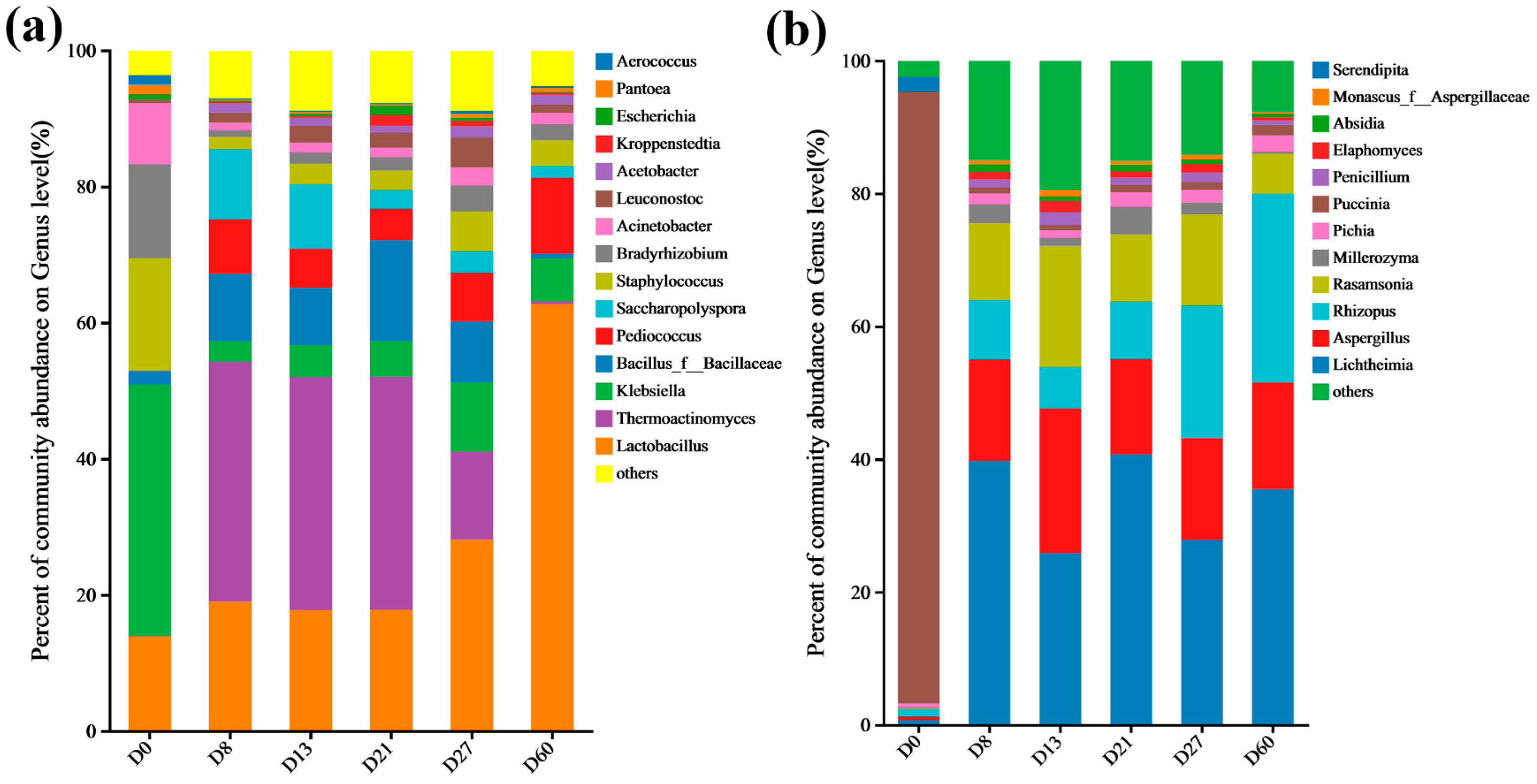
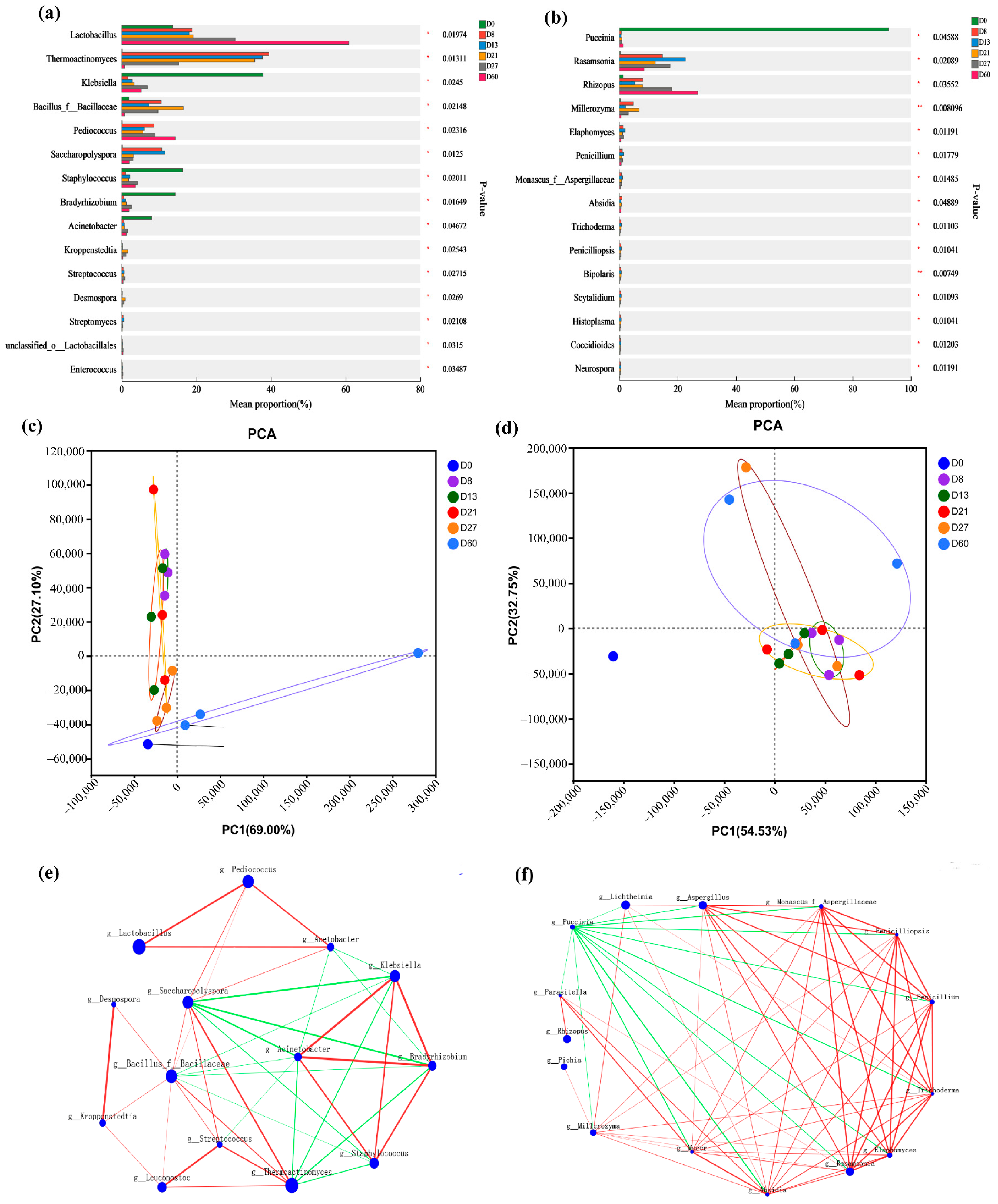
3.4. Analysis of Species and Functional Differences in Daqu Fermentation Process
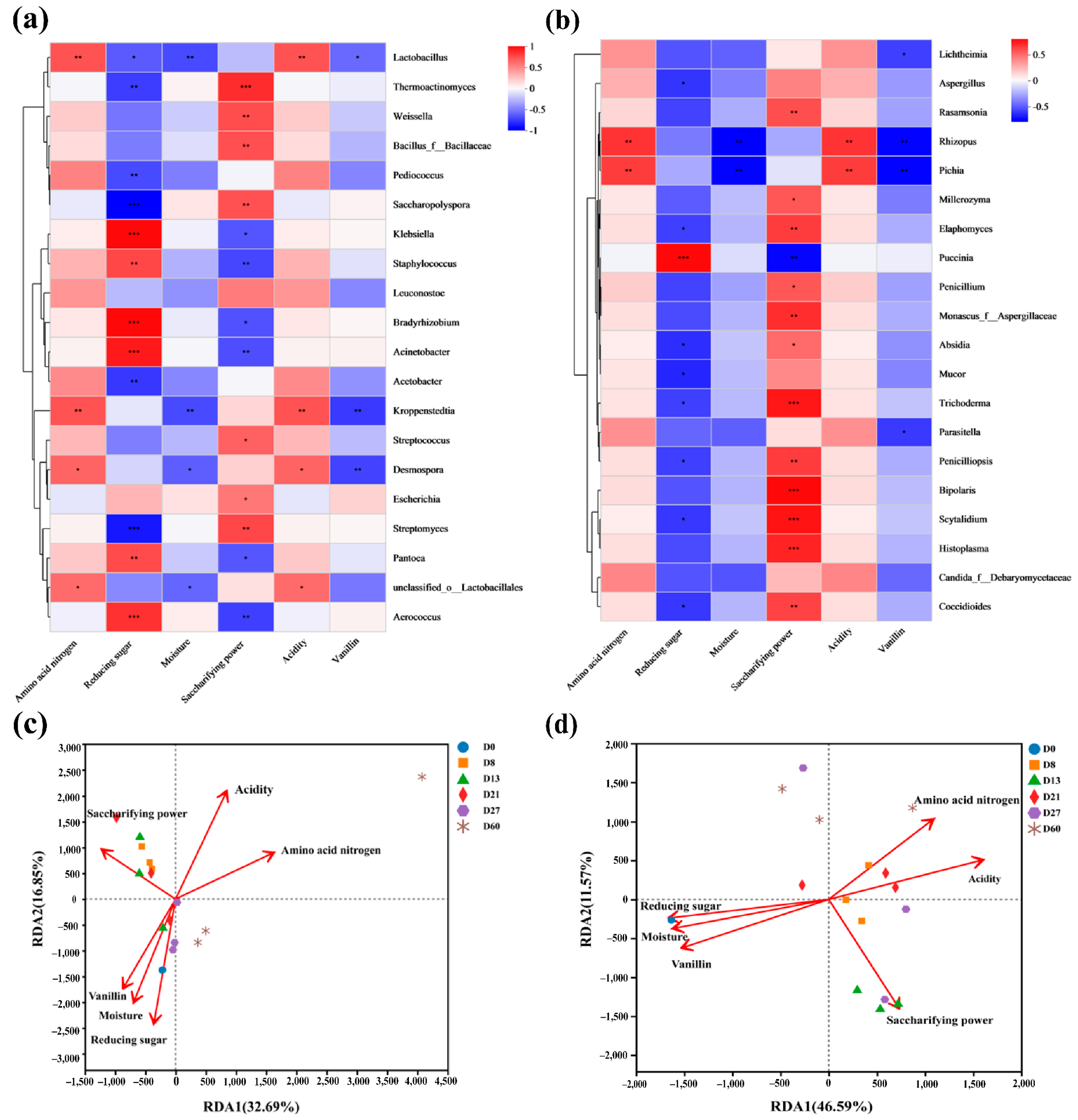
3.5. Correlation Analysis between Microorganisms and Physicochemical Properties
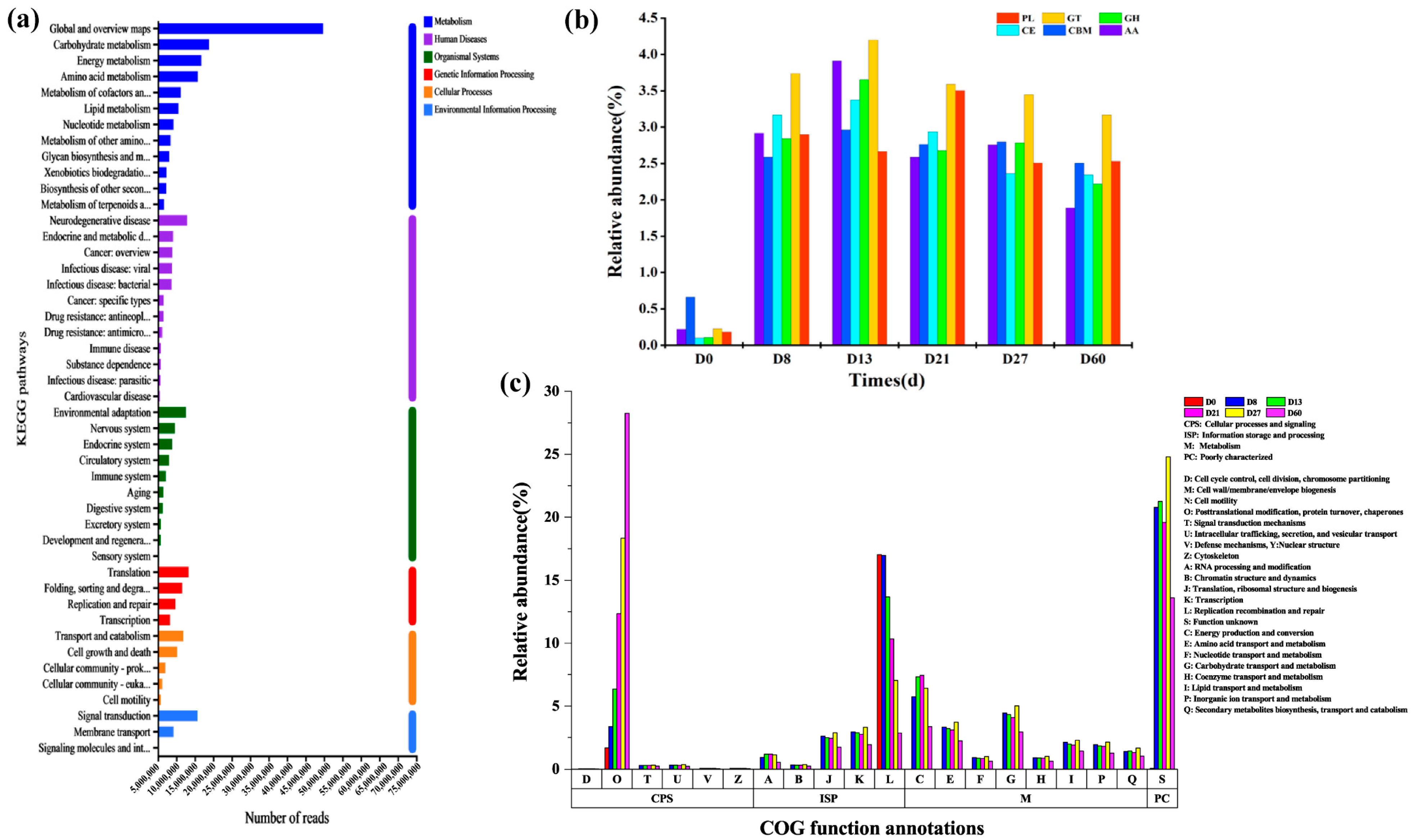
3.6. Annotations on Species and Functions during the Fermentation Process of Daqu
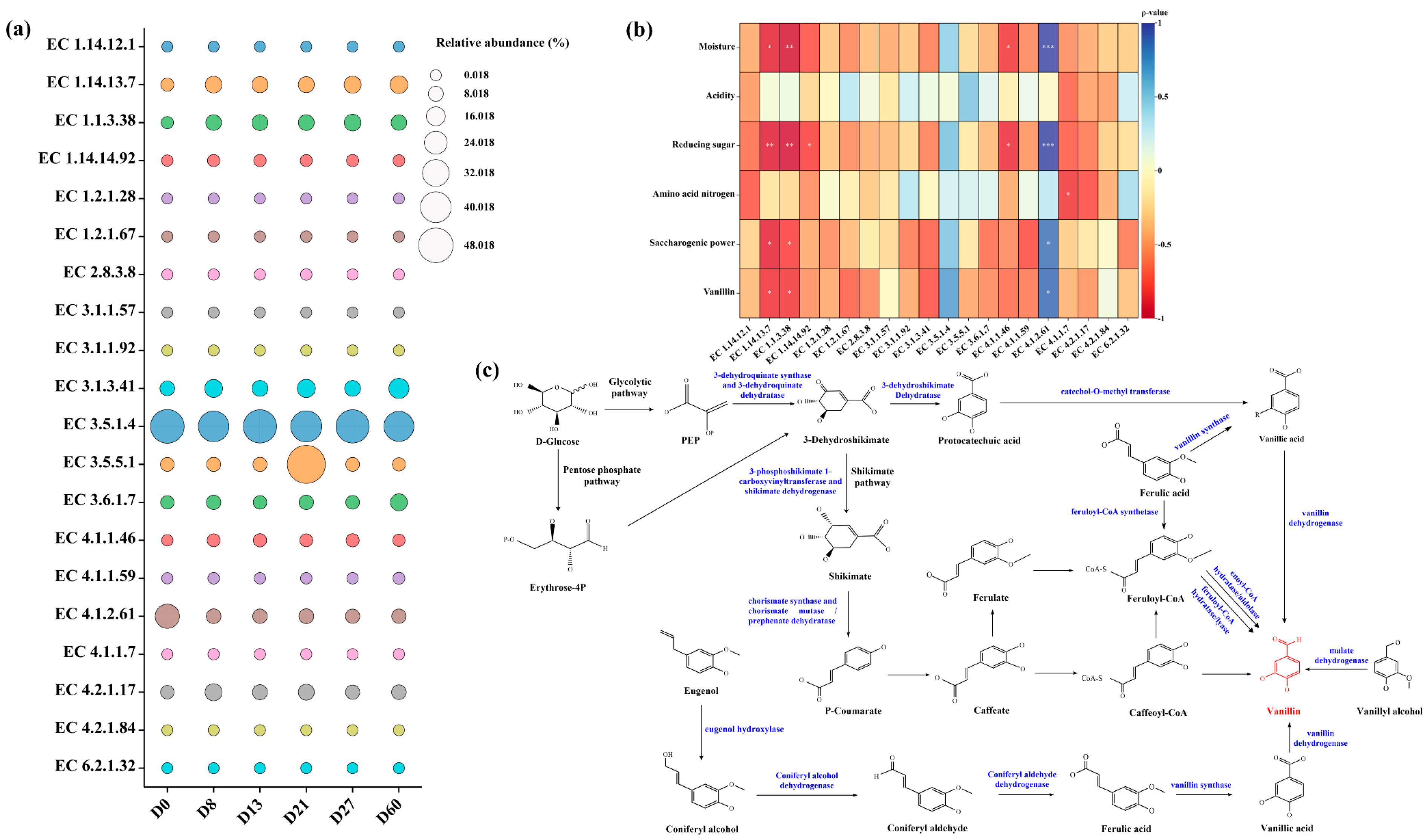
3.7. Analysis of Enzymes and Related Factors Involved in Vanillin Metabolism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ma, Y.-C.; Zheng, Y.; Wang, L.-H.; Sun, B.-G.; Zhao, M.-M.; Huang, M.-Q.; Wu, J.-H.; Li, H.-H.; Sun, X.-T. Integrated distilled spent grain with husk utilization: Current situation, trend, and design. Renew. Sustain. Energy Rev. 2023, 179, 113275. [Google Scholar] [CrossRef]
- Du, P.; Jiao, G.; Zhang, Z.; Wang, J.; Li, P.; Dong, J.; Wang, R. Relationship between Representative Trace Components and Health Functions of Chinese Baijiu: A Review. Fermentation 2023, 9, 658. [Google Scholar] [CrossRef]
- Zhang, W.-x.; Qiao, Z.-w.; Shigematsu, T.; Tang, Y.-q.; Hu, C.; Morimura, S.; Kida, K. Analysis of the Bacterial Community in Zaopei during Production of Chinese Luzhou-flavor Liquor. J. Inst. Brew. 2005, 111, 215–222. [Google Scholar] [CrossRef]
- Hao, F.; Tan, Y.; Lv, X.; Chen, L.; Yang, F.; Wang, H.; Du, H.; Wang, L.; Xu, Y. Microbial Community Succession and Its Environment driving Factors During Initial Fermentation of Maotai-Flavor Baijiu. Front. Microbiol. 2021, 12, 669201. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Jin, Y.; Zhou, R.; Zhao, D.; Zheng, J.; Wu, C. Dynamic succession of microbial community in Nongxiangxing daqu and microbial roles involved in flavor formation. Food Res. Int. 2022, 159, 111559. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Zeng, Y.; Cui, Y.; Chen, P.; Cai, K.; Guo, T.; Tan, G.; Peng, N.; Liang, Y.; Zhao, S. RETRACTED: Unraveling the composition and succession of microbial community and its relationship to flavor substances during Xin-flavor baijiu brewing. Int. J. Food Microbiol. 2022, 372, 109679. [Google Scholar] [CrossRef]
- Tu, W.; Cao, X.; Cheng, J.; Li, L.; Zhang, T.; Wu, Q.; Xiang, P.; Shen, C.; Li, Q. Chinese Baijiu: The perfect works of microorganisms. Front. Microbiol. 2022, 13, 919044. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, M.; Niu, J.; Lin, M.; Zhu, H.; Wang, K.; Li, X.; Sun, B. Characteristics and correlation of the microbial communities and flavor compounds during the first three rounds of fermentation in Chinese sauce-flavor Baijiu. Foods 2023, 12, 207. [Google Scholar] [CrossRef]
- Cai, W.; Wang, Y.; Ni, H.; Liu, Z.; Liu, J.; Hou, Q.; Shan, C.; Yang, X.; Guo, Z. Diversity of microbiota, microbial functions, and flavor in different types of low-temperature Daqu. Food Res. Int. 2021, 150, 110734. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, S.-T.; Lu, Z.-M.; Zhang, X.-J.; Chai, L.-J.; Shen, C.-H.; Shi, J.-S.; Xu, Z.-H. Metagenomics unveils microbial roles involved in metabolic network of flavor development in medium-temperature daqu starter. Food Res. Int. 2021, 140, 110037. [Google Scholar] [CrossRef]
- Fu, G.; Cai, W.; Dong, B.; Wan, Y.; Pan, F.; Zheng, F.; Chen, Y.; Deng, M.; Huang, B. Effects of bio-augmented Daqu on microbial community, aroma compounds and physicochemical parameters of fermented grains during the brewing of Chinese special-flavor baijiu. J. Sci. Food Agric. 2023, 103, 273–282. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, C.; Xiang, X.; Xu, H.; Han, G. Analysis of microbial diversity and succession during Xiaoqu Baijiu fermentation using high-throughput sequencing technology. Eng. Life Sci. 2022, 22, 495–504. [Google Scholar] [CrossRef]
- Ren, T.; Su, W.; Mu, Y.; Qi, Q.; Zhang, D. Study on the correlation between microbial communities with physicochemical properties and flavor substances in the Xiasha round of cave-brewed sauce-flavor Baijiu. Front. Microbiol. 2023, 14, 1124817. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ao, Z.; Chui, W.; Shen, C.; Tao, W.; Zhang, S. Characterization of the aroma-active compounds in Daqu: A tradition Chinese liquor starter. Eur. Food Res. Technol. 2012, 234, 69–76. [Google Scholar] [CrossRef]
- Wang, C.-l.; Shi, D.-j.; Gong, G.-l. Microorganisms in Daqu: A starter culture of Chinese Maotai-flavor liquor. World J. Microbiol. Biotechnol. 2008, 24, 2183–2190. [Google Scholar] [CrossRef]
- Xie, M.; Lv, F.; Ma, G.; Farooq, A.; Li, H.; Du, Y.; Liu, Y. High throughput sequencing of the bacterial composition and dynamic succession in Daqu for Chinese sesame flavour liquor. J. Inst. Brew. 2020, 126, 98–104. [Google Scholar] [CrossRef]
- Hong, J.; Zhao, D.; Sun, B. Research Progress on the Profile of Trace Components in Baijiu. Food Rev. Int. 2023, 39, 1666–1693. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Zhang, S.; Liu, T.; Qin, H.; Shen, C.; Liu, H.; Yang, F.; Yang, C.; Yin, Q.; et al. Spatiotemporal distribution of environmental microbiota in spontaneous fermentation workshop: The case of Chinese Baijiu. Food Res. Int. 2022, 156, 111126. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Li, H.; Zhang, H.; Shen, X.; Zhang, L.; Han, S.; Pan, C. Taorong-type Baijiu starter: Analysis of fungal community and metabolic characteristics of middle-temperature Daqu and high-temperature Daqu. PLoS ONE 2022, 17, e0274881. [Google Scholar] [CrossRef]
- Li, D.; Jia, F.; Wang, L.; Chang, F. The initial composition and structure of microbial community determined the yield and quality of Baijiu during the spontaneous fermentation. Int. Microbiol. 2023, 26, 1–12. [Google Scholar] [CrossRef]
- Cheng, W.; Chen, X.; Zeng, H.; Xue, X. Association between microbial community composition and quality indicators of strong-flavor Daqu of different producing regions in China. CyTA J. Food 2023, 21, 82–92. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Yang, J.-G.; Zhao, Q.-S.; Zhang, K.-Z.; Su, C. Research Progress on Flavor Compounds and Microorganisms of Maotai Flavor Baijiu. J. Food Sci. 2019, 84, 6–18. [Google Scholar] [CrossRef]
- Sun, B.; Wu, J.; Huang, M.; Sun, J.; Zheng, F. Recent advances of flavor chemistry in Chinese liquor spirits (Baijiu). J. Chin. Inst. Food Sci. Technol. 2015, 15, 1–8. [Google Scholar] [CrossRef]
- Zhao, D.; Sun, J.; Sun, B.; Zhao, M.; Zheng, F.; Huang, M.; Sun, X.; Li, H. Intracellular antioxidant effect of vanillin, 4-methylguaiacol and 4-ethylguaiacol: Three components in Chinese Baijiu. RSC Adv. 2017, 7, 46395–46405. [Google Scholar] [CrossRef]
- Squina, F. Ferulic acid and derivatives: Molecules with potential application in the pharmaceutical field. Braz. J. Pharm. Sci. 2013, 49, 298. [Google Scholar]
- Sun, X.; Qian, Q.; Xiong, Y.; Xie, Q.; Yue, X.; Liu, J.; Wei, S.; Yang, Q. Characterization of the key aroma compounds in aged Chinese Xiaoqu Baijiu by means of the sensomics approach. Food Chem. 2022, 384, 132452. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Chattopadhyay, P. Vanillin biotechnology: The perspectives and future. J. Sci. Food Agric. 2019, 99, 499–506. [Google Scholar] [CrossRef]
- Anand, A.; Khurana, R.G.; Wahal, N.; Mahajan, S.; Mehta, M.; Satija, S.; Sharma, N.; Vyas, M.; Khurana, N. Vanillin: A Comprehensive Review of Pharmacological Activities. Plant Arch. 2019, 19, 1000–1004. [Google Scholar]
- Sinha, A.K.; Sharma, U.K.; Sharma, N. A comprehensive review on vanilla flavor: Extraction, isolation and quantification of vanillin and others constituents. Int. J. Food Sci. Nutr. 2008, 59, 299–326. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, X.; Feng, Y.; Sun, J.; Jiang, Y.; Zhang, W.; Xin, F.; Jiang, M. Current Status, Challenges, and Prospects for the Biological Production of Vanillin. Fermentation 2023, 9, 389. [Google Scholar] [CrossRef]
- Su, Y.; Yang, L.; Hui, L.; Yuan-Yuan, G.; Ming-Juan, Z.; Chun-Hui, X.; Ling, X.; Chi, C. Bacterial communities during the process of high-temperature Daqu production of roasted sesame-like flavour liquor. J. Inst. Brew. 2015, 121, 440–448. [Google Scholar] [CrossRef]
- Dong, W.; Yang, Q.; Liao, Y.; Liu, Y.; Hu, Y.; Peng, N.; Liang, Y.; Zhao, S. Characterisation and comparison of the microflora of traditional and pure culture xiaoqu during the baijiu liquor brewing process. J. Inst. Brew. 2020, 126, 213–220. [Google Scholar] [CrossRef]
- QB/T 4257-2011; General methods of analysis for Daqu. China Research Institute of Food and Fermentation Industries: Beijing, China, 2011.
- Deng, L.; Mao, X.; Liu, D.; Ning, X.-Q.; Shen, Y.; Chen, B.; Nie, H.-F.; Huang, D.; Luo, H.-B. Comparative Analysis of Physicochemical Properties and Microbial Composition in High-Temperature Daqu with Different Colors. Front. Microbiol. 2020, 11, 588117. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Chen, T.; Chen, X.; Zhang, S.; Zhu, J.; Tang, B.; Wang, A.; Dong, L.; Zhang, Z.; Yu, C.; Sun, Y.; et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar] [CrossRef]
- Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [CrossRef]
- Liu, H.; Sun, B. Effect of Fermentation Processing on the Flavor of Baijiu. J. Agric. Food Chem. 2018, 66, 5425–5432. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, J.; Xie, J.; Zhao, D.; Qiao, Z.-W.; Huang, D.; Luo, H.-B. Effects of environmental factors on the microbial community changes during medium-high temperature Daqu manufacturing. Food Res. Int. 2022, 153, 110955. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Liu, L.; Zhao, S.; Huang, Z.; Li, C.; Jiang, S.; Li, Q.; Gu, P. Biosynthesis of vanillin by different microorganisms: A review. World J. Microbiol. Biotechnol. 2022, 38, 40. [Google Scholar] [CrossRef]
- Lesage-Meessen, L.; Delattre, M.; Haon, M.; Thibault, J.-F.; Ceccaldi, B.C.; Brunerie, P.; Asther, M. A two-step bioconversion process for vanillin production from ferulic acid combining Aspergillus niger and Pycnoporus cinnabarinus. J. Biotechnol. 1996, 50, 107–113. [Google Scholar] [CrossRef]
- Shimoni, E.; Ravid, U.; Shoham, Y. Isolation of a Bacillus sp. capable of transforming isoeugenol to vanillin. J. Biotechnol. 2000, 78, 1–9. [Google Scholar] [CrossRef]
- Breton, C.; Mucha, J.; Jeanneau, C. Structural and functional features of glycosyltransferases. Biochimie 2001, 83, 713–718. [Google Scholar] [CrossRef]
- Wang, J.; Hou, B. Glycosyltransferases: Key players involved in the modification of plant secondary metabolites. Front. Biol. China 2009, 4, 39–46. [Google Scholar] [CrossRef]
- Zou, Y.; Yuan, Y.; Liu, M.; Li, X.; Lai, Y.; Liu, X.; Tan, L.; Tang, Q.; Chen, W.; Li, D.; et al. Metagenomics reveal the role of microorganism and GH genes contribute to Sichuan South-road dark tea quality formation during pile fermentation. LWT 2023, 178, 114618. [Google Scholar] [CrossRef]
- Barba-Cedillo, V.; Montanier, C.Y. Effect of multimodularity and spatial organization of glycoside hydrolases on catalysis. Essays Biochem. 2023, 67, 629–638. [Google Scholar] [CrossRef]
- Ma, S.; Shang, Z.; Chen, J.; Shen, Y.; Li, Z.; Huang, D.; Luo, H. Differences in structure, volatile metabolites, and functions of microbial communities in Nongxiangxing daqu from different production areas. LWT 2022, 166, 113784. [Google Scholar] [CrossRef]
- Toushik, S.H.; Lee, K.-T.; Lee, J.-S.; Kim, K.-S. Functional Applications of Lignocellulolytic Enzymes in the Fruit and Vegetable Processing Industries. J. Food Sci. 2017, 82, 585–593. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, S.; Sun, H.; Jiang, Z.; Xu, Y.; Mao, J.; Qian, B.; Wang, L.; Mao, J. Metagenomics-based insights into the microbial community profiling and flavor development potentiality of baijiu Daqu and huangjiu wheat Qu. Food Res. Int. 2022, 152, 110707. [Google Scholar] [CrossRef]
- Liu, H.M.; Zou, Y.; Yao, C.Y.; Yang, Z. Enzymatic synthesis of vanillin and related catalytic mechanism. Flavour Fragr. J. 2020, 35, 51–58. [Google Scholar] [CrossRef]
- Paul, V.; Rai, D.C.; T.S, R.L.; Srivastava, S.K.; Tripathi, A.D. A comprehensive review on vanillin: Its microbial synthesis, isolation and recovery. Food Biotechnol. 2021, 35, 22–49. [Google Scholar] [CrossRef]
- Xu, L.; Liaqat, F.; Sun, J.; Khazi, M.I.; Xie, R.; Zhu, D. Advances in the vanillin synthesis and biotransformation: A review. Renew. Sustain. Energy Rev. 2024, 189, 113905. [Google Scholar] [CrossRef]
- Mortzfeld, F.B.; Hashem, C.; Vranková, K.; Winkler, M.; Rudroff, F. Pyrazines: Synthesis and industrial application of these valuable flavor and fragrance compounds. Biotechnol. J. 2020, 15, 2000064. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year of Publication | Related Colonies | Research Results |
|---|---|---|
| 2020 [17] | Firmicutes, Proteobacteria, Actinobacteria, Kroppenstedtia, Lactobacillus, Weissella, Lentibacillus, Bacillus, Saccharopolyspora. | Firmicutes have significant advantages in the fermentation stage, Kroppenstedtia, Lactobacillus, Weissella, Lentibacillus, Bacillus, and Saccharopolyspora were detected as the main bacterial groups in the high-temperature Daqu of Chinese sesame-flavor liquor. |
| 2021 [9] | Lactic acid bacteria, Pichia pastoris. | The results showed that microorganisms were obviously enriched, and the diversity of bacteria and fungi showed a downward trend during the heap fermentation process of Maotai-flavor Baijiu. However, the diversity of fungi in the pit fermentation process increased. |
| 2021 [18] | Thermoactinomyces, Lactobacillus, Saccharopolyspora, Bacillus, Streptomyces, Saccharomycopsis, Thermoascus. | The different types of low-temperature Daqu had distinct flavor profiles, and the differences in the taste profiles were more significant. Dominated by Thermoactinomyces and Lactobacillus, together with Saccharopolyspora, Bacillus, Streptomyces, Saccharomycopsis, and Thermoascus, they formed the core microbiota that influenced the flavor of low-temperature Daqu. The bacteria mainly influenced the taste of low-temperature Daqu, whereas the fungi mainly influenced the aroma. |
| 2022 [19] | Saccharomycopsis, fibuligera, Debaryomyces hansenii, Lichtheimia ramosa, Lichtheimia corymbifera, Pichia kudriavzevii. | The composition of the fungal community was similar, with Saccharomycopsis fibuligera, Debaryomyces hansenii, Lichtheimia ramosa, Lichtheimia corymbifera, and Pichia kudriavzevii being the most abundant and detected in most samples. |
| 2022 [20] | Lactococcus, Enterobacteriaceae, Lactobacillus, Weiss. | The results showed that 39 different microbial genera were detected from Daqu samples of three storage periods, among which the dominant genera were Lactococcus, Enterobacteriaceae, Lactobacillus, and Weiss. The dominant microbial communities of Daqu vary greatly with different storage periods. |
| 2023 [21] | Lactobacillus, Stenotrophomonas, Firmicutes, Gammaproteobacteria, Actinobacteria, Streptomyces, Bordetella, Olivibacter | During the initial fermentation stage, the bacterial community exerted a more pronounced effect on Baijiu quality than the fungal community. Lactobacillus was the dominant genus and biomarker in high-yield pit mud, and it constituted the only genus within the bacterial association network during the late fermentation stage. |
| 2023 [22] | Weissella, Pediococcus, Leuconostoc, Rhizopus, Staphylococcus, Alternaria, Weissella, Pediococcus, Burkholderia. | In different sample groups of surface and central parts, the differential fungal genus was Alternaria, whereas differential bacteria genera were Weissella, Pediococcus, and Leuconostoc. |
| Samples | Bacteria | Fungi | ||
|---|---|---|---|---|
| Chao1 | Shannon | Chao1 | Shannon | |
| D0 | 190.67 | 1.9763 | 124.00 | 0.4793 |
| D8 | 789.67 | 2.1744 | 461.33 | 2.5040 |
| D13 | 770.00 | 2.2579 | 459.00 | 2.7354 |
| D21 | 754.67 | 2.2459 | 459.33 | 2.5659 |
| D27 | 696.00 | 2.4055 | 453.00 | 2.4620 |
| D60 | 666.00 | 1.7792 | 453.67 | 1.9592 |
| Bacteria | Fungi | |||||||
|---|---|---|---|---|---|---|---|---|
| RDA1 | RDA2 | R2 | p | RDA1 | RDA2 | R2 | p | |
| Amino acid nitrogen | 0.9661 | 0.2580 | 0.1846 | 0.2290 | 0.7781 | 0.6281 | 0.6455 | 0.0010 |
| Reducing sugar | 0.1665 | −0.9860 | 0.3759 | 0.0450 | −0.9773 | −0.2117 | 0.8265 | 0.0010 |
| Moisture | −0.0549 | −0.9985 | 0.2516 | 0.1920 | −0.9618 | −0.2738 | 0.8138 | 0.0010 |
| Saccharifying power | −0.7716 | 0.6362 | 0.4191 | 0.0160 | 0.4824 | −0.8759 | 0.5482 | 0.0030 |
| Acidity | 0.1261 | 0.9920 | 0.2763 | 0.1490 | 0.9434 | 0.3316 | 0.8402 | 0.0010 |
| Vanillin | −0.2448 | −0.9696 | 0.1961 | 0.2270 | −0.9217 | −0.3878 | 0.8119 | 0.0020 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tong, W.; Wang, S.; Yang, Y.; Huang, Z.; Li, Y.; Huang, D.; Luo, H.; Zhao, L. Insights into the Dynamic Succession of Microbial Community and Related Factors of Vanillin Content Change Based by High-Throughput Sequencing and Daqu Quality Drivers. Foods 2023, 12, 4312. https://doi.org/10.3390/foods12234312
Tong W, Wang S, Yang Y, Huang Z, Li Y, Huang D, Luo H, Zhao L. Insights into the Dynamic Succession of Microbial Community and Related Factors of Vanillin Content Change Based by High-Throughput Sequencing and Daqu Quality Drivers. Foods. 2023; 12(23):4312. https://doi.org/10.3390/foods12234312
Chicago/Turabian StyleTong, Wenhua, Shuqin Wang, Ying Yang, Zhijiu Huang, Yiyun Li, Dan Huang, Huibo Luo, and Liming Zhao. 2023. "Insights into the Dynamic Succession of Microbial Community and Related Factors of Vanillin Content Change Based by High-Throughput Sequencing and Daqu Quality Drivers" Foods 12, no. 23: 4312. https://doi.org/10.3390/foods12234312