
Linking Microbial Functional Gene Abundance and Daqu Extracellular Enzyme Activity: Implications for Carbon Metabolism during Fermentation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Extracellular Enzyme Properties of Daqu
2.3. DNA Extraction, Amplicon Sequencing, and Shotgun Metagenomic Sequencing
2.4. Amplicon Data Processing
2.5. Metagenomic Data Processing
2.6. Statistical Analysis
3. Results
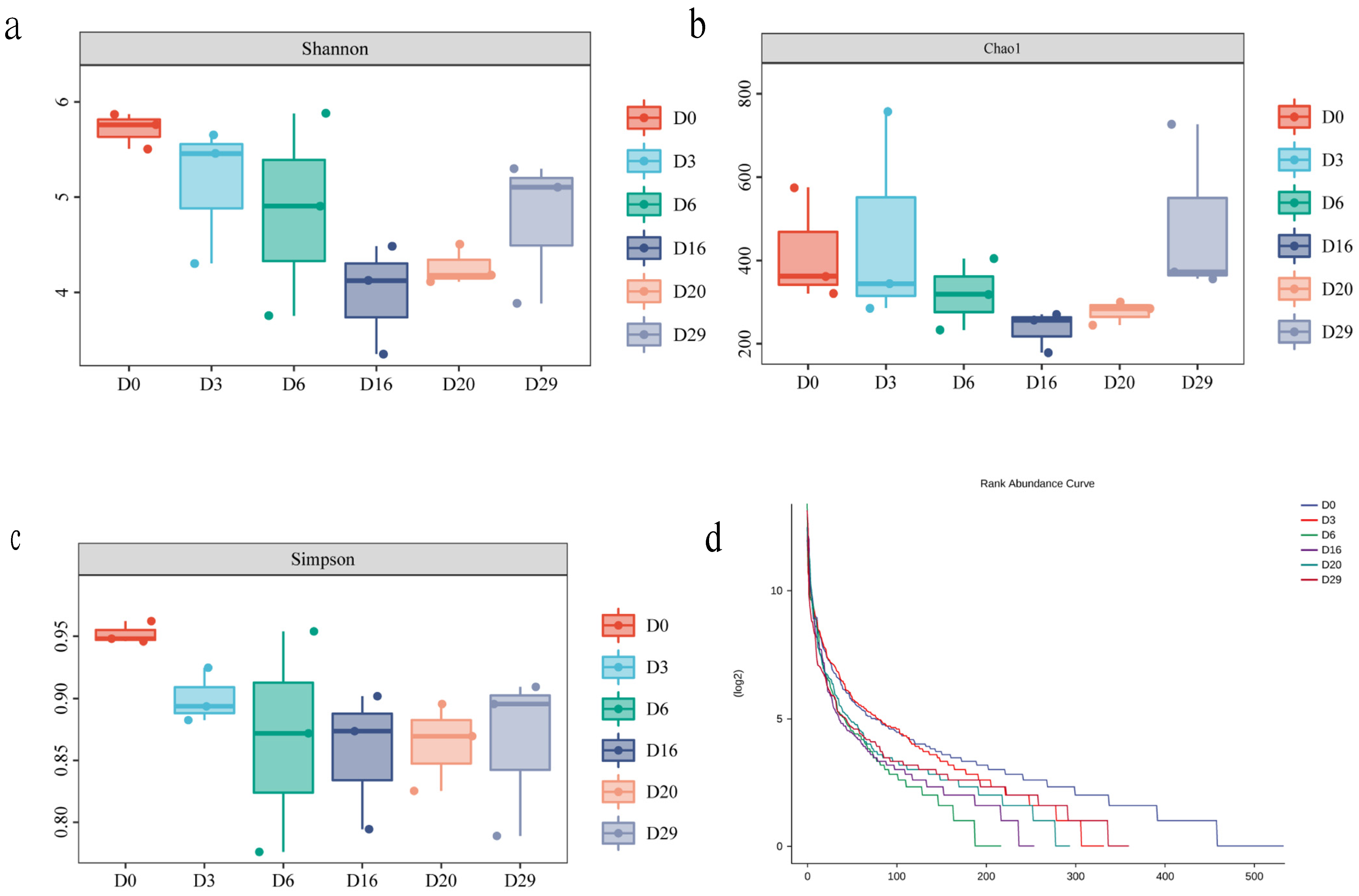
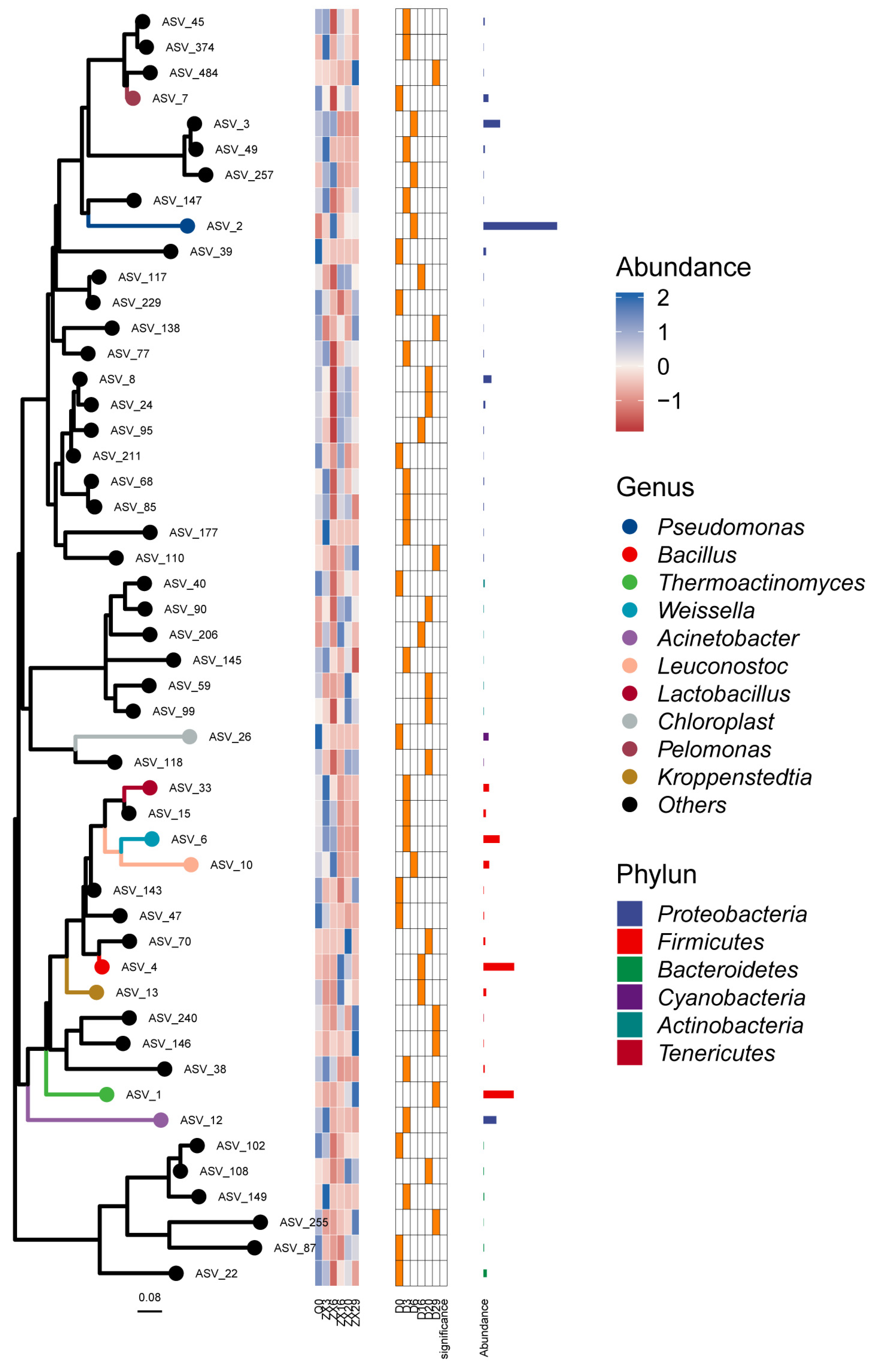
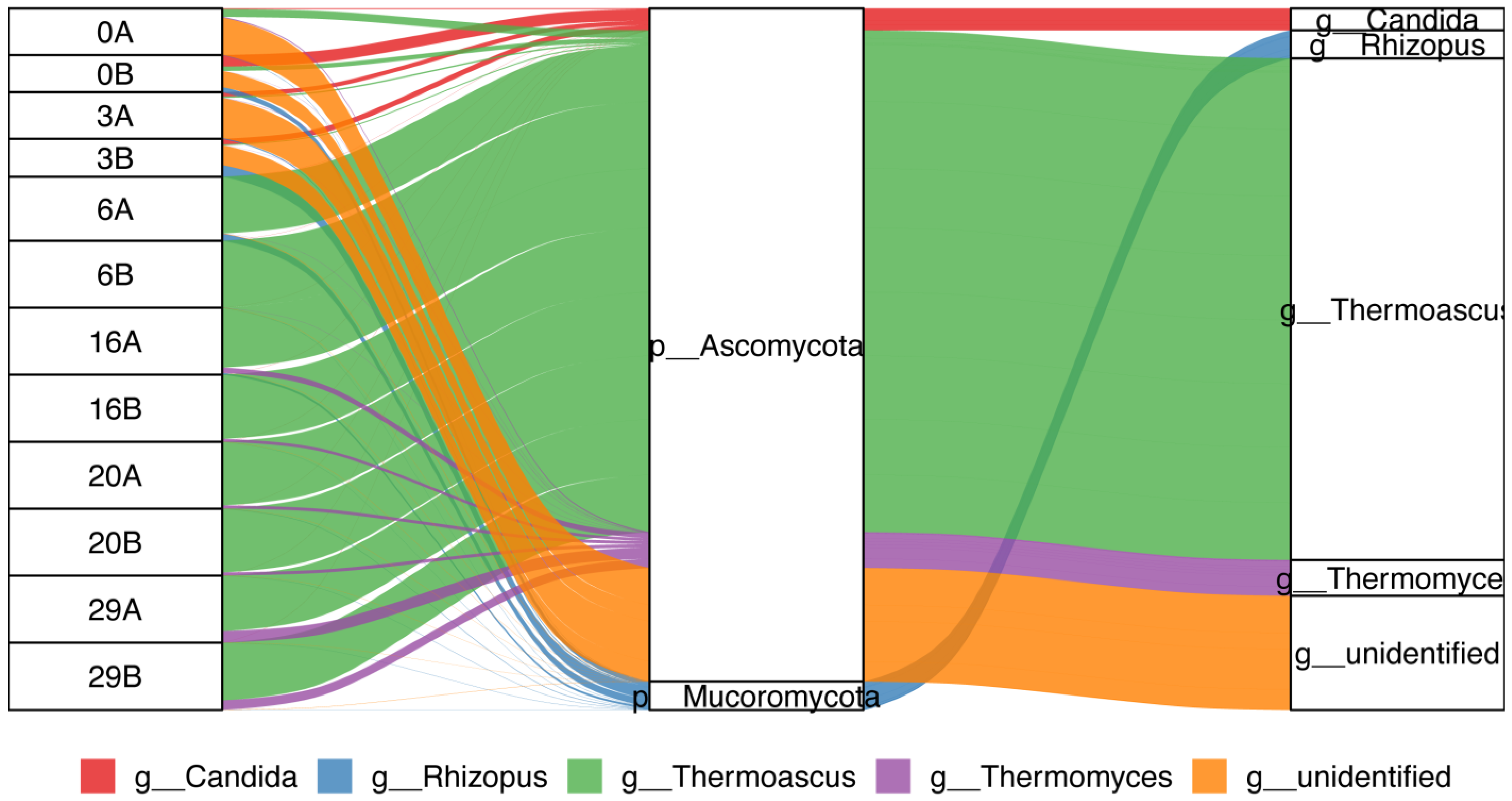
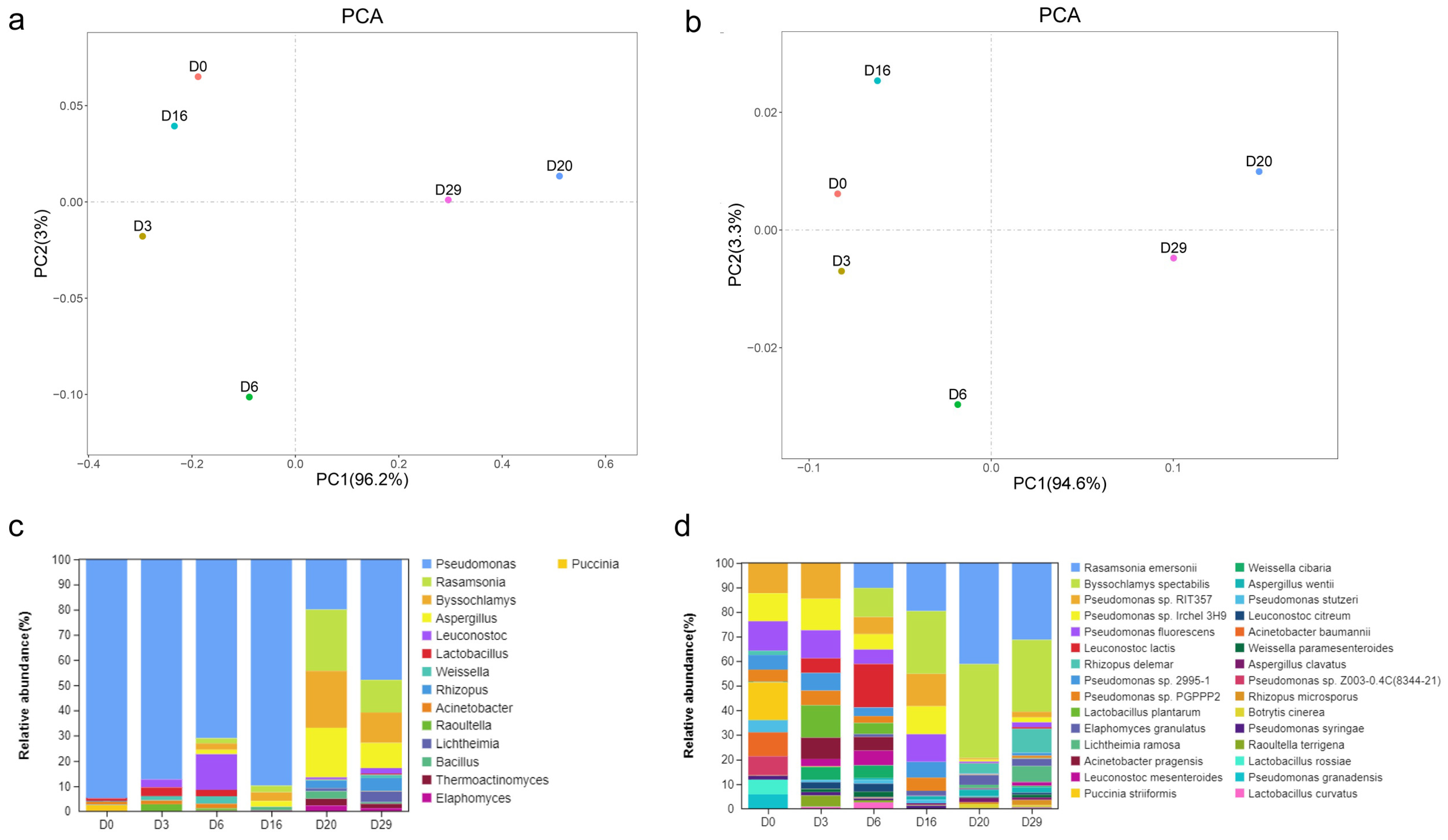
3.1. Composition of the Microbial Community in Fermented Daqu
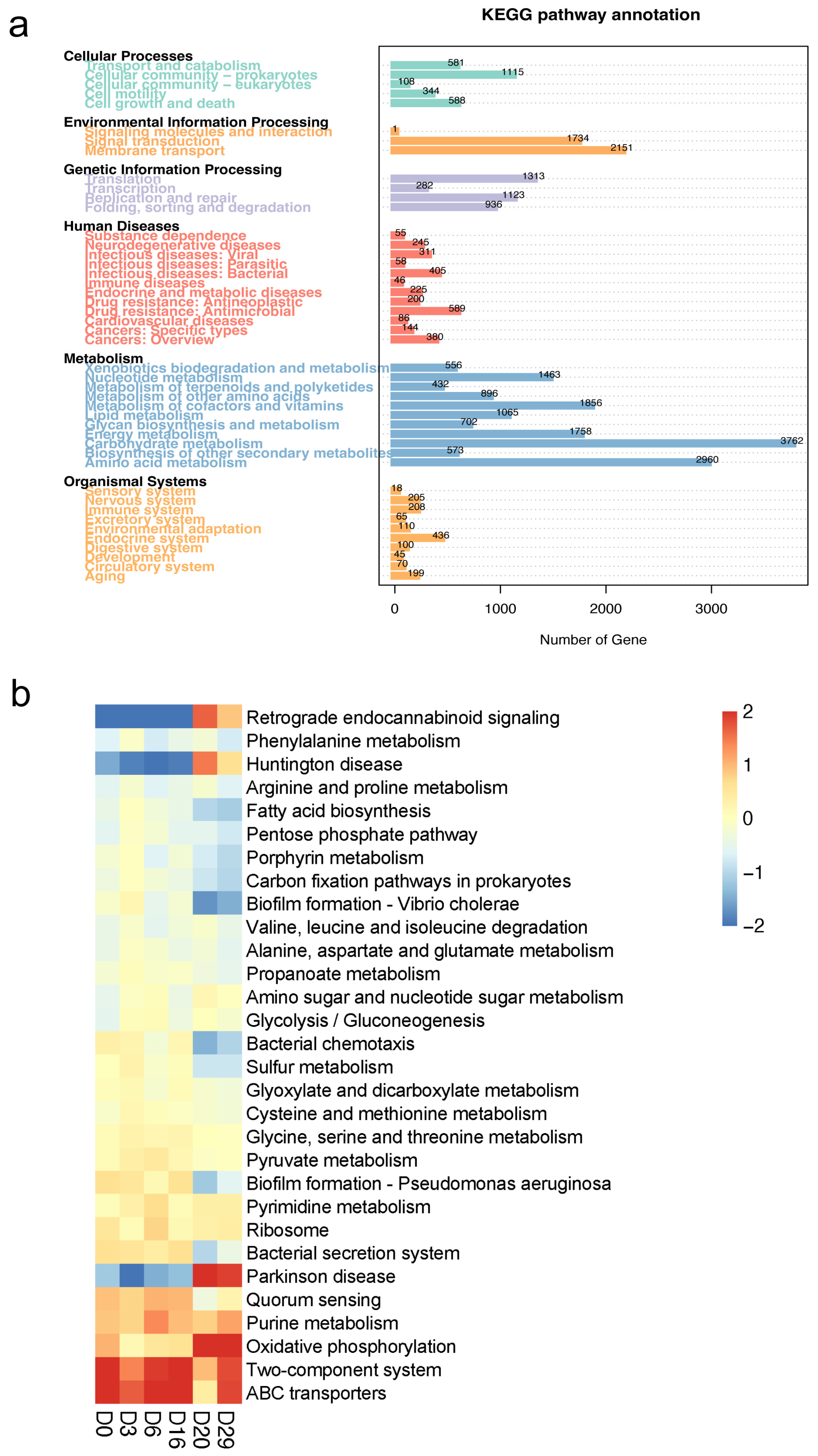
3.2. Functional Characteristics of Daqu Microorganisms at Different Fermentation Stages
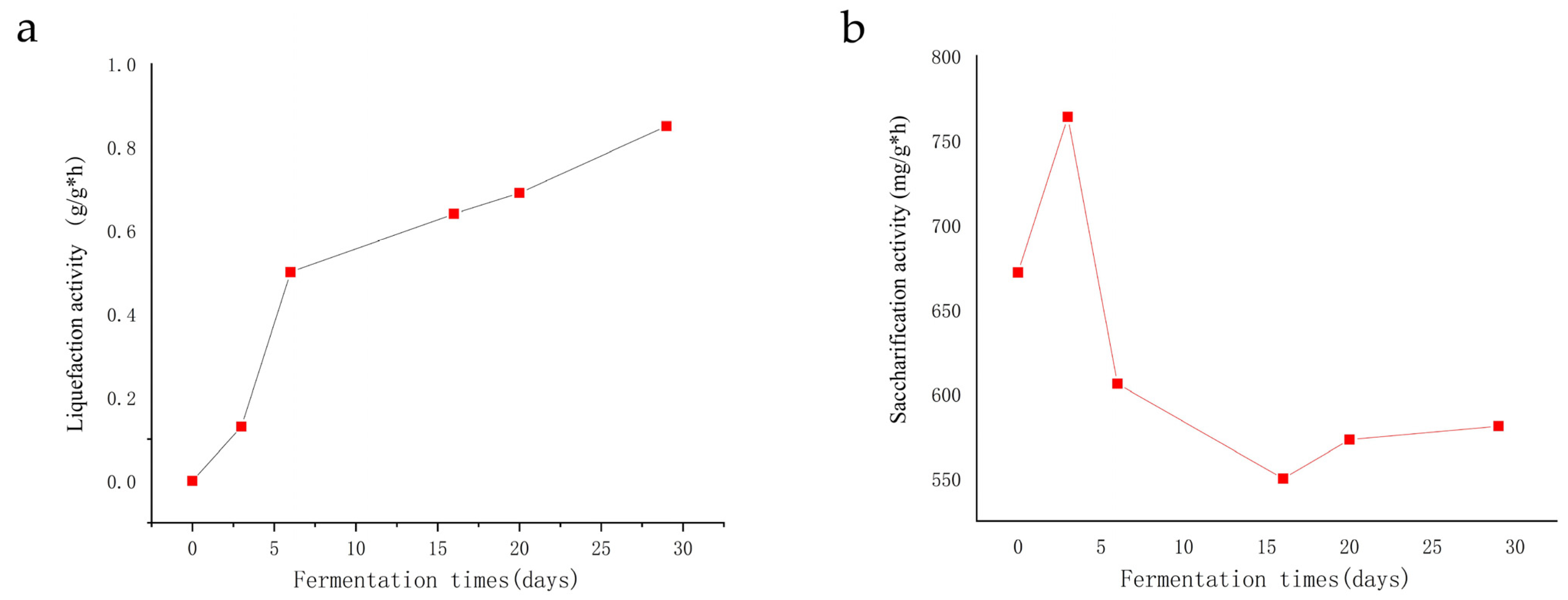
3.3. Changes in Enzyme Activity
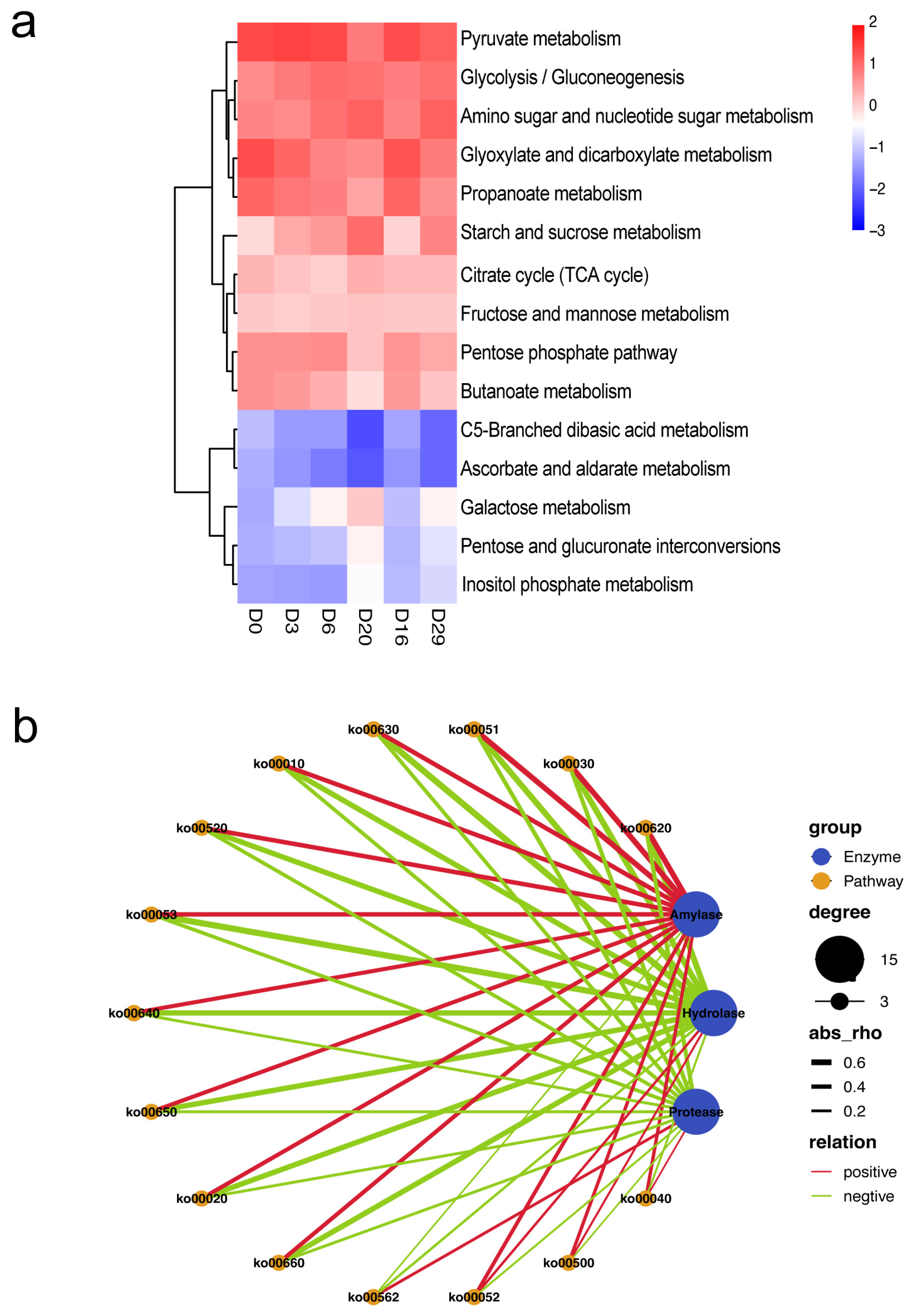
3.4. Correlation between Extracellular Enzyme Activity and Functional Genes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Du, Y.K.; Xin, W.; Xia, Y.; Zhu, M.; Qin, J.L.; Pan, Z.F.; Wu, R.F.; Luo, G.R.; Wu, P.S.; Wu, Z.Y.; et al. Analysis of fermentation control factors on volatile compounds of primary microorganisms in Jiang-flavor Daqu. J. Food Biochem. 2022, 46, e14277. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Tong, Q.; Guang, J. Yeast dynamics and changes in volatile compounds during the fermentation of the traditional Chinese strong-flavor Daqu. LWT 2019, 106, 57–63. [Google Scholar] [CrossRef]
- Awasthi, M.K.; Wong, J.W.C.; Kumar, S.; Awasthi, S.K.; Wang, Q.; Wang, M.; Ren, X.; Zhao, J.; Chen, H.; Zhang, Z. Biodegradation of food waste using microbial cultures producing thermostable α-amylase and cellulase under different pH and temperature. Bioresour. Technol. 2018, 248, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.H.; Yang, F.; Sahu, S.K.; Luo, R.Y.; Liao, S.L.; Wang, H.Y.; Jin, T.; Wang, L.; Zhang, P.F.; Liu, X.; et al. Deciphering the Composition and Functional Profile of the Microbial Communities in Chinese Moutai Liquor Starters. Front. Microbiol. 2019, 10, 1540. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Luo, Y.; García-Palacios, P.; Cao, J.; Dacal, M.; Zhou, X.; Li, J.; Xia, J.; Niu, S.; Yang, H.; et al. Differential responses of carbon-degrading enzyme activities to warming: Implications for soil respiration. Glob. Chang. Biol. 2018, 24, 4816–4826. [Google Scholar] [CrossRef]
- Ma, S.; Luo, H.; Zhao, D.; Qiao, Z.; Zheng, J.; An, M.; Huang, D. Environmental factors and interactions among microorganisms drive microbial community succession during fermentation of Nongxiangxing daqu. Bioresour. Technol. 2022, 345, 126549. [Google Scholar] [CrossRef]
- Guan, T.; Lin, Y.; Chen, K.; Ou, M.; Zhang, J. Physicochemical Factors Affecting Microbiota Dynamics During Traditional Solid-State Fermentation of Chinese Strong-Flavor Baijiu. Front. Microbiol. 2020, 11, 2090. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Ping, Q.; Zheng, M.; Dai, X.; Li, Y. Metagenomic characterization of the enhanced performance of anaerobic fermentation of waste activated sludge with CaO(2) addition at ambient temperature: Fatty acid biosynthesis metabolic pathway and CAZymes. Water Res. 2020, 170, 115309. [Google Scholar] [CrossRef]
- Chen, L.; Yi, Z.; Fang, Y.; Jin, Y.; He, K.; Xiao, Y.; Zhao, D.; Luo, H.; He, H.; Sun, Q.; et al. Biochemical and synergistic properties of a novel alpha-amylase from Chinese nong-flavor Daqu. Microb. Cell Fact. 2021, 20, 80. [Google Scholar] [CrossRef]
- Ma, S.; Shang, Z.; Chen, J.; Shen, Y.; Li, Z.; Huang, D.; Luo, H. Differences in structure, volatile metabolites, and functions of microbial communities in Nongxiangxing daqu from different production areas. LWT 2022, 166, 113784. [Google Scholar] [CrossRef]
- Verce, M.; De Vuyst, L.; Weckx, S. Shotgun Metagenomics of a Water Kefir Fermentation Ecosystem Reveals a Novel Oenococcus Species. Front. Microbiol. 2019, 10, 479. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Huang, J.; Wu, C.; Jin, Y.; Zhou, R. Bioturbation effect of fortified Daqu on microbial community and flavor metabolite in Chinese strong-flavor liquor brewing microecosystem. Food Res. Int. 2020, 129, 108851. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Mao, X.; Liu, D.; Ning, X.Q.; Shen, Y.; Chen, B.; Nie, H.F.; Huang, D.; Luo, H.B. Comparative Analysis of Physicochemical Properties and Microbial Composition in High-Temperature Daqu With Different Colors. Front. Microbiol. 2020, 11, 588117. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Xu, W.; Dou, W.; Xu, H.; Xu, Z. Comparison of total microbial DNA extraction methods from solid-culture of Zhenjiang vinegar. Acta Microbiol. Sin. 2010, 50, 119–125. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Stackebrandt, E.; Goebel, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Bacteriol. 1994, 44, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Lin, W.; Liu, X.; Wang, X.; Gan, X.; Luo, L.; Lin, W.T. Effect of bioaugmented inoculation on microbiota dynamics during solid-state fermentation of Daqu starter using autochthonous of Bacillus, Pediococcus, Wickerhamomyces and Saccharomycopsis. Food Microbiol. 2017, 61, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Xi, B.; He, X.; Dang, Q.; Yang, T.; Li, M.; Wang, X.; Li, D.; Tang, J. Effect of multi-stage inoculation on the bacterial and fungal community structure during organic municipal solid wastes composting. Bioresour. Technol. 2015, 196, 399–405. [Google Scholar] [CrossRef]
- Jiang, L.; Su, W.; Mu, Y.; Mu, Y. Major Metabolites and Microbial Community of Fermented Black Glutinous Rice Wine with Different Starters. Front. Microbiol. 2020, 11, 593. [Google Scholar] [CrossRef]
- Hou, X.; Hui, M.; Sun, Z.; Li, X.; Shi, X.; Xiao, R.; Wang, J.; Pan, C.; Li, R. Comparative analysis of the microbiotas and physicochemical properties inside and outside medium-temperature Daqu during the fermentation and storage. Front. Microbiol. 2022, 13, 934696. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, J.; Xie, J.; Zhao, D.; Qiao, Z.W.; Huang, D.; Luo, H.B. Effects of environmental factors on the microbial community changes during medium-high temperature Daqu manufacturing. Food Res. Int. 2022, 153, 110955. [Google Scholar] [CrossRef]
- Alonso, S.; Rendueles, M.; Díaz, M. Simultaneous production of lactobionic and gluconic acid in cheese whey/glucose co-fermentation by Pseudomonas taetrolens. Bioresour. Technol. 2015, 196, 314–323. [Google Scholar] [CrossRef]
- Chacón-Vargas, K.; Torres, J.; Giles-Gómez, M.; Escalante, A.; Gibbons, J.G. Genomic profiling of bacterial and fungal communities and their predictive functionality during pulque fermentation by whole-genome shotgun sequencing. Sci. Rep. 2020, 10, 15115. [Google Scholar] [CrossRef]
- Fan, Y.; Huang, X.; Chen, J.; Han, B. Formation of a Mixed-Species Biofilm Is a Survival Strategy for Unculturable Lactic Acid Bacteria and Saccharomyces cerevisiae in Daqu, a Chinese Traditional Fermentation Starter. Front. Microbiol. 2020, 11, 138. [Google Scholar] [CrossRef] [PubMed]
- Thanh, V.N.; Mai, T.L.; Tuan, D.A. Microbial diversity of traditional Vietnamese alcohol fermentation starters (banh men) as determined by PCR-mediated DGGE. Int. J. Food Microbiol. 2008, 128, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, R.; Ishii, K.; Maeda, I.; Kozaki, T.; Iwabuchi, K.; Saito, T. Evaluation of bacterial communities by bacteriome analysis targeting 16S rRNA genes and quantitative analysis of ammonia monooxygenase gene in different types of compost. J. Biosci. Bioeng. 2016, 121, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, R.; Shen, Y.; Lian, B. The Potential Correlation Between Bacterial Sporulation and the Characteristic Flavor of Chinese Maotai Liquor. Front. Microbiol. 2018, 9, 1435. [Google Scholar] [CrossRef]
- Nakamura, S.; Machida, K.; Ohtsubo, K. Search for cell-wall-degrading enzymes of world-wide rice grains by PCR and their effects on the palatability of rice. Biosci. Biotechnol. Biochem. 2012, 76, 1645–1654. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Zhang, K.Z.; Cao, X.Z.; Yang, J.G. Effects of Saccharomycopsis fibuligera and Saccharomyces cerevisiae inoculation on small fermentation starters in Sichuan-style Xiaoqu liquor. Food Res. Int. 2020, 137, 109425. [Google Scholar] [CrossRef]
- Moore, J.A.M.; Anthony, M.A.; Pec, G.J.; Trocha, L.K.; Trzebny, A.; Geyer, K.M.; van Diepen, L.T.A.; Frey, S.D. Fungal community structure and function shifts with atmospheric nitrogen deposition. Glob. Chang. Biol. 2021, 27, 1349–1364. [Google Scholar] [CrossRef]
- Guo, X.; Gao, Q.; Yuan, M.; Wang, G.; Zhou, X.; Feng, J.; Shi, Z.; Hale, L.; Wu, L.; Zhou, A.; et al. Gene-informed decomposition model predicts lower soil carbon loss due to persistent microbial adaptation to warming. Nat. Commun. 2020, 11, 4897. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.-T.; Deng, Y.-K.; Zou, Y.-F.; Han, B.-L.; Pu, J.-Z.; Rao, J.-Q.; Huang, D.; Luo, H.-B. Linking Microbial Functional Gene Abundance and Daqu Extracellular Enzyme Activity: Implications for Carbon Metabolism during Fermentation. Foods 2022, 11, 3623. https://doi.org/10.3390/foods11223623
Zhang Y-T, Deng Y-K, Zou Y-F, Han B-L, Pu J-Z, Rao J-Q, Huang D, Luo H-B. Linking Microbial Functional Gene Abundance and Daqu Extracellular Enzyme Activity: Implications for Carbon Metabolism during Fermentation. Foods. 2022; 11(22):3623. https://doi.org/10.3390/foods11223623
Chicago/Turabian StyleZhang, Yu-Ting, Yu-Ke Deng, Yong-Fang Zou, Bao-Lin Han, Ji-Zhou Pu, Jia-Quan Rao, Dan Huang, and Hui-Bo Luo. 2022. "Linking Microbial Functional Gene Abundance and Daqu Extracellular Enzyme Activity: Implications for Carbon Metabolism during Fermentation" Foods 11, no. 22: 3623. https://doi.org/10.3390/foods11223623