
The Application of Metagenomics to Study Microbial Communities and Develop Desirable Traits in Fermented Foods

 , ,
, ,
Abstract
:1. Introduction
2. Microbial DNA Extraction
3. Host Depletion
4. Differentiating between Live and Dead Bacteria
5. Sequencing Platforms
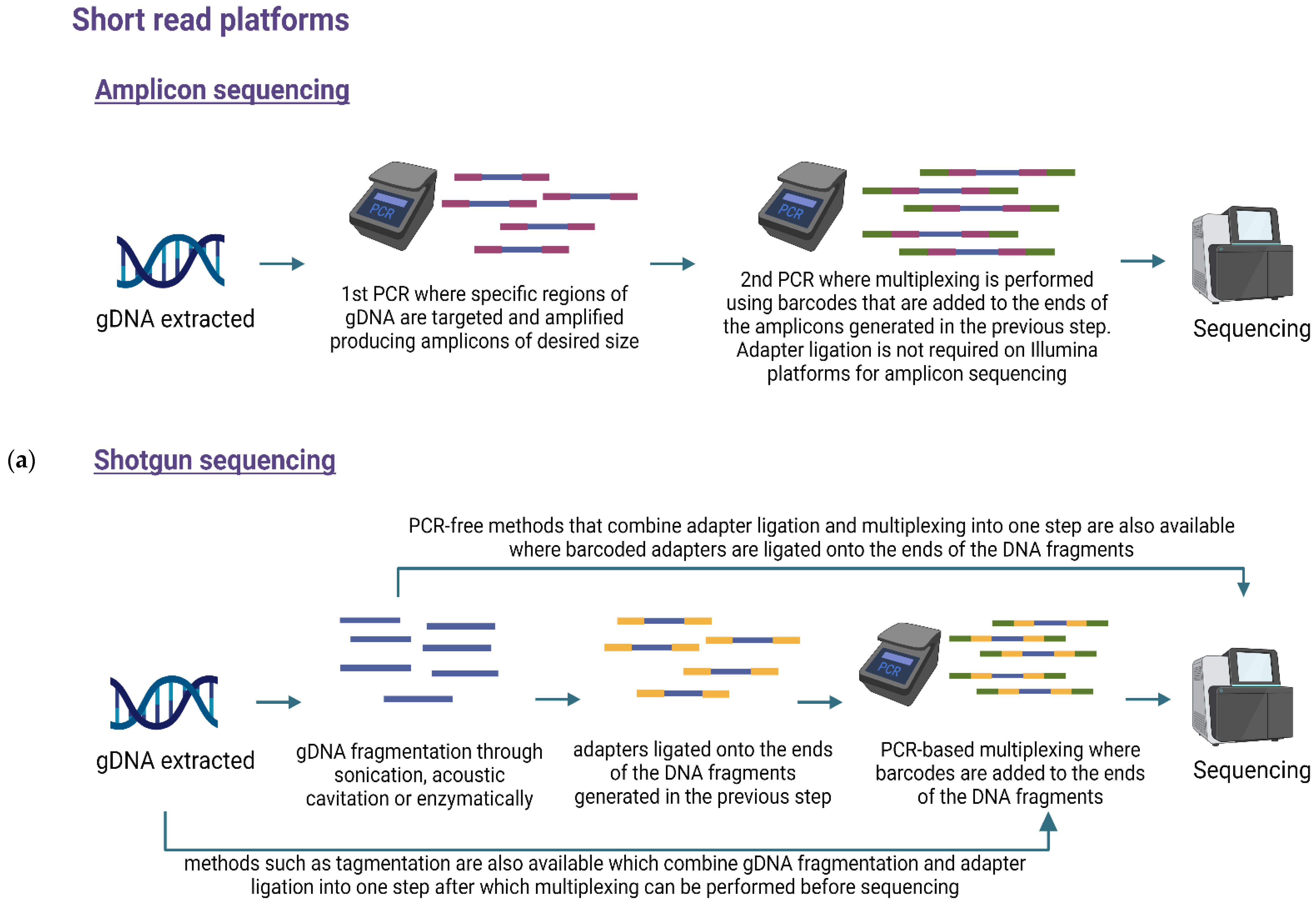
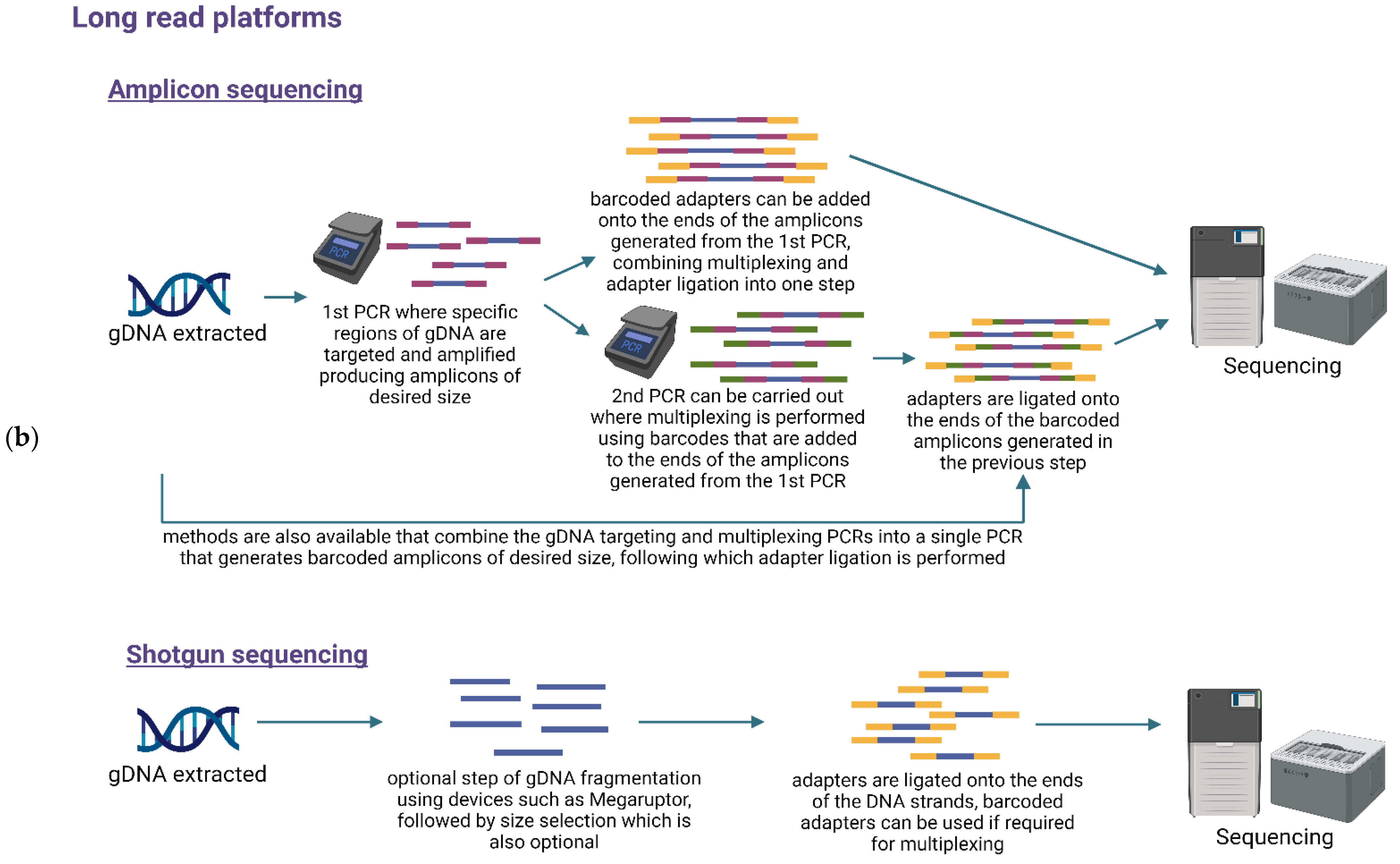
6. Library Preparation and Multiplexing
7. Sequencing Methods
7.1. Targeted or Amplicon-Based Sequencing
7.2. Untargeted or Shotgun Metagenomic Sequencing

{kind=link}
{kind=link}
{kind=link}
| Factors | Amplicon Sequencing | Shotgun Sequencing | References |
|---|---|---|---|
| Cost and speed of analysis | Advantages: (1) Requires less sequencing per sample (2) Faster and financially feasible when many samples are to be analysed or when only taxonomic profiling is required (3) Bioinformatic analysis is relatively easier with many GUI-based software freely available, thereby reducing computational costs Disadvantages: Less data/information obtained on microbial communities | Advantages: Untargeted sequencing of metagenomic samples generates large amounts of data useful for functional profiling Disadvantages: Analysis methods involved can be time consuming and computationally heavy often requiring complex and expensive network infrastructures | [133,143] |
| Library prep | Advantages: (1) PCR-involving library preparation steps can increase template DNA numbers for low microbial populations, thereby improving their representation in the sequencing data generated (2) Improves microbial sequencing from host-derived samples Disadvantages: (1) PCR related biases apply such as differences in: (i) ease or rate of amplification (ii) variation in GC content (iii) copy number of 16S gene (iv) sequence variation between 16S copies within a bacterial genome (v) selection of targeted region (2) More susceptible to biasing microbial community representations in the presence of contaminating microbial strains such as those introduced into libraries from kit reagents used | Advantages: (1) PCR related biases also apply, but can be reduced using PCR-free library prep methods (2) Less susceptible to biasing microbial community representations in the presence of kitome contaminants Disadvantages: Host-derived samples need to be depleted for host DNA before sequencing, if not sequencing resources will be wasted on sequencing large proportions of host DNA and can lead to under/mis-representations of microbial communities | [16,72,82,118] |
| Microbial community profiling | Advantages: (1) Taxonomic classification possible for which computational processing and analysis is relatively simple and quick (2) For functional classification tools such as PICURSt2 and Tax4Fun exist that functionally assign species detected in a community through metabarcoding to predict microbial functional abilities Disadvantages: Functional profiles can only be predicted from amplicon data but is difficult for highly diverse and complex samples. The resulting profiles are often of low resolution and do not account for mobile genetic elements such as Horizontal Gene Transfers (HGT) and pathogenicity islands | Advantages: (1) The large amounts of sequencing data generated through shotgun metagenomics allows better functional profiling than metabarcoding (2) Better resolution of microbial community, even at strain level, can be obtained Disadvantages: (1) The extent and quality of the functional profiles obtained depend on the complexity of the sample community and the sequencing depth (2) Computational analysis is time consuming and requires complex network infrastructure to be set up and maintained which is expensive | [19,97,133,159,160] |
| Detection and classification of previously unidentified or uncharacterised genomes in a community | Disadvantages: Dependent on existing databases, making classification of new species and strains difficult | Advantages: Performance of de novo assembly allows characterisation of new species and strains and their addition to databases Disadvantages: MAG assembly for new species and strains can be very difficult for low abundance microbial populations and highly diverse microbial communities | [125,144] |
| Fungal or viral profiling | Advantages: (1) ITS-based fungal metabarcoding is relatively well characterized (2) PCR-based library prep can improve sequencing of low abundance viral microbial community members Disadvantages: (1) Requires different primers for fungal and viral community members and cannot be identified from a single library (2) PCR-based approaches for viral sequencing is restricted to similar or closely related viral families and can fail to detect new viral families | Advantages: Bacterial, fungal and viral sequences can be identified from a single library Disadvantages: (1) Fungal sub-populations or secondary symbionts are difficult to sequence (2) Only DNA-encoded viruses can be identified | [161,162,163,164] |
| Extra-chromosomal DNA profiling | Disadvantages: Plasmidome study is not possible | Advantages: Plasmidomes can be characterised along with gDNA Disadvantages: It is difficult to extract plasmid and genomic DNA together, and to computationally process and assemble reads. However, Hi-C approaches developed are allowing the linkage of plasmids to their carriage strains | [165,166,167] |
8. New Technologies
8.1. Synthetic Long Read (SLR) Sequencing
8.2. Hi-C
9. Applications of Metagenomics in the Fermented Food Industry
| Area | Application | References |
|---|---|---|
| Health promotion | Screening for health promoting bacteria Understanding the gut-brain axis Identifying prebiotics and their effect on host gut microbiota and health | [24,187,195] [222,223] [224,225,226,227] |
| Characterising fermentations | Organoleptic quality assessment through fermentation microbiome and volatile profiling Bacteriophage:
| [6,162,228,229,230,231,232] [233,234,235] [5,236] |
| Food safety | Detection and prediction of foodborne pathogens and spoilage microbes Screening for bacteriocin gene clusters Checking for the presence of antibiotic resistance genes (ARGs) | [176,237,238] [239,240,241] [185,242,243] |
| Food fraud | Fingerprinting plant, animal and microbial components of food, determining food authenticity, and detection of contaminants and adulterants | [244,245,246,247] |
| Production analysis | Accessing the effect of the following factors on fermentations:
| [101,248,249,250,251,252] [230] [204,230,253] |
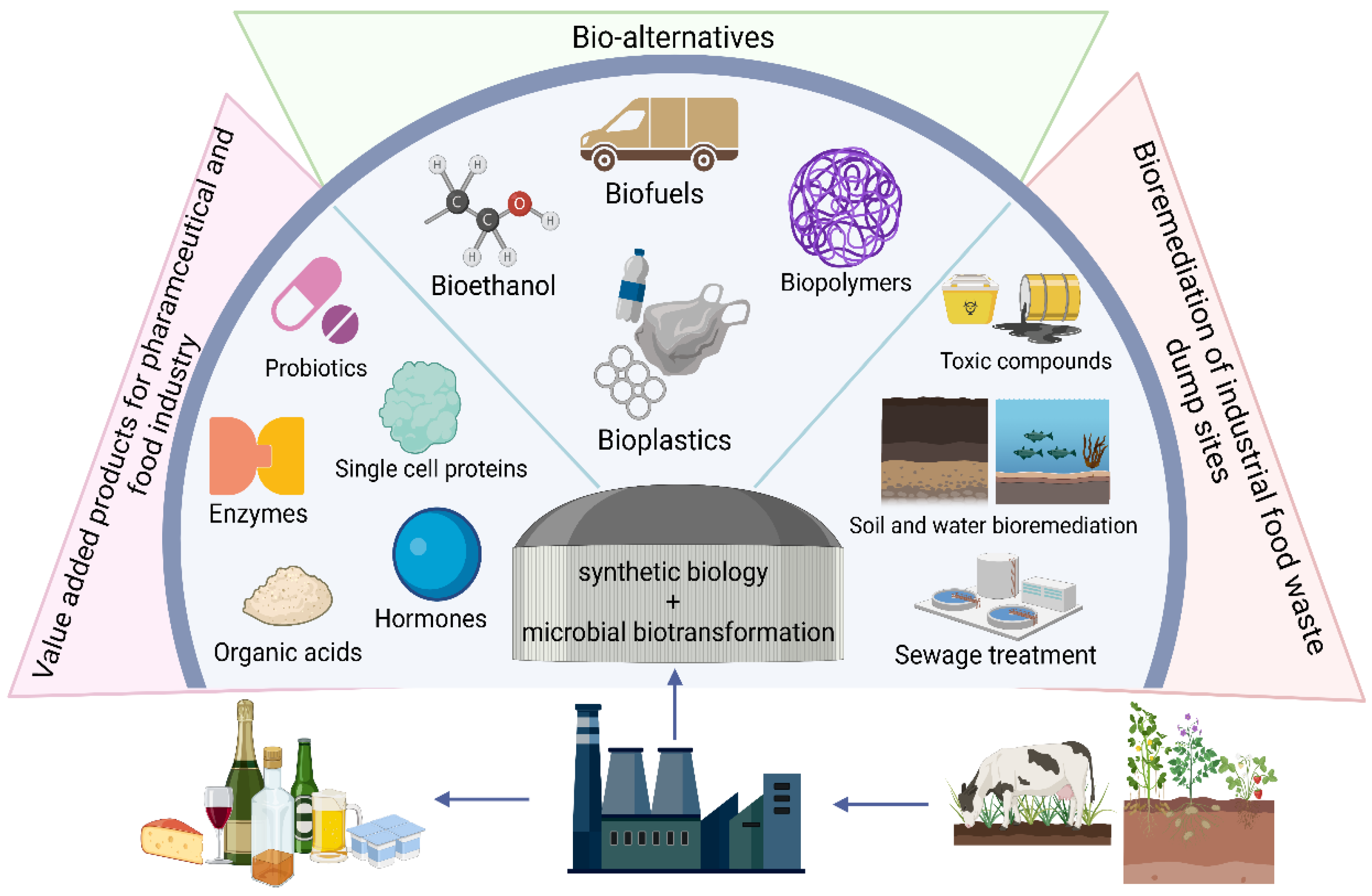
10. Synthetic Biology
11. Food Waste Valorisation
12. Future of Molecular Biology in Fermented Foods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tamang, J.P.; Cotter, P.D.; Endo, A.; Han, N.S.; Kort, R.; Liu, S.Q.; Mayo, B.; Westerik, N.; Hutkins, R. Fermented Foods in a Global Age: East Meets West. Compr. Rev. Food Sci. Food Saf. 2020, 19, 184–217. [Google Scholar] [CrossRef] [Green Version]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The Gut Microbiota–Brain Axis in Behaviour and Brain Disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Obafemi, Y.D.; Oranusi, S.U.; Ajanaku, K.O.; Akinduti, P.A.; Leech, J.; Cotter, P.D. African Fermented Foods: Overview, Emerging Benefits, and Novel Approaches to Microbiome Profiling. Npj Sci. Food 2022, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, D.; Su, X.; Duan, S.; Wan, J.; Yuan, W.; Liu, B.; Ma, Y.; Pan, Y. An Integrated Metagenomics/Metaproteomics Investigation of the Microbial Communities and Enzymes in Solid-State Fermentation of Pu-Erh Tea. Sci. Rep. 2015, 5, 10117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somerville, V.; Berthoud, H.; Schmidt, R.S.; Bachmann, H.-P.; Meng, Y.H.; Fuchsmann, P.; von Ah, U.; Engel, P. Functional Strain Redundancy and Persistent Phage Infection in Swiss Hard Cheese Starter Cultures. ISME J. 2022, 16, 388–399. [Google Scholar] [CrossRef]
- Ferrocino, I.; Rantsiou, K.; Cocolin, L. Investigating Dairy Microbiome: An Opportunity to Ensure Quality, Safety and Typicity. Curr. Opin. Biotechnol. 2022, 73, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Dysvik, A.; La Rosa, S.L.; De Rouck, G.; Rukke, E.-O.; Westereng, B.; Wicklund, T. Microbial Dynamics in Traditional and Modern Sour Beer Production. Appl. Environ. Microbiol. 2020, 86, e00566-20. [Google Scholar] [CrossRef] [PubMed]
- Mateus, D.; Sousa, S.; Coimbra, C.; Rogerson, S.F.; Simões, J. Identification and Characterization of Non-Saccharomyces Species Isolated from Port Wine Spontaneous Fermentations. Foods 2020, 9, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.S.; Lee, S.H.; Ahn, S.W.; Kim, J.Y.; Rhee, J.-K.; Roh, S.W. Effects of the Main Ingredients of the Fermented Food, Kimchi, on Bacterial Composition and Metabolite Profile. Food Res. Int. 2021, 149, 110668. [Google Scholar] [CrossRef] [PubMed]
- Tlais, A.Z.A.; Lemos Junior, W.J.F.; Filannino, P.; Campanaro, S.; Gobbetti, M.; Di Cagno, R. How Microbiome Composition Correlates with Biochemical Changes during Sauerkraut Fermentation: A Focus on Neglected Bacterial Players and Functionalities. Microbiol. Spectr. 2022, 10, e00168-22. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Tamang, J.P. Changes in Microbial Communities and Their Predictive Functionalities during Fermentation of Toddy, an Alcoholic Beverage of India. Microbiol. Res. 2021, 248, 126769. [Google Scholar] [CrossRef] [PubMed]
- Ashaolu, T.J.; Khalifa, I.; Mesak, M.A.; Lorenzo, J.M.; Farag, M.A. A Comprehensive Review of the Role of Microorganisms on Texture Change, Flavor and Biogenic Amines Formation in Fermented Meat with Their Action Mechanisms and Safety. Crit Rev. Food Sci. Nutr. 2021, 1–18. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, L.; Wen, R.; Chen, Q.; Kong, B. Role of Lactic Acid Bacteria in Flavor Development in Traditional Chinese Fermented Foods: A Review. Crit. Rev. Food Sci. Nutr. 2022, 62, 2741–2755. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Wu, R.; Wu, C.; Wang, R.; Wu, J.; Shi, H. Multi-Omics Approaches to Elucidate the Role of Interactions between Microbial Communities in Cheese Flavor and Quality. Food Rev. Int. 2022, 1–13. [Google Scholar] [CrossRef]
- Techtmann, S.M.; Hazen, T.C. Metagenomic Applications in Environmental Monitoring and Bioremediation. J. Ind. Microbiol. Biotechnol. 2016, 43, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A Review of Methods and Databases for Metagenomic Classification and Assembly. Brief Bioinform. 2019, 20, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Prayogo, F.A.; Budiharjo, A.; Kusumaningrum, H.P.; Wijanarka, W.; Suprihadi, A.; Nurhayati, N. Metagenomic Applications in Exploration and Development of Novel Enzymes from Nature: A Review. J. Genet. Eng. Biotechnol. 2020, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A Review on the Applications of Next Generation Sequencing Technologies as Applied to Food-Related Microbiome Studies. Front. Microbiol. 2017, 8, 1829. [Google Scholar]
- De Filippis, F.; Valentino, V.; Alvarez-Ordóñez, A.; Cotter, P.D.; Ercolini, D. Environmental Microbiome Mapping as a Strategy to Improve Quality and Safety in the Food Industry. Curr. Opin. Food Sci. 2021, 38, 168–176. [Google Scholar] [CrossRef]
- Yap, M.; Feehily, C.; Walsh, C.J.; Fenelon, M.; Murphy, E.F.; McAuliffe, F.M.; van Sinderen, D.; O’Toole, P.W.; O’Sullivan, O.; Cotter, P.D. Evaluation of Methods for the Reduction of Contaminating Host Reads When Performing Shotgun Metagenomic Sequencing of the Milk Microbiome. Sci. Rep. 2020, 10, 21665. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Zepeda, A.; Sanchez-Flores, A.; Quirasco Baruch, M. Metagenomic Analysis of a Mexican Ripened Cheese Reveals a Unique Complex Microbiota. Food Microbiol. 2016, 57, 116–127. [Google Scholar] [CrossRef]
- Diaz, M.; Kellingray, L.; Akinyemi, N.; Adefiranye, O.O.; Olaonipekun, A.B.; Bayili, G.R.; Ibezim, J.; du Plessis, A.S.; Houngbédji, M.; Kamya, D.; et al. Comparison of the Microbial Composition of African Fermented Foods Using Amplicon Sequencing. Sci. Rep. 2019, 9, 13863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, W.; Humblot, C.; Guyot, J.-P. Genetic Screening of Functional Properties of Lactic Acid Bacteria in a Fermented Pearl Millet Slurry and in the Metagenome of Fermented Starchy Foods. Appl. Environ. Microbiol. 2011, 77, 8722–8734. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Cai, J.; Zhang, C.; Guo, Z.; Lu, W.; Yang, B.; Tian, F.-W.; Liu, X.-M.; Zhang, H.; Chen, W. Suitability of Various DNA Extraction Methods for a Traditional Chinese Paocai System. Bioengineered 2017, 8, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Keisam, S.; Romi, W.; Ahmed, G.; Jeyaram, K. Quantifying the Biases in Metagenome Mining for Realistic Assessment of Microbial Ecology of Naturally Fermented Foods. Sci. Rep. 2016, 6, 34155. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, J.P.; Carpenter, C.S.; Martino, C.; Salido, R.A.; Minich, J.J.; Bryant, M.; Sanders, K.; Schwartz, T.; Humphrey, G.; Swafford, A.D.; et al. A Comparison of Six DNA Extraction Protocols for 16S, ITS, and Shotgun Metagenomic Sequencing of Microbial Communities. BioTechniques 2022, 73, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.pacb.com/wp-content/uploads/Technical-Note-Preparing-DNA-for-PacBio-HiFi-Sequencing-Extraction-and-Quality-Control.pdf (accessed on 29 July 2022).
- Cai, W.; Wang, Y.; Hou, Q.; Zhang, Z.; Tang, F.; Shan, C.; Yang, X.; Guo, Z. PacBio Sequencing Combined with Metagenomic Shotgun Sequencing Provides Insight into the Microbial Diversity of Zha-Chili. Food BioSci. 2021, 40, 100884. [Google Scholar] [CrossRef]
- Quijada, N.M.; Schmitz-Esser, S.; Zwirzitz, B.; Guse, C.; Strachan, C.R.; Wagner, M.; Wetzels, S.U.; Selberherr, E.; Dzieciol, M. Austrian Raw-Milk Hard-Cheese Ripening Involves Successional Dynamics of Non-Inoculated Bacteria and Fungi. Foods 2020, 9, 1851. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhao, F.; Hou, Q.; Wang, J.; Li, M.; Sun, Z. PacBio Sequencing Reveals Bacterial Community Diversity in Cheeses Collected from Different Regions. J. Dairy Sci. 2020, 103, 1238–1249. [Google Scholar] [CrossRef]
- Jones, A.; Torkel, C.; Stanley, D.; Nasim, J.; Borevitz, J.; Schwessinger, B. High-Molecular Weight DNA Extraction, Clean-up and Size Selection for Long-Read Sequencing. PLoS ONE 2021, 16, e0253830. [Google Scholar] [CrossRef] [PubMed]
- Mayjonade, B.; Gouzy, J.; Donnadieu, C.; Pouilly, N.; Marande, W.; Callot, C.; Langlade, N.; Muños, S. Extraction of High-Molecular-Weight Genomic DNA for Long-Read Sequencing of Single Molecules. BioTechniques 2016, 61, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Ganda, E.; Beck, K.L.; Haiminen, N.; Silverman, J.D.; Kawas, B.; Cronk, B.D.; Anderson, R.R.; Goodman, L.B.; Wiedmann, M. DNA Extraction and Host Depletion Methods Significantly Impact and Potentially Bias Bacterial Detection in a Biological Fluid. mSystems 2021, 6, e00619-21. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Song, E.-J.; Kim, S.H.; Lee, J.; Nam, Y.-D. Comparison of DNA Extraction Methods for Human Gut Microbial Community Profiling. Syst. Appl. Microbiol. 2018, 41, 151–157. [Google Scholar] [CrossRef]
- Werner, D.; Acharya, K.; Blackburn, A.; Zan, R.; Plaimart, J.; Allen, B.; Mgana, S.M.; Sabai, S.M.; Halla, F.F.; Massawa, S.M.; et al. MinION Nanopore Sequencing Accelerates Progress towards Ubiquitous Genetics in Water Research. Water 2022, 14, 2491. [Google Scholar] [CrossRef]
- Tighe, S.; Afshinnekoo, E.; Rock, T.M.; McGrath, K.; Alexander, N.; McIntyre, A.; Ahsanuddin, S.; Bezdan, D.; Green, S.J.; Joye, S.; et al. Genomic Methods and Microbiological Technologies for Profiling Novel and Extreme Environments for the Extreme Microbiome Project (XMP). J. Biomol. Tech. 2017, 28, 31–39. [Google Scholar] [CrossRef]
- Feehery, G.R.; Yigit, E.; Oyola, S.O.; Langhorst, B.W.; Schmidt, V.T.; Stewart, F.J.; Dimalanta, E.T.; Amaral-Zettler, L.A.; Davis, T.; Quail, M.A.; et al. A Method for Selectively Enriching Microbial DNA from Contaminating Vertebrate Host DNA. PLoS ONE 2013, 8, e76096. [Google Scholar] [CrossRef] [PubMed]
- McHugh, A.J.; Feehily, C.; Hill, C.; Cotter, P.D. Detection and Enumeration of Spore-Forming Bacteria in Powdered Dairy Products. Front. Microbiol. 2017, 8, 109. [Google Scholar] [PubMed] [Green Version]
- Schuele, L.; Cassidy, H.; Peker, N.; Rossen, J.W.A.; Couto, N. Future Potential of Metagenomics in Microbiology Laboratories. Expert Rev. Mol. Diagn. 2021, 21, 1273–1285. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Lau, H.C.-H.; Yu, J. Metagenomic Sequencing for Microbial DNA in Human Samples: Emerging Technological Advances. Int. J. Mol. Sci. 2022, 23, 2181. [Google Scholar] [CrossRef]
- McHugh, A.J.; Feehily, C.; Fenelon, M.A.; Gleeson, D.; Hill, C.; Cotter, P.D. Tracking the Dairy Microbiota from Farm Bulk Tank to Skimmed Milk Powder. mSystems 2020, 5, e00226-20. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Marques, J.; Hout, A.; Ferreira, R.M.; Weber, M.; Pinto-Ribeiro, I.; van Doorn, L.-J.; Knetsch, C.W.; Figueiredo, C. Impact of Host DNA and Sequencing Depth on the Taxonomic Resolution of Whole Metagenome Sequencing for Microbiome Analysis. Front. Microbiol. 2019, 10, 1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubiola, S.; Chiesa, F.; Dalmasso, A.; Di Ciccio, P.; Civera, T. Detection of Antimicrobial Resistance Genes in the Milk Production Environment: Impact of Host DNA and Sequencing Depth. Front. Microbiol. 2020, 11, 1983. [Google Scholar] [CrossRef] [PubMed]
- Heravi, F.S.; Zakrzewski, M.; Vickery, K.; Hu, H. Host DNA Depletion Efficiency of Microbiome DNA Enrichment Methods in Infected Tissue Samples. J. Microbiol. Methods 2020, 170, 105856. [Google Scholar] [CrossRef] [PubMed]
- Marotz, C.A.; Sanders, J.G.; Zuniga, C.; Zaramela, L.S.; Knight, R.; Zengler, K. Improving Saliva Shotgun Metagenomics by Chemical Host DNA Depletion. Microbiome 2018, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Mo, L.; Yu, J.; Jin, H.; Hou, Q.; Yao, C.; Ren, D.; An, X.; Tsogtgerel, T.; Zhang, H. Investigating the Bacterial Microbiota of Traditional Fermented Dairy Products Using Propidium Monoazide with Single-Molecule Real-Time Sequencing. J. Dairy Sci. 2019, 102, 3912–3923. [Google Scholar] [CrossRef]
- Tantikachornkiat, M.; Sakakibara, S.; Neuner, M.; Durall, D.M. The Use of Propidium Monoazide in Conjunction with QPCR and Illumina Sequencing to Identify and Quantify Live Yeasts and Bacteria. Int. J. Food Microbiol. 2016, 234, 53–59. [Google Scholar] [CrossRef]
- Thoendel, M.; Jeraldo, P.R.; Greenwood-Quaintance, K.E.; Yao, J.Z.; Chia, N.; Hanssen, A.D.; Abdel, M.P.; Patel, R. Comparison of Microbial DNA Enrichment Tools for Metagenomic Whole Genome Sequencing. J. Microbiol. Methods 2016, 127, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Marquet, M.; Zöllkau, J.; Pastuschek, J.; Viehweger, A.; Schleußner, E.; Makarewicz, O.; Pletz, M.W.; Ehricht, R.; Brandt, C. Evaluation of Microbiome Enrichment and Host DNA Depletion in Human Vaginal Samples Using Oxford Nanopore’s Adaptive Sequencing. Sci. Rep. 2022, 12, 4000. [Google Scholar] [CrossRef]
- Erkus, O.; de Jager, V.C.L.; Geene, R.T.C.M.; van Alen-Boerrigter, I.; Hazelwood, L.; van Hijum, S.A.F.T.; Kleerebezem, M.; Smid, E.J. Use of Propidium Monoazide for Selective Profiling of Viable Microbial Cells during Gouda Cheese Ripening. Int. J. Food Microbiol. 2016, 228, 1–9. [Google Scholar] [CrossRef]
- Cangelosi, G.A.; Meschke, J.S. Dead or Alive: Molecular Assessment of Microbial Viability. Appl. Environ. Microbiol. 2014, 80, 5884–5891. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Tun, H.M.; Jahan, M.; Zhang, Z.; Kumar, A.; Dilantha Fernando, W.G.; Farenhorst, A.; Khafipour, E. Comparison of DNA-, PMA-, and RNA-Based 16S RRNA Illumina Sequencing for Detection of Live Bacteria in Water. Sci. Rep. 2017, 7, 5752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancabelli, L.; Milani, C.; Anzalone, R.; Alessandri, G.; Lugli, G.A.; Tarracchini, C.; Fontana, F.; Turroni, F.; Ventura, M. Free DNA and Metagenomics Analyses: Evaluation of Free DNA Inactivation Protocols for Shotgun Metagenomics Analysis of Human Biological Matrices. Front. Microbiol. 2021, 12, 749373. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; McFarland, A.G.; Young, V.B.; Hayden, M.K.; Hartmann, E.M. Toward Accurate and Robust Environmental Surveillance Using Metagenomics. Front. Genet. 2021, 12, 600111. [Google Scholar] [CrossRef]
- Emerson, J.B.; Adams, R.I.; Román, C.M.B.; Brooks, B.; Coil, D.A.; Dahlhausen, K.; Ganz, H.H.; Hartmann, E.M.; Hsu, T.; Justice, N.B.; et al. Schrödinger’s Microbes: Tools for Distinguishing the Living from the Dead in Microbial Ecosystems. Microbiome 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinson, L.F.; Trevenen, M.L.; Geddes, D.T. The Viable Microbiome of Human Milk Differs from the Metataxonomic Profile. Nutrients 2021, 13, 4445. [Google Scholar] [CrossRef]
- Chen, W. Demystification of Fermented Foods by Omics Technologies. Curr. Opin. Food Sci. 2022, 46, 100845. [Google Scholar] [CrossRef]
- Balkir, P.; Kemahlioglu, K.; Yucel, U. Foodomics: A New Approach in Food Quality and Safety. Trends Food Sci. Technol. 2021, 108, 49–57. [Google Scholar] [CrossRef]
- Okeke, E.S.; Ita, R.E.; Egong, E.J.; Udofia, L.E.; Mgbechidinma, C.L.; Akan, O.D. Metaproteomics Insights into Fermented Fish and Vegetable Products and Associated Microbes. Food Chem. 2021, 3, 100045. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, Z.; Miyao, S.; Zhang, W. Unraveling the Flavor Profile and Microbial Roles during Industrial Sichuan Radish Paocai Fermentation by Molecular Sensory Science and Metatranscriptomics. Food Biosci. 2022, 48, 101815. [Google Scholar] [CrossRef]
- Heather, J.M.; Chain, B. The Sequence of Sequencers: The History of Sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Allali, I.; Arnold, J.W.; Roach, J.; Cadenas, M.B.; Butz, N.; Hassan, H.M.; Koci, M.; Ballou, A.; Mendoza, M.; Ali, R.; et al. A Comparison of Sequencing Platforms and Bioinformatics Pipelines for Compositional Analysis of the Gut Microbiome. BMC Microbiol. 2017, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Gupta, U. Chapter 20—Next Generation Sequencing and Its Applications. In Animal Biotechnology, 2nd ed.; Verma, A.S., Singh, A., Eds.; Academic Press: Boston, MA, USA, 2020; pp. 395–421. ISBN 978-0-12-811710-1. [Google Scholar]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A Guide from Sampling to Data Analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Shao, Y.; Tian, L.; Flasch, D.A.; Mulder, H.L.; Edmonson, M.N.; Liu, Y.; Chen, X.; Newman, S.; Nakitandwe, J.; et al. Analysis of Error Profiles in Deep Next-Generation Sequencing Data. Genome Biol. 2019, 20, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoler, N.; Nekrutenko, A. Sequencing Error Profiles of Illumina Sequencing Instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, N.; Blanco, C.; Chen, I.A.; Seelig, B. PacBio Sequencing Output Increased through Uniform and Directional Fivefold Concatenation. Sci. Rep. 2021, 11, 18065. [Google Scholar] [CrossRef] [PubMed]
- Aunin, E.; Böhme, U.; Blake, D.; Dove, A.; Smith, M.; Corton, C.; Oliver, K.; Betteridge, E.; Quail, M.A.; McCarthy, S.A.; et al. The Complete Genome Sequence of Eimeria Tenella (Tyzzer 1929), a Common Gut Parasite of Chickens. Wellcome Open Res. 2021, 6, 225. [Google Scholar] [CrossRef]
- Kenny, N.J.; McCarthy, S.A.; Dudchenko, O.; James, K.; Betteridge, E.; Corton, C.; Dolucan, J.; Mead, D.; Oliver, K.; Omer, A.D.; et al. The Gene-Rich Genome of the Scallop Pecten Maximus. GigaScience 2020, 9, giaa037. [Google Scholar] [CrossRef]
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards Complete and Error-Free Genome Assemblies of All Vertebrate Species. Nature 2021, 592, 737–746. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-Generation Sequencing Technologies: An Overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Zhang, H.; Jain, C.; Aluru, S. A Comprehensive Evaluation of Long Read Error Correction Methods. BMC Genom. 2020, 21, 889. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohm, J.C.; Peters, P.; Stralis-Pavese, N.; Himmelbauer, H. Benchmarking of Long-Read Correction Methods. NAR Genom. Bioinform. 2020, 2, lqaa037. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S. Forensic Lab-on-a-Chip DNA Analysis. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2019. [Google Scholar]
- Lin, B.; Hui, J.; Mao, H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.pacb.com/wp-content/uploads/Baybayan-PAG-2017-Best-Practices-for-Whole-Genome-Sequencing-Using-the-Sequel-System.pdf (accessed on 18 July 2022).
- Kim, K.E.; Peluso, P.; Babayan, P.; Yeadon, P.J.; Yu, C.; Fisher, W.W.; Chin, C.-S.; Rapicavoli, N.A.; Rank, D.R.; Li, J.; et al. Long-Read, Whole-Genome Shotgun Sequence Data for Five Model Organisms. Sci. Data 2014, 1, 140045. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.pacb.com/wp-content/uploads/Procedure-Checklist-%E2%80%93-Preparing-10-kb-Library-Using-SMRTbell-Express-Template-Prep-Kit-2.0-for-Metagenomics-Shotgun-Sequencing.pdf (accessed on 29 July 2022).
- Kircher, M.; Sawyer, S.; Meyer, M. Double Indexing Overcomes Inaccuracies in Multiplex Sequencing on the Illumina Platform. Nucleic Acids Res. 2012, 40, e3. [Google Scholar] [CrossRef] [Green Version]
- Aigrain, L. Beginner’s Guide to next-Generation Sequencing. Biochem 2021, 43, 58–64. [Google Scholar] [CrossRef]
- MacConaill, L.E.; Burns, R.T.; Nag, A.; Coleman, H.A.; Slevin, M.K.; Giorda, K.; Light, M.; Lai, K.; Jarosz, M.; McNeill, M.S.; et al. Unique, Dual-Indexed Sequencing Adapters with UMIs Effectively Eliminate Index Cross-Talk and Significantly Improve Sensitivity of Massively Parallel Sequencing. BMC Genom. 2018, 19, 30. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.; Stanley, G.; Gulati, G.S.; Ezran, C.; Travaglini, K.J.; Wei, E.; Chan, C.K.F.; Nabhan, A.N.; Su, T.; Morganti, R.M.; et al. Index Switching Causes “Spreading-of-Signal” among Multiplexed Samples in Illumina HiSeq 4000 DNA Sequencing. BioRxiv 2017. [Google Scholar] [CrossRef]
- Ros-Freixedes, R.; Battagin, M.; Johnsson, M.; Gorjanc, G.; Mileham, A.J.; Rounsley, S.D.; Hickey, J.M. Impact of Index Hopping and Bias towards the Reference Allele on Accuracy of Genotype Calls from Low-Coverage Sequencing. Genet. Sel. Evol. 2018, 50, 64. [Google Scholar] [CrossRef] [Green Version]
- Van der Valk, T.; Vezzi, F.; Ormestad, M.; Dalén, L.; Guschanski, K. Index Hopping on the Illumina HiseqX Platform and Its Consequences for Ancient DNA Studies. Mol. Ecol. Resour. 2020, 20, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.S.; Vetsigian, K.H. Quality Filtering of Illumina Index Reads Mitigates Sample Cross-Talk. BMC Genom. 2016, 17, 876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenay-Greunke, Y.; Bohan, D.A.; Traugott, M.; Wallinger, C. Handling of Targeted Amplicon Sequencing Data Focusing on Index Hopping and Demultiplexing Using a Nested Metabarcoding Approach in Ecology. Sci. Rep. 2021, 11, 19510. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, X.; Zhang, W.; Wang, L.; Wang, J.; Xu, D.; Mei, Z.; Liu, Q.; Du, S.; Li, Z.; et al. Reliable Multiplex Sequencing with Rare Index Mis-Assignment on DNB-Based NGS Platform. BMC Genom. 2019, 20, 215. [Google Scholar] [CrossRef] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and Laboratory Contamination Can Critically Impact Sequence-Based Microbiome Analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.-C.; Needham, D.M.; Sieradzki, E.T.; Fuhrman, J.A. Taxon Disappearance from Microbiome Analysis Reinforces the Value of Mock Communities as a Standard in Every Sequencing Run. mSystems 2018, 3, e00023-18. [Google Scholar] [CrossRef] [Green Version]
- Frau, A.; Kenny, J.G.; Lenzi, L.; Campbell, B.J.; Ijaz, U.Z.; Duckworth, C.A.; Burkitt, M.D.; Hall, N.; Anson, J.; Darby, A.C.; et al. DNA Extraction and Amplicon Production Strategies Deeply Inf Luence the Outcome of Gut Mycobiome Studies. Sci. Rep. 2019, 9, 9328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedersoo, L.; Bahram, M.; Zinger, L.; Nilsson, R.H.; Kennedy, P.G.; Yang, T.; Anslan, S.; Mikryukov, V. Best Practices in Metabarcoding of Fungi: From Experimental Design to Results. Mol. Ecol. 2022, 31, 2769–2795. [Google Scholar] [CrossRef]
- Jay, Z.J.; Inskeep, W.P. The Distribution, Diversity, and Importance of 16S RRNA Gene Introns in the Order Thermoproteales. Biol. Direct 2015, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The Effect of 16S RRNA Region Choice on Bacterial Community Metabarcoding Results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A Detailed Analysis of 16S Ribosomal RNA Gene Segments for the Diagnosis of Pathogenic Bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Stefanini, I.; Cavalieri, D. Metagenomic Approaches to Investigate the Contribution of the Vineyard Environment to the Quality of Wine Fermentation: Potentials and Difficulties. Front. Microbiol. 2018, 9, 991. [Google Scholar] [CrossRef] [PubMed]
- Amrouche, T.; Mounier, J.; Pawtowski, A.; Thomas, F.; Picot, A. Microbiota Associated with Dromedary Camel Milk from Algerian Sahara. Curr. Microbiol. 2020, 77, 24–31. [Google Scholar] [CrossRef]
- Maillet, A.; Bouju-Albert, A.; Roblin, S.; Vaissié, P.; Leuillet, S.; Dousset, X.; Jaffrès, E.; Combrisson, J.; Prévost, H. Impact of DNA Extraction and Sampling Methods on Bacterial Communities Monitored by 16S RDNA Metabarcoding in Cold-Smoked Salmon and Processing Plant Surfaces. Food Microbiol. 2021, 95, 103705. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, S.; Petrovits, G.E.; Kyritsi, M.; Argiriou, A. Amplicon Metabarcoding Data of Prokaryotes and Eukaryotes Present in ‘Kalamata’ Table Olives Packaged under Modified Atmosphere. Data Brief 2021, 38, 107314. [Google Scholar] [CrossRef]
- Penland, M.; Mounier, J.; Pawtowski, A.; Tréguer, S.; Deutsch, S.-M.; Coton, M. Use of Metabarcoding and Source Tracking to Identify Desirable or Spoilage Autochthonous Microorganism Sources during Black Olive Fermentations. Food Res. Int. 2021, 144, 110344. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Beiko, R.G. 16S RRNA Gene Analysis with QIIME2. In Microbiome Analysis: Methods and Protocols; Beiko, R.G., Hsiao, W., Parkinson, J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 113–129. ISBN 978-1-4939-8728-3. [Google Scholar]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D. Reintroducing Mothur: 10 Years Later. Appl. Environ. Microbiol. 2020, 86, e02343-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The Metagenomics RAST Server—A Public Resource for the Automatic Phylogenetic and Functional Analysis of Metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open Software Development for Computational Biology and Bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A Practical Guide to Amplicon and Metagenomic Analysis of Microbiome Data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Chi, L.; Zhu, Y.; Shi, X.; Tu, P.; Li, B.; Yin, J.; Gao, N.; Shen, W.; Schnabl, B. An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies. Biomolecules 2021, 11, 530. [Google Scholar] [CrossRef] [PubMed]
- Wajid, B.; Anwar, F.; Wajid, I.; Nisar, H.; Meraj, S.; Zafar, A.; Al-Shawaqfeh, M.K.; Ekti, A.R.; Khatoon, A.; Suchodolski, J.S. Music of Metagenomics—A Review of Its Applications, Analysis Pipeline, and Associated Tools. Funct. Integr. Genom. 2022, 22, 3–26. [Google Scholar] [CrossRef]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of Genome Sequencing by Long-Read Sequencer Using SMRT Technology in Medical Area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Mo, L.; Pan, L.; Hou, Q.; Li, C.; Darima, I.; Yu, J. Using PacBio Sequencing to Investigate the Bacterial Microbiota of Traditional Buryatian Cottage Cheese and Comparison with Italian and Kazakhstan Artisanal Cheeses. J. Dairy Sci. 2018, 101, 6885–6896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Cao, J.; Xu, H.; Hou, Q.; Yu, Z.; Zhang, H.; Sun, Z. Bacterial Diversity and Community Structure in Chongqing Radish Paocai Brines Revealed Using PacBio Single-Molecule Real-Time Sequencing Technology. J. Sci. Food Agric. 2018, 98, 3234–3245. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, A.; Catozzi, C.; Viñes, J.; Sanchez, A.; Francino, O. Microbiota Profiling with Long Amplicons Using Nanopore Sequencing: Full-Length 16S RRNA Gene and the 16S-ITS-23S of the rrn Operon. F1000Research 2019, 7, 1755. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Martins, L.; Page, A.J.; Mather, A.E.; Charles, I.G. Taxonomic Resolution of the Ribosomal RNA Operon in Bacteria: Implications for Its Use with Long-Read Sequencing. NAR Genom. Bioinform. 2020, 2, lqz016. [Google Scholar] [CrossRef] [Green Version]
- Gehrig, J.L.; Portik, D.M.; Driscoll, M.D.; Jackson, E.; Chakraborty, S.; Gratalo, D.; Ashby, M.; Valladares, R. Finding the Right Fit: Evaluation of Short-Read and Long-Read Sequencing Approaches to Maximize the Utility of Clinical Microbiome Data. Microb. Genom. 2022, 8, 000794. [Google Scholar] [CrossRef] [PubMed]
- Kerkhof, L.J.; Dillon, K.P.; Häggblom, M.M.; McGuinness, L.R. Profiling Bacterial Communities by MinION Sequencing of Ribosomal Operons. Microbiome 2017, 5, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, Y.; Niwa, H.; Uchida-Fujii, E.; Nukada, T. Establishment and Assessment of an Amplicon Sequencing Method Targeting the 16S-ITS-23S RRNA Operon for Analysis of the Equine Gut Microbiome. Sci. Rep. 2021, 11, 11884. [Google Scholar] [CrossRef] [PubMed]
- Martijn, J.; Lind, A.E.; Schön, M.E.; Spiertz, I.; Juzokaite, L.; Bunikis, I.; Pettersson, O.V.; Ettema, T.J.G. Confident Phylogenetic Identification of Uncultured Prokaryotes through Long Read Amplicon Sequencing of the 16S-ITS-23S RRNA Operon. Environ. Microbiol. 2019, 21, 2485–2498. [Google Scholar] [CrossRef]
- Karst, S.M.; Ziels, R.M.; Kirkegaard, R.H.; Sørensen, E.A.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. High-Accuracy Long-Read Amplicon Sequences Using Unique Molecular Identifiers with Nanopore or PacBio Sequencing. Nat. Methods 2021, 18, 165–169. [Google Scholar] [CrossRef]
- Brewer, T.E.; Albertsen, M.; Edwards, A.; Kirkegaard, R.H.; Rocha, E.P.C.; Fierer, N. Unlinked RRNA Genes Are Widespread among Bacteria and Archaea. ISME J. 2020, 14, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bik, H.M. Just Keep It Simple? Benchmarking the Accuracy of Taxonomy Assignment Software in Metabarcoding Studies. Mol. Ecol. Resour. 2021, 21, 2187–2189. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.K.; Schmidt, T.M. RrnDB: Improved Tools for Interpreting RRNA Gene Abundance in Bacteria and Archaea and a New Foundation for Future Development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.pacb.com/wp-content/uploads/Driscoll-ASM-Microbe-2019-Microbiome-Profiling-at-the-Strain-Level-Using-rRNA-Amplicons.pdf (accessed on 18 July 2022).
- Seol, D.; Lim, J.S.; Sung, S.; Lee, Y.H.; Jeong, M.; Cho, S.; Kwak, W.; Kim, H. Microbial Identification Using RRNA Operon Region: Database and Tool for Metataxonomics with Long-Read Sequence. Microbiol. Spectr. 2022, 10, e02017–e02021. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Páez, A.; Portune, K.J.; Sanz, Y. Species-Level Resolution of 16S RRNA Gene Amplicons Sequenced through the MinIONTM Portable Nanopore Sequencer. GigaScience 2016, 5, s13742-016-0111-z. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciuffreda, L.; Rodríguez-Pérez, H.; Flores, C. Nanopore Sequencing and Its Application to the Study of Microbial Communities. Comput. Struct. Biotechnol. J. 2021, 19, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting Functional Profiles from Metagenomic 16S RRNA Data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [Green Version]
- Zotta, T.; Ricciardi, A.; Condelli, N.; Parente, E. Metataxonomic and Metagenomic Approaches for the Study of Undefined Strain Starters for Cheese Manufacture. Crit. Rev. Food Sci. Nutr. 2022, 62, 3898–3912. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and Sensitive Taxonomic Classification for Metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic Microbial Community Profiling Using Unique Clade-Specific Marker Genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Tovo, A.; Menzel, P.; Krogh, A.; Cosentino Lagomarsino, M.; Suweis, S. Taxonomic Classification Method for Metagenomics Based on Core Protein Families with Core-Kaiju. Nucleic Acids Res. 2020, 48, e93. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef]
- Nasko, D.J.; Koren, S.; Phillippy, A.M.; Treangen, T.J. RefSeq Database Growth Influences the Accuracy of K-Mer-Based Lowest Common Ancestor Species Identification. Genome Biol. 2018, 19, 165. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liu, M.; Yang, J. Recovering Metagenome-Assembled Genomes from Shotgun Metagenomic Sequencing Data: Methods, Applications, Challenges, and Opportunities. Microbiol. Res. 2022, 260, 127023. [Google Scholar] [CrossRef]
- Ayling, M.; Clark, M.D.; Leggett, R.M. New Approaches for Metagenome Assembly with Short Reads. Brief Bioinform. 2020, 21, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. Meta-IDBA: A de Novo Assembler for Metagenomic Data. Bioinformatics 2011, 27, i94–i101. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an Efficient Tool for Accurately Reconstructing Single Genomes from Complex Microbial Communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [Green Version]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Loman, N.J.; Andersson, A.F.; Quince, C. CONCOCT: Clustering CONtigs on COverage and ComposiTion. arXiv 2013, arXiv:1312.4038. [Google Scholar]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Tang, Y.-H.; Tringe, S.G.; Simmons, B.A.; Singer, S.W. MaxBin: An Automated Binning Method to Recover Individual Genomes from Metagenomes Using an Expectation-Maximization Algorithm. Microbiome 2014, 2, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, F.; Jia, B.; Gray, K.L.; Lau, W.Y.V.; Beiko, R.G.; Brinkman, F.S.L.Y. Metagenome-Assembled Genome Binning Methods with Short Reads Disproportionately Fail for Plasmids and Genomic Islands. Microb. Genom. 2020, 6, e000436. [Google Scholar] [CrossRef]
- Xie, H.; Yang, C.; Sun, Y.; Igarashi, Y.; Jin, T.; Luo, F. PacBio Long Reads Improve Metagenomic Assemblies, Gene Catalogs, and Genome Binning. Front. Genet. 2020, 11, 516269. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; Weissensteiner, M.H.; Sedlazeck, F.J. Towards Population-Scale Long-Read Sequencing. Nat. Rev. Genet. 2021, 22, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Albertsen, M.; Anslan, S.; Callahan, B. Perspectives and Benefits of High-Throughput Long-Read Sequencing in Microbial Ecology. Appl. Environ. Microbiol. 2021, 87, e00626-21. [Google Scholar] [CrossRef]
- Hu, Y.; Fang, L.; Nicholson, C.; Wang, K. Implications of Error-Prone Long-Read Whole-Genome Shotgun Sequencing on Characterizing Reference Microbiomes. iScience 2020, 23, 101223. [Google Scholar] [CrossRef]
- Portik, D.M.; Brown, C.T.; Pierce-Ward, N.T. Evaluation of Taxonomic Profiling Methods for Long-Read Shotgun Metagenomic Sequencing Datasets. bioRxiv 2022. [Google Scholar] [CrossRef]
- Fu, S.; Wang, A.; Au, K.F. A Comparative Evaluation of Hybrid Error Correction Methods for Error-Prone Long Reads. Genome Biol. 2019, 20, 26. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Parente, E.; Ercolini, D. Recent Past, Present, and Future of the Food Microbiome. Available online: https://www.annualreviews.org/doi/epdf/10.1146/annurev-food-030117-012312 (accessed on 18 July 2022).
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S RRNA and Shotgun Sequencing Data for the Taxonomic Characterization of the Gut Microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef] [PubMed]
- Arıkan, M.; Mitchell, A.L.; Finn, R.D.; Gürel, F. Microbial Composition of Kombucha Determined Using Amplicon Sequencing and Shotgun Metagenomics. J. Food Sci. 2020, 85, 455–464. [Google Scholar] [CrossRef]
- You, L.; Yang, C.; Jin, H.; Kwok, L.-Y.; Sun, Z.; Zhang, H. Metagenomic Features of Traditional Fermented Milk Products. LWT 2022, 155, 112945. [Google Scholar] [CrossRef]
- Maske, B.L.; de Melo Pereira, G.V.; da Silva Vale, A.; Marques Souza, D.S.; De Dea Lindner, J.; Soccol, C.R. Viruses in Fermented Foods: Are They Good or Bad? Two Sides of the Same Coin. Food Microbiol. 2021, 98, 103794. [Google Scholar] [CrossRef] [PubMed]
- Tagirdzhanova, G.; Saary, P.; Tingley, J.P.; Díaz-Escandón, D.; Abbott, D.W.; Finn, R.D.; Spribille, T. Predicted Input of Uncultured Fungal Symbionts to a Lichen Symbiosis from Metagenome-Assembled Genomes. Genome Biol. Evol. 2021, 13, evab047. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Raiko, M.; Lapidus, A.; Pevzner, P.A. Plasmid Detection and Assembly in Genomic and Metagenomic Data Sets. Genome Res. 2019, 29, 961–968. [Google Scholar] [CrossRef] [Green Version]
- Beaulaurier, J.; Zhu, S.; Deikus, G.; Mogno, I.; Zhang, X.-S.; Davis-Richardson, A.; Canepa, R.; Triplett, E.W.; Faith, J.J.; Sebra, R.; et al. Metagenomic Binning and Association of Plasmids with Bacterial Host Genomes Using DNA Methylation. Nat. Biotechnol. 2018, 36, 61–69. [Google Scholar] [CrossRef]
- Hilpert, C.; Bricheux, G.; Debroas, D. Reconstruction of Plasmids by Shotgun Sequencing from Environmental DNA: Which Bioinformatic Workflow? Brief Bioinform. 2021, 22, bbaa059. [Google Scholar] [CrossRef]
- Callahan, B.J.; Grinevich, D.; Thakur, S.; Balamotis, M.A.; Yehezkel, T.B. Ultra-Accurate Microbial Amplicon Sequencing with Synthetic Long Reads. Microbiome 2021, 9, 130. [Google Scholar] [CrossRef]
- Liu, S.; Wu, I.; Yu, Y.-P.; Balamotis, M.; Ren, B.; Ben Yehezkel, T.; Luo, J.-H. Targeted Transcriptome Analysis Using Synthetic Long Read Sequencing Uncovers Isoform Reprograming in the Progression of Colon Cancer. Commun Biol. 2021, 4, 506. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hsieh, C.-L.; Young, A.; Zhang, Z.; Ren, X.; Zhao, Z. Illumina Synthetic Long Read Sequencing Allows Recovery of Missing Sequences Even in the “Finished” C. Elegans Genome. Sci. Rep. 2015, 5, 10814. [Google Scholar] [CrossRef]
- Burton, J.N.; Liachko, I.; Dunham, M.J.; Shendure, J. Species-Level Deconvolution of Metagenome Assemblies with Hi-C–Based Contact Probability Maps. G3 2014, 4, 1339–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbers, J.P.; Rogers, M.F.; Perelman, P.L.; Proskuryakova, A.A.; Serdyukova, N.A.; Johnson, W.E.; Horin, P.; Corander, J.; Murphy, D.; Burger, P.A. Improving Illumina Assemblies with Hi-C and Long Reads: An Example with the North African Dromedary. Mol. Ecol. Resour. 2019, 19, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Zhang, Y. Deciphering Hi-C: From 3D Genome to Function. Cell Biol. Toxicol. 2019, 35, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Ning, D.-L.; Wu, T.; Xiao, L.-J.; Ma, T.; Fang, W.-L.; Dong, R.-Q.; Cao, F.-L. Chromosomal-Level Assembly of Juglans Sigillata Genome Using Nanopore, BioNano, and Hi-C Analysis. GigaScience 2020, 9, giaa006. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Kolmogorov, M.; Tseng, E.; Portik, D.M.; Korobeynikov, A.; Tolstoganov, I.; Uritskiy, G.; Liachko, I.; Sullivan, S.T.; Shin, S.B.; et al. Generating Lineage-Resolved, Complete Metagenome-Assembled Genomes from Complex Microbial Communities. Nat. Biotechnol. 2022, 40, 711–719. [Google Scholar] [CrossRef]
- Jagadeesan, B.; Gerner-Smidt, P.; Allard, M.W.; Leuillet, S.; Winkler, A.; Xiao, Y.; Chaffron, S.; Van Der Vossen, J.; Tang, S.; Katase, M.; et al. The Use of next Generation Sequencing for Improving Food Safety: Translation into Practice. Food Microbiol. 2019, 79, 96–115. [Google Scholar] [CrossRef]
- Bao, Y.; Wadden, J.; Erb-Downward, J.R.; Ranjan, P.; Zhou, W.; McDonald, T.L.; Mills, R.E.; Boyle, A.P.; Dickson, R.P.; Blaauw, D.; et al. SquiggleNet: Real-Time, Direct Classification of Nanopore Signals. Genome Biol. 2021, 22, 298. [Google Scholar] [CrossRef]
- Cao, M.D.; Ganesamoorthy, D.; Elliott, A.G.; Zhang, H.; Cooper, M.A.; Coin, L.J.M. Streaming Algorithms for Identification Pathogens and Antibiotic Resistance Potential from Real-Time MinIONTM Sequencing. GigaScience 2016, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Juul, S.; Izquierdo, F.; Hurst, A.; Dai, X.; Wright, A.; Kulesha, E.; Pettett, R.; Turner, D.J. What’s in My Pot? Real-Time Species Identification on the MinIONTM. bioRxiv 2015. [Google Scholar] [CrossRef] [Green Version]
- Edwards, H.S.; Krishnakumar, R.; Sinha, A.; Bird, S.W.; Patel, K.D.; Bartsch, M.S. Real-Time Selective Sequencing with RUBRIC: Read Until with Basecall and Reference-Informed Criteria. Sci. Rep. 2019, 9, 11475. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.J.; Loose, M. Readfish Enables Targeted Nanopore Sequencing of Gigabase-Sized Genomes. Nat. Biotechnol. 2021, 39, 442–450. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Biological Hazards (BIOHAZ); Koutsoumanis, K.; Allende, A.; Álvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Herman, L.; et al. Role Played by the Environment in the Emergence and Spread of Antimicrobial Resistance (AMR) through the Food Chain. EFSA J. 2021, 19, e06651. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Macori, G.; Kilcawley, K.N.; Cotter, P.D. Meta-Analysis of Cheese Microbiomes Highlights Contributions to Multiple Aspects of Quality. Nat. Food 2020, 1, 500–510. [Google Scholar] [CrossRef]
- Devirgiliis, C.; Barile, S.; Perozzi, G. Antibiotic Resistance Determinants in the Interplay between Food and Gut Microbiota. Genes Nutr. 2011, 6, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.; Hu, M.; Li, X.; Pan, Z.; Li, M.; Li, L.; Zheng, Z.; Yang, M. Metagenomics Reveals the Diversity and Taxonomy of Antibiotic Resistance Genes in Sufu Bacterial Communities. Food Control 2021, 121, 107641. [Google Scholar] [CrossRef]
- Song, Q.; Wang, B.; Han, Y.; Zhou, Z. Metagenomics Reveals the Diversity and Taxonomy of Carbohydrate-Active Enzymes and Antibiotic Resistance Genes in Suancai Bacterial Communities. Genes 2022, 13, 773. [Google Scholar] [CrossRef]
- Leech, J.; Cabrera-Rubio, R.; Walsh, A.M.; Macori, G.; Walsh, C.J.; Barton, W.; Finnegan, L.; Crispie, F.; O’Sullivan, O.; Claesson, M.J.; et al. Fermented-Food Metagenomics Reveals Substrate-Associated Differences in Taxonomy and Health-Associated and Antibiotic Resistance Determinants. mSystems 2020, 5, e00522-20. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [Green Version]
- Florensa, A.F.; Kaas, R.S.; Clausen, P.T.L.C.; Aytan-Aktug, D.; Aarestrup, F.M. ResFinder—An Open Online Resource for Identification of Antimicrobial Resistance Genes in next-Generation Sequencing Data and Prediction of Phenotypes from Genotypes. Microb. Genom. 2022, 8, 000748. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Crispie, F.; Kilcawley, K.; O’Sullivan, O.; O’Sullivan, M.G.; Claesson, M.J.; Cotter, P.D. Microbial Succession and Flavor Production in the Fermented Dairy Beverage Kefir. mSystems 2016, 1, e00052-16. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chen, C.; Lei, Z. Meta-Omics Insights in the Microbial Community Profiling and Functional Characterization of Fermented Foods. Trends Food Sci. Technol. 2017, 65, 23–31. [Google Scholar] [CrossRef]
- Dimidi, E.; Cox, S.R.; Rossi, M.; Whelan, K. Fermented Foods: Definitions and Characteristics, Impact on the Gut Microbiota and Effects on Gastrointestinal Health and Disease. Nutrients 2019, 11, 1806. [Google Scholar] [CrossRef] [Green Version]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Scientific Opinion on the Substantiation of Health Claims Related to Live Yoghurt Cultures and Improved Lactose Digestion (ID 1143, 2976) Pursuant to Article 13(1) of Regulation (EC) No 1924/2006. EFSA J. 2010, 8, 1763. [Google Scholar] [CrossRef]
- De Filippis, F.; Pasolli, E.; Ercolini, D. The Food-Gut Axis: Lactic Acid Bacteria and Their Link to Food, the Gut Microbiome and Human Health. FEMS Microbiol. Rev. 2020, 44, 454–489. [Google Scholar] [CrossRef]
- Kok, C.R.; Hutkins, R. Yogurt and Other Fermented Foods as Sources of Health-Promoting Bacteria. Nutr. Rev. 2018, 76, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Aslam, H.; Green, J.; Jacka, F.N.; Collier, F.; Berk, M.; Pasco, J.; Dawson, S.L. Fermented Foods, the Gut and Mental Health: A Mechanistic Overview with Implications for Depression and Anxiety. Nutr. Neurosci. 2020, 23, 659–671. [Google Scholar] [CrossRef]
- Wang, D.H.; Yang, Y.; Wang, Z.; Lawrence, P.; Worobo, R.W.; Brenna, J.T. High Levels of Branched Chain Fatty Acids in Nātto and Other Asian Fermented Foods. Food Chem. 2019, 286, 428–433. [Google Scholar] [CrossRef]
- Hati, S.; Patel, M.; Mishra, B.K.; Das, S. Short-Chain Fatty Acid and Vitamin Production Potentials of Lactobacillus Isolated from Fermented Foods of Khasi Tribes, Meghalaya, India. Ann. Microbiol. 2019, 69, 1191–1199. [Google Scholar] [CrossRef]
- Barbara, G.; Feinle-Bisset, C.; Ghoshal, U.C.; Santos, J.; Vanner, S.J.; Vergnolle, N.; Zoetendal, E.G.; Quigley, E.M. The Intestinal Microenvironment and Functional Gastrointestinal Disorders. Gastroenterology 2016, 150, 1305–1318.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, A.; Naghibi, M.M.; Garcha, D. The Role of Bacteria, Probiotics and Diet in Irritable Bowel Syndrome. Foods 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Pollard, K.S. Toward Accurate and Quantitative Comparative Metagenomics. Cell 2016, 166, 1103–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Whon, T.W.; Roh, S.W.; Jeon, C.O. Unraveling Microbial Fermentation Features in Kimchi: From Classical to Meta-Omics Approaches. Appl. Microbiol. Biotechnol. 2020, 104, 7731–7744. [Google Scholar] [CrossRef]
- Blasche, S.; Kim, Y.; Mars, R.A.T.; Machado, D.; Maansson, M.; Kafkia, E.; Milanese, A.; Zeller, G.; Teusink, B.; Nielsen, J.; et al. Metabolic Cooperation and Spatiotemporal Niche Partitioning in a Kefir Microbial Community. Nat. Microbiol. 2021, 6, 196–208. [Google Scholar] [CrossRef]
- Kamilari, E.; Tomazou, M.; Antoniades, A.; Tsaltas, D. High Throughput Sequencing Technologies as a New Toolbox for Deep Analysis, Characterization and Potentially Authentication of Protection Designation of Origin Cheeses? Int. J. Food Sci. 2019, 2019, e5837301. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.J.; Cotter, P.D.; O’Sullivan, O.; Giblin, L.; McSweeney, P.L.H.; Sheehan, J.J. Temporal and Spatial Differences in Microbial Composition during the Manufacture of a Continental-Type Cheese. Appl. Environ. Microbiol. 2015, 81, 2525–2533. [Google Scholar] [CrossRef] [Green Version]
- Pierce, E.C.; Morin, M.; Little, J.C.; Liu, R.B.; Tannous, J.; Keller, N.P.; Pogliano, K.; Wolfe, B.E.; Sanchez, L.M.; Dutton, R.J. Bacterial–Fungal Interactions Revealed by Genome-Wide Analysis of Bacterial Mutant Fitness. Nat. Microbiol. 2021, 6, 87–102. [Google Scholar] [CrossRef]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese Rind Communities Provide Tractable Systems for In Situ and In Vitro Studies of Microbial Diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Paillet, T.; Dugat-Bony, E. Bacteriophage Ecology of Fermented Foods: Anything New under the Sun? Curr. Opin. Food Sci. 2021, 40, 102–111. [Google Scholar] [CrossRef]
- Roux, S.; Matthijnssens, J.; Dutilh, B.E. Metagenomics in Virology. Encycl. Virol. 2021, 1, 133–140. [Google Scholar] [CrossRef]
- Tamang, J.P.; Das, S.; Kharnaior, P.; Pariyar, P.; Thapa, N.; Jo, S.-W.; Yim, E.-J.; Shin, D.-H. Shotgun Metagenomics of Cheonggukjang, a Fermented Soybean Food of Korea: Community Structure, Predictive Functionalities and Amino Acids Profile. Food Res. Int. 2022, 151, 110904. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Sharma, N.; Kaushal, G.; Samurailatpam, S.; Sahoo, D.; Rai, A.K.; Singh, S.P. Metagenomic Insights Into the Taxonomic and Functional Features of Kinema, a Traditional Fermented Soybean Product of Sikkim Himalaya. Front. Microbiol. 2019, 10, 1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, F.; Oppelt, J.; Maragkakis, M.; Mourelatos, Z. TERA-Seq: True End-to-End Sequencing of Native RNA Molecules for Transcriptome Characterization. Nucleic Acids Res. 2021, 49, e115. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. A Decade of RNA Virus Metagenomics Is (Not) Enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Van Reckem, E.; De Vuyst, L.; Weckx, S.; Leroy, F. Next-Generation Sequencing to Enhance the Taxonomic Resolution of the Microbiological Analysis of Meat and Meat-Derived Products. Curr. Opin. Food Sci. 2021, 37, 58–65. [Google Scholar] [CrossRef]
- Suminda, G.G.D.; Bhandari, S.; Won, Y.; Goutam, U.; Kanth Pulicherla, K.; Son, Y.-O.; Ghosh, M. High-Throughput Sequencing Technologies in the Detection of Livestock Pathogens, Diagnosis, and Zoonotic Surveillance. Comput. Struct. Biotechnol. J. 2022, 20, 5378–5392. [Google Scholar] [CrossRef] [PubMed]
- Gołębiewski, M.; Tretyn, A. Generating Amplicon Reads for Microbial Community Assessment with Next-Generation Sequencing. J. Appl. Microbiol. 2020, 128, 330–354. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; McLaren, M.; Callahan, B. Understanding and Interpreting Community Sequencing Measurements of the Vaginal Microbiome. BJOG 2020, 127, 139–146. [Google Scholar] [CrossRef]
- Weinroth, M.D.; Belk, A.D.; Dean, C.; Noyes, N.; Dittoe, D.K.; Rothrock, M.J., Jr.; Ricke, S.C.; Myer, P.R.; Henniger, M.T.; Ramírez, G.A.; et al. Considerations and Best Practices in Animal Science 16S Ribosomal RNA Gene Sequencing Microbiome Studies. J. Anim. Sci. 2022, 100, skab346. [Google Scholar] [CrossRef] [PubMed]
- Delbeke, H.; Younas, S.; Casteels, I.; Joossens, M. Current Knowledge on the Human Eye Microbiome: A Systematic Review of Available Amplicon and Metagenomic Sequencing Data. Acta Ophthalmol. 2021, 99, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-Generation Sequencing: Insights to Advance Clinical Investigations of the Microbiome. J. Clin. Investig. 2022, 132, e154944. [Google Scholar] [CrossRef] [PubMed]
- Joseph, T.A.; Pe’er, I. An Introduction to Whole-Metagenome Shotgun Sequencing Studies. In Deep Sequencing Data Analysis; Shomron, N., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; pp. 107–122. ISBN 978-1-07-161103-6. [Google Scholar]
- Casertano, M.; Fogliano, V.; Ercolini, D. Psychobiotics, Gut Microbiota and Fermented Foods Can Help Preserving Mental Health. Food Res. Int. 2022, 152, 110892. [Google Scholar] [CrossRef] [PubMed]
- Van de Wouw, M.; Walsh, A.M.; Crispie, F.; van Leuven, L.; Lyte, J.M.; Boehme, M.; Clarke, G.; Dinan, T.G.; Cotter, P.D.; Cryan, J.F. Distinct Actions of the Fermented Beverage Kefir on Host Behaviour, Immunity and Microbiome Gut-Brain Modules in the Mouse. Microbiome 2020, 8, 67. [Google Scholar] [CrossRef]
- Dai, S.; Pan, M.; El-Nezami, H.S.; Wan, J.M.F.; Wang, M.F.; Habimana, O.; Lee, J.C.Y.; Louie, J.C.Y.; Shah, N.P. Effects of Lactic Acid Bacteria-Fermented Soymilk on Isoflavone Metabolites and Short-Chain Fatty Acids Excretion and Their Modulating Effects on Gut Microbiota. J. Food Sci. 2019, 84, 1854–1863. [Google Scholar] [CrossRef]
- Shimizu, H.; Masujima, Y.; Ushiroda, C.; Mizushima, R.; Taira, S.; Ohue-Kitano, R.; Kimura, I. Dietary Short-Chain Fatty Acid Intake Improves the Hepatic Metabolic Condition via FFAR3. Sci. Rep. 2019, 9, 16574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vital, M.; Howe, A.; Bergeron, N.; Krauss, R.M.; Jansson, J.K.; Tiedje, J.M. Metagenomic Insights into the Degradation of Resistant Starch by Human Gut Microbiota. Appl. Environ. Microbiol. 2018, 84, e01562-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut Bacteria Selectively Promoted by Dietary Fibers Alleviate Type 2 Diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-Hernández, C.; Mota-Gutierrez, J.; Ferrocino, I.; Hernández-Estrada, Z.J.; González-Ríos, O.; Cocolin, L.; Suárez-Quiroz, M.L. The Challenges and Perspectives of the Selection of Starter Cultures for Fermented Cocoa Beans. Int. J. Food Microbiol. 2019, 301, 41–50. [Google Scholar] [CrossRef]
- Ianni, A.; Di Domenico, M.; Bennato, F.; Peserico, A.; Martino, C.; Rinaldi, A.; Candeloro, L.; Grotta, L.; Cammà, C.; Pomilio, F.; et al. Metagenomic and Volatile Profiles of Ripened Cheese Obtained from Dairy Ewes Fed a Dietary Hemp Seed Supplementation. J. Dairy Sci. 2020, 103, 5882–5892. [Google Scholar] [CrossRef]
- Landis, E.A.; Oliverio, A.M.; McKenney, E.A.; Nichols, L.M.; Kfoury, N.; Biango-Daniels, M.; Shell, L.K.; Madden, A.A.; Shapiro, L.; Sakunala, S.; et al. The Diversity and Function of Sourdough Starter Microbiomes. Elife 2021, 10, e61644. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Fontana, F.; Alessandri, G.; Mancabelli, L.; Lugli, G.A.; Longhi, G.; Anzalone, R.; Viappiani, A.; Duranti, S.; Turroni, F.; et al. Ecology of Lactobacilli Present in Italian Cheeses Produced from Raw Milk. Appl. Environ. Microbiol. 2020, 86, e00139-20. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Montealegre, M.E.; Dávila-Mora, L.L.; Botero-Rute, L.M.; Reyes, A.; Caro-Quintero, A. Fine Resolution Analysis of Microbial Communities Provides Insights Into the Variability of Cocoa Bean Fermentation. Front. Microbiol. 2020, 11, 650. [Google Scholar] [CrossRef] [Green Version]
- Casey, E.; McDonnell, B.; White, K.; Stamou, P.; Crowley, T.; O’Neill, I.; Lavelle, K.; Hayes, S.; Lugli, G.A.; Arboleya, S.; et al. Needle in a Whey-Stack: PhRACS as a Discovery Tool for Unknown Phage-Host Combinations. mBio 2022, 13, e03334-21. [Google Scholar] [CrossRef]
- Mahony, J.; van Sinderen, D. Virome Studies of Food Production Systems: Time for ‘Farm to Fork’ Analyses. Curr. Opin. Biotechnol. 2022, 73, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.K.; Kot, W.; Neve, H.; Mahony, J.; Castro-Mejía, J.L.; Krych, L.; Hansen, L.H.; Nielsen, D.S.; Sørensen, S.J.; Heller, K.J.; et al. Metagenomic Analysis of Dairy Bacteriophages: Extraction Method and Pilot Study on Whey Samples Derived from Using Undefined and Defined Mesophilic Starter Cultures. Appl. Environ. Microbiol. 2017, 83, e00888-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, P.; Vindeløv, J.; Arneborg, N.; Brockmann, E. Development of Quantitative PCR and Metagenomics-Based Approaches for Strain Quantification of a Defined Mixed-Strain Starter Culture. Syst. Appl. Microbiol. 2014, 37, 186–193. [Google Scholar] [CrossRef]
- Hussain, B.; Chen, J.-S.; Hsu, B.-M.; Chu, I.-T.; Koner, S.; Chen, T.-H.; Rathod, J.; Chan, M.W.Y. Deciphering Bacterial Community Structure, Functional Prediction and Food Safety Assessment in Fermented Fruits Using Next-Generation 16S RRNA Amplicon Sequencing. Microorganisms 2021, 9, 1574. [Google Scholar] [CrossRef]
- Walsh, A.M.; Crispie, F.; Daari, K.; O’Sullivan, O.; Martin, J.C.; Arthur, C.T.; Claesson, M.J.; Scott, K.P.; Cotter, P.D. Strain-Level Metagenomic Analysis of the Fermented Dairy Beverage Nunu Highlights Potential Food Safety Risks. Appl. Environ. Microbiol. 2017, 83, e01144-17. [Google Scholar] [CrossRef] [Green Version]
- Illeghems, K.; Weckx, S.; De Vuyst, L. Applying Meta-Pathway Analyses through Metagenomics to Identify the Functional Properties of the Major Bacterial Communities of a Single Spontaneous Cocoa Bean Fermentation Process Sample. Food Microbiol. 2015, 50, 54–63. [Google Scholar] [CrossRef]
- O’Connor, P.M.; Kuniyoshi, T.M.; Oliveira, R.P.; Hill, C.; Ross, R.P.; Cotter, P.D. Antimicrobials for Food and Feed; a Bacteriocin Perspective. Curr. Opin. Biotechnol. 2020, 61, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Suárez, N.; Weckx, S.; Minahk, C.; Hebert, E.M.; Saavedra, L. Metagenomics-Based Approach for Studying and Selecting Bioprotective Strains from the Bacterial Community of Artisanal Cheeses. Int. J. Food Microbiol. 2020, 335, 108894. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-G.; Huang, Q.; Yin, X.; Zhang, T. Source Tracking of Antibiotic Resistance Genes in the Environment—Challenges, Progress, and Prospects. Water Res. 2020, 185, 116127. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Al-Zahrani, I.A.; Bibi, F.; Abd El Ghany, M.; Azhar, E.I. New Insights of Bacterial Communities in Fermented Vegetables from Shotgun Metagenomics and Identification of Antibiotic Resistance Genes and Probiotic Bacteria. Food Res. Int. 2022, 157, 111190. [Google Scholar] [CrossRef]
- Haiminen, N.; Edlund, S.; Chambliss, D.; Kunitomi, M.; Weimer, B.C.; Ganesan, B.; Baker, R.; Markwell, P.; Davis, M.; Huang, B.C.; et al. Food Authentication from Shotgun Sequencing Reads with an Application on High Protein Powders. Npj Sci. Food 2019, 3, 24. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Xu, S.-F.; Tang, T.-S.; Miao, L.; Luo, B.-Z.; Ni, Y.; Kong, F.-D.; Liu, C. Development and Evaluation of a Meat Mitochondrial Metagenomic (3MG) Method for Composition Determination of Meat from Fifteen Mammalian and Avian Species. BMC Genom. 2022, 23, 36. [Google Scholar] [CrossRef]
- Kobus, R.; Abuín, J.M.; Müller, A.; Hellmann, S.L.; Pichel, J.C.; Pena, T.F.; Hildebrandt, A.; Hankeln, T.; Schmidt, B. A Big Data Approach to Metagenomics for All-Food-Sequencing. BMC Bioinform. 2020, 21, 102. [Google Scholar] [CrossRef]
- Voorhuijzen-Harink, M.M.; Hagelaar, R.; van Dijk, J.P.; Prins, T.W.; Kok, E.J.; Staats, M. Toward On-Site Food Authentication Using Nanopore Sequencing. Food Chem. X 2019, 2, 100035. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial Biogeography of Wine Grapes Is Conditioned by Cultivar, Vintage, and Climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef] [Green Version]
- Gul, O.; Atalar, I.; Mortas, M.; Dervisoglu, M. Rheological, Textural, Colour and Sensorial Properties of Kefir Produced with Buffalo Milk Using Kefir Grains and Starter Culture: A Comparison with Cows’ Milk Kefir. Int. J. Dairy Technol. 2018, 71, 73–80. [Google Scholar] [CrossRef]
- Vermote, L.; Verce, M.; De Vuyst, L.; Weckx, S. Amplicon and Shotgun Metagenomic Sequencing Indicates That Microbial Ecosystems Present in Cheese Brines Reflect Environmental Inoculation during the Cheese Production Process. Int. Dairy J. 2018, 87, 44–53. [Google Scholar] [CrossRef]
- Yang, X.; Hu, W.; Xiu, Z.; Jiang, A.; Yang, X.; Saren, G.; Ji, Y.; Guan, Y.; Feng, K. Microbial Community Dynamics and Metabolome Changes During Spontaneous Fermentation of Northeast Sauerkraut From Different Households. Front. Microbiol. 2020, 11, 1878. [Google Scholar] [CrossRef] [PubMed]
- Hananiah, N.; Rahim, A.A. The Application of Hurdle Technology in Extending the Shelf Life and Improving the Quality of Fermented Freshwater Fish (Pekasam): A Review. MJoSHT 2022, 8, 44–54. [Google Scholar] [CrossRef]
- Kazou, M.; Grafakou, A.; Tsakalidou, E.; Georgalaki, M. Zooming Into the Microbiota of Home-Made and Industrial Kefir Produced in Greece Using Classical Microbiological and Amplicon-Based Metagenomics Analyses. Front. Microbiol. 2021, 12, 621069. [Google Scholar] [CrossRef]
- Katz, L.; Chen, Y.Y.; Gonzalez, R.; Peterson, T.C.; Zhao, H.; Baltz, R.H. Synthetic Biology Advances and Applications in the Biotechnology Industry: A Perspective. J. Ind. Microbiol. Biotechnol. 2018, 45, 449–461. [Google Scholar] [CrossRef]
- Son, J.; Jeong, K. Recent Advances in Synthetic Biology for the Engineering of Lactic Acid Bacteria. Biotechnol. Bioprocess. Eng. 2020, 25, 962–973. [Google Scholar] [CrossRef]
- Sambyal, K.; Singh, R.V. Production Aspects of Testosterone by Microbial Biotransformation and Future Prospects. Steroids 2020, 159, 108651. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, P.; Singh, J.; Singh, S.; Nain, L. Prospecting the Potential of Agroresidues as Substrate for Microbial Flavor Production. Front. Sustain. Food Syst. 2020, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Amicarelli, V.; Lagioia, G.; Bux, C. Global Warming Potential of Food Waste through the Life Cycle Assessment: An Analytical Review. Environ. Impact. Assess Rev. 2021, 91, 106677. [Google Scholar] [CrossRef]
- Wesana, J.; Gellynck, X.; Dora, M.K.; Pearce, D.; De Steur, H. Measuring Food and Nutritional Losses through Value Stream Mapping along the Dairy Value Chain in Uganda. Resour. Conserv. Recycl. 2019, 150, 104416. [Google Scholar] [CrossRef]
- Calvete-Torre, I.; Sabater, C.; Antón, M.J.; Moreno, F.J.; Riestra, S.; Margolles, A.; Ruiz, L. Prebiotic Potential of Apple Pomace and Pectins from Different Apple Varieties: Modulatory Effects on Key Target Commensal Microbial Populations. Food Hydrocoll. 2022, 133, 107958. [Google Scholar] [CrossRef]
- Hyun Chung, T.; Ranjan Dhar, B. A Multi-Perspective Review on Microbial Electrochemical Technologies for Food Waste Valorization. Bioresour. Technol. 2021, 342, 125950. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, T.; Nadeem, F.; Bilal, M.; Iqbal, H.M.N. 8—Recent Trends on the Food Wastes Valorization to Value-Added Commodities. In Advanced Technology for the Conversion of Waste into Fuels and Chemicals; Khan, A., Jawaid, M., Pizzi, A., Azum, N., Asiri, A., Isa, I., Eds.; Woodhead Publishing: Sawston, UK, 2021; pp. 171–196. ISBN 978-0-12-823139-5. [Google Scholar]
- Sabater, C.; Calvete-Torre, I.; Villamiel, M.; Moreno, F.J.; Margolles, A.; Ruiz, L. Vegetable Waste and By-Products to Feed a Healthy Gut Microbiota: Current Evidence, Machine Learning and Computational Tools to Design Novel Microbiome-Targeted Foods. Trends Food Sci. Technol. 2021, 118, 399–417. [Google Scholar] [CrossRef]
- Socas-Rodríguez, B.; Álvarez-Rivera, G.; Valdés, A.; Ibáñez, E.; Cifuentes, A. Food By-Products and Food Wastes: Are They Safe Enough for Their Valorization? Trends Food Sci. Technol. 2021, 114, 133–147. [Google Scholar] [CrossRef]
- Talan, A.; Tiwari, B.; Yadav, B.; Tyagi, R.D.; Wong, J.W.C.; Drogui, P. Food Waste Valorization: Energy Production Using Novel Integrated Systems. Bioresour. Technol. 2021, 322, 124538. [Google Scholar] [CrossRef]
- Iquebal, M.A.; Jagannadham, J.; Jaiswal, S.; Prabha, R.; Rai, A.; Kumar, D. Potential Use of Microbial Community Genomes in Various Dimensions of Agriculture Productivity and Its Management: A Review. Front. Microbiol. 2022, 13, 708335. [Google Scholar] [CrossRef]
- Eckstrom, K.; Barlow, J.W. Resistome Metagenomics from Plate to Farm: The Resistome and Microbial Composition during Food Waste Feeding and Composting on a Vermont Poultry Farm. PLoS ONE 2019, 14, e0219807. [Google Scholar] [CrossRef] [Green Version]
- Bianco, A.; Budroni, M.; Zara, S.; Mannazzu, I.; Fancello, F.; Zara, G. The Role of Microorganisms on Biotransformation of Brewers’ Spent Grain. Appl. Microbiol. Biotechnol. 2020, 104, 8661–8678. [Google Scholar] [CrossRef]
- Crognale, S.; Braguglia, C.M.; Gallipoli, A.; Gianico, A.; Rossetti, S.; Montecchio, D. Direct Conversion of Food Waste Extract into Caproate: Metagenomics Assessment of Chain Elongation Process. Microorganisms 2021, 9, 327. [Google Scholar] [CrossRef]
- Zhang, L.; Loh, K.-C.; Kuroki, A.; Dai, Y.; Tong, Y.W. Microbial Biodiesel Production from Industrial Organic Wastes by Oleaginous Microorganisms: Current Status and Prospects. J. Hazard. Mater. 2021, 402, 123543. [Google Scholar] [CrossRef]
- Javourez, U.; O’Donohue, M.; Hamelin, L. Waste-to-Nutrition: A Review of Current and Emerging Conversion Pathways. Biotechnol. Adv. 2021, 53, 107857. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Wu, Y.; Gong, M.; Deng, J.; Gu, Y.; Liu, Y.; Li, J.; Du, G.; Ledesma-Amaro, R.; Liu, L.; et al. Synthetic Biology for Future Food: Research Progress and Future Directions. Future Foods 2021, 3, 100025. [Google Scholar] [CrossRef]
- Galimberti, A.; Bruno, A.; Agostinetto, G.; Casiraghi, M.; Guzzetti, L.; Labra, M. Fermented Food Products in the Era of Globalization: Tradition Meets Biotechnology Innovations. Curr. Opin. Biotechnol. 2021, 70, 36–41. [Google Scholar] [CrossRef]
- Branduardi, P. Closing the Loop: The Power of Microbial Biotransformations from Traditional Bioprocesses to Biorefineries, and Beyond. Microb. Biotechnol. 2021, 14, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Ubando, A.T.; Felix, C.B.; Chen, W.-H. Biorefineries in Circular Bioeconomy: A Comprehensive Review. Bioresour. Technol. 2020, 299, 122585. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.; Yadav, B.; Atmakuri, A.; Tyagi, R.D.; Wong, J.W.C.; Drogui, P. Bioconversion of Organic Wastes into Value-Added Products: A Review. Bioresour. Technol. 2022, 344, 126398. [Google Scholar] [CrossRef]
- Jayasekara, S.; Dissanayake, L.; Jayakody, L.N. Opportunities in the Microbial Valorization of Sugar Industrial Organic Waste to Biodegradable Smart Food Packaging Materials. Int. J. Food Microbiol. 2022, 377, 109785. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srinivas, M.; O’Sullivan, O.; Cotter, P.D.; Sinderen, D.v.; Kenny, J.G. The Application of Metagenomics to Study Microbial Communities and Develop Desirable Traits in Fermented Foods. Foods 2022, 11, 3297. https://doi.org/10.3390/foods11203297
Srinivas M, O’Sullivan O, Cotter PD, Sinderen Dv, Kenny JG. The Application of Metagenomics to Study Microbial Communities and Develop Desirable Traits in Fermented Foods. Foods. 2022; 11(20):3297. https://doi.org/10.3390/foods11203297
Chicago/Turabian StyleSrinivas, Meghana, Orla O’Sullivan, Paul D. Cotter, Douwe van Sinderen, and John G. Kenny. 2022. "The Application of Metagenomics to Study Microbial Communities and Develop Desirable Traits in Fermented Foods" Foods 11, no. 20: 3297. https://doi.org/10.3390/foods11203297