
Structural Characteristics and Formation Mechanism of Microbiota Related to Fermentation Ability and Alcohol Production Ability in Nongxiang Daqu
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Physicochemical Properties
2.3. DNA Extraction and Processing
2.4. Processing of Sequencing Data
2.5. Statistical Analysis
3. Results and Discussion
3.1. Variations in the Fermentation Ability and Alcohol Production Ability
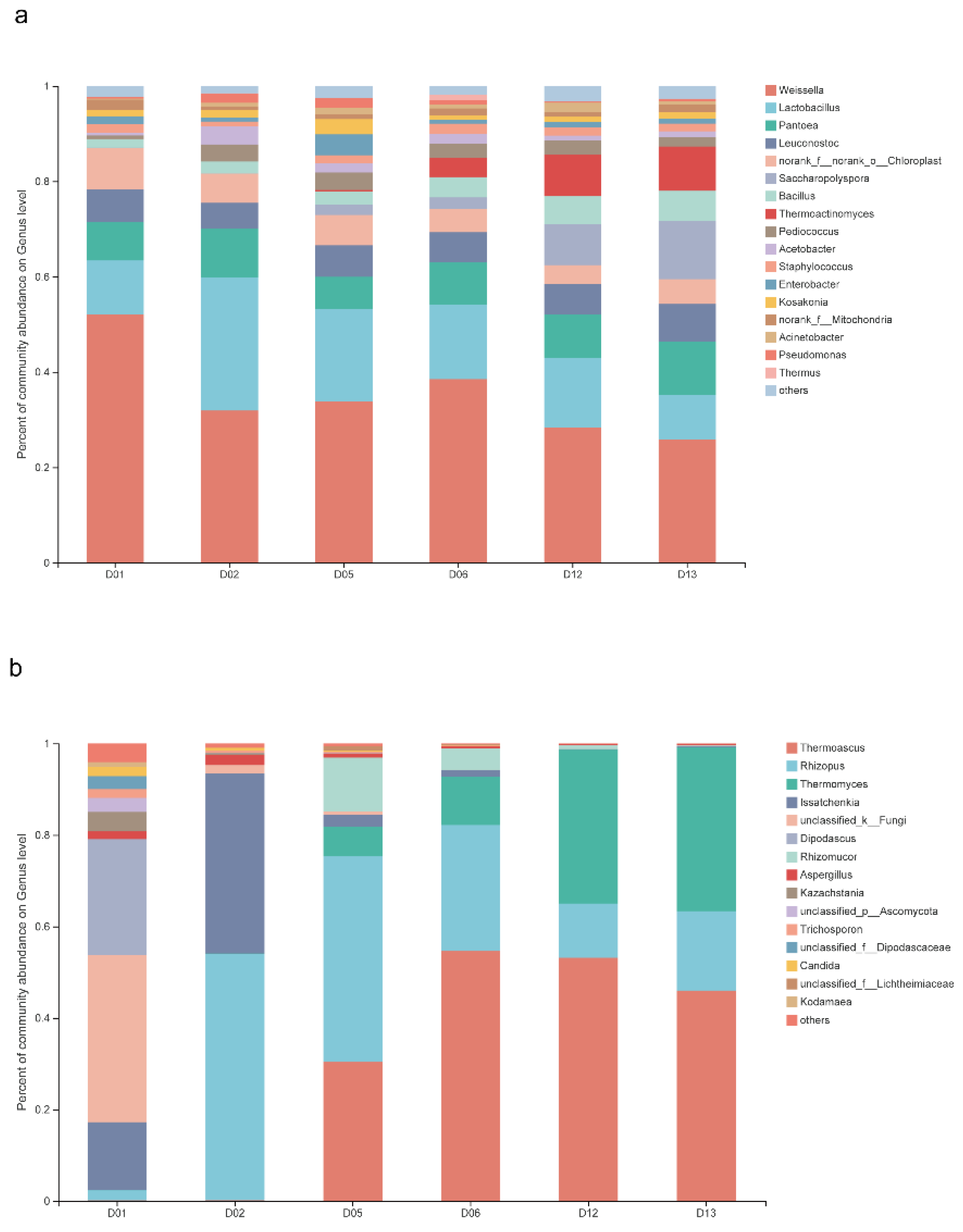
3.2. Structural Characteristics of Functional Microbiota
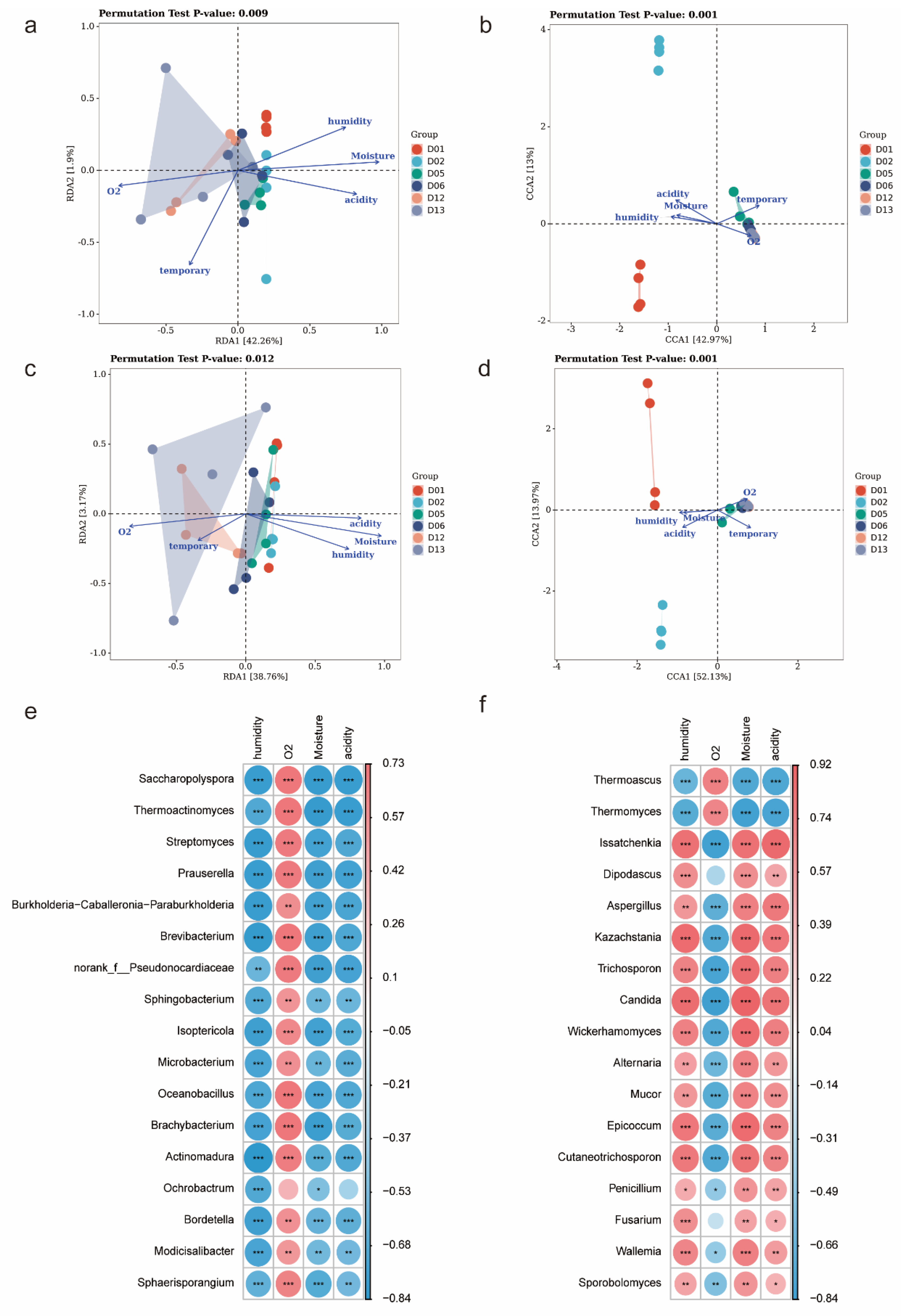
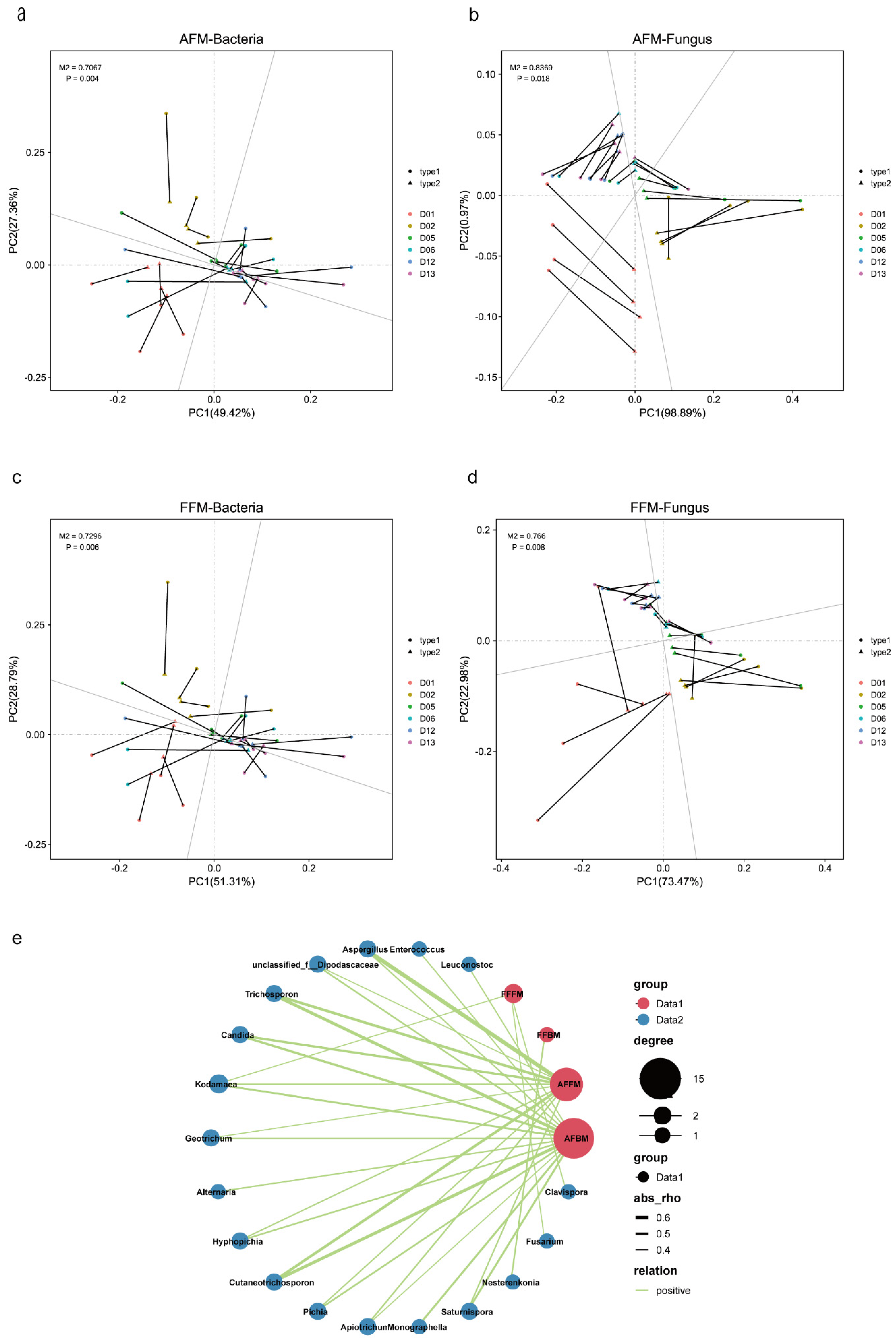
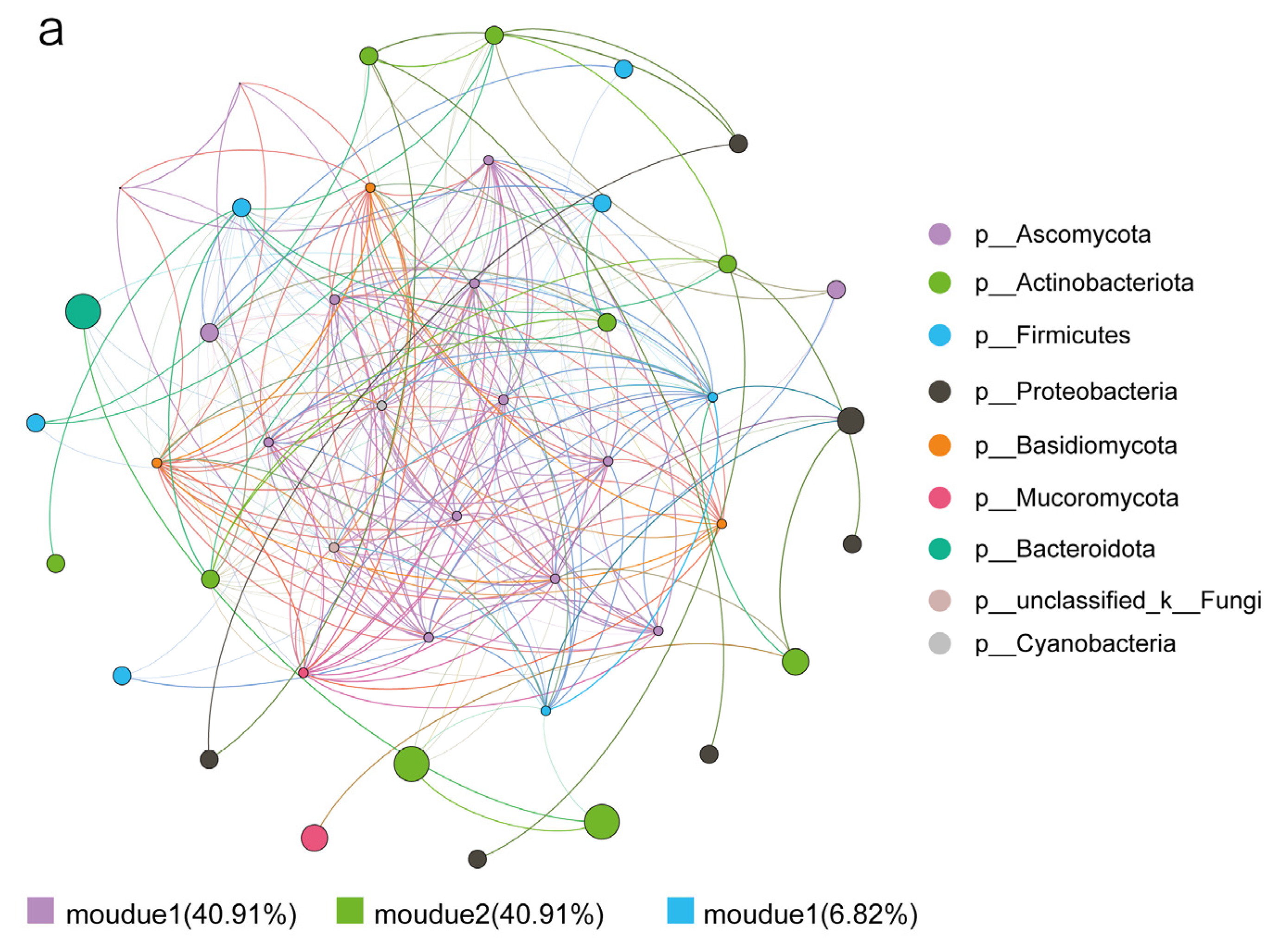
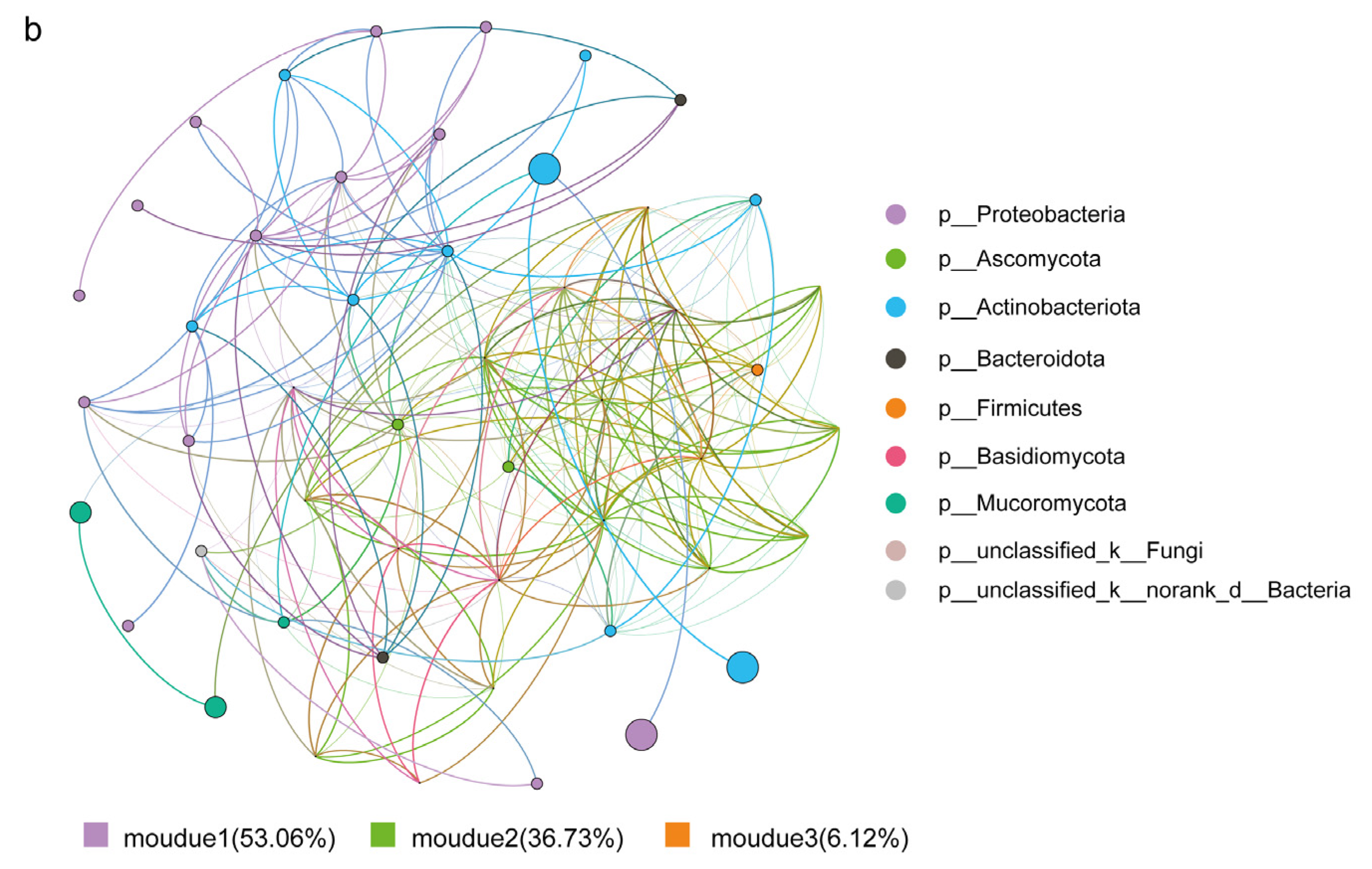
3.3. Effects of Biotic and Abiotic Factors on Functional Microbial Communities
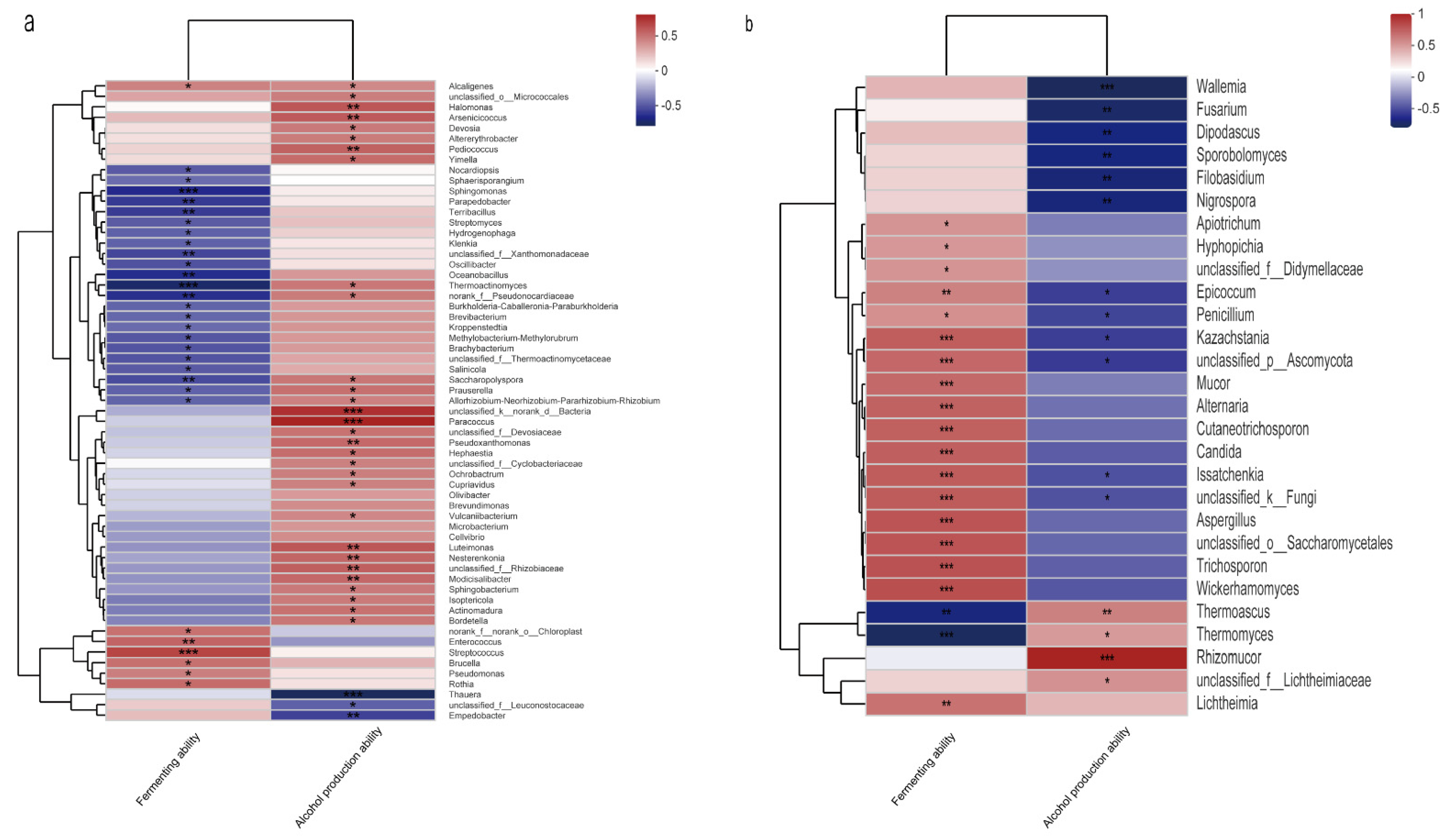
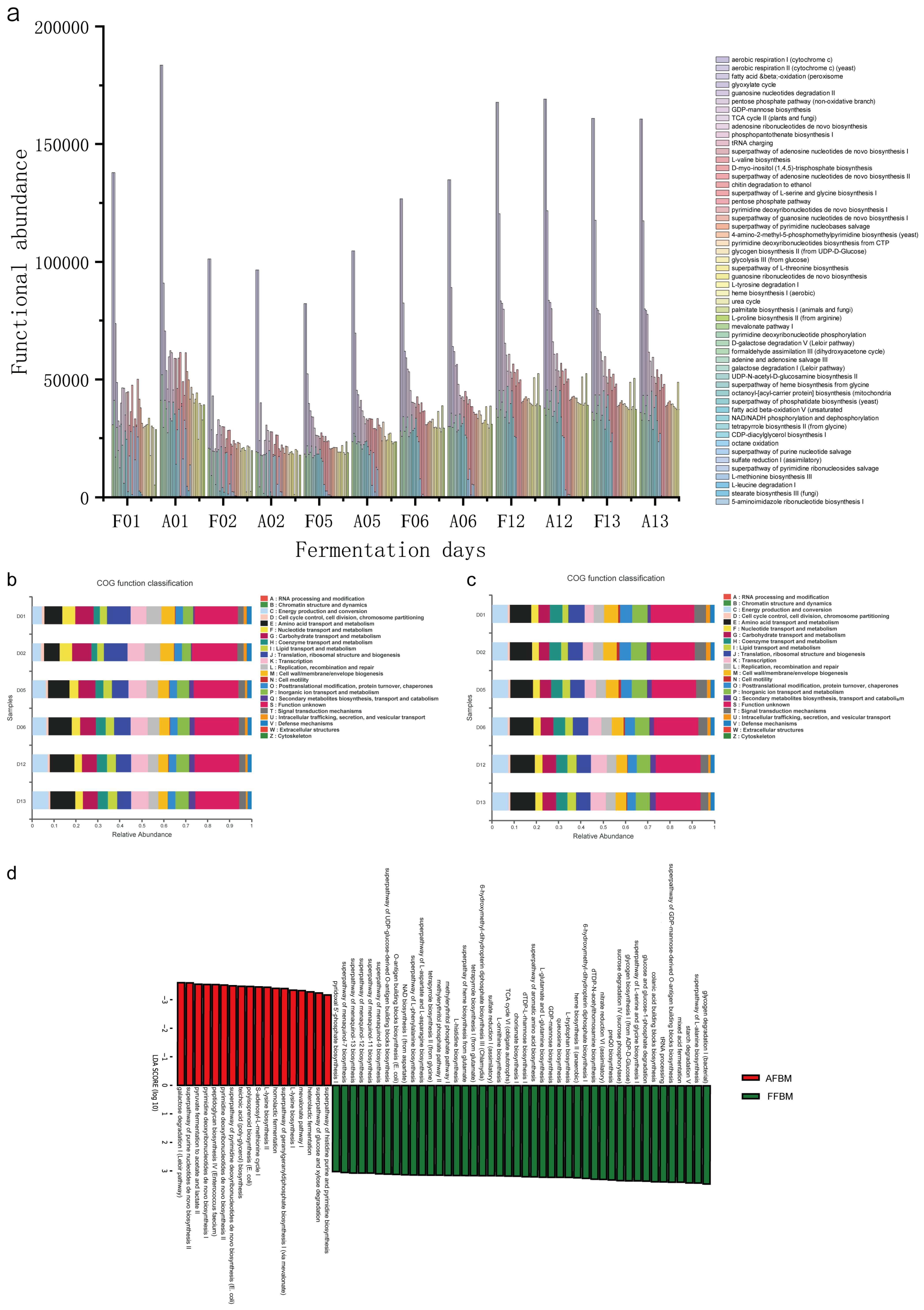
3.4. Potential Functions of the Two Microbial Communities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cao, Z.; Shen, C.; Qin, H.; Wang, S.; Wang, X.; Shen, C.; Wu, J.; Ao, Z.; Wang, C.; Liu, Q.; et al. Relationships between the Aroma of Nongxiang daqu and Its Microbiological, Physiological and Biochemical Indexes. Liquor-Mak. Sci. Technol. 2016, 261, 42–44+48. [Google Scholar] [CrossRef]
- Fan, G.; Fu, Z.; Teng, C.; Wu, Q.; Liu, P.; Yang, R.; Minhazul, K.A.H.M.; Li, X. Comprehensive analysis of different grades of roasted-sesame-like flavored Daqu. Int. J. Food Prop. 2019, 22, 1205–1222. [Google Scholar] [CrossRef]
- Qiao, Z.; Zheng, X.; Shi, S.; Tu, F. Change of physicochemical indexes in the cultivation process of Daqu with different sensory quality. China Brew. 2016, 35, 116–119. [Google Scholar] [CrossRef]
- Deng, L.; Mao, X.; Liu, D.; Ning, X.Q.; Shen, Y.; Chen, B.; Nie, H.F.; Huang, D.; Luo, H.B. Comparative Analysis of Physicochemical Properties and Microbial Composition in High-Temperature Daqu with Different Colors. Front. Microbiol. 2020, 11, 588117. [Google Scholar] [CrossRef]
- He, G.; Dong, Y.; Huang, J.; Wang, X.; Zhang, S.; Wu, C.; Jin, Y.; Zhou, R. Alteration of microbial community for improving flavor character of Daqu by inoculation with Bacillus velezensis and Bacillus subtilis. LWT 2019, 111, 1–8. [Google Scholar] [CrossRef]
- Song, J.; Tang, H.; Liang, H.; Luo, L.; Lin, W. Effect of bioaugmentation on biochemical characterisation and microbial communities in Daqu using Bacillu, Saccharomycopsis and Absidia. Int. J. Food Sci. Technol. 2019, 54, 2639–2651. [Google Scholar] [CrossRef]
- Whittaker, R.A. consideration of climax theory: The climax as a population and pattern. Ecol. Monogr. 1953, 23, 41–78. [Google Scholar] [CrossRef]
- Zhou, N.Y.; Li, L.C. An Ecosocial Climax Model Based on Concepts of Climax Community for Analyzing Communities in Silas Marner to Ensure Protection of Ecosystem. J. Environ. Inform. 2020, 35, 45–55. [Google Scholar] [CrossRef]
- Ma, S.; Luo, H.; Zhao, D.; Qiao, Z.; Zheng, J.; An, M.; Huang, D. Environmental factors and interactions among microorganisms drive microbial community succession during fermentation of Nongxiangxing daqu. Bioresour. Technol. 2021, 345, 126549. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, J.; Xie, J.; Zhao, D.; Qiao, Z.W.; Huang, D.; Luo, H.B. Effects of environmental factors on the microbial community changes during medium-high temperature Daqu manufacturing. Food Res. Int. 2022, 153, 110955. [Google Scholar] [CrossRef]
- Mao, J.; Liu, X.; Gao, T.; Gu, S.; Wu, Y.; Zhao, L.; Ma, J.; Li, X.; Zhang, J. Unraveling the correlations between bacterial diversity, physicochemical properties and bacterial community succession during the fermentation of traditional Chinese strong-flavor Daqu. LWT 2022, 154, 112764. [Google Scholar] [CrossRef]
- Shen, Y. Whole Book of Baijiu Production Technology; China Light Industry Press: Beijing, China, 1998; p. 654. [Google Scholar]
- Ni, Z.; Xu, W.; Dou, W.; Xu, H.; Xu, Z. Comparison of Total Microbial DNA Extraction Methods from Solid-culture of Zhenjiang Vinegar. Weishengwu Xuebao 2010, 50, 119–125. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- STACKEBRANDT, E.; GOEBEL, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Bacteriol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Lu, Q.; He, X.; Wang, Y.; Li, H.; Xiao, Q.; Zheng, X.; Lin, R. Pseudomonas sp. TCd-1 significantly alters the rhizosphere bacterial community of rice in Cd contaminated paddy field. Chemosphere 2021, 290, 133257. [Google Scholar] [CrossRef] [PubMed]
- Tachihara, T.; Hashimoto, H.; Ishizaki, S.; Komai, T.; Fujita, A.; Ishikawa, M.; Kitahara, T. Microbial resolution of 2-methylbutyric acid and its application to several chiral flavour compounds. In Flavour Science: Recent Advances and Trends; Bredie, W.L.P., Petersen, M.A., Eds.; Developments in Food Science; Elsevier: Amsterdam, The Netherlands, 2006; Volume 43, pp. 97–100. [Google Scholar]
- Zhang, Y.; Shen, Y.; Cheng, W.; Wang, X.; Xue, Y.; Chen, X.; Han, B.Z. Understanding the Shifts of Microbial Community and Metabolite Profile from Wheat to Mature Daqu. Front. Microbiol. 2021, 12, 714726. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Miao, L. Multiple Batches of Fermentation Promote the Formation of Functional Microbiota in Chinese Miscellaneous-Flavor Baijiu Fermentation. Front. Microbiol. 2020, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Xu, Y.; Xin, C.; Xu, L.; Liu, Y.; Li, H.; Li, J.; Zhao, J.; Cheng, C. Genome Sequence of Thermoactinomyces daqus H-18, a Novel Thermophilic Species Isolated from High-Temperature Daqu. Genome 2015, 3, e01394-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.P.; Mao, J.; Liu, Y.Y.; Meng, X.Y.; Ji, Z.W.; Zhou, Z.L.; Ai-lati, A. Bacterial succession and the dynamics of volatile compounds during the fermentation of Chinese rice wine from Shaoxing region. World J. Microbiol. Biotechnol. 2015, 31, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Xue, Y.; Wang, Y.; Wang, W.; Shu, N.; Zhao, H.; Tang, F.; Yang, X.; Guo, Z.; Shan, C. The Fungal Communities and Flavor Profiles in Different Types of High-Temperature Daqu as Revealed by High-Throughput Sequencing and Electronic Senses. Front. Microbiol. 2021, 12, 784651. [Google Scholar] [CrossRef]
- Wang, H.; Huang, Y.; Huang, Y. Microbiome diversity and evolution in stacking fermentation during different rounds of Jiang-flavoured Baijiu brewing. LWT 2021, 143, 111119. [Google Scholar] [CrossRef]
- Xiao, C.; Yang, Y.; Lu, Z.M.; Chai, L.J.; Zhang, X.J.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Daqu microbiota exhibits species-specific and periodic succession features in Chinese baijiu fermentation process. Food Microbiol. 2021, 98, 103766. [Google Scholar] [CrossRef]
- McClendon, S.D.; Batth, T.; Petzold, C.J.; Adams, P.D.; Simmons, B.A.; Singer, S.W. Thermoascus aurantiacus is a promising source of enzymes for biomass deconstruction under thermophilic conditions. Biotechnol. Biofuels 2012, 5, 54. [Google Scholar] [CrossRef]
- Jin, Y.; Li, D.; Ai, M.; Tang, Q.; Huang, J.; Ding, X.; Wu, C.; Zhou, R. Correlation between volatile profiles and microbial communities: A metabonomic approach to study Jiang-flavor liquor Daqu. Food Res. Int. 2019, 121, 422–432. [Google Scholar] [CrossRef]
- Chen, B.; Wu, Q.; Xu, Y. Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 2014, 179, 80–84. [Google Scholar] [CrossRef]
- Yi, Z.; Jin, Y.; Xiao, Y.; Chen, L.; Tan, L.; Du, A.; He, K.; Liu, D.; Luo, H.; Fang, Y.; et al. Unraveling the Contribution of High Temperature Stage to Jiang-Flavor Daqu, a Liquor Starter for Production of Chinese Jiang-Flavor Baijiu, With Special Reference to Metatranscriptomics. Front. Microbiol. 2019, 10, 472. [Google Scholar] [CrossRef]
- Jood, I.; Hoff, J.W.; Setati, M.E. Evaluating fermentation characteristics of Kazachstania spp. and their potential influence on wine quality. World J. Microbiol. Biotechnol. 2017, 33, 129. [Google Scholar] [CrossRef]
- Wang, X.; Du, H.; Zhang, Y.; Xu, Y. Environmental Microbiota Drives Microbial Succession and Metabolic Profiles during Chinese Liquor Fermentation. Appl. Environ. Microbiol. 2018, 84, e02369-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Wang, S.; Zhou, R.; Hu, X.; Chu, Y.; Wang, T. Characteristics of yeast flora in Chinese strong-flavoured liquor fermentation in the Yibin region of China. J. Inst. Brew. 2016, 122, 517–523. [Google Scholar] [CrossRef]
- Ma, S.; Shang, Z.; Chen, J.; Shen, Y.; Li, Z.; Huang, D.; Luo, H. Differences in structure, volatile metabolites, and functions of microbial communities in Nongxiangxing daqu from different production areas. LWT 2022, 166, 113784. [Google Scholar] [CrossRef]
- Hong, M.; Li, J.; Chen, Y.; Qi, B.; Huang, Y.; Wu, J.; Yue, H.; Tong, Z.; Liu, Y.; Wang, F. Impact of mixed non-Saccharomyces yeast during fermentation on volatile aroma compounds of Vidal blanc icewine. LWT 2021, 145, 111342. [Google Scholar] [CrossRef]
- Martins, M.; Henriques, M.; Azeredo, J.; Rocha, S.M.; Coimbra, M.A.; Oliveira, R. Candida species extracellular alcohols: Production and effect in sessile cells. J. Basic Microbiol. 2010, 50 (Suppl. S1), S89–S97. [Google Scholar] [CrossRef]
- Li, P.; Lin, W.; Liu, X.; Wang, X.; Luo, L. Environmental Factors Affecting Microbiota Dynamics during Traditional Solid-state Fermentation of Chinese Daqu Starter. Front. Microbiol. 2016, 7, 1237. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, W.; Wang, W.; Shu, N.; Zhang, Z.; Hou, Q.; Shan, C.; Guo, Z. Analysis of microbial diversity and functional differences in different types of high-temperature Daqu. Food Sci. Nutr. 2021, 9, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.M.; Dourado, M.N.; Araujo, W.L. Microbial interactions: Ecology in a molecular perspective. Braz. J. Microbiol. 2016, 47 (Suppl. S1), 86–98. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Chen, J.; Chen, D.; Li, Z.; Huang, D.; Luo, H. Structural Characteristics and Formation Mechanism of Microbiota Related to Fermentation Ability and Alcohol Production Ability in Nongxiang Daqu. Foods 2022, 11, 2602. https://doi.org/10.3390/foods11172602
Tang J, Chen J, Chen D, Li Z, Huang D, Luo H. Structural Characteristics and Formation Mechanism of Microbiota Related to Fermentation Ability and Alcohol Production Ability in Nongxiang Daqu. Foods. 2022; 11(17):2602. https://doi.org/10.3390/foods11172602
Chicago/Turabian StyleTang, Jie, Jie Chen, Deming Chen, Zijian Li, Dan Huang, and Huibo Luo. 2022. "Structural Characteristics and Formation Mechanism of Microbiota Related to Fermentation Ability and Alcohol Production Ability in Nongxiang Daqu" Foods 11, no. 17: 2602. https://doi.org/10.3390/foods11172602