
Review of the Hydrogen Evolution Reaction—A Basic Approach



Department of Engineering Science, University of Agder, 4879 Grimstad, Norway
*
Author to whom correspondence should be addressed.
Energies 2021, 14(24), 8535; https://doi.org/10.3390/en14248535
Submission received: 27 November 2021
/
Revised: 14 December 2021
/
Accepted: 15 December 2021
/
Published: 17 December 2021
(This article belongs to the Special Issue Recent Advances of Catalysts for Hydrogen Evolution Reaction in Water Splitting)
Abstract
:An increasing emphasis on energy storage has resulted in a surge of R&D efforts into producing catalyst materials for the hydrogen evolution reaction (HER) with emphasis on decreasing the usage of platinum group metals (PGMs). Alkaline water electrolysis holds promise for satisfying future energy storage demands, however the intrinsic potential of this technology is impeded by sluggish reaction kinetics. Here, we summarize the latest efforts within alkaline HER electrocatalyst design, where these efforts are divided between three catalyst design strategies inspired by the three prevailing theories describing the pH-dependence of the HER activity. Modifying the electronic structure of a host through codoping and creating specific sites for hydrogen/hydroxide adsorption stand out as promising strategies. However, with the vast amount of possible combinations, emphasis on screening parameters is important. The authors predict that creating a codoped catalyst using the first strategy by screening materials based on their hydrogen, hydroxide and water binding energies, and utilizing the second and third strategies as optimization parameters might yield both active and stable HER catalyst materials. This strategy has the potential to greatly advance the current status of alkaline water electrolysis as an energy storage option.
1. Introduction
The hydrogen evolution reaction is an important electrochemical process with its critical roles in water electrolyzers, the chlor-alkali industry and in chlorate cells [1,2], and while it has been studied for over a century, many of its intrinsic qualities remain elusive.
This review will explore the realm of catalyst design pertaining to the alkaline hydrogen evolution reaction. The current trend within both acidic and alkaline water electrolysis research is to move away from using traditional platinum group metals (PGMs) to using more cost-efficient options employing earth-abundant elements. This review will focus on existing efforts applied to creating such materials, with emphasis on activity and stability. There are three main strategies for creating HER catalysts, namely the modification of a material’s electronic structure, creating specific sites for hydroxide and hydrogen adsorption, and altering the surface of a catalyst [3]. Existing efforts are presented and divided between the aforementioned categories followed by a perspective and conclusion, where the future of catalyst design pertaining to the HER is briefly discussed and summarized.
The commonly accepted pathway for the evolution of hydrogen involves the Volmer reaction in Equation (1), followed either by the Heyrovsky or the Tafel reaction, as seen in Equations (2) and (3) respectively, and illustrated in Figure 1 [4]. Water molecules are reduced in alkaline solutions to produce adsorbed hydrogen atoms () on active metal sites (M) and hydroxide ions.
Depending on low/high coverage of adsorbed hydrogen (), the reaction proceeds through either the Heyrovsky/Tafel reaction, respectively. At low values, an adsorbed hydrogen atom reacts with a proton or a water molecule and an electron to create molecular hydrogen and free up one active site (M).
High values increase the probability that two hydrogen atoms are adsorbed onto neighboring sites, resulting in subsequent recombination to form molecular hydrogen and free up two metal sites, as shown in Equation (3) and schematically exhibited in Figure 1.
Studies on both the HER and the complimentary hydrogen oxidation reaction (HOR) revealed notably slower reaction kinetics in alkaline electrolytes compared to that of acidic ones. Using the exchange current density (ECD) as a measuring factor, the alkaline HER was found to lag by approximately two orders of magnitude [6].
A study on the pH dependence of the HER/HOR investigated the fundamental electrochemistry and physics of each part of the two reactions as a function of pH [7]. At equilibrium, the thermochemistry of protons and electrons are assumed to be independent of coverage, adsorption energy, and the entropy of adsorbed hydrogen (supported by cyclic voltammograms of under-potential deposited hydrogen the {111} facet of platinum), implying that the hydrogen-binding energy (relative to ) is almost unaffected by electrode potential and is possibly only weakly dependent on the electrical field (supported by density functional theory (DFT) simulations). At a set potential, a change in the pH would be analogous to a change in the dipole and the associated field, as it changes to match the solvation energy of the protons in the bulk solution. This means that the proton transfer’s pH dependency must originate from some intermediary state between when the solvated proton is located in the bulk of the electrolyte and when it becomes adsorbed onto the metal surface. This intermediary position is labeled the outer Helmholtz layer (OHL), where the proton is neither in the bulk or nor on the metal surface. Atomic vibrations in the OHL are independent of the pH, a relation which extends to the configurational entropy; thus, a majority or part of the entropy of the proton must be lost prior to reaching this intermediary stage, as shown in Figure 2a, where the -TS term increases. While in this intermediary stage, the change in the electrostatic potential is not yet sensed by the proton, which results in a free energy that is greater in the OHL than in the bulk/on the surface (Figure 2b). This increase originates in the large potential difference located in the confined space between the first layer of water molecules and the metal surface. The free energy in the OHL is interpreted as an estimate of the transition state energy. Considering that a greater entropic stabilization is required for at high pHs to surmount this barrier, the protons will lose more entropy when approaching the OHL in alkaline situations and gain more enthalpy in the proton-surface interaction when exiting the OHL. The barrier in the transition state shown in Figure 2b is primarily summarized by the prefactor from the Arrhenius equation.
The hydrogen-binding energy (HBE) calculated for a series of common PGM catalysts (Rh/C, Pd/C, Ir/C and Pt/C) were linearly correlated over a wide pH-range, causing the authors to suggest that the pH-dependence of the HBE was independent of the metal [8]. Indications that the four catalysts followed the same fundamental- and rate-limiting steps were established based on the correlation between the ECD and the HBE, as seen in Figure 2c, signifying that the HBE may be used as a descriptor of HER/HOR activity.
A third theory is that the dissociation of water carries an addition energy barrier that determines the rate of the HER [9,10]. The exact cause of the poor HER kinetics under alkaline conditions is still unknown, and experimental/theoretical findings support all three theories, hence there is uncertainty as to which of these theories comes closest to describing the cause.
Technology associated with anion exchange membrane (AEM) fuel cells and electrolyzers experienced a great progression over the last 10 years, making the lack of corresponding progress in the development of the associated catalytic materials a major disappointment [11,12]. The lack of breakthrough performances in HER/HOR catalysis can somewhat be attributed to a trial and error-based selection of materials [10]. A methodical study of trends in fundamental properties at the atomic scale of these catalytic environments on thoroughly characterized materials was lacking until fairly recently.
This might seem contradictory to what one usually reads in various papers reporting HER catalyst materials, as many report performances equal to or greater than the platinum standard, thereby implying the imminence of a turn-key solution to the replacement of platinum and a complete elucidation of the alkaline HER. However, there is much work left to do, as will be illustrated here [13].
The three preceding theories represent efforts of such methodical studies. Following these theories are three strategies for creating HER catalyst materials and also the structure this review adopted. There are many methods available to create novel catalyst materials, such as “Modifying the Electronic Structure” (elemental doping, alloying), “Creating Specific Sites for Hydroxide and Hydrogen Adsorption” (phase regulation and hybridising 2D substrates) and “Altering the Surface of a Catalyst” (defect, facet, and interface engineering and use of coatings) [3,14]. These three strategies may be realized by following the more specific routes parenthesized, however, none of these methods are one-way tickets to achieving the unparenthesized goal. Electrocatalysis is highly nonlinear in the sense that a route or procedure more commonly applied to, e.g., changing the electronic structure of a material can also be applied to changing surface-related aspects, or more likely, both simultaneously. While the three ensuing sections “Modifying the Electronic Structure”, “Creating Specific Sites for Hydroxide and Hydrogen Adsorption”, and “Altering the Surface of a Catalyst” refer to specific aspects of HER catalysis, the resulting performance of the catalyst will to some degree be a function of changes involving all three aspects.
2. Modifying the Electronic Structure
The electronic structure of a catalyst is of principal importance as it is pivotal in determining many material characteristics, such as the adsorption potential for possible intermediates and the strength of the resulting bond [7,15]. For the HER, the usual intention for modifying the electronic structure of a catalyst lies in changing its HBE.
Modern techniques such as elemental doping revealed several transition metal-based groups with a favorable electronic structure for the HER, such as phosphides, nitrides, chalcogenides, and dichalcogenides [16,17,18,19]. Single heteroatoms were widely employed with various materials to create the aforementioned material groups, however doping with multiple elements (codoping) represents a new group with exciting results [16]. These groups all opened up many possibilities, where previously defined material groups are being combined with one another to create amalgamations, thereby erasing former boundaries.
2.1. Alloys and Oxides
“A shift in the materials d-band …” and other similarly phrased sentences are a common sight in research papers featuring analysis of catalyst materials. The state of the d-band in a metal catalyst is highly associated with its hydrogen adsorption ability, where an almost empty d-band implies a strong hydrogen adsorption bond, and likewise, the opposite [15,20].
Experimentally measured ECDs as a function of hydrogen bond strengths calculated through density functional theory (DFT) produced the volcano plot in Figure 3. This shows how hydrogen evolution activity depends on the hydrogen bond strength. The left-side, containing typical transitional metals (Ni, Mo, Co, W), binds hydrogen too strongly causing the surface to fill up and a lack of available sites for further adsorption of hydrogen, thereby resulting in a decline in reaction speed.
Conversely, noble metals (Au and Ag) bind hydrogen too weakly. As the hydrogen-metal bond strength decreases, the difficulty in transferring proton increases due to the unstable bond in the surface-adsorbed hydrogen. Pt and Ir reside at the top because all steps of the HER proceed at an almost thermoneutral overpotential. Generally, one can summarize that most feasible materials would produce poor catalysts for the alkaline HER.
To circumvent this issue, one can combine two or more elements, in which the strain effect due to modified bond lengths and the ligand effect arising from the forming of heteroatom bonds contributes to improved electrocatalytic activity and stability [21,22]. From Figure 3, it follows that combining two metals from opposite sides of the plot may result in an alloy placed in the center around the likes of Pt and Ir in terms of HBE, which was proven true [23]. This is likely due to a mass electron transfer between active sites on the two metal atoms, making this a highly efficient way of modifying the electronic structure of a catalyst. Such a catalyst was predicted and created by optimizing the Ti-content in a CuTi alloy, where the optimized HBE caused by the alloying effect was key to its improved performance [23]. DFT calculations revealed 5 at.% Ti results in near-optimum HBEs at two Cu-Cu-Ti hollow sites, where further increments/reductions of the Ti-percentage lead to the introduction of inactive Cu-Ti-Ti sites and a lower quantity of the active Cu-Cu-Ti sites, as shown in Figure 4.
An alloy of nickel and cobalt was electrodeposited onto titanium foil for varied time intervals resulting in several HER alloy catalysts with varied composition, fair activity, and stability [24]. While falling short of matching the 40 wt.% Pt/C benchmark in terms of activity, the alloy succeeded in displaying satisfactory stability as exhibited through a 150 h chronoamperometry test at 10 mA cm. This caused a 12% loss of performance, which was attributed to passivation and a loss of the metallic oxidation state as determined through X-ray diffraction (XRD). A different report on the creation of a nickel-based catalyst underlined the importance of the substrate, as the oxidized carbon nanotube (CNT) substrate interacted with the precursor during thermal decomposition, which retarded the oxidation process of nickel, which in turn slowed down the reduction of nickel agglomeration through Ostwald ripening [25]. As shown in Figure 5a, there are a variety of catalyst decomposition processes, and all featured materials will to some degree be subject to these. Poor interaction between catalyst and substrate/support can result in agglomeration and detachment, whereas the intrinsic properties of the material will determine the degree to which dissolution may come into play. Figure 5b shows a Pourbaix diagram of some common PGM and non-PGM catalyst materials in typical water electrolyzer conditions for both the oxygen evolution reaction (OER) and the HER. The use of Pourbaix diagrams are paramount for evaluating thermodynamic stability as a function of key variables in electrochemistry.
A series of transition metal alloys (MNi (M = Cr, Mo and W; x ≈ 0.2)) were created and examined in both acidic and alkaline conditions, revealing surprisingly good activity for the CrNi alloy under alkaline conditions [26]. The activity of the catalysts followed the order CrNi> MoNi> WNi, where the activity of the chrome nickel alloy was partially attributed to the HBE influencing the binding energy of hydrogen. This work alluded to the possibility that the alkaline HER is primarily a function of the binding energy of hydrogen and hydroxide in addition to the water dissociation energy.
A variation of the ever-popular NiMo alloy was created by Faid et al. [27] through an ambient temperature chemical reduction process of precursors containing nickel and molybdenum in presence of sodium borohydride and tested in an AEM electrolysis cell. The increase of catalytic activity corresponded with the increment of molybdenum, and the reaction proceeded through the Volmer–Heyrovsky pathway. While the catalyst failed to match the activity of the 60 wt.% Pt/C benchmark by factor of ∼5, the calculated cost reduction for replacing the platinum (× mg cm) electrode with with the NiMo catalyst (5× mg cm) would amount to a 60× cost reduction based off 2018 material prices. Short-term stability was assessed through an ex situ 20 h chronoamperometry (CA) at −0.20 V (∼14 mA cm) where the NiMo exhibited good activity with no clear sign of the instabilities that infamously plague this material combination [28,29].
The stability of a NiMo combination (NiMo) was elucidated in a recent theoretical study [28], where the initial framework [30] for theoretically predicting the aqueous stability of solids was constructed by employing the strongly constrained and appropriately normed (SCAN) functional in tandem with data from “The Materials Project” [31]. Accelerating the progress of material science was the motivation for starting up this project back in 2011. By giving public access to computational tools and creating a data depository for both theoretical and experimental material properties, numerous materials were created and many advances were made. Moreover, such large datasets are highly useful as training sets for machine learning [32]. The NiMo alloy was seen to decompose to MoO and Ni(s) in the potential range of −0.10–0 V, and MoO and Ni(OH) at potentials >0 V as seen in Figure 6a. Moreover, the Pourbaix decomposition energy () which measures material stability increases drastically at positive potentials, where this thermodynamic driving force was key to the leaching issue of the alloy together with the intrinsic instability of the material; however, the latter was most influential at open circuit potential. The dissolution of the alloy was verified by analyzing the content of the electrolyte using inductively coupled plasma mass spectrometry (ICP-MS), which corroborates other experimental work [29]. The complete SCAN-determined Pourbaix diagram is shown in Figure 6b, where the difference between the experimental and the SCAN-determined diagram was minimal, further proving the accuracy of this functional over the popular Perdew–Burke–Ernzerhof (PBE) functional and the possibilities affiliated with this methodology.
Nickel is a versatile material, and due to favorable interactions with lanthanum cerium oxide, the resulting material was found to be active towards the evolution of hydrogen [33]. The addition of LaCeO increased the active area by halving the nickel particle size and also the intrinsic activity as measured by linear sweep voltammetry (LSV) normalized by the electrochemical active surface area (ECSA). The enhancement of intrinsic activity was brought by the interfacial interactions between the nickel, the oxide, and the carbon support, where nickel provides active sites for hydrogen adsorption and the LaCe oxide supplies active sites for hydrogen desorption while saving the nickel sites from OH poisoning by preferential adsorption. Testing in a complete AEM water electrolyzer system operating at 45 °C, 500 mA cm (∼1.9V) with a 0.1 M KOH electrolyte for 100 h revealed that the catalyst has real world potential by displaying a promising degradation rate of ∼0.28 mV h. Another example where lanthanum was employed to increase the performance of a transition metal (CoO) is given by Zhao et al. [34], where various percentages of lanthanum doped into CoO were investigated for several annealing temperatures. Similarly to the work by Jang et al. [33], the usage of lanthanum resulted in increased ECSA through decreased morphology in addition to increasing the intrinsic activity of the cobalt oxide. The optimized 2 wt.%La-CoO annealed at 350 °C displayed good stability when measured against the 20 wt.% Pt/C benchmark during a 3.3 h chronopotentiometry test. Stability was assessed through both chronopotentiometry and an accelerated stress test (cyclic voltammetry (CV), 3000 scans), where the benchmark was most notably affected by the former and the investigated catalyst the latter, thereby exemplifying how different materials respond to different types of stability tests.
Surface sulfurized cobalt oxide nanosheets (NSs) on nickel foam (NF) (CoS NS/NF) displayed a morphology of vertically grown NSs, thereby imparting a large ECSA [35]. Falling short of matching the activity of the 40 wt.% Pt/C, the catalyst displayed good stability over 220 h of chronopotentiometry. Moreover, when employed as the cathode catalyst in a single cell AEM water electrolyzer, the material remained stable for 12 h in a 1.0 M KOH electrolyte at 45–48 °C, 500 mA cm (∼2.1V), which illustrates the potential of the catalyst with a degradation rate of 4.2 mV h. The sulfurization process was crucial to the activity of the catalyst, as the sulfur content and the activity increased jointly. Sulfur is a heteroatom [36] which adds electronegativity to the cobalt oxide, thereby modifying the electronic structure of the material by providing active sites for hydrogen adsorption. The use of heteroatoms as dopants and their effects will be outlined in several coming subsections on phosphides, nitrides, and codoping. Mesoporous MoO (mMoO) with oxygen vacancies was reported as an efficient HER catalyst, where the material displayed both activity and stability as measured by Tafel slopes, LSV, electrochemical impedance spectroscopy (EIS) and chronoamperometry (CA) [37]. Displaying a Tafel slope of 56 mV dec and stability over 12 h of CA (@ 174 mV), the mMoO outperformed the commercial MoO benchmark (130 mV dec) and approached that of commercial 20 wt.% Pt/C (30 mV dec). Oxygen vacancies adjacent to Mo sites were active towards hydrogen evolution as determined from DFT calculations, where the vacancies contributed to lowering the kinetic boundary for water adsorption () and inducing H-H bond formation, thus easing hydrogen desorption.
Thousands of 2D MXO, M-MXO and M’-MXO (M = 3d, 4d, 5d transition metals, X = nitrogen or carbon) catalysts were thoroughly theoretically investigated through combining DFT with a machine learning scheme to rapidly classify materials in regard to activity and stability [38]. Finding key descriptors for a good HER catalyst was one of the primary goals, and through the analysis the initial 63 descriptors were whittled down to 5. These descriptors, in descending order of importance, are the O-M bond length, the O-O bond length, the difference in ionization energy, and the average affinity energy of the alloy elements and the valence electrons of X (nitrogen or carbon).
These descriptors were explained by the following: Adsorption of hydrogen onto an oxygen site causes the hybridized H 1s and O 2p orbitals (hybridized due to alloying) to divide into filled bonding orbitals and the partially filled antibonding orbitals . Secondary order charge density difference and Bader analysis exhibited that the alloying effect in addition to the adsorbed hydrogen atom induced a charge transfer from the d-block metal (M) to both the carbon and oxygen, proving that alloying can be used to tailor charge transfer characteristics. The varied atomic radii of the various elements comprising MXO, M-MXO and M’-MXO strains the lattice, thereby affecting the O-M and O-O bond strengths. Furthermore, alloying causes a difference in the hybridization of the metal d-orbital and the oxygen p-orbital, which in turn influences the state of the oxygen antibonding orbital . Weak binding strength is a result of filled orbitals. Projected density of state (PDOS) analysis alludes to the possibility that the alloy d-bands and the oxygen p-bands adjust both shape and width of the energy band, in addition to the charge density near the Fermi level. Additionally, comparing the charge transfers between M-C and M-N materials reveals that the process is more efficient in the latter, meaning that a change of activity is easier experienced with nitrogen than carbon, an effect which is partially attributed to the effect of the valence electrons.
The predictions from this work were experimentally corroborated for both mono-metallic TiCO ( = 0.013 eV, 0.5 H-coverage) and the binary alloy MoTiCO ( = 0.058 eV, 0.25 H-coverage). One-hundred-and-ten catalysts that exceeded the performance of platinum were predicted from this work, displaying the great potential associated with such strategies.
While perovskites are traditionally associated with OER catalysts, an emerging research trend proved they are also active towards the HER [39,40]. While the perovskite (Pr(BaSr)Co-FeO (PBSCF)) did not come close to matching the activity of the 20 wt.% Pt/C, the stability was significantly greater as displayed by 50 h of chronopotentiometry at 50 mA cm and 1000 CVs. The stability of the perovskite PBSCF was attributed a greater ECSA, more efficient electron transfer, the partially oxidized cobalt on the catalyst surface and the augmented aggregation of lattice-located oxygen, all of which was partially affiliated with the praseodymium doping.
2.2. Phosphides
Another group of materials that garnered attention due to their electronic structure is transition metal phosphides (TMPs) [16].
The stability and activity of two nickel phosphide catalysts (NiP and NiP) was thoroughly researched both experimentally and theoretically as the preconceived notion of poor activity from a NiP catalyst was disproved [41]. The ionic Ni-P bonding influenced material characteristics to overcome issues related to activity and stability. Experimental stability evaluated by chronopotentiometry (CP) and ICP-MS show that NiP is stable in alkaline media with no discernible leaching issues. The stability and activity was also analyzed theoretically by examining the electron localization function (ELF) as determined through DFT, where a delocalized cloud corresponding to a metallic bonding was seen in the majority of the areas between atoms as seen in Figure 7a, corroborating the metallic conductivity indicated by the measured resistivity of NiP (4 × 10 m). An ionic contribution was also determined through Bader charge analysis, which may substantiate the increased stability of NiP over Ni(s) in both alkaline and acid media.
Furthermore, the point wave function was calculated to define which electronic states would become occupied upon polarization of the electrode. The electronic energies of 0.1 and −5 eV relative to the Fermi level were chosen as they coincide with where the phosphorous 3p and nickel 3d states overlap both spatially and energetically, as determined by calculating the density of states (DOS). Additionally, 0.1 eV corresponds to the lowest unoccupied molecular orbital, i.e., the first electronic states to be filled upon polarization. As seen in Figure 7b, positive and negative wave functions extend from nickel and phosphorous respectively, revealing the nature of the state as localized and antibonding. When these antibonding states are filled during polarization, the Ni-P bond is weakened and the density of electrons on both atoms is increased, which in turn creates additional hydrogen adsorption sites. The calculated HBE at these sites was near the thermoneutral potential with = 0.04 eV. The band at −5 eV vs. the Fermi level displays an in-phase interaction between two nickel atoms and a phosphorous atom that is delocalized, which confirms the delocalized metallic bond foreseen by the aforementioned ELF results. Generally, the bonding between the nickel and phosphorous was predominantly metallic with influences of ionic bonding which contributed to the stability of the NiP catalyst in both alkaline and acidic media. These bonds were weakened during HER conditions, thereby allowing the HBE to approach thermoneutrality. While the content of phosphorous in NiP was notably lower than in NiP, the material still retained approximately the same stability vs. Ni(s) as defined by comparing their respective enthalpies of formation.
The use of in situ X-ray absorption spectroscopy (XAS), in situ Raman spectroscopy and liquid-phase transmission electron microscopy (LP-TEM) was employed to describe the highly dynamic structural changes occurring during the alkaline HER on both a P-doped CoSe and a CoSe catalyst [42]. The former was unstable in 1.0 M KOH, where the formation of Co(OH) and dissolution of the catalyst was clearly visible in both images and video from in situ scanning transmission electron microscopy (STEM) as seen in Figure 8a. Small nanoparticles around 10 nm wide corresponding to Co(OH) (confirmed using high-resolution transmission electron microscopy (HRTEM)) are formed around the catalyst center. The video of the CoSe catalyst displays significantly greater stability, however, the affiliated activity is low. The rapid (<60 s) structural conversion of the P-doped CoSe during alkaline submersion was possibly due to the preferential dissolution of phosphorous on the CoP phase in alkaline electrolyte. Moreover, X-ray absorption near edge structure (XANES) spectra of both materials in Figure 8b display an increase in energy after contact with KOH, indicative of partial oxidation of the cobalt, which corresponds well the STEM images below and in situ Raman spectroscopy. Additionally, the resulting XANES spectra from increased cathodic polarization shows a more aggressive decline where the oxidation states of the P-doped material resemble that of cobalt foil. This suggests that the real active sites are not in P-CoSe, but rather in metallic cobalt. The phosphidation process introduces a number of defects and vacancies into the CoSe, which accelerates the conversion from the “pre-catalyst” form P-CoSe to the active metallic cobalt state. Such use of in situ spectroscopic techniques can be most useful to quantify the origin of degradation mechanisms and the sources of catalytic activity, where the continued development of these methods will be highly influential.
Another great example of a phosphide is provided by the (FeNi)P nanosheets created by Yang et al. [43]. The optimized (FeNi)P nanosheets displayed great activity partially due to the small amounts of iron which increased the charge accumulation around the phosphorus centers. The difference between calculated charge densities around a phosphorus center is displayed by Figure 9a,b. These figures illustrate that the accumulation of charge (yellow) around hydrogen and depletion of charge (cyan) around phosphorus is greater in the iron-doped catalyst shown in Figure 9b, compared to that of the nondoped NiP catalyst in Figure 9a. In other words, iron-doping increased the charge transfer rate between the active phosphorus-centers and the adsorbed hydrogen atom. Optimized amounts of iron in the phosphide catalyst greatly improved the electronic structure of the nanosheets, thereby decreasing both the hydrogen adsorption energy and the dissociation energy of water (Figure 9c).
While some research groups attempted to create a novel catalyst without the use of PGMs, others compromised and attempted to lower the PGM-loading. An example of this is where Pt was doped into the interior of CoP to create a Pt-CoP catalyst [44]. The addition of Pt in CoP modified its electronic structure and created near-thermoneutral sites for HER. Displaying a hydrogen evolution onset potential (the potential required to reach a current density of 10 mA cm) of only 2 mV (with iR-compensation) and a low Tafel slope of 44 mV dec, where the activity of the catalyst and the influence of the PGM is notable. Similar performances were noted in another recent publication noting the creation of a RuP nanofiber catalyst [45]. The low onset potential of 9 mV (with iR-compensation) and a Tafel slope of 38 mV dec are certainly great arguments when debating catalytic activity, however the acumen becomes difficult when the associated stability reflects otherwise. The X-ray photoelectron spectroscopy (XPS) results in Figure 10 shows a notably greater presence of adsorbed hydroxides on the catalyst surface following the stability test, where this likely contributed to the loss of performance. Additionally, the use of PGMs is inadvisable when the intended goal is mass-production given the affiliated scarcity. An AEM electrolysis cell was constructed using the RuP nanofiber catalyst, yielding a performance of 1000 and 2000 mA cm at ∼1.8V and ∼2.1V, respectively.
The use of PGMs extended from platinum to several other PGM alternatives, where a recent publication employed a rhodium precursor (tris(triphenylphosphine)rhodium(I) chloride) in a pyrolysis method resulting in defected RhP nanoparticles, which exhibited activity and stability in an alkaline environment [46]. The P-defected RhP catalyst exhibited lower overpotentials at 10 mA cm compared to that of the commercial Pt/C catalyst (4.3 mV vs. 31.5 mV), despite similar metal loadings (0.111 mg cm vs. 0.114 mg cm). This is in part due to the defect of the catalyst in the form of a missing phosphorus atom, where these specific sites can preferentially adsorb hydrogen atoms at near-optimal conditions. Additionally, the very small proximity between the adsorbed hydrogen atoms at the aforementioned sites allows the hydrogen desorption step to be carried out via the Tafel step, thereby greatly accelerating the HER.
Phosphorus was employed as a dopant in a two-faced molybdenum catalyst, where the resulting 2D nanosheet (NS) morphology increased the number of accessible active sites thereby increasing activity and mass transport characteristics [47]. Additionally, the hydrogen binding energy was improved due to the strong interfacial conductivity of the P-doped material and the engineered porous morphology which increased the number of such sites. The role of morphology was also influential in the beneficial results obtained by Lu et al. [48], where 3D NiCoP nanocubes on 2D NiCoP NSs were created. The strong interfacial contact indicated by shifts in binding energies determined by XPS enabled fast charge transfer processes, and the phosphidation process orchestrated a redistribution of charge, thus endowing the transition metal catalyst with improved hydrogen binding and water dissociation energies.
A series of transition metal-doped (V, Cr(Mo), Mn, Fe, Co, Ni, Cu) cobalt phosphides were created, where a linear correlation was established between catalyst activity and the degree of vacancy in the d-orbitals of the transition metal dopants as displayed in Figure 11a [49]. The water dissociation energy calculated through crystal orbital Hamilton population (COHP) analysis as a function of d-orbital vacancy is shown in Figure 11b, which displays the same linear correlation. This exhibits that the performance increment is predominantly due to increased water dissociation efficiency. No such correlation was established for hydrogen adsorption/desorption characteristics. Additionally, shown in Figure 11c is the energetic pathway for the reaction on simplified CoP {111} and Cr-CoP {111} surfaces for the Heyrovsky route with a preadsorbed hydrogen atom, as determined by COHP analysis.
The stability of a cobalt phosphide prepared through thermal phosphidisation was investigated in both acidic and alkaline electrolytes through subjecting the electrodes to 2000 LSVs [50]. Material stability or lack thereof was confirmed to be pH-dependent where alkaline conditions induced the selective precipitation of phosphorus and the formation of surface cobalt hydroxide, whereas both metals were leached out under acidic conditions. The ECSA of the two electrodes were compared before and after the stability test, and a modest increment was noted for the acidic electrode, however the ECSA of its alkaline counterpart was doubled due to changes in surface composition as revealed by XPS. Normalizing the catalytic activity by the ECSA of the catalysts before and after the stability tests reveals a significant loss of performance. The performance loss for the acidic electrode was partially accredited chemical decomposition in the electrolyte, an aspect which did not notably affect the alkaline electrode. The notable loss experienced by the electrode submerged in the alkaline electrolyte was attributed the stability test, as adsorbed hydroxides can poison a catalyst [22]. Zhang et al. [50] emphasizes the correct use of ECSA normalized material performances, i.e., measuring this aspect both before and after stability assessments to correctly reflect the true activity of a catalyst.
Two different core-shell structured nickel phosphide catalysts (NiP/Ni(PO)) were created through a water in oil microemulsion method, composed of either hollow spheres or solid spheres [51]. While both displayed adequate activity and fair stability when tested in standard ex situ LSV and CP analysis, the activity of the hollow sphere catalyst surpassed that of its more replete counterpart, however, these roles were reversed once the current was normalized for the ECSA. The solid sphere version was speculated to contain a greater degree of NiP, which was determined as the predominant source of catalytic activity. The hollow sphere catalyst was employed as both anode and cathode catalyst in an AEM electrolysis cell, delivering 358 mA cm at 1.8 V and tested at 300 mA cm, 50 °C in 1.0 M KOH for 110 h yielding a degradation rate of ∼0.74 mV h. Post-electrolysis XPS revealed negligible changes in the binding energies of nickel and phosphorous in the cathode catalyst, alluding to the possibility that the majority of the performance loss was due to the anode side.
2.3. Nitrides
Transition metal nitrides represent another group of catalysts that attracted attention as possible non-PGM HER catalysts [52], where an excellent example is given in [53] where self-supported nickel nitride on NF (NF = nickel foam) (NiN/NF) displayed both activity and stability. The excellent stability of the unsupported catalyst was ascertained through CA, cyclic voltammetry and LSVs at 25 °C/60 °C, yielding promising results as displayed in Table 1. Performance loss after potential cycling through 1000 CV scans was ascribed the partial formation of a thin Ni(OH) film replacing the NiN layer, thereby blocking active sites. The film was only detectable through XPS and not XRD, revealing the low thickness of the film. The effect of oxygen and hydroxide adsorption on nitrides was detailed in a recent theoretical publication [54], where it was revealed that these adsorptions caused a downshift in the d-band center of top-layer molybdenum atoms of MoN, thereby decreasing its hydrogen adsorption capabilities.
A second example of is given by Lv et al. [55], where molybdenum nitrides on a nitrogen-doped carbon matrix (MoN@NC) catalyst is reported and displays great activity and solid stability. Molybdenum nitrides have similar electronic structures to noble metals due to the electronegativity of nitrogen, which modifies the d-band density of molybdenum. The MoN@NC catalyst was compared against mixed nitride/carbide compounds and incomplete nitrides, and surpassed their performance in regard to Tafel slopes, exchange current density and charge transfer resistance measured by EIS. Probing the electronic structure with XPS showed successful doping of nitrogen into the carbon matrix and a strong d-band was revealed by Raman spectroscopy. This was due to a notable disorder caused by lattice defects from the nitrogen doping.
N-rich CoS@NC arrays on Ti foil (N-CoS@NC/Ti) were created by Wang et al. [56] where XPS analysis revealed that the addition of nitrogen affected the electron density of cobalt resulting in increased HER activity. Similar conclusions were found using EIS, where nitrogen-rich samples yielded lower charge transfer resistances, implying faster proton/electron transfer rates. Using heteroatoms such as nitrogen, boride, and sulfur as dopants in both catalyst and catalyst support became an established practice in electrolysis and fuel cell electrocatalysis, including the HER/HOR and likewise for oxygen reactions [36,57]. This approach can enhance material characteristics towards adsorption/desorption of reaction intermediates, manage charge density, and increase HER performance [58].
A series of two-phased catalysts composed of intimately coupled MoC and MoN on nitrogen-doped carbon were created by Tang et al. [59] and displayed both activity and stability in an alkaline electrolyte. Distinguished by the molar weight of ammonium molybdate employed in the synthesis process, these materials showed varied activity, though the material composed of 0.5 mmol ammonium molybdate (Mo(CN)) surpassed the activity of 20 wt.% Pt/C benchmark at high current densities (>50 mA cm). This was ascribed to the ubiquitous presence of fine MoC and MoN nanoparticles, which exposed a great number of active sites that were intrinsically more active compared to that of materials with other compositions. The two materials (Mo(CN) and Mo(CN)) with the greatest intrinsic activity displayed XPS peaks corresponding to MoN species, where the second of the two (Mo(CN)) displays the greatest content of this species, alluding to the possibility that the increase of intrinsic activity may be causal to an increase in the content of nitrogen.
Self-supported nickel nitride nanorods on nickel foam (NiN/NF) were created through a simple process where the nanorods were grown directly from the NF with ammonia as a source of nitrogen [60]. HAADF images in Figure 12a show an even distribution of nickel and nitrogen in the nanorods, proving the growth of nanorods was realized. According to DFT calculations, the addition of nitrogen had a notable impact upon the HBE by lowering it from 0.14 (Ni) to 0.08 eV (NiN) as seen in Figure 12b, which was corroborated by ultraviolet photoelectron spectroscopy (UPS) data that displayed a downshift of the d-band energy of NiN relative to Ni.
Guided by theoretical predictions, a catalyst comprising edge-rich cobalt nanoparticles supported on nitrogen-doped reduced graphene oxide (Co-NPs/N-rGO) was created [61]. The material exhibited great stability during chronopotentiometry, by performing at a stable level for 480 h at 10 mA cm and 200 h at 500 mA cm. Additionally, the data of extended X-ray absorption fine structure (EXAFS) spectra were fitted to a DFT model by employing an artificial neural network, allowing the performance of the catalyst to be primarily attributed to the plane model (Co-4N-P). Another cobalt-based single atomic site M-N-C catalyst was investigated, this time employing in situ X-ray absorption fine structure (XAFS) analysis to reveal the active structure of the material during alkaline hydrogen evolution [62]. The valence state of Co increased from +2 to +2.2 and +2.4 during open circuit and cathodic polarisation respectively, a consequence of the preferential adsorption of hydroxide (OH-Co-N) and water (HO-(OH-Co-N)) where the origin of the OH-Co-N sites were determined as the probable source of catalytic activity.
Nickel nanoparticles on vanadium nitride were prepared through thermal ammonolysis, resulting in a highly efficient non-PGM catalyst which matched the activity of the 20 wt.% Pt/C benchmark [63]. The stability of the catalyst was not akin to its activity, as a significant loss of performance was noted during 20 h of chronopotentiometry at 20 mA cm, possibly due to catalyst detachment. Postexperiment XPS displayed little change in the state of the surface valence. The notable activity was attributed to the enhanced ECSA as determined through the double layer capacitance which increased six-fold relative to nickel nanoparticles and vanadium nitride separately.
The stability of M-N-C (M = any metal) catalyst was systematically assessed by Yang et al. [64] for the HER, the OER, the oxygen reduction reaction, the nitrogen reduction reaction and the carbon dioxide reduction reaction. The effects of varying operational parameters such as temperature, pH and overpotential were illuminated together with the associated effects of changing material parameters such as M valence states, porosity, edge density, etc. Loss of metal atoms through leaching and oxidation of the carbon substrate were named as the two principal degradation mechanisms, where they can work in a symbiotic manner. Leaching causes a loss of active sites while accelerating the oxidation of the ionomer, membrane, and carbon support in an MEA, where oxidation of the latter in turn stimulates the loss of active sites. These active sites are primarily composed of MNC moieties, whose stability is affiliated with the durability of the M ions in the actual moieties and their activity in a Fenton-like reaction once leached out of the material [64,65,66].
Describing the in situ stability of such materials is complex, however, the influence of thickness is considerable as the total thickness of the membrane electrode assembly (MEA) is usually at least an entire magnitude greater (∼100 μm) than the thickness of catalyst surfaces in standard ex situ tests on a rotating disc electrode (RDE) set-up, thereby opening up for mass transportation overpotential and gas bubble formation issues. The current density and temperature is usually far greater than those common to ex situ testing (10 mA cm at room temperature), thereby enhancing the affect of both the aforementioned issues. Employing 3D structures such as metal (Ni, Cu, etc.) foams can aid in mitigating such issues [22,64].
2.4. Co-Doping
A multifaceted heteroatom-doped nickel catalyst was first created by Qiao et al. [67], employing both nitrogen and phosphorus simultaneously as dopants (N-P-Ni/C). The electronic structure was tuned by utilizing the difference in electronegativity between the dopants, where phosphorus (electronegativity = 2.19) supplies electrons to nitrogen (electronegativity = 3.04) thereby decreasing the N-Ni interaction. The valence state of the host metal (Ni) decreased relative to the single heteroatom doped N-Ni catalyst due to the additional electrons added by phosphorus into the co-doped catalyst as confirmed through XPS and X-ray adsorption near edge structure (XANES). These interactions resulted in lower HBEs and fast kinetics as exhibited through Tafel slopes and LSV. Moreover, the same codoping synthesis route was employed with cobalt as the host metal and yielded similar results, which proved the potential of this strategy.
A similar design strategy was realized employing both hetero- and metal atoms as codopants, which resulted in a Zn,S-CoP NRCs/CP catalyst (NRCs/CP = nanorod clusters grown on carbon paper) [68]. The material displayed notable potential as a catalyst for hydrogen and oxygen evolution in both alkaline and neutral electrolytes. XPS and XRD in conjuction with DFT calculations elucidated that the dopants enhanced the electrical conductivity and modulated the electronic structure of the CoP host, thus placing the HBE close to the thermoneutral value of 0 eV. Defect sites were created when zinc substituted cobalt, because the larger atomic size of the dopant caused an increment in the lattice parameters relative to the sulfur doped CoP catalyst. Sulfur replaced phosphorus which also introduced defect sites due to a doping defect, where both of these aspects contributed to improving the electronic configuration of CoP.
Combining metal and non-metal dopants in transition metals resulted in a multifaceted performance increment where multiple aspects played an active role in the describing the performance of the oxygen and copper co-doped CoP catalyst developed by Fan et al. [69]. Lattice distortion due to the atomically smaller additions of oxygen and copper was detected through XRD, XPS and HRTEM, implying a greater number of exposed active sites. This was confirmed by measuring the ECSA through CV. The codoping also greatly affected the electronic structure of the material, where the associated water dissociation energy () was halved relative to undoped CoP in addition to reducing the HBE () tenfold to 0.04 eV. These extrinsic and intrinsic changes notably affected the activity relative to pristine CoP as evidenced by Tafel slopes, electrochemical impedance spectroscopy (EIS) spectra, and LSV. Similar efforts and results have also been reported in recent years, where one publication reported the use of vanadium and nitrogen as codopants in CoP on carbon cloth [70]. The collegial effects of codoping with vanadium and nitrogen yielding (V, N)-CoP/CC (CC = carbon cloth) induced a change in the electronic structure of CoP by moving the hydrogen binding energy closer to the thermoneutral value. Moreover, codoping caused small deformities in the lattice, exposing additional active sites for hydrogen evolution. All these aspects contributed to increasing hydrogen evolution activity.
Another example of an efficient codoped catalyst is CoS/P-CNT, where the combined use of both sulfur and phosphorus imparted both activity and stability onto the resulting material [71]. The combination of the heteroatoms phosphorus and sulfur is heavily associated with stability, as they are effective corrosion inhibitors [36,72]. Liu et al. [71] also emphasized the importance of substituting some of the sulfur with phosphorus in regard to stability, as the activity resulted from the conversion of CoO to CoS nanoparticles. While the material reported by Liu et al. [71] was investigated under acidic conditions, the effects reported can also apply to materials intended for alkaline solutions.
By implanting Ni-O-VO sites into nickel doped with copper on nickel foam (Ni(Cu)VO/NF) through a simple electrodeposition process, a very efficient catalyst was created [73]. Hydrogen adsorption/desorption characteristics were compared between several facets, as seen in Figure 13a–c, and were significantly altered by the addition of the Ni-O-VO sites, which caused a charge redistribution. Electrons congregated around VO clusters instead of Ni atoms as seen in Figure 13e,f, causing adsorbed hydrogen atoms to attach to two nickel atoms and one copper atom, as opposed to tree nickel atoms. Copper has a lower HBE relative to nickel, so the copper dopant in the Ni(Cu)VO/NF contributed together with the VO clusters to lower the HBE as seen in Figure 13d. The resulting activity is expressed through the low Tafel slope of 28 mV dec, very close to the 20 wt.% Pt/C benchmark (25 mV dec).
The stability of the material was evaluated through a 100 h ex situ CP test at 100 mA cm, where the material remained fairly stable after an initial voltage increment during the first 20 h. TEM images taken after testing revealed little change in structure and elemental distribution of the material. Comparing XRD spectra before and after testing reveals virtually no change in the crystallinity in the catalyst as seen in Figure 14b, however, XPS analysis shows a decline in the V2p-signal indicating partial dissolution of VO as shown in Figure 14b. This is likely causal to the initial voltage increment in the first 20 h, as the concentration of VO in the electrolyte was constant for the last 80 h as determined through inductively coupled plasma optical emission spectroscopy (ICP-OES).
Codoping was also utilized to enhance PGMs, where a new publication reported the results of a ruthenium doped ReP catalyst supported on a nitrogen and phosphorus doped carbon [74]. The catalyst exhibited activity and stability due to the ruthenium doping and the porous nanostructure which revealed active sites while enabling effective mass transportation. Moreover, the doped support negated the leaching effect, allowing the Ru-doped rhenium phosphate to operate smoothly with little degradation.
Another nickel-based catalyst employed both nitrogen and phosphorus as heteroatom dopants resulting in nickel phosphorus nanorods encapsulated with nitrogen-doped carbon (NiP@NC/NF) [75]. Similar to the results reported by Gao et al. [74], the heteroatom-doped carbon aided in protecting the nanorods from dissolution, resulting in a fairly stable catalyst material as determined by inductively coupled plasma mass spectrometry results where the content of nickel leached was indeed low. While failing to match or surpass the 20 wt.% Pt/C benchmark, the catalyst did display solid activity and presented a comparable performances at current densities ∼100 mA cm.
Theoretical predictions based of DFT calculations such as partial density of states and Bader charge analysis indicated the great possible improvements of adding copper to a phosphorous-doped nickel iron catalyst (NiP-FeP) [76]. Such calculations indicated most efficient adsorption/desorption of hydrogen at the P-site of NiP–FeP/Cu (G = 0.03 eV), in addition to very efficient water dissociation characteristics (G = 0.16 eV) in the interface. Motivated by these theoretical predictions, such a catalyst was created by growing copper nanorods directly from copper foam (CF), adding nickel and iron through electrodeposition followed by a phosphidation process. XPS results revealed the presence of the theoretically predicted P-sites necessary for attracting hydrogen, in addition to the charge redistribution brought by the phosphidation process. Most stability-tests are conducted at low current densities, quite far from what may be considered realistic operating conditions, however, this is not so for the present case, as fair stability was displayed through a 50 h chronoamperometry test at 1000 mA cm.
The activity of a transition metal oxide (CoO) was notably augmented through dual doping with nickel and zinc [77], where the former aggregated around O-vacancies on the surface and decreased the hydrogen binding energy in addition to the OH-H cleavage abilities associated with O-vacancies on transition metal oxides [9,77]. As witnessed by the increase in turnover frequency, the zinc dopant increased the electrical conductivity of the oxide by altering the electronic structure of the material, which resulted in a greater amount of electrons interacting with active sites.
The aforementioned successes achieved through codoping, through the many combinations between dopants and hosts, added a vast expanse of options into an already nigh on infinite set of possibilities. To sift through these options without having to physically create and characterize each material, high throughput analysis, machine learning, and statistical methods are likely to be useful [78,79].
2.5. Chalcogenides
Chalcogenides are chalcogens (group 16 of the periodic table) with the form MX, the first being one electropositive element (M = Ni, Ti, Nb, Ta, W, Fe, Mo, V and Hf) with one electronegative anion (X = oxygen, sulfur, selenium, tellurium, and polonium). These represent a class of exciting materials with several subcategories including sulfides, selenides, and tellurides, all of which will be covered here [17,80].
2.5.1. Sulfides
Nickel sulfides historically received more attention compared to nickel selenides and nickel tellurides, though early research efforts yielded materials requiring high overpotentials in the range of ∼250 mV up to 400 mV to achieve a current density of 10 mA cm. These materials were NiS as thin films, polymorphs and other variations without adding other elements. Combining nickel sulfides with transitional metals such as cobalt, iron, molybdenum, etc. has resulted in a greater performance [17,81]. A MoSNiSMoO nanowire catalyst was created [81] which displayed good activity and great stability. The synergistic use of both molybdenum oxides altered the electronic structure of the nickel sulfide, thus changing the hydrogen binding energy of the resulting material as determined using XRD, XPS, and HRTEM.
The aforementioned nickel sulfide was the most efficient catalyst of its kind prior to the arrival of vanadium-doped NiS nanowires [82]. The doping of vanadium had a notable impact in regard to both activity and stability, where specifically the doping increased the density of charge carriers near the Fermi level, where the d-band electrons of vanadium are key. A greater density of electrons near the Fermi level enhances the electronic structure of the nanowires yielding most effective electron transfer processes and greater HER activity.
Another example of sulfides is given by Li et al. [83], who created the nitrogen-doped sulfide heterostructure NiS/MoS on nickel foam (NiS/ MoS/NF) via a hydro-thermal synthesis followed by annealing. The electronegativity provided by the heteroatom nitrogen effectively lowered the electron density of nickel, thereby increasing the catalytic performance of the catalyst. Moreover, DFT calculations comparing NiS and N-NiS surfaces showed that water dissociates and hydrogen adsorbs/desorbs with a much lower kinetic energy barrier on the former relative to the undoped NiS.
2.5.2. Selenides
Nickel selenides are semiconductors, the majority of which are naturally nonstoichiometric and form structures filled with defects. The catalytic activity is highly dependent on the local surface electronic structure and similarly sensitive to the electrode substrate. Furthermore, this type of chalcogenide has a higher electrical conductivity than nickel sulfides due to the more prominent metallic characteristic of its anion (selenium), resulting in nickel selenides generating greater HER activities than nickel sulfides [17]. A good example of a nickel selenide was created by Liu et al. [84], where a CoNiSe/Ti catalyst displayed both activity and stability in an alkaline electrolyte. The novel catalyst was compared against a NiSe/Ti and the increased performance was attributed fast kinetics as determined through EIS, and a larger ECSA affording a greater number of active sites for HER activity. Additionally, the supplement of cobalt decreased the kinetic barrier for H formation through inducing H-H bond formation between adjacently adsorbed hydrogen atoms, i.e., hydrogen desorption kinetics were improved.
2.5.3. Tellurides
Nickel sulfides and nickel selenides are two fairly well-investigated chalcogenides compared to nickel tellurides. This class of chalcogenide is yet more electrically conductive, as the anion (tellurium) has a higher degree of metallic characteristics relative to both sulfur and selenium. Nickel tellurides possess greater stability than the other two chalcogenides, while the their associated activity does not follow this trend. This is due to the comparatively low electronegativity of tellurium rendering materials less able to attract protons [17].
Ananthara et al. created NiTe nanowires (NWs) which exhibited passable activity and good stability [85]. The NWs were outperformed by the 20 wt.% Pt/C benchmark at low overpotentials, however, these roles were reversed once the overpotential was increased to the range of ∼400 mV, which was found by comparing both LSV curves and Tafel curves. The exact reason for the significant activity at such high overpotentials was unspecified, however, the high tellurium:nickel ratio in the NWs is likely a principal component. Somewhat similarly, another group synthesized a heterostructured NiP/NiTe catalyst, where the crystalline phase of NiTe contributed to great alkaline HER activity [86]. The NiP/NiTe interface produced additional active sites according to both DFT calculations and atomic force microscopy (AFM) results, where the former indicated that the kinetic barrier for water dissociation was lowered and adsorbed hydrogen atoms could bind to the nickel in both NiP and NiTe simultaneously. The resulting activity is listed in Table 1.
2.6. Dichalcogenides
Dichalcogenides may generally be split between transition metal dichalcogenides (TMDCs) and noble metal dichalcogenides [18,19]. TMDCs are a fairly new group of 2D-nanosheets usually in the form of MX, where M is the transition metal (Mo, W, Nb, Re, Ni, Hf, Ta or V) and X is the chalcogen (S, Se, or Te) [18,80,87]. Noble metal dichalcogenides are basically the same as TMDCs structurally speaking as the transition metal is replaced with a noble metal (Pt, Pd, Ir), though these are less frequently studied as of late due to increasing emphasis on non-PGM materials [19]. A series of TMDCs (MoX; X = S, Se, Te) were investigated, and the general results based on LSV, chronopotentiometry (CP), and EIS follow the general trends in acitivity/stability found in chalcogenides, as seen in Table 1 [88]. While MoS displays the smallest overpotential at 10 mA cm, the stability is poor compared to that of both MoSe and MoTe. The MoSe displays the best all-round performance, however all of these fail to come close to any reasonable PGM benchmark in terms of activity with onset potentials over the range of ∼100 mV.
A single molybdenum atom on a cogenetic monolayer of MoS catalyst was created, where DFT calculations revealed that an increase in hydrogen bond strength between the adsorbed hydrogen atoms and substrate was induced by single, unsaturated, and coordinated molybdenum atoms on top of the MoS[89]. This lead to augmented HER activity and improved adsorption/desorption kinetics. The hybrid of molybdenum and hydrogen orbitals decreased the Fermi level compared to pristine MoS and increased chemical bonding. Both of these aspects played a part in lowering Gibb’s free energy of hydrogen adsorption () towards zero, thereby increasing HER activity.
Systematic high-throughput calculations were employed by combining machine learning methods and density functional theory (DFT) to reveal several promising materials by using metallic electron conductivity, good HER catalytic activity from thorough composition, low energy phase, and local structure spaces as screening parameters [90]. These materials include X-vacant MnS, CrSe, TiTe, VSe, and FeS, M-vacant ZrTe and PdTe and perfect monolayer NiS and VS. Model validity was proven through a favourable comparison between calculated overpotentials and experimental results. Furthermore, a quantitative expression for the hydrogen binding energy was determined as a function of electronegativity of the local structure (first and second neighboring atoms) and the valence electron number.
3. Creating Specific Sites for Hydroxide and Hydrogen Adsorption
Similarly to the preceding catalyst design strategy, this one also involves specific changes to the catalyst electronic structure. Creating specific separate sites for the adsorption of hydrogen atoms and hydroxide ions has proved to be a highly efficient design strategy, as creating single sites with favorable adsorption/desorption characteristics for both intermediates is difficult due to the incomplete understanding of how the binding energies of the two intermediates scale relative to each other. The general idea is to harmonize the associated properties of hydrogen adsorption/desorption and those of water dissociation/OH-H bond cleavage [3].
A recent paper attributed the rate of the HER to both the oxophilic and the electronic effect [94,95] where the authors noted that the OH-binding energy had a great influence on the HER rate. Therefore, alloying Pt with an oxophilic transition metal such as Ni resulted in additional active sites for OH adsorption, effectively freeing up sites for hydrogen adsorption and increasing the rate of the HER [94]. This caused several research groups to suggest that both the hydrogen and hydroxide binding energy should be employed as descriptors of HER activity [94,96].
Such a result was obtained by Sheng et al. [94] where covering an increasing surface percentage of a Pt disc with Ni(OH) resulted in lowering the hydrogen binding energy and thereby increasing its HER activity. This was confirmed by examining the cyclic voltammograms of the three Pt-based nanocatalyst materials (Pt/C, PtNi/C and acid-PtNi/C). Furthermore, the potential of adsorbed CO oxidation was proposed as a descriptor for HER activity based on a linear correlation between the potential at which CO oxidizes and the ECD. The nickel contribution in the PtNi/C and acid-PtNi/C lowered the HBEs and created distinct adsorption sites for hydroxide species (oxophilic effect). This positively affects the oxidation of CO by lowering the potential at which it occurs.
A NiW-decorated WO on NF was synthesized using a hydrothermal method, followed by hydrogen reduction creating two distinct surfaces for hydrogen adsorption and water dissociation [97]. The NiW alloy-amorphous NiWO interface cut the OH-H bond with OH attaching itself to the amorphous side and hydrogen adsorption/desorption occurring on NiW alloy particles. Moreover, the size and coverage of the aforementioned particles was highly influential in defining the interplay between chemical oxidation of the particles caused by the alkaline electrolyte and electrochemical reduction of oxides due to cathodic potentials. Stable surfaces were achieved by implementing large NiW particles at great coverages.
Such characteristics were also displayed by an edge-rich 1T–MoS/Ni(OH) catalyst [98], where MoS in the 1T phase acts as a substitute for the platinum in the work of Sheng et al. [94]. The edges of Ni(OH) encourage water dissociation and the edges of 1T–MoS hydrogen adsorption, thus edge/edge sites stimulate overall great HER activity. The energetics of these edge/edge sites were compared against three other site possibilities as displayed in Figure 15a, where edge/edge sites were found to have an incredibly low energy barrier for water dissociation () and an equally low one for the adsorption/desorption of hydrogen ().
The 1T–MoS monolayers of variable thicknesses were created through cathodic electrodeposition on Ni(OH), resulting in both edge-rich quantum sheets (QSs ∼ 6 nm) of 1T–MoS and edge-poor large sheets (LSs ∼ 145 nm) of 1T–MoS. XPS results show that both share comparable interfacial heterostructures, though the edge-rich 1T–MoS QS/Ni(OH) exhibited far greater HER activity which was attributed to a greater total length of exposed edges, as these were far more active than in-plane sites (Figure 15a). The 1T–MoS QS/Ni(OH) displayed a very low Tafel slope of 30 mV dec, almost identical to the 20 wt.% Pt/C benchmark (29 mV dec), indicating that the Tafel step was rate determining and that Ni(OH) has a high efficiency as a cleaver of OH-H bonds. Moreover, stability was tested via CP at 10, 100 and 500 mA cm for 100 h where the increase in overpotential measured only 40 mV (∼21% increase) for a current density of 500 mA cm.
This strategy was also applied earlier by Jiang et al. [99], though to a lesser effect compared to [98]. Jiang et al. created a 3-D Ni(OH)/MoS hybrid catalyst, and again the Ni(OH) provided sites for water dissociation and MoS yielded sites for hydrogen adsorption/desorption, where the result of this synergy was an improvement in HER kinetics relative to both components on their own in the same role.
The performance of a PtNi/NiS nanowire compared most favorably against commercial 20 wt.% Pt/C by delivering a current density of 37.2 mA cm at 70 mV, 9.7 times that of the commercial catalyst [100]. DFT calculations revealed the nanowire provided distinct sites for water dissociation and hydrogen adsorption/desorption, where nickel sulfide nanoparticles catered to the former and the platinum nickel alloy handled the latter. While such design strategies are effective for creating efficient catalyst materials, the analysis is difficult due to the complex electrocatalytic mechanisms at play. The authors suggested the use of in situ spectroscopic techniques to combat this issue.
An impressive Co-doped WO/amorphous CoW hybrid electrocatalyst was created through a one-step process by an in situ decomposition method [101]. The low Tafel slope of 20 mV dec implies the Tafel step was rate-determining, which was supported by the low DFT-determined value for the kinetic energy barrier for water dissociation. This aspect was handled by the amorphous CoW, with hydrogen adsorption/desorption occurring very efficiently on cobalt-doped WO with a near-optimum Gibb’s free energy for hydrogen adsorption ( = −0.06). This latter aspect was calculated to be more efficient on the Co-doped WO/amorphous CoW hybrid than the commercial 20 wt.% Pt/C (−0.09 eV).
The water dissociation ability of nickel and the hydrogen adsorption characteristics of molybdenum were combined in a paper by Feng et al. [102] where the resulting MoNi catalyst on MoO cuboids on nickel foam (MoNi/MoO@NF) displayed outstanding activity. According to DFT calculations, the MoNi {200} facet provides very efficient sites for water adsorption () in addition to hydrogen adsorption sites with a lower kinetic barrier () than MoO. This explains the efficiency of the water dissociation step (Volmer), and how the Tafel step became rate determining. Requiring only 15 and 44 mV to deliver 10 and 200 mA cm and showing good stability at 10, 100, and 200 mA cm, this catalyst displays potential as a catalyst for alkaline water electrolysis.
A most recent effort exhibits the creation of a tungsten carbide on nickel hydroxide catalyst material, where similar trends to those previously described reemerge [103]. Both interfaces provide a surface for the adsorption of a reaction intermediate, where the tungsten carbide caters to the adsorption of hydrogen, and the nickel hydroxide to the cleavage of the OH-H bond. Moreover, the strong interfacial contact between the nickel hydroxide and the tungsten carbide aids in optimizing the hydrogen binding energy of the latter, which unaided would bind hydrogen too strongly as displayed in Figure 3. This was confirmed by comparing the performance of the WC–Ni(OH) catalyst to that of WC.
NiMoN/NiN heterostructures were investigated as a function of heating time by Hua et al. [104], where the activity of the catalyst materials peaked at 12 h. While the NiMoN/NiN-12 catalyst displayed the greatest surface area as determined by double layer capacitance, the inherent catalyst activity measured by ECSA-normalized LSVs revealed that the material was also intrinsically greater than catalysts with other heating periods. The respectable activity was ascribed to the synergy between the two interfaces, which aided in modifying the electronic structure of both NiMoN and NiN, yielding specific sites for hydrogen adsorption/desorption and water dissociation respectively.
The collective effect of most catalyst preparation methods featured in this review were employed in a paper detailing the creation of a CoO-Ag catalyst resembling a strawberry when inspected through spectroscopic techniques [105]. The combination of a metal (Ag) and a metal oxide (CoO) yielded specific sites for hydrogen adsorption/desorption and water dissociation respectively. Adding CoO to the silver substrate increased the tensile strain and lowered the coordination number of the substrate, as indicated by X-ray adsorption spectroscopy (XAS) and DFT-calculations, thereby optimizing the hydrogen adsorption characteristics of CoO. Simultaneously as the activity of the commercial Pt/C benchmark was outperformed, the CoO-Ag catalyst also displayed solid durability both as a single HER catalyst and as HER/OER catalyst over 50 h.
A bimetallic nitride (NiFeN/NiFe) displayed fair activity, where the NiFe interface facilitated water dissociation most efficiently (−0.06 eV) and NiFeN catered to hydrogen adsorption desorption (−0.08 eV) [106]. Compared against that of its principal components NiFe and NiFeN, the formation of the hybrid was most beneficial for the energetics of both water dissociation and the hydrogen adsorption, as the prior values for the aforementioned surfaces were 0.18 eV and −0.15 eV respectively. Moreover, multiple theoretical approaches including crystal-field diagrams, electron density difference and d-orbital center reveal that the enhanced metallic characteristic of the hybrid interface was the progenitor to much of the increase in activity and stability.
To assist both the initial process of water dissociation and the subsequent adsorption of hydrogen, the ratio of Ni/NiO was carefully managed to optimize the alliance between the two interfaces handling these processes [107]. The use of high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) confirmed the formation of Ni/NiO heterostructures of various crystal sizes, the majority of which possessed a core-shell architecture. DFT studies show that the most efficient reaction pathway is through water dissociation occurring on the NiO {111} surface, leaving OH to adsorb onto the oxygen-vacancy of NiO {111} and the hydrogen atom to adsorb onto the Ni {111} site, as schematically illustrated in Figure 16a–c. The activity of the material was highly correlated to the ratio of Ni/NiO, where it peaked around ∼0.2 and aggressively degraded upon further rise/fall as shown in Figure 16d.
A final example is given by Li et al. [108] where copper nanodots (NDs)-embellished with NiS nanotubes (NTs) supported on carbon fibers (CFs) (Cu NDs/NiS NTs-CFs) were reported. The electronic interactions between copper and NiS contribute positively by charging the copper NDs, thus enhancing water adsorption and OH-H bond cleavage. Additionally, the negatively charged NiS NTs can weaken bonds formed between sulfur and adsorbed hydrogen on the catalyst surface, hence the hybrid catalyst enhanced hydrogen adsorption/desorption kinetics and advanced both Volmer and Heyrovsky steps of the HER. The synergistic cooperation of Cu and NiS produced a very adept catalyst based on its activity measured through Tafel slopes and onset potential (potential at 10 mA cm), and stability measured through CP at potentials between 100–250 mV.
4. Altering the Surface of a Catalyst
Catalyst activity is highly sensitive to surface conditions, where early studies of the HER on various crystal facets of Pt displayed this effect [109]. The activity of the platinum crystal facets followed the sequence {111} < {100} < {110}, which was attributed to the distinct states of the adsorbed hydrogen and to various effects these states had on the reaction as a whole. Adsorbed hydrogen was observed in two states, strongly adsorbed (i.e., underpotential deposited hydrogen, H) and weakly adsorbed (i.e., overpotential deposited hydrogen, H), where the former had a retarding effect on the HER by blocking active sites. The sites required for hydrogen recombination of H are only available if the H moves to a subsurface state when the negative potential is increased. The Pt {111} facet created the least amount of subsurface hydrogen, an aspect which was deemed key to its lack of activity compared to the two other investigated surfaces. Many studies followed the work of Markovic et al. [109], however, the exact explanation for the surface sensitivity of the alkaline HER remains hidden [3].
4.1. Strain
Altering the structure of a catalyst through, e.g., lattice-strain engineering is a straightforward way of influencing the surface of a catalyst to enhance its catalytic characteristics [110,111,112,113]. The effect of strain engineering on the activity of a metal catalyst has several explanations, where the d-band theory proved illustrative in describing certain cases. Without the influence of strain, the d-band is predominantly affected by changes in the local coordination environment of the metal, i.e., an alteration in the d-band is caused by fluctuations in the number of adjacent metal atoms [111,114]. Decreasing the local coordination by imposing tensile strain results in a smaller local bandwidth and an increase in the d-band center, as illustrated in Figure 17. The tensile strain causes the interatomic spacing of a material to increase which causes a smaller d-orbital overlay between atoms and therefore a decreased d-band width. Given that the total number of electrons in the d-band remains the same, the partial filling of the d-band will also be the same though the d-band center (the central moment of the d-band) will move upwards as a result of the narrowing. A high d-band center in this sense implies a stronger interaction between an adsorbing species and the surface.
Increasing the width of the d-band and decreasing its center energy is achieved through compressive strain. When the energy of the d-band center is lowered, more antibonding states will be below the Fermi level (), which increases their interaction with intermediates and lowers binding energies.
Inducing strain may be realized externally by applying a mechanical loading or internally through lattice mismatch, which involves utilizing core-shell structures or sparsely coating metallic alloy surfaces, where the lattice mismatch will strain the catalyst surface [111,112]. A core-shell Ru-Pt catalyst was employed to separate and quantify the influence of strain and the ligand-effect on the activity of the catalyst material [21]. It was theorized that interfacial platinum-ruthenium interactions caused the growth of the face-centered cubic (fcc)-structured ruthenium core, which imposed a notable compressing strain on the surrounding platinum shell, causing interfacial lattice mismatch between the core (Ru) and shell (Pt). The strain affected the icosehedral (i.e., a polyhedron with 20 faces) platinum shell quite significantly due to the small width of the shell (∼2 nm). Aided by the aforementioned facets, the core-shell catalyst exhibited a good HBE and a great hydroxide binding energy (as measured through CO oxidation), all of which contributed to increasing HER activity.
While d-band theory proved useful, several publications concluded that the strain-HBE relationship is more complex [112,115]. Motivated by the incomplete theoretical description provided by the aforementioned d-band theory, a recent publication detailed how the usual scaling relationships between initial and transition states in heterogeneous catalysis may be broken by altering the catalyst structure through strain engineering [112]. Khorshidi et al. [112] employed a mechanics-based eigenstress model to analyze the extent to which the catalyst-intermediate bond strength is affected by strain. Using the dimerization of nitrogen to N on a Pt {100} facet as an example, uniaxial compression was applied to the catalyst. An expansive Poisson response occurs due to the uniaxial compression, where this response lowers the energy barrier for the transition state and boosts the energy of the initial state, thereby violating the Bell–Evans–Polanyi principle. Such exceptions from scaling relationships are beneficial when designing catalyst materials, as they promote the conception of novel materials and better understanding of the subject [116].
The use of strain was also applied with great effect to a group of materials previously inactive towards the HER, namely transition metal oxides. A recent publication reported how the activity of CoO was improved when 3% tensile strain was exerted on an {111}-O surface with abundant O-vacancies, which resulted in a hydrogen binding energy () of −0.10 eV [117]. Water is adsorbed effectively onto a site with such a O-vacancy, where the OH-H bond is cut which allows the hydrogen atom to adsorb onto the closest oxygen atom to form adsorbed OH and the remaining OH to fill the O-vacancy, as illustrated in Figure 18a. Through increasing the percentage of exerted strain from 0–3%, the binding energy of hydrogen was increased and 3% was deemed the optimal value based on the small value (Figure 18b). Additionally, the strain caused the oxygen 2p band of CoO to shift upwards, resulting in a greater convalency between cobalt and oxygen and thereby decreasing the HBE. Surface oxygen can strongly bind to adjacent Co atoms, thus lowering its ability to acquire electrons from adsorbing hydrogen atoms, resulting in a decreased HBE as shown in Figure 18. Moreover, the increased activity due to electron migration from the Co d-band (i.e., greater d-band vacancy) to O 2p-band (Figure 18c) agrees with the d-band vacancy-HER activity correlation described above for transition metal doped cobalt phosphides [49]. Furthermore, the adsorption of atomic hydrogen was assumed to occur through the coupling of the hydrogen 1s band to the occupied oxygen 2p-band, supporting the descriptors determined by machine learning and DFT as mentioned earlier [38].
Analysis of non-PGM catalyst materials created through lattice-strain engineering is limited due to the difficulty in separating the effect of the strain on catalyst activity from complementary factors such as substrate, vacancy, or heterostructure, which also affect the activity of the catalyst [113]. A NiSSe core-shell catalyst was developed by Han et al. [113] and displayed both activity and stability as a result of the lattice-strain alone. Both experimental and DFT calculations alluded that this technique resulted in a lower degree of metal d-orbital overlap, inducing a closer d-band center towards the Fermi level. This improved the intrinsic electrical conductivity and electrocatalytic properties of the catalyst and enhanced the reaction pathway.
4.2. Additional Geometrical Modification Methods
Oxygen plasma engraving is another approach to modify a catalyst surface, where Wu et al. [118] utilized this approach to great effect. The oxygen plasma engraving method (OPEM) prompted the formation of CoO species on a CoP nanowire on carbon cloth (CoP NWs/CC). Engraving time was varied and optimized for the CoP NWs/CC catalyst, where 40 s of engraving resulted in a highly active and stable catalyst material. OPEM effectively reorganized the surface of the CoP NWs/CC catalyst, forming a CoP/CoO interface with specific sites for both hydrogen adsorption/desorption and water dissociation. The latter was facilitated on CoO, where the hydroxide species generated from the dissociation of water adsorbed onto this side of the interface due to the powerful electrostatic forces in CoO which originate from the unfilled and highly positively charged d-orbital of Co. Hydrogen adsorption/desorption was achieved on the adjacent CoP sites. This synergy resulted in a HER catalyst producing a Tafel slope of 42.8 mV dec and the low overpotential of 65 mV at 10 mA cm (both iR corrected).
Laser ablation was utilized to create RhO clusters embedded in the surface layer of rhodium nanoparticles where strain was caused by the lattice mismatch between the clusters and the substrate, which stabilized the RhO nanoparticles [119]. Moreover, in situ X-ray absorption spectroscopy (XAS) measurements were conducted through use of a custom three-electrode cell setup as seen in Figure 19a. As seen in Figure 19b, in situ XAS measurements during CA display a small negative shift in the energy of the adsorption edge after 1 h which indicates a partial reduction of the catalyst. Moreover, the Fourier transformed (FT) k-weighted (k)-function of EXAFS spectra revealed an increase in the Rh-O bond length, from 1.59 Å to 1.60 Å after 1 h CA. Both of these aspects are related to the initial performance loss, indicating the stabilizing effect of the compressing strain. The material was stabilized following the initial shift, where no additional change in the bond length was discovered. The material was subject to increasing degrees of reductive polarization, where the material remained stable up to 0.3V after the initial performance decline at 0.1V as seen in Figure 19c which increased the Rh-O bond length from 1.59 Å to 1.61 Å. A great decline in performance was noted at 0.4V, where the EXAFS spectra reveal that the aforementioned bonds disappeared together with the compressing strain, while the ratio of Rh-O/Rh-Rh intensity from the FT k-weighted (k)-function dropped from 0.3 to 0.
Similarly, plasma treatment under argon and nitrogen atmospheres was utilized to modify MoP on carbon cloth (N-MoP/CC) [120]. Both atmospheres resulted in smaller particle sizes where the latter also doped the MoP/CC with nitrogen (N-MoP/CC), in which this addition imparted active sites for hydroxide desorption. The addition of nitrogen caused a redistribution of charge due to its electronegativity, thereby improving the active sites of MoP. Evidenced by Tafel slopes, EIS, LSV and DFT calculations, the plasma treatment under a nitrogen atmosphere had a large impact upon catalyst activity.
Electrochemical surface techniques can also be employed, where the work of Anantharaj et al. [121] exemplifies the usage of anodic potential cycling. This process was applied to a Ni-S/NF catalyst, resulting in an amorphous and hydroxylated surface. These qualities imparted a greater ECSA as determined by the double layer capacitance but also improved charge transferal as determined by EIS. Furthermore, a mere 100 anodic cycles was enough to notably increase the activity of the nickel sulfide catalyst, a trend which persisted until approximately 500 cycles. Despite the inferior low-potential efficiency compared against the commercial 20 wt.% Pt/C benchmark, the hydroxylated nickel sulfide on nickel foam catalyst exhibited great improvement at higher current densities, yielding 230 and 305 mV overpotential at 100 mA cm, respectively. Oxidation can affect even cathodic reductive environments [97,122], where a recent publication discovered that etching their FeP electrode with hydrochloric acid was found to replenish the activity of the electrode [122]. Oxygen was labeled as source of the corrosion, where simply immersing the electrode into 1.0 M KOH turned the electrode from black to yellow. Diffraction peaks from XRD measurements show that FeP peaks disappear completely after 12 h of immersion as seen in Figure 20a, where the yellow film was confirmed to be FeOOH. Etching the electrode in hydrochloric acid replenished the performance, as shown in Figure 20b. The influence of oxygen was also confirmed by repeating the measurements in a solution bubbled with argon gas in a sealed chamber, where no degradation was observed.
The effect of carbon-coating was highlighted in a recent publication where an iron-doped NiS/NiP heterojunction with carbon-coating on nickel foam (NPZFNS@C/NF) catalyst was created [123]. The coating altered the electronic structure of the catalyst to a metallic state, thereby increasing the charge transfer efficiency and interfacial electronic contact and improving the hydrogen adsorption energy. Moreover, comparing the results of SEM, XRD, and XPS before and after the 25 h chronoamperometry test illustrated excellent catalyst stability, and inductively coupled plasma mass spectrometry (ICPMS) was utilized to show negligible nickel/iron leaching.
More detailed information on strain-controlled catalysts is beyond the scope of this review, though the following reviews and their sources may satisfy such in-depth inquiries [92,111,124]. Shown in Table 1 are some key metrics to the majority of the featured catalyst materials which summarize their performance. The table is divided into the same three categories seen in this review, as indicated in bold.

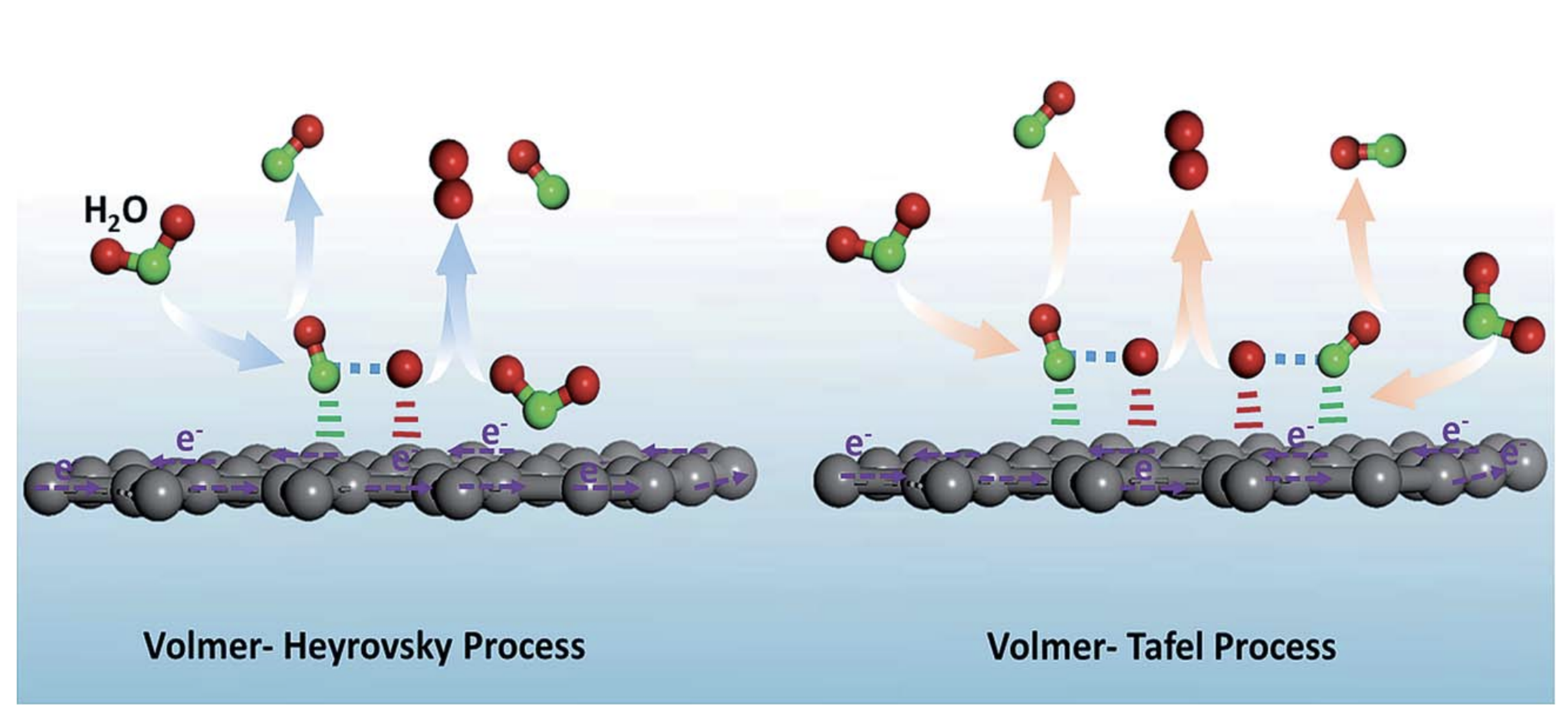{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Key characteristics of featured catalyst materials for hydrogen evolution reaction. Some PGM reference performances were also added to provide perspective into individual catalyst performances. Stability is assessed by calculating change in performance after chronoamperometry (CA)/chronopotentiometry (CP) and assigning either ↑/↓ to indicate stability/a systematic decline. Stability is also evaluated through cyclic voltammetry (CV), where latter usually involves ≥1000 scans within appropriate range. * Indicates values were iR-compensated. ** Indicates stability encompasses both the HER and OER.
Table 1.
Key characteristics of featured catalyst materials for hydrogen evolution reaction. Some PGM reference performances were also added to provide perspective into individual catalyst performances. Stability is assessed by calculating change in performance after chronoamperometry (CA)/chronopotentiometry (CP) and assigning either ↑/↓ to indicate stability/a systematic decline. Stability is also evaluated through cyclic voltammetry (CV), where latter usually involves ≥1000 scans within appropriate range. * Indicates values were iR-compensated. ** Indicates stability encompasses both the HER and OER.
| Catalyst Material | Overpotential | Tafel Slope | Stability | Stability From |
|---|---|---|---|---|
| at 10 mA cm | [mV dec] | from CA/CP [h] | CVs [% of Current] | |
| Electronic | ||||
| Structure | ||||
| Alloys and oxides | ||||
| Np-CuTi [23] | 50 mV * | 110 * | - | ∼98% (5000) |
| (@ 30 mA cm) | ||||
| Ni-Co-Ti-2 [24] | 125 mV * | 47 * | 150 (↓88.4%@ 10 mA cm) | - |
| (CP) | ||||
| NiO/Ni/CNT [25] | 80 mV * | 82 * | 2 (↑91.1%@ 20 mA cm) | - |
| (CP) | ||||
| CrNi [26] | 106 mV | 71 | 6 (↑99%@ 10 mA cm) | - |
| (CA) | ||||
| NiMo [27] | 185 mV * | 120 * | 20 (↑98.5%@ ∼14 mA cm) | - |
| (CA) | ||||
| Ni-LaCeO/C [33] | 114 mV * | 72.7 * | 24 (↑76.2%@ 10 mA cm) | ∼88% (500) |
| (CP) | (@ 25 mA cm) | |||
| 2%La-CoO-350 [34] | 98 mV | 72.8 | 3.3 (↓44.7%@ 10 mA cm) | ∼ 93% (3000) |
| (CP) | (@ 100 mA cm) | |||
| CoS NS/NF [35] | 93 mV | 55.1 | 220 (↑93%@ 10 mA cm) | - |
| (CP) | ||||
| mMoO [37] | 138 mV * | 56 * | 12 (↓96%@ ∼23 mA cm) | - |
| (CA) | ||||
| PBSCF [39] | 237 mV * | 45 * | 25 (↑99%@ 50 mA cm) | 100% (1000) |
| (CP) | (@ 100 mA cm) | |||
| SrNbCoFeO [40] | 262 mV * | 135 * | 10 (↑100%@ 10 mA cm) | ∼99% (1000) |
| (CP) | (@ 40 mA cm) | |||
| Phosphides | ||||
| NiP NP [41] | 291 mV * | 119 * | 16 (↑99%@ 10 mA cm) | - |
| (CP) | ||||
| (FeNi)P [43] | 103 mV | 77 | 20 (↑95.2%@ 100 mA cm) | ∼99% (1000) |
| (CA) | (@ 100 mA cm) | |||
| Pt-CoP [44] | 2.0 mV * | 44 * | 200 ** (↓@ 100 mA cm) | - |
| (CA) | ||||
| 10 wt.% Pt/C [44] | 20 mV * | 65 * | ∼55 ** (↓@ 100 mA cm) | - |
| (CA) | ||||
| RuP NF [45] | 9 mV * | 38 * | - | ∼88% (10,000) |
| (@ 100 mA cm) | ||||
| RhP-500 [46] | 4.3 mV * | 35.2 * | 16.7 (↓81.8%@ 10 mA cm) | ∼99% (2000) |
| (CA) | (@ 50 mA cm) | |||
| MoP/MoO [47] | 79 mV * | 41 * | 24 (↑98.2%@ 10 mA cm) | ∼84% (1000) |
| (CP) | (@ 100 mA cm) | |||
| 2D/3D NiCoP/NF [48] | 37 mV * | 50.8 * | 50 (↑81.8%@ 10 mA cm) | ∼97% (5000) |
| (CP) | (@ 200 mA cm) | |||
| Cr-CoP [49] | 36 mV * | 54 * | 514 (↓65.8%@ 10 mA cm) | ∼98% (3000) |
| (CP) | (@ 200 mA cm) | |||
| NiP/Ni(PO) HSs [51] | 114 mV * | 93.1 * | 10 (↑100%@ 10 mA cm) | ∼99% (1000) |
| (CP) | (@ 37.5 mA cm) | |||
| Nitrides | ||||
| (NiN/NF) [53] | 121 mV * | 109 * | 200 (↑92.6%@ 200 mA cm) | ∼97% (1000) |
| (CA) | (@ 360 mA cm) | |||
| MoN@NC [55] | 85 mV | 54 | 10 (↑89.7%@ 10 mA cm) | 100% (1000) |
| (CA) | (@ 80 mA cm) | |||
| N-CoS@NC/Ti [56] | 140 mV | 92 | 10 (↑98.1%@ 30 mA cm) | 96% (1000) |
| (CP) | (@ 100 mA cm) | |||
| Mo(CN) [59] | 80 mV * | 40 * | 24 (↓91.1%@ 18–19 mA cm) | ∼98% (5000) |
| (CA) | @ 300 mA cm) | |||
| NiN/NF [60] | 45 mV | 97 | 20 (↓78.2%@ ∼20 mA cm) | ∼91% (1000) |
| (CA) | (@ 80 mA cm) | |||
| Co-NPs/N-rGO [61] | 59 mV * | 105 * | 480 (↑100%@ 10 mA cm) | - |
| (CP) | ||||
| Co-NPs/N-rGO [61] | 200 (↑100%@ 500 mA cm) | - | ||
| (CP) | ||||
| Ni/V-N [63] | 43 mV * | 33 * | 20 (↓87.4%@ 20 mA cm) | - |
| (CP) | ||||
| Co-doping | ||||
| N-P-Ni/C [67] | 26 mV | 34 | 50 (↑96.8%@ 12 mA cm) | ∼85% (1000) |
| (CA) | @ 10 mA cm) | |||
| Zn,S-CoP | 49 mV * | 42 * | 20 (↑100%@ 10 mA cm) | ∼94% (1000) |
| NRCs/CP [68] | (CP) | (@ 100 mA cm) | ||
| O, Cu-CoP-2 [69] | 72 mV * | 58 * | 24 (↑100%@ 50 mA cm) | ∼98% (5000) |
| (CP) | (@ 200 mA cm) | |||
| V, N-CoP [70] | 57 mV | 51 | 50 (↑94.1%@ 10 mA cm) | ∼99% (1000) |
| (CA) | (@ 100 mA cm) | |||
| Ni(Cu)VO/NF [73] | 21 mV | 28 | 100 (↓55.5%@ 100 mA cm) | - |
| (CP) | ||||
| Ni(Cu)VO/NF [73] | 10 mV * | - | - | - |
| Ru-ReP/NPC [74] | 39 mV * | 29.9 * | 12 (↑100%@ 10 mA cm) | - |
| (CP) | ||||
| NiP@NC/NF [75] | 93 mV * | 77.8 * | 30 (↓83.3%@ 20 mA cm) | ∼97% (3000) |
| (CP) | (@ 120 mA cm) | |||
| (NiP-FeP)/C/CF [76] | 23.6 mV * | 52 * | 50 (↓62.9%@ 1000 mA cm) | - |
| (CP) | ||||
| (Ni, Zn)-CoO NR [77] | 53 mV | 47 | 24 ** (↑96%@ 10 mA cm) | - |
| (CP) (in 6.0M KOH) | ||||
| Chalcogenides | ||||
| MoSNiSMoO [81] | 91 mV * | 54 * | 20 (↑86.4%@ ∼10 mA cm) | 96.5% (1000) |
| (CP) | (@ 100 mA cm) | |||
| V-NiS [82] | 68 mV | 112 | 12 (↑100%@ 20 mA cm) | - |
| (CP) | ||||
| NiS/MoS/NF [83] | 71 mV | 79 | 20 (↑96.7%@ 30 mA cm) | - |
| (CA) | ||||
| CoNiSe/Ti [84] | 64 mV * | 63 * | 60 (↓66.4%@ 10–30 mA cm) | ∼95% (5000) |
| (CP) | (@ 90 mA cm) | |||
| Chalcogenides | ||||
| NiTe NW [85] | 113 mV * | 69 * | 16.5 (↑98.3%@ ∼50 mA cm) | 86.7% (1000) |
| (CP) | (@ 100 mA cm) | |||
| NiP/NiTe [86] | 63 mV * | 80 * | 50 (↑98%@ 50 mA cm) | ∼91% after 50 h CA |
| (CA) | (@ 100 mA cm) | |||
| Te@NiTe/NiS | 101 mV | 118 | 10 (↑100%@ 50 mA cm) | ∼99% (1000) |
| /AB [87] | (CA) | (@ 350 mA cm) | ||
| MoS [88] | 173 mV * | 110 * | 12 (↓89%@ 50 mA cm) | - |
| (CP) | ||||
| MoSe [88] | 208 mV * | 66 * | 12 (↓91.8%@ 50 mA cm) | - |
| (CP) | ||||
| MoTe [88] | 283 mV * | 102 * | 12 (↑100%@ 50 mA cm) | - |
| (CP) | ||||
| Mo SAs/MLMoS [89] | 209 mV | 35 | - | - |
| Creating | ||||
| sites | ||||
| NiW-WO/NF [97] | 48 mV | 33 | 14 (↑87.3%@ ∼20 mA cm) | - |
| (CA) | ||||
| 1T–MoS QS/Ni(OH) | 57 mV | 30 | 100 (↓85.8%@ 100 mA cm) | ∼99% (1000) |
| [98] | (CP) | (@ 100 mA cm) | ||
| Ni(OH)/MoS [99] | 80 mV * | 60 * | 16 (↓84.9%@ 10 mA cm) | - |
| (CP) | ||||
| PtNi/NiS NW [100] | 42 mV | - | 5 (↓63.5%@ 5 mA cm) | - |
| Co-WO/CoW | 25 mV * | 20 * | 100 (↑99%@ 10 mA cm) | ∼92% (5000) |
| /NF-1/3 [101] | (CP) | (@ 200 mA cm) | ||
| MoNi/MoO@NF [102] | 15 mV | 30 | 10 (↑96.3%@ 100 mA cm) | ∼99% (2000) |
| (CP) | (@ 600 mA cm) | |||
| WC–Ni(OH) [103] | 60 mV | 55.9 | 20+10 (↑100% and ↓94.7% | - |
| @ 10 and 100 mA cm)(CP) | ||||
| NiMoN/NiN [104] | 28 mV * | 49 * | 24 (↓87.5%@ 10 mA cm) | ∼99% (2000) |
| (CP) | (@ 200 mA cm) | |||
| CoO-Ag [105] | 51 mV | 49 | 100+100 (↑100% and ↓96.8% | - |
| @ 10 and 50 mA cm)(CP) | ||||
| NiFeN/NiFe [106] | 74 mV | 54 | 12 (↑100%@ 10 mA cm) | - |
| (CP) | ||||
| Ni/NiO-3.8 [107] | 90 mV * | 41 * | - | ∼98% (5000) |
| (@ 60 mA cm) | ||||
| Cu NDs/NiS | 128 mV | 76 | 30 (↑99%@ 87 mA cm) | - |
| NTs-CF [108] | (CP) | |||
| Altering | ||||
| surface | ||||
| NiSSe [113] | 70 mV | 78 | 300 (↓73.5%@ 100 mA cm) | ∼99% (10,000) |
| (CP) | (@ 300 mA cm) | |||
| 3.0% S-CoO NRs [117] | 73 mV * | 82 * | 28 (↓91.7%@ 9-10 mA cm) | ∼93% (1000) |
| (CA) | (@ 200 mA cm) | |||
| CoP NWs 40s/CC [118] | 65 mV * | 43 * | 35 (↓95.2%@ 110 mA cm) | ∼94% (5000) |
| (CA) | (@ 100 mA cm) | |||
| N-MoP/CC [120] | 70 mV | 55 | 36 (↑100%@ ∼15 mA cm) | - |
| (CA) | ||||
| Ni-S-OH/NF [121] | 179 mV * | 120 * | 25+25 (↑100% and ↓96.9% | - |
| @ 100 and 200 mA cm)(CP) | ||||
| Fe-(NiS/NiP)@C/NF [123] | 129 mV | 66.8 | 25 (↑100%@ ∼10 mA cm) | ∼84% (3000) |
| (CP) | (@ 150 mA cm) | |||
| NF-NiS-A [125] | 67 mV * | 63 * | 40 (↓100%@ 20 mA cm) | - |
| (CP) |
While Table 1 displays the performance of all featured HER catalyst materials, there are several additional factors which may influence the true usefulness of a material. Factors such as catalyst loading and the scalability of the synthesis process will all contribute to the potential of a material, and the variation in these aspects is notable from publication to publication. Moreover, results from in situ experiments can significantly vary from the anticipated performance gleaned from ex situ tests as discussed in Section 2.3 on Nitrides [13,126,127]. Stability is usually tested through either x thousand CV scans, continuous CA/CP over <100 h or both, however these tests measure different types of stability. Furthermore, their results will usually fail to yield a true indication of the operational stability of a material save those who employ realistic operating conditions in the ballpark of 1000 mA cm and 50 °C [13]. This makes it difficult to compare various classes of catalyst and there are few practical solutions to overcome such difficulties, hence catalyst performance should not be judged solely on one-dimensional metrics.
5. Perspective
Design strategies aiming to alter the electronic structure through, e.g., elemental doping or alloying represents the largest category. This strategy produced a great number of materials that returned respectable results under the generally mild conditions they were subject to as displayed in Table 1. Recent efforts with codoping also produced materials with encouraging performances. This methodology introduced a substantial amount of options due to the promising results from combining heteroatoms with other elements. Given the vast amount of options, establishing effective screening parameters will be key. High-throughput analysis employing such parameters could prove invaluable when searching for a combination of dopants and hosts which are able to rival the likes of PGMs in terms of activity while retaining an equally great stability. The second strategy of creating specific sites for the adsorption of hydroxide and hydrogen has also proven to be effective. By addressing each step of the HER separately, it created some efficient catalysts, and future improvements of this strategy are likely to yield both high-performing catalyst materials and intrinsic understanding of the alkaline HER. The same applies to the third strategy, altering the structure of a catalyst, and both of these strategies could serve as ways to optimize already high-performing materials, e.g., straining codoped catalysts. However, the actual creation of such materials is nontrivial and the road towards creating the materials’ models is long.
Practical employment of the materials detailed in this review is a topic which curiously enough remains concealed. The number of papers employing realistic testing conditions when assessing activity and stability is dwarfed by those claiming “outstanding stability” for a material which remained stable during an ex situ 24 h chronopotentiometry test at only 10 mA cm, 25 °C. This marks a stark difference to the more realistic conditions of performing the same test at 1000 mA cm, 50 °C for 1000 h in situ, and also underlines the lack of focus on catalyst degradation. There are several studies which test their materials in actual AEM electrolysis cells with MEAs, AEMs and all the relevant hardware [24,27,33,35,45,51], however the majority employ either simplified two-cell tests where the electrodes are submerged in separate beakers and separated by a membrane [47,73] or some other variation such as half-cell tests [23,25,26,34,37,39,40,43,44,46,48,49,53,55,56,59,60,61,63,67,68,69,70,74,75,76,77,81,82,83,84,85,86,87,88,89,98,99,100,101,102,103,104,105,106,107,108,113,117,118,120,121,123,125]. Moreover, the vast majority of the catalysts displayed in Table 1 display a systematic decline in the CA/CP tests when subject to current densities greater than 100 mA cm, which underlines how much work there is left before a material with large-scale potential can be announced. A greater focus on classifying catalyst instability and identifying facile mitigation strategies is required.
Catalyst development is almost solely based on usage of non-PGMs, however, the emphasis on avoiding the use of critical raw materials such as cobalt, molybdenum, and tungsten has yet to gain traction. Further work on creating catalysts solely comprising abundant materials through scalable methods represents another challenge [128,129].
DFT-studies evolved significantly over the past decade, though the majority employ simplified versions of the system they intend to study and there is a great variety in methodology. Specifically, choice of pseudopotentials, exchange correlation functions and physical model will greatly impact the conclusion of the calculations. Standardized methodologies are necessary if comparative work is to serve any purpose. Additionally, there is still difficulty in determining the mechanics of systems with multifunctional sites. These aspects represent current challenges within computational electrochemistry. The advent of machine learning was most advantageous in combination with DFT studies, as this decreased computational cost from days to minutes [32,130]. Moreover, multiple examples through this paper underlined the great potential of this combination [38,78,79,90], however, the current lack of large reliable data sets limits this potential. Initiatives such as “The Materials Project” [30,31], which recently celebrated its 10-year anniversary represents efforts targeted at accelerating the development of novel materials and shortening their time to deployment by making great amounts of data and computation tools widely available. Continuation of work along these lines is highly likely to play a decisive role in advancing the current state of the art in electrocatalysis.
Spectroscopic techniques is an aspect of catalyst analysis one cannot avoid discussing. The incomplete understanding of the HER was notoriously underlined, and the use of in situ spectroscopic techniques will likely aid in shining some light on this dark spot [42,119]. Many effects both beneficial and detrimental to catalyst stability and activity occur in situ, rendering the function of ex situ test results notably diminished. Even the comparison of, e.g., SEM or XPS results before and after prolonged polarization, returns an incomplete picture, as many dynamic processes such as surface reconstruction, bonding, dissolution, etc., occurs only during operation. Several common spectroscopic techniques were already mentioned, though it is the continued development of novel in situ testing methods which will likely contribute to unraveling the obscurities of the alkaline hydrogen evolution reaction.
Generally, the theoretical understanding of the alkaline HER is incomplete, however, its completion is likely to come in the near future. Given the continuation of the revolutionary progress of computational science, the days marking the survival of such mysteries are numbered.
Future aspects will likely include more specialized attempts where several of these strategies are combined. Synthesis routes are also becoming more specialized in the search of active and stable materials, however, the end goal to increase the production to an industrial scale should not be forgotten. Utilizing the binding energy of hydrogen, hydroxide, and water (, and ) as screening parameters for catalyst creation (Figure 21) will likely yield active and stable catalyst materials for the hydrogen evolution.
6. Conclusions
This review detailed the development of catalyst materials for the hydrogen evolution reaction in alkaline electrolytes. The development was described in three categories, inspired by the three prevailing theories describing the origin of the pH dependence of the HER/HOR activity. From these three categories, it is the usage of dual dopants as a means of optimizing the electronic structure that stands out as one of the most efficient. Additionally, creating specific sites for the adsorption of the various intermediate states of the HER also resulted in many exciting materials. The continued progress of density functional theory (DFT) played a great contribution towards the invention of these strategies, and the merger between DFT and machine learning reverberated throughout the realm of electrocatalysis. The road ahead will likely contain many contributions from this duo, yielding both theoretical descriptions and hopefully materials of practical use.
A great many materials were discussed in this review, however, only a handful showed true potential towards use in industrial applications, where the origin of this lies predominantly in the lack of stability. The use of mild testing conditions is prevalent in literature, however this is likely to change as the blooming emphasis on catalyst stability spreads. The usage of what is deemed “realistic” operating conditions has increased with time, hence, a greater degree of recently published works are expected to positively contribute to this trend. While the topic of catalyst degradation could easily be a review in itself, this review laid emphasis on the most commonly observed degradation mechanisms, methods of detection and some mitigation strategies.
Finally, numerous voids in literature were specified throughout this review, giving those who wish to contribute to the field easy access to research areas in need of further work. It is the goal of this review to accentuate these voids so that their status as such ceases. Accordingly, the symbiotic relationship between machine learning and DFT, in combination with large datasets based off experimental work employing realistic testing conditions, will aid in unraveling the mysteries of the alkaline HER.
Author Contributions
Conceptualization, T.B.F.; methodology, T.B.F., P.H.M. and M.L.K.; validation, T.B.F., P.H.M. and M.L.K.; formal analysis, T.B.F.; investigation, T.B.F.; resources, P.H.M.; data curation, T.B.F.; writing—original draft preparation, T.B.F.; writing—review and editing, T.B.F.; visualization, T.B.F.; supervision, P.H.M. and M.L.K.; project administration, P.H.M. and M.L.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AEM | Anion Exchange Membrane |
| AFM | Atomic Force Microscopy |
| CA | Chronoamperometry |
| CC | Carbon Cloth |
| CF | Copper Foam |
| CNT | Carbon Nanotubes |
| COHP | Crystal Orbital Hamilton Population |
| CP | Chronopotentiometry |
| CV | Cyclic Voltammetry |
| DFT | Density Functional Theory |
| DOS | Density Of States |
| ECD | Exchange Current Density |
| ECSA | Electrochemical Surface Area |
| EIS | Electrochemical Impedance Spectroscopy |
| ELF | Electron Localisation Function |
| EXAFS | Extended X-ray Adsorption Fine Structure |
| FCC | Face centerd Cubic |
| HAADF | High-Angle Annular Dark-Field |
| HBE | Hydrogen Binding Energy |
| HER | Hydrogen Evolution Reaction |
| HOR | Hydrogen Oxidation Reaction |
| HRTEM | High Resolution Transmission Electron Spectroscopy |
| ICPMS | Inductively Coupled Plasma Mass Spectrometry |
| ICP-OES | Inductively Coupled Plasma Optical Emission Spectroscopy |
| LSV | Linear Sweep Voltammetry |
| MEA | Membrane Electrode Assembly |
| NF | Nickel Foam |
| NP | Nano Particle |
| NR | Nanorod |
| NS | Nanosheet |
| NW | Nanowire |
| OER | Oxygen Evolution Reaction |
| OPEM | Oxygen Plasma Engraving Method |
| PGM | Platinum Group Metal |
| RHE | Reversible Hydrogen Electrode |
| SCAN | Strongly Constrained and Appropriately Normed |
| SEM | Scanning Electron Spectroscopy |
| SHE | Standard Hydrogen Electrode |
| TEM | Transmission Electron Spectroscopy |
| TMDC | Transition Metal Dichalcogenides |
| UPS | Ultraviolet Photoelectron Spectroscopy |
| XANES | X-ray Adsorption Near Edge Spectroscopy |
| XAS | X-ray Adsorption Spectroscopy |
| XPS | X-ray Photoelectron Spectroscopy |
| XRD | X-ray Diffraction |
References
- Tilak, B.; Ramamurthy, A.; Conway, B. High performance electrode materials for the hydrogen evolution reaction from alkaline media. In Proceedings of the Indian Academy of Sciences-Chemical Sciences; Springer: Berlin/Heidelberg, Germany, 1986; Volume 97, pp. 359–393. [Google Scholar]
- Li, S.; Kang, Q.; Baeyens, J.; Zhang, H.; Deng, Y. Hydrogen production: State of technology. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Vienna, Austria, 18–21 May 2020; Volume 544, p. 012011. [Google Scholar]
- Wang, X.; Zheng, Y.; Sheng, W.; Xu, Z.; Jaroniec, M.; Qiao, S.Z. Strategies for design of electrocatalysts for hydrogen evolution under alkaline conditions. Mater. Today 2020, 36, 125–138. [Google Scholar] [CrossRef]
- Gerischer, H.; Mehl, W. Zum mechanismus der kathodischen wasserstoffabscheidung an quecksilber, silber und kupfer. Z. Elektrochem. Berichte Bunsenges. Phys. Chem. 1955, 59, 1049–1059. [Google Scholar]
- Chen, Z.; Duan, X.; Wei, W.; Wang, S.; Ni, B.J. Recent advances in transition metal-based electrocatalysts for alkaline hydrogen evolution. J. Mater. Chem. A 2019, 7, 14971–15005. [Google Scholar] [CrossRef]
- Firouzjaie, H.; Mustain, W. Catalytic Advantages, Challenges, and Priorities in Alkaline Membrane Fuel Cells; American Chemical Society: Washington, DC, USA, 2019. [Google Scholar]
- Rossmeisl, J.; Chan, K.; Skulason, E.; Björketun, M.; Tripkovic, V. On the pH dependence of electrochemical proton transfer barriers. Catal. Today 2016, 262, 36–40. [Google Scholar] [CrossRef]
- Zheng, J.; Sheng, W.; Zhuang, Z.; Xu, B.; Yan, Y. Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy. Sci. Adv. 2016, 2, e1501602. [Google Scholar] [CrossRef] [Green Version]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.C.; Uchimura, M.; Paulikas, A.; Stamenkovic, V.; Markovic, N. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Subbaraman, R.; Tripkovic, D.; Chang, K.C.; Strmcnik, D.; Paulikas, A.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N. Trends in activity for the water electrolyzer reactions on 3d-M(Ni, Co, Fe, Mn)-hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef]
- Mustain, W. Understanding how high-performance anion exchange membrane fuel cells were achieved: Component, interfacial, and cell-level factors. Curr. Opin. Electrochem. 2018, 12, 233–239. [Google Scholar] [CrossRef]
- Ferriday, T.; Middleton, P. Alkaline fuel cell technology—A review. Int. J. Hydrogen Energy 2021, 46, 18489–18510. [Google Scholar] [CrossRef]
- Gutić, S.; Dobrota, A.; Fako, E.; Skorodumova, N.; López, N.; Pašti, I. Hydrogen evolution reaction-from single crystal to single atom catalysts. Catalysts 2020, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Qi, J.; Lei, F.; Xie, Y. Modulation of electronic structures in two-dimensional electrocatalysts for the hydrogen evolution reaction. Chem. Commun. 2020, 56, 11910–11930. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.; Bligaard, T.; Logadottir, A.; Kitchin, J.; Chen, J.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef] [Green Version]
- Ge, Z.; Fu, B.; Zhao, J.; Li, X.; Ma, B.; Chen, Y. A review of the electrocatalysts on hydrogen evolution reaction with an emphasis on Fe, Co and Ni-based phosphides. J. Mater. Sci. 2020, 55, 14081–14104. [Google Scholar] [CrossRef]
- Anantharaj, S.; Kundu, S.; Noda, S. Progress in nickel chalcogenide electrocatalyzed hydrogen evolution reaction. J. Mater. Chem. A 2020, 8, 4174–4192. [Google Scholar] [CrossRef]
- Xu, M.; Liang, T.; Shi, M.; Chen, H. Graphene-Like Two-Dimensional Materials. Chem. Rev. 2013, 113, 3766–3798. [Google Scholar] [CrossRef] [PubMed]
- Chia, X.; Sofer, Z.; Luxa, J.; Pumera, M. Layered Noble Metal Dichalcogenides: Tailoring Electrochemical and Catalytic Properties. ACS Appl. Mater. Interfaces 2017, 9, 25587–25599. [Google Scholar] [CrossRef]
- Nørskov, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhu, Y.; Vasileff, A.; Jiao, Y.; Chen, S.; Song, L.; Zheng, B.; Zheng, Y.; Qiao, S.Z. Strain effect in bimetallic electrocatalysts in the hydrogen evolution reaction. ACS Energy Lett. 2018, 3, 1198–1204. [Google Scholar] [CrossRef]
- Jin, H.; Ruqia, B.; Park, Y.; Kim, H.; Oh, H.S.; Choi, S.I.; Lee, K. Nanocatalyst Design for Long-Term Operation of Proton/Anion Exchange Membrane Water Electrolysis. Adv. Energy Mater. 2021, 11, 2003188. [Google Scholar] [CrossRef]
- Lu, Q.; Hutchings, G.; Yu, W.; Zhou, Y.; Forest, R.; Tao, R.; Rosen, J.; Yonemoto, B.; Cao, Z.; Zheng, H.; et al. Highly porous non-precious bimetallic electrocatalysts for efficient hydrogen evolution. Nat. Commun. 2015, 6, 6567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, P.; Sivanantham, A.; Shanmugam, S. Nanostructured Nickel–Cobalt–Titanium Alloy Grown on Titanium Substrate as Efficient Electrocatalyst for Alkaline Water Electrolysis. ACS Appl. Mater. Interfaces 2017, 9, 12416–12426. [Google Scholar] [CrossRef]
- Gong, M.; Zhou, W.; Tsai, M.C.; Zhou, J.; Guan, M.; Lin, M.C.; Zhang, B.; Hu, Y.; Wang, D.Y.; Yang, J.; et al. Nanoscale nickel oxide/nickel heterostructures for active hydrogen evolution electrocatalysis. Nat. Commun. 2014, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ji, S.J.; Xue, H.G.; Suen, N.T. HER activity of MxNi1-x (M = Cr, Mo and W; x ≈ 0.2) alloy in acid and alkaline media. Int. J. Hydrogen Energy 2020, 45, 17533–17539. [Google Scholar] [CrossRef]
- Faid, A.; Oyarce Barnett, A.; Seland, F.; Sunde, S. Highly active nickel-based catalyst for hydrogen evolution in anion exchange membrane electrolysis. Catalysts 2018, 8, 614. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zheng, Y.R.; Montoya, J.; Hochfilzer, D.; Cao, A.; Kibsgaard, J.; Chorkendorff, I.; Nørskov, J. Origins of the Instability of Nonprecious Hydrogen Evolution Reaction Catalysts at Open-Circuit Potential. ACS Energy Lett. 2021, 6, 2268–2274. [Google Scholar] [CrossRef]
- Schalenbach, M.; Speck, F.; Ledendecker, M.; Kasian, O.; Goehl, D.; Mingers, A.; Breitbach, B.; Springer, H.; Cherevko, S.; Mayrhofer, K. Nickel-molybdenum alloy catalysts for the hydrogen evolution reaction: Activity and stability revised. Electrochim. Acta 2018, 259, 1154–1161. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, X.; Montoya, J.; Nørskov, J. Predicting aqueous stability of solid with computed Pourbaix diagram using SCAN functional. Npj Comput. Mater. 2020, 6, 1–7. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.; Hautier, G.; Chen, W.; Richards, W.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gao, Y.; Kong, H.; Kim, J.; Choi, S.; Ciucci, F.; Hao, Y.; Yang, S.; Shao, Z.; Lim, J. Non-precious-metal catalysts for alkaline water electrolysis: Operando characterizations, theoretical calculations, and recent advances. Chem. Soc. Rev. 2020, 49, 9154–9196. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Yang, J.; Jeong, J.; Kim, G.; Kwon, C.; Myung, N.; Lee, K.; Choi, S. Promotion Effect of Modified Ni/C by La-Ce Oxide for Durable Hydrogen Evolution Reaction. ACS Sustain. Chem. Eng. 2021, 9, 12508–12513. [Google Scholar] [CrossRef]
- Zhao, X.; Yin, F.; He, X.; Chen, B.; Li, G. Enhancing hydrogen evolution reaction activity on cobalt oxide in alkaline electrolyte by doping inactive rare-earth metal. Electrochim. Acta 2020, 363, 137230. [Google Scholar] [CrossRef]
- Park, Y.; Lee, J.; Jang, M.; Jeong, J.; Park, S.; Choi, W.S.; Kim, Y.; Yang, J.; Choi, S. Co3S4 nanosheets on Ni foam via electrodeposition with sulfurization as highly active electrocatalysts for anion exchange membrane electrolyzer. Int. J. Hydrogen Energy 2020, 45, 36–45. [Google Scholar] [CrossRef]
- Paraknowitsch, J.; Thomas, A. Doping carbons beyond nitrogen: An overview of advanced heteroatom doped carbons with boron, sulfur and phosphorus for energy applications. Energy Environ. Sci. 2013, 6, 2839–2855. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Miao, R.; Huan, T.D.; Mosa, I.; Poyraz, A.; Zhong, W.; Cloud, J.; Kriz, D.; Thanneeru, S.; He, J.; et al. Mesoporous MoO3-x material as an efficient electrocatalyst for hydrogen evolution reactions. Adv. Energy Mater. 2016, 6, 1600528. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Ci, S.; Ma, Y.; Liu, T.; Gao, L.; Qian, P.; Ji, C.; Su, Y. Accelerating 2D MXenes Catalyst Discovery for Hydrogen Evolution Reaction by Computer-Driven Workflow and Ensemble Learning Strategy. J. Mater. Chem. A 2020, 8, 23488–23497. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Zhou, W.; Zhu, Z.; Su, C.; Liu, M.; Shao, Z. A Perovskite Electrocatalyst for Efficient Hydrogen Evolution Reaction. Adv. Mater. 2016, 28, 6442–6448. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Zhong, Y.; Bu, Y.; Chen, X.; Zhong, Q.; Liu, M.; Shao, Z. A Perovskite Nanorod as Bifunctional Electrocatalyst for Overall Water Splitting. Adv. Energy Mater. 2017, 7, 1602122. [Google Scholar] [CrossRef]
- Laursen, A.; Wexler, R.; Whitaker, M.; Izett, E.; Calvinho, K.; Hwang, S.; Rucker, R.; Wang, H.; Li, J.; Garfunkel, E.; et al. Climbing the Volcano of Electrocatalytic Activity while Avoiding Catalyst Corrosion: Ni3P, a Hydrogen Evolution Electrocatalyst Stable in Both Acid and Alkali. ACS Catal. 2018, 8, 4408–4419. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, H.C.; Hsu, C.S.; Lin, T.S.; Chang, C.J.; Chang, S.C.; Tsai, L.D.; Chen, H.M. Operando unraveling of the structural and chemical stability of P-substituted CoSe2 electrocatalysts toward hydrogen and oxygen evolution reactions in alkaline electrolyte. ACS Energy Lett. 2019, 4, 987–994. [Google Scholar] [CrossRef]
- Zhang, W.; Zou, Y.; Liu, H.; Chen, S.; Wang, X.; Zhang, H.; She, X.; Yang, D. Single-crystalline (FexNi1-x)2P nanosheets with dominant {011¯1¯} facets: Efficient electrocatalysts for hydrogen evolution reaction at all pH values. Nano Energy 2019, 56, 813–822. [Google Scholar] [CrossRef]
- Li, Z.; Niu, W.; Yang, Z.; Kara, A.; Wang, Q.; Wang, M.; Gu, M.; Feng, Z.; Du, Y.; Yang, Y. Boosting alkaline hydrogen evolution: The dominating role of interior modification in surface electrocatalysis. Energy Environ. Sci. 2020, 13, 3110–3118. [Google Scholar] [CrossRef]
- Kim, J.C.; Kim, J.; Park, J.; Ahn, S.; Kim, D.W. Ru2P Nanofibers for High-Performance Anion Exchange Membrane Water Electrolyzer. Chem. Eng. J. 2021, 420, 130491. [Google Scholar] [CrossRef]
- Xin, H.; Dai, Z.; Zhao, Y.; Guo, S.; Sun, J.; Luo, Q.; Zhang, P.; Sun, L.; Ogiwara, N.; Kitagawa, H.; et al. Recording the Pt-beyond Hydrogen Production Electrocatalysis by Dirhodium Phosphide with an Overpotential of Only 4.3 mV in Alkaline Electrolyte. Appl. Catal. B Environ. 2021, 297, 120457. [Google Scholar] [CrossRef]
- Ding, M.; Xu, H.; Liu, M.; Wang, Y.; Wang, A.; Lin, T.; Zhang, L.; Zhang, K. Interface construction and porosity engineering to trigger efficient hydrogen evolution of two-dimensional porous molybdenum phosphide/dioxide heterojunction nanosheets in acidic and alkaline electrolytes. Chem. Eng. J. 2022, 430, 132674. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, C.; Xu, S.; Liu, Y.; Jiang, D.; Zhu, J. Facile synthesis of hierarchical NiCoP nanosheets/NiCoP nanocubes homojunction electrocatalyst for highly efficient and stable hydrogen evolution reaction. Appl. Surf. Sci. 2021, 565, 150537. [Google Scholar] [CrossRef]
- Men, Y.; Li, P.; Zhou, J.; Chen, S.; Luo, W. Trends in Alkaline Hydrogen Evolution Activity on Cobalt Phosphide Electrocatalysts Doped with Transition Metals. Cell Rep. Phys. Sci. 2020, 1, 100136. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, L.; Hensen, E.; Hofmann, J. Evaluating the stability of Co2P electrocatalysts in the hydrogen evolution reaction for both acidic and alkaline electrolytes. ACS Energy Lett. 2018, 3, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lv, Q.; Li, G.; Ge, J.; Liu, C. and Xing, W. Core-shell structured Ni12P5/Ni3(PO4)2 hollow spheres as difunctional and efficient electrocatalysts for overall water electrolysis. Appl. Catal. B Environ. 2017, 204, 486–496. [Google Scholar] [CrossRef]
- Sinha, A.; Ojha, U. Recent Trends in Development of Metal Nitride Nanocatalysts for Water Electrolysis Application. In Electrocatalysis; IntechOpen: London, UK, 2021. [Google Scholar]
- Xing, Z.; Li, Q.; Wang, D.; Yang, X.; Sun, X. Self-supported nickel nitride as an efficient high-performance three-dimensional cathode for the alkaline hydrogen evolution reaction. Electrochim. Acta 2016, 191, 841–845. [Google Scholar] [CrossRef]
- Liao, J.W.; Lu, X.; Huang, B.Y.; Yu, G.Q.; Li, X.B. Hydrogen evolution on different facets of δ1-MoN and δ3-MoN: Considering the adsorbed oxygen and hydroxyl by Surface Pourbaix diagrams. Int. J. Hydrogen Energy 2021, 46, 9077–9086. [Google Scholar] [CrossRef]
- Lv, Z.; Tahir, M.; Lang, X.; Yuan, G.; Pan, L.; Zhang, X.; Zou, J.J. Well-dispersed molybdenum nitrides on a nitrogen-doped carbon matrix for highly efficient hydrogen evolution in alkaline media. J. Mater. Chem. A 2017, 5, 20932–20937. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Li, Y.; He, B.; Wang, R.; Gong, Y. MOFs-derived hybrid nanosheet arrays of nitrogen-rich CoS2 and nitrogen-doped carbon for efficient hydrogen evolution in both alkaline and acidic media. Int. J. Hydrogen Energy 2018, 43, 23319–23326. [Google Scholar] [CrossRef]
- Hanif, S.; Iqbal, N.; Shi, X.; Noor, T.; Ali, G.; Kannan, A. NiCo-N-doped carbon nanotubes based cathode catalyst for alkaline membrane fuel cell. Renew. Energy 2020, 154, 508–516. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, T.; Chen, M.; Tong, Y.; Zhang, N.; Peng, X.; Chu, W.; Wu, X.; Wu, C.; Xie, Y. Enhanced catalytic activity in nitrogen-anion modified metallic cobalt disulfide porous nanowire arrays for hydrogen evolution. ACS Catal. 2017, 7, 7405–7411. [Google Scholar] [CrossRef]
- Tang, C.; Hu, Q.; Li, F.; He, C.; Chai, X.; Zhu, C.; Liu, J.; Zhang, Q.; Zhu, B.; Fan, L. Coupled molybdenum carbide and nitride on carbon nanosheets: An efficient and durable hydrogen evolution electrocatalyst in both acid and alkaline media. Electrochim. Acta 2018, 280, 323–331. [Google Scholar] [CrossRef]
- Liu, W.; Xia, T.; Ye, Y.; Wang, H.; Fang, Z.; Du, Z.; Hou, X. Self-supported Ni3N nanoarray as an efficient nonnoble-metal catalyst for alkaline hydrogen evolution reaction. Int. J. Hydrogen Energy 2021, 46, 27037–27043. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Han, C.; Zong, H.; Yang, G.; Lin, S.; Kumar, A.; Jadhav, A.; Tran, N.; Hwang, Y.; et al. Identifying the Activity Origin of a Cobalt Single-Atom Catalyst for Hydrogen Evolution Using Supervised Learning. Adv. Funct. Mater. 2021, 31, 2100547. [Google Scholar] [CrossRef]
- Cao, L.; Luo, Q.; Liu, W.; Lin, Y.; Liu, X.; Cao, Y.; Zhang, W.; Wu, Y.; Yang, J.; Yao, T.; et al. Identification of single-atom active sites in carbon-based cobalt catalysts during electrocatalytic hydrogen evolution. Nat. Catal. 2019, 2, 134–141. [Google Scholar] [CrossRef]
- Shen, H.; Liang, S.; Adimi, S.; Guo, X.; Zhu, Y.; Guo, H.; Thomas, T.; Attfield, T.; Yang, M. Supporting Nickel on Vanadium Nitride for Platinum Comparable Hydrogen Evolution in Alkaline Solution. J. Mater. Chem. 2021, 9, 19669–19674. [Google Scholar] [CrossRef]
- Yang, Y.; Lai, L.; Wei, L.; Chen, Y. Degradation: A Critical Challenge for M–N–C Electrocatalysts. Available online: https://www.sciencedirect.com/science/article/abs/pii/S2095495621005714 (accessed on 15 November 2021).
- Xie, X.; He, C.; Li, B.; He, Y.; Cullen, D.; Wegener, E.; Kropf, A.; Martinez, U.; Cheng, Y.; Engelhard, M.; et al. Performance enhancement and degradation mechanism identification of a single-atom Co-N-C catalyst for proton exchange membrane fuel cells. Nat. Catal. 2020, 3, 1044–1054. [Google Scholar] [CrossRef]
- Kim, J.; Yoo, J.; Lee, H.; Sung, Y.E.; Hyeon, T. Single-atom M–N–C catalysts for oxygen reduction electrocatalysis. Trends Chem. 2021, 3, 779–794. [Google Scholar] [CrossRef]
- Jin, H.; Liu, X.; Chen, S.; Vasileff, A.; Li, L.; Jiao, Y.; Song, L.; Zheng, Y.; Qiao, S.Z. Heteroatom-Doped Transition Metal Electrocatalysts for Hydrogen Evolution Reaction. ACS Energy Lett. 2019, 4, 805–810. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, B.; Zhu, J.; Liu, Z.; Zhang, H.; Li, Y. Callistemon-like Zn and S co-doped CoP nanorod clusters as highly efficient electrocatalysts for neutral-pH overall water splitting. J. Mater. Chem. A 2019, 7, 22453–22462. [Google Scholar] [CrossRef]
- Xu, K.; Sun, Y.; Sun, Y.; Zhang, Y.; Jia, G.; Zhang, Q.; Gu, L.; Li, S.; Li, Y.; Fan, H. Yin-Yang Harmony: Metal and Non-metal Dual-Doping Boosts Electrocatalytic Activity for Alkaline Hydrogen Evolution. ACS Energy Lett. 2018, 3, 2750–2756. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Liu, Q.; Guo, J.; Zhang, X. Vanadium and nitrogen co-doped CoP nanoleaf array as pH-universal electrocatalyst for efficient hydrogen evolution. J. Alloys Compd. 2019, 791, 1070–1078. [Google Scholar] [CrossRef]
- Liu, W.; Hu, E.; Jiang, H.; Xiang, Y.; Weng, Z.; Li, M.; Fan, Q.; Yu, X.; Altman, E.; Wang, H. A highly active and stable hydrogen evolution catalyst based on pyrite-structured cobalt phosphosulfide. Nat. Commun. 2016, 7, 10771. [Google Scholar] [CrossRef]
- Verma, C.; Verma, D.; Ebenso, E.; Quraishi, M. Sulfur and phosphorus heteroatom-containing compounds as corrosion inhibitors: An overview. Heteroat. Chem. 2018, 29, e21437. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tan, X.; Hocking, R.; Bo, X.; Ren, H.; Johannessen, B.; Smith, S.; Zhao, C. Implanting Ni-O-VOx sites into Cu-doped Ni for low-overpotential alkaline hydrogen evolution. Nat. Commun. 2020, 11, 2720. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Y.; Liu, H.; Shao, M.; Chen, Z.; Ma, T.; Wu, Z.; Wang, L. N, P-doped carbon supported ruthenium doped Rhenium phosphide with porous nanostructure for hydrogen evolution reaction using sustainable energies. J. Colloid Interface Sci. 2022, 606, 1874–1881. [Google Scholar] [CrossRef]
- Zhang, H.; Li, W.; Feng, X.; Zhu, L.; Fang, Q.; Li, S.; Wang, L.; Li, Z.; Kou, Z. A chainmail effect of ultrathin N-doped carbon shell on Ni2P nanorod arrays for efficient hydrogen evolution reaction catalysis. J. Colloid Interface Sci. 2022, 607, 281–289. [Google Scholar] [CrossRef]
- Kumar, A.; Bui, V.; Lee, J.; Jadhav, A.; Hwang, Y.; Kim, M.; Kawazoe, Y.; Lee, H. Modulating Interfacial Charge Density of NiP2-FeP2 via Coupling with Metallic Cu for Accelerating Alkaline Hydrogen Evolution. ACS Energy Lett. 2021, 6, 354–363. [Google Scholar] [CrossRef]
- Ling, T.; Zhang, T.; Ge, B.; Han, L.; Zheng, L.; Lin, F.; Xu, Z.; Hu, W.B.; Du, X.W.; Davey, K.; et al. Well-dispersed nickel-and zinc-tailored electronic structure of a transition metal oxide for highly active alkaline hydrogen evolution reaction. Adv. Mater. 2019, 31, 1807771. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.; Bonde, J.; Chorkendorff, I.; Nørskov, J. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef]
- Mamun, O.; Winther, K.; Boes, J.; Bligaard, T. A bayesian framework for adsorption energy prediction on bimetallic alloy catalysts. Npj Comput. Mater. 2020, 6, 177. [Google Scholar] [CrossRef]
- Jung, Y.; Zhou, Y.; Cha, J. Intercalation in two-dimensional transition metal chalcogenides. Inorg. Chem. Front. 2016, 3, 452–463. [Google Scholar] [CrossRef]
- Wang, C.; Tian, B.; Wu, M.; Wang, J. Revelation of the excellent intrinsic activity of MoS2|NiS|MoO3 nanowires for hydrogen evolution reaction in alkaline medium. ACS Appl. Mater. Interfaces 2017, 9, 7084–7090. [Google Scholar] [CrossRef]
- Qu, Y.; Yang, M.; Chai, J.; Tang, Z.; Shao, M.; Kwok, C.; Yang, M.; Wang, Z.; Chua, D.; Wang, S.; et al. Facile Synthesis of Vanadium-Doped Ni3S2 Nanowire Arrays as Active Electrocatalyst for Hydrogen Evolution Reaction. ACS Appl. Mater. Interfaces 2017, 9, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Jiang, Y.; Liu, S.; Zhang, M.; Guo, Q.; Shen, W.; He, R.; Su, W.; Li, M. Regulating the electron density of dual transition metal sulfide heterostructures for highly efficient hydrogen evolution in alkaline electrolytes. Nanoscale 2019, 11, 14016–14023. [Google Scholar] [CrossRef]
- Liu, T.; Asiri, A.; Sun, X. Electrodeposited Co-doped NiSe2 nanoparticles film: A good electrocatalyst for efficient water splitting. Nanoscale 2016, 8, 3911–3915. [Google Scholar] [CrossRef]
- Anantharaj, S.; Karthick, K.; Kundu, S. NiTe2 Nanowire Outperforms Pt/C in High-Rate Hydrogen Evolution at Extreme pH Conditions. Inorg. Chem. 2018, 57, 3082–3096. [Google Scholar] [CrossRef]
- Li, Y.; Tan, X.; Tan, H.; Ren, H.; Chen, S.; Yang, W.; Smith, S.; Zhao, C. Phosphine vapor-assisted construction of heterostructured Ni2P/NiTe2 catalysts for efficient hydrogen evolution. Energy Environ. Sci. 2020, 13, 1799–1807. [Google Scholar] [CrossRef]
- Mingli, F.; Xue, L.; Dandan, W.; Yinling, W.; Maoguo, L. Fabrication of Te@NiTe2/NiS heterostructures for electrocatalytic hydrogen evolution reaction. Electrochim. Acta 2019, 328, 135075. [Google Scholar] [CrossRef]
- Bhat, K.S.; Nagaraja, H. Performance evaluation of molybdenum dichalcogenide (MoX2; X = S, Se, Te) nanostructures for hydrogen evolution reaction. Int. J. Hydrogen Energy 2019, 44, 17878–17886. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, S.; Pan, H.; Xiao, S.; Guo, Z.; Tang, L.; Khan, U.; Ding, B.F.; Li, M.; Cai, Z.; et al. Unsaturated Single Atoms on Monolayer Transition Metal Dichalcogenides for Ultrafast Hydrogen Evolution. ACS Nano 2020, 14, 767–776. [Google Scholar]
- Ran, N.; Sun, B.; Qiu, W.; Song, E.; Chen, T.; Liu, J. Identifying Metallic Transition-Metal Dichalcogenides for Hydrogen Evolution through Multilevel High-Throughput Calculations and Machine Learning. J. Phys. Chem. Lett. 2021, 12, 2102–2111. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, D.; Gong, L.; Zhang, L.; Xia, Z. Catalytic Activity Origin and Design Principles of Graphitic Carbon Nitride Electrocatalysts for Hydrogen Evolution. Front. Mater. 2019, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Ling, T.; Jaroniec, M.; Qiao, S.Z. Recent Progress in Engineering the Atomic and Electronic Structure of Electrocatalysts via Cation Exchange Reactions. Adv. Mater. 2020, 32, 2001866. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhou, M.; Long, A.; Xue, Y.; Liao, H.; Wei, C.; Xu, Z. Heterostructured electrocatalysts for hydrogen evolution reaction under alkaline conditions. Nano-Micro Lett. 2018, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.F.; Lu, B.A.; Qu, X.M.; Ye, J.Y.; Zhang, J.M.; Yin, S.H.; Wu, Q.H.; Wang, R.X.; Shen, S.Y.; Sheng, T.; et al. Does the oxophilic effect serve the same role for hydrogen evolution/oxidation reaction in alkaline media? Nano Energy 2019, 62, 601–609. [Google Scholar] [CrossRef]
- Strmcnik, D.; Uchimura, M.; Wang, C.; Subbaraman, R.; Danilovic, N.; Van Der Vliet, D.; Paulikas, A.; Stamenkovic, V.; Markovic, N. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 2013, 5, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Jia, Q.; Bates, M.; Li, J.; Xu, C.; Gath, K.; Yang, J.; Waldecker, J.; Che, H.; Liang, W.; et al. Tuning Nb-Pt Interactions to Facilitate Fuel Cell Electrocatalysis. ACS Catal. 2017, 7, 4936–4946. [Google Scholar] [CrossRef]
- Xu, W.; Wang, B.; Ni, X.; Liu, H.; Wang, W.; Zhang, L.; Zhang, H.; Peng, Z.; Liu, Z. Heterogeneous Synergetic Effect of Metal–Oxide Interfaces for Efficient Hydrogen Evolution in Alkaline Solutions. ACS Appl. Mater. Interfaces 2021, 13, 13838–13847. [Google Scholar] [CrossRef]
- Chen, W.; Gu, J.; Du, Y.; Song, F.; Bu, F.; Li, J.; Yuan, Y.; Luo, R.; Liu, Q.; Zhang, D. Achieving Rich and Active Alkaline Hydrogen Evolution Heterostructures via Interface Engineering on 2D 1T-MoS2 Quantum Sheets. Adv. Funct. Mater. 2020, 30, 2000551. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Wang, J.; Ruan, Y.; Ji, X.; Xu, K.; Chen, C.; Wan, H.; Miao, L.; Jiang, J. Interface engineering: The Ni(OH)2/MoS2 heterostructure for highly efficient alkaline hydrogen evolution. Nano Energy 2017, 37, 74–80. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, X.; Zhang, J.; Wan, S.; Guo, S.; Lu, G.; Yao, J.; Huang, X. Precise tuning in platinum-nickel/nickel sulfide interface nanowires for synergistic hydrogen evolution catalysis. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Chen, J.; Jin, Q.; Li, Y.; Li, Y.; Cui, H.; Wang, C. Design Superior Alkaline Hydrogen Evolution Electrocatalyst by Engineering Dual Active Sites for Water Dissociation and Hydrogen Desorption. ACS Appl. Mater. Interfaces 2019, 11, 38771–38778. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Liu, P.; Liao, Z.; Liu, S.; Zhuang, X.; Chen, M.; Zschech, E.; Feng, X. Efficient hydrogen production on MoNi4 electrocatalysts with fast water dissociation kinetics. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wang, X.; Chen, X.; Zhang, Q.; Li, N.; Luo, H. Interfacial engineering of Ni(OH)2 on W2C for remarkable alkaline hydrogen production. Appl. Catal. Environ. 2021, 540, 120818. [Google Scholar] [CrossRef]
- Hua, W.; Sun, H.; Liu, H.; Li, Y.; Wang, J.G. Interface engineered NiMoN/Ni3N heterostructures for enhanced alkaline hydrogen evolution reaction. Appl. Surf. Sci. 2021, 540, 148407. [Google Scholar] [CrossRef]
- Feng, Y.; Li, Z.; Cheng, C.Q.; Kang, W.J.; Mao, J.; Shen, G.R.; Yang, J.; Dong, C.K.; Liu, H.; Du, X.W. Strawberry-like Co3O4-Ag bifunctional catalyst for overall water splitting. Appl. Catal. B Environ. 2021, 299, 120658. [Google Scholar] [CrossRef]
- Hu, Y.; Xiong, T.; Balogun Tang, M.-S.J.; Huang, Y.; Adekoya, D.; Zhang, S.; Tong, Y. Enhanced metallicity boosts hydrogen evolution capability of dual-bimetallic Ni–Fe nitride nanoparticles. Mater. Today Phys. 2020, 15, 100267. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Y.; Zhao, Z.; Zhang, Q.H.; Huang, L.B.; Gu, L.; Lu, G.; Hu, J.S.; Wan, L.J. Steering elementary steps towards efficient alkaline hydrogen evolution via size-dependent Ni/NiO nanoscale heterosurfaces. Natl. Sci. Rev. 2020, 7, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.X.; Wu, J.Q.; Tong, Y.X.; Li, G.R. Efficient Hydrogen Evolution on Cu Nanodots-Decorated Ni3S2 Nanotubes by Optimizing Atomic Hydrogen Adsorption and Desorption. J. Am. Chem. Soc. 2018, 140, 610–617. [Google Scholar] [CrossRef]
- Markovića, N.; Sarraf, S.; Gasteiger, H.; Ross, P. Hydrogen electrochemistry on platinum low-index single-crystal surfaces in alkaline solution. J. Chem. Soc. Faraday Trans. 1996, 92, 3719–3725. [Google Scholar] [CrossRef]
- Voiry, D.; Yamaguchi, H.; Li, J.; Silva, R.; Alves, D.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.; Eda, G.; et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 2013, 12, 850–855. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Tang, M.; Tsai, C.; Abild-Pedersen, F.; Zheng, X.; Li, H. Enhancing electrocatalytic water splitting by strain engineering. Adv. Mater. 2019, 31, 1807001. [Google Scholar] [CrossRef]
- Khorshidi, A.; Violet, J.; Hashemi, J.; Peterson, A. How strain can break the scaling relations of catalysis. Nat. Catal. 2018, 1, 263–268. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Zhang, M.; Zhou, Y.; Rao, D.; Zhong, C.; Zhang, J.; Han, X.; Hu, W.; Zhang, Y.; et al. Lattice-Strain Engineering of Homogeneous NiS0.5Se0.5 Core-Shell Nanostructure as a Highly Efficient and Robust Electrocatalyst for Overall Water Splitting. Adv. Mater. 2020, 32, 2000231. [Google Scholar] [CrossRef] [PubMed]
- Schnur, S.; Groß, A. Strain and coordination effects in the adsorption properties of early transition metals: A density-functional theory study. Phys. Rev. B 2010, 81, 033402. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Wu, C.; Yang, G.; Yang, S. CO oxidation over strained Pt (100) surface: A DFT study. J. Phys. Chem. C 2015, 119, 15500–15505. [Google Scholar] [CrossRef]
- Rosen, A.; Notestein, J.; Snurr, R. Structure-Activity Relationships That Identify Metal-Organic Framework Catalysts for Methane Activation. ACS Catal. 2019, 9, 3576–3587. [Google Scholar] [CrossRef]
- Ling, T.; Yan, D.Y.; Wang, H.; Jiao, Y.; Hu, Z.; Zheng, Y.; Zheng, L.; Mao, J.; Liu, H.; Du, X.W.; et al. Activating cobalt (II) oxide nanorods for efficient electrocatalysis by strain engineering. Nat. Commun. 2017, 8, 1509. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Cheng, H.; Lv, H.; Wang, J.; Liu, L.; Liu, S.; Wu, X.; Chu, W.; Wu, C.; Xie, Y. Controllable Surface Reorganization Engineering on Cobalt Phosphide Nanowire Arrays for Efficient Alkaline Hydrogen Evolution Reaction. Adv. Mater. 2018, 30, 1703322. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Feng, Y.; Liang, Y.L.; Cheng, C.Q.; Dong, C.K.; Liu, H.; Du, X.W. Stable Rhodium (IV) Oxide for Alkaline Hydrogen Evolution Reaction. Adv. Mater. 2020, 32, 1908521. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, W.; Zeng, J.; He, L.; Li, D.; Gao, Q. Plasma-Engineered MoP with nitrogen doping: Electron localization toward efficient alkaline hydrogen evolution. Appl. Catal. B Environ. 2020, 268, 118441. [Google Scholar] [CrossRef]
- Anantharaj, S.; Sugime, H.; Noda, S. Surface amorphized nickel hydroxy sulfide for efficient hydrogen evolution reaction in alkaline medium. Chem. Eng. J. 2021, 408, 127275. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Z.; Cao, X.; Hu, J.; Wu, X.; Ng, A.; Lu, G.P.; Chen, Z. Elucidating the sources of activity and stability of FeP electrocatalyst for hydrogen evolution reactions in acidic and alkaline media. Appl. Catal. B Environ. 2020, 260, 118156. [Google Scholar] [CrossRef]
- Chen, W.Z.; Liu, P.Y.; Zhang, L.; Liu, Y.; Liu, Z.; He, J.; Wang, Y.Q. High-Efficient and Durable Overall Water Splitting Performance by Interfacial Engineering of Fe-Doped Urchin-like Ni2P/Ni3S2 Heterostructure. Chem. Eng. J. 2021, 424, 130434. [Google Scholar] [CrossRef]
- Luo, M.; Guo, S. Strain-controlled electrocatalysis on multimetallic nanomaterials. Nat. Rev. Mater. 2017, 2, 17059. [Google Scholar] [CrossRef]
- Ma, Q.; Hu, C.; Liu, K.; Hung, S.F.; Ou, D.; Chen, H.; Fu, G.; Zheng, N. Identifying the electrocatalytic sites of nickel disulfide in alkaline hydrogen evolution reaction. Nano Energy 2017, 41, 148–153. [Google Scholar] [CrossRef]
- Liu, L.; Chu, X.; Liao, J.; Huang, Y.; Li, Y.; Ge, Z.; Hickner, M.; Li, N. Tuning the properties of poly (2,6-dimethyl-1,4-phenylene oxide) anion exchange membranes and their performance in H2/O2 fuel cells. Energy Environ. Sci. 2018, 11, 435–446. [Google Scholar] [CrossRef]
- He, Y.; Liu, S.; Priest, C.; Shi, Q.; Wu, G. Atomically dispersed metal-nitrogen-carbon catalysts for fuel cells: Advances in catalyst design, electrode performance, and durability improvement. Chem. Soc. Rev. 2020, 49, 3484–3524. [Google Scholar] [CrossRef] [PubMed]
- Tkaczyk, A.; Bartl, A.; Amato, A.; Lapkovskis, V.; Petranikova, M. Sustainability evaluation of essential critical raw materials: Cobalt, niobium, tungsten and rare earth elements. J. Phys. D Appl. Phys. 2018, 51, 203001. [Google Scholar] [CrossRef] [Green Version]
- Entr, D. Report on Critical Raw Materials for the EU; European Commission: Brussels, Belgium, 2014. [Google Scholar]
- Faber, F.; Hutchison, L.; Huang, B.; Gilmer, J.; Schoenholz, S.; Dahl, G.; Vinyals, O.; Kearnes, S.; Riley, P.; Von Lilienfeld, O. Prediction errors of molecular machine learning models lower than hybrid DFT error. J. Chem. Theory Comput. 2017, 13, 5255–5264. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The two ways the hydrogen evolution reaction may proceed illustrated. Reproduced from [5] with permission from The Royal Society of Chemistry.
Figure 1.
The two ways the hydrogen evolution reaction may proceed illustrated. Reproduced from [5] with permission from The Royal Society of Chemistry.

Figure 2.
(a) Schematic illustrations exhibiting the entropy/enthalpy changes and (b) the entropic barrier at low and high pH [7]. Reprinted from [7], with permission from Elsevier. (c) The exchange current densities of HER/HOR on Rh/C, Pd/C, Ir/C and Pt/C as a function of the lowest underpotential deposited hydrogen () desorption peak potential, where the gray dashed line represents linear fitting of all data points [8].
Figure 2.
(a) Schematic illustrations exhibiting the entropy/enthalpy changes and (b) the entropic barrier at low and high pH [7]. Reprinted from [7], with permission from Elsevier. (c) The exchange current densities of HER/HOR on Rh/C, Pd/C, Ir/C and Pt/C as a function of the lowest underpotential deposited hydrogen () desorption peak potential, where the gray dashed line represents linear fitting of all data points [8].

Figure 3.
Exchange current densities (ECDs) measured experimentally for the HER on various metal surfaces as a function of the DFT calculated hydrogen chemisorption energy per atom () (upper axis). Single crystal data are indicated by open circles. Gibbs free energy for hydrogen adsorption () determined from a simple kinetic model (bottom axis), = + eV. Used with permission of Electrochemical Society, from [15].
Figure 3.
Exchange current densities (ECDs) measured experimentally for the HER on various metal surfaces as a function of the DFT calculated hydrogen chemisorption energy per atom () (upper axis). Single crystal data are indicated by open circles. Gibbs free energy for hydrogen adsorption () determined from a simple kinetic model (bottom axis), = + eV. Used with permission of Electrochemical Society, from [15].

Figure 4.
(a) Possible bimetallic sites on the surface of copper modified with titanium. (b) Corresponding HBEs showing the activity of the different CuTi sites relative to other common elements [23].
Figure 4.
(a) Possible bimetallic sites on the surface of copper modified with titanium. (b) Corresponding HBEs showing the activity of the different CuTi sites relative to other common elements [23].

Figure 5.
(a) Schematic of common catalyst degradation mechanisms. (b) Pourbaix diagram displaying typical PGM and non–PGM HER catalyst materials in standard water electrolysis ranges, where colored ovals signify deactivation. Both reproduced from [22] with permission from Wiley–VCH. Copyright 2021.
Figure 5.
(a) Schematic of common catalyst degradation mechanisms. (b) Pourbaix diagram displaying typical PGM and non–PGM HER catalyst materials in standard water electrolysis ranges, where colored ovals signify deactivation. Both reproduced from [22] with permission from Wiley–VCH. Copyright 2021.

Figure 6.
(a) Plot of the Pourbaix decomposition energy () of NiMo in an alkaline (pH = 14) aqueous solution with an ion concentration of 10 M. (b) The SCAN determined Pourbaix diagram for NiMo. Reprinted with permission from [30]. Copyright 2021 American Chemical Society.
Figure 6.
(a) Plot of the Pourbaix decomposition energy () of NiMo in an alkaline (pH = 14) aqueous solution with an ion concentration of 10 M. (b) The SCAN determined Pourbaix diagram for NiMo. Reprinted with permission from [30]. Copyright 2021 American Chemical Society.

Figure 7.
(a) Electron localization function (ELF) for plane coincident with two nickel-phosphorous bonds, where a value of 1 (red) corresponds to localized electrons, and 0.5 (green) to delocalized electrons. (b) Point wave function calculated on nickel-phosphorous bond, with red corresponding to a positive wave sign and blue a negative wave sign. Adapted with permission from [41]. Copyright 2021 American Chemical Society.
Figure 7.
(a) Electron localization function (ELF) for plane coincident with two nickel-phosphorous bonds, where a value of 1 (red) corresponds to localized electrons, and 0.5 (green) to delocalized electrons. (b) Point wave function calculated on nickel-phosphorous bond, with red corresponding to a positive wave sign and blue a negative wave sign. Adapted with permission from [41]. Copyright 2021 American Chemical Society.

Figure 8.
(a) In situ STEM images of CoSeP at several times during submersion in 1.0 M KOH. (b) In situ Co K-edge X-ray absorption near edge structure (XANES) spectra of c-CoSe and CoSeP at varied degrees of polarization. Both adapted with permission from [42]. Copyright 2019 American Chemical Society.
Figure 8.
(a) In situ STEM images of CoSeP at several times during submersion in 1.0 M KOH. (b) In situ Co K-edge X-ray absorption near edge structure (XANES) spectra of c-CoSe and CoSeP at varied degrees of polarization. Both adapted with permission from [42]. Copyright 2019 American Chemical Society.

Figure 9.
(1): Illustrations of calculated charge densities of a hydrogen atom adsorbed onto (a) NiP and (b) (FeNi)P. Yellow represents the charge accumulation and cyan charge depletion in the space. (2): A magnified part along the c axis showing the adsorption sites from (1). (3): The adsorption sites from (1) magnified. The nickel, phosphorus, iron, hydrogen, and oxygen atoms are represented by blue, pink, brown, white, and red spheres. (c) Water dissociation energy and hydrogen adsorption free energy on a phosphorus site on an exposed facet. Both reprinted from [43], with permission from Elsevier.
Figure 9.
(1): Illustrations of calculated charge densities of a hydrogen atom adsorbed onto (a) NiP and (b) (FeNi)P. Yellow represents the charge accumulation and cyan charge depletion in the space. (2): A magnified part along the c axis showing the adsorption sites from (1). (3): The adsorption sites from (1) magnified. The nickel, phosphorus, iron, hydrogen, and oxygen atoms are represented by blue, pink, brown, white, and red spheres. (c) Water dissociation energy and hydrogen adsorption free energy on a phosphorus site on an exposed facet. Both reprinted from [43], with permission from Elsevier.

Figure 10.
X-ray photoelectron spectroscopy (XPS) spectra of RuP nanofibers before and after stability test. Reprinted from [45], Copyright 2021, with permission from Elsevier.
Figure 10.
X-ray photoelectron spectroscopy (XPS) spectra of RuP nanofibers before and after stability test. Reprinted from [45], Copyright 2021, with permission from Elsevier.

Figure 11.
(a) Electrocatalytic activity as a function of d-orbital vacancy for CoP doped with V, Cr(Mo), Mn, Fe, Co, Ni, or Cu. (b) Water dissociation barrier as a function of d-orbital vacancy for doped materials, displaying primary origin of augmented activity. (c) Energetics of HER calculated from COHP analysis on simplified surfaces of CoP {111} and Cr-CoP {111} for Heyrovsky route with a preadsorbed hydrogen atom. Reprinted from [49], with permission from Elsevier.
Figure 11.
(a) Electrocatalytic activity as a function of d-orbital vacancy for CoP doped with V, Cr(Mo), Mn, Fe, Co, Ni, or Cu. (b) Water dissociation barrier as a function of d-orbital vacancy for doped materials, displaying primary origin of augmented activity. (c) Energetics of HER calculated from COHP analysis on simplified surfaces of CoP {111} and Cr-CoP {111} for Heyrovsky route with a preadsorbed hydrogen atom. Reprinted from [49], with permission from Elsevier.

Figure 12.
(a) High-angle annular dark-field (HAADF) elemental mapping of nickel nitride nanorods displaying homogenous distribution. (b) DFT determined energetic steps of HER on different surfaces. Both reprinted from [60], with permission from Elsevier.
Figure 12.
(a) High-angle annular dark-field (HAADF) elemental mapping of nickel nitride nanorods displaying homogenous distribution. (b) DFT determined energetic steps of HER on different surfaces. Both reprinted from [60], with permission from Elsevier.

Figure 13.
Comparison of H adsorbed onto optimized structures of bare (a) Ni(111), (b) V–Ni(111), and (c) V/Cu–Ni(111). (d) Free energy diagram of calculated values in conjunction with HER at equilibrium potential for three compared catalysts and Pt(111) benchmark. (e,f) Charge redistribution determined from deformation electronic density calculations of V–Ni(111), where yellow and cyan refer to electron-rich and electron-deficient areas, respectively [73].
Figure 13.
Comparison of H adsorbed onto optimized structures of bare (a) Ni(111), (b) V–Ni(111), and (c) V/Cu–Ni(111). (d) Free energy diagram of calculated values in conjunction with HER at equilibrium potential for three compared catalysts and Pt(111) benchmark. (e,f) Charge redistribution determined from deformation electronic density calculations of V–Ni(111), where yellow and cyan refer to electron-rich and electron-deficient areas, respectively [73].

Figure 14.
(a) XRD spectra taken before and after 100 h ex situ CP test at 100 mA cm, and (b) displays decreased V2p-signal following test [73].
Figure 14.
(a) XRD spectra taken before and after 100 h ex situ CP test at 100 mA cm, and (b) displays decreased V2p-signal following test [73].

Figure 15.
(a) DFT-based analysis of four different sites on catalyst: In-plane, edge, in-plane/edge, and edge/edge sites in 1T–MoS/Ni(OH) nanohybrid catalyst. (a) A free energy diagram displays wide variation of energy barriers associated with HER on these four sites and (a) a comparison of energy barriers , , and associated with dissociation of water, adsorption/desorption of hydrogen and chemisorption of hydroxide species for all sites considered. (b) Illustrations of both edge-rich 1T–MoS QS/Ni(OH) and edge-poor 1T–MoS LS/Ni(OH) catalysts. Both reproduced from [98], with permission from John Wiley and Sons.
Figure 15.
(a) DFT-based analysis of four different sites on catalyst: In-plane, edge, in-plane/edge, and edge/edge sites in 1T–MoS/Ni(OH) nanohybrid catalyst. (a) A free energy diagram displays wide variation of energy barriers associated with HER on these four sites and (a) a comparison of energy barriers , , and associated with dissociation of water, adsorption/desorption of hydrogen and chemisorption of hydroxide species for all sites considered. (b) Illustrations of both edge-rich 1T–MoS QS/Ni(OH) and edge-poor 1T–MoS LS/Ni(OH) catalysts. Both reproduced from [98], with permission from John Wiley and Sons.

Figure 16.
DFT calculated reaction energies for (a) dissociation of water and (b) adsorption of hydrogen atoms on Ni and NiO surfaces. (c) A schematic of complete reaction on dual Ni/NiO surfaces and (d) catalytic activity as a function of Ni/NiO ratio [107].
Figure 16.
DFT calculated reaction energies for (a) dissociation of water and (b) adsorption of hydrogen atoms on Ni and NiO surfaces. (c) A schematic of complete reaction on dual Ni/NiO surfaces and (d) catalytic activity as a function of Ni/NiO ratio [107].

Figure 17.
Effect of tensile strain, which lowers coordination on width and position of d-band of an early transition metal: (a) less than half-filled d-band; (b) reduced bandwidth due to decreased overlap or lower coordination; and (c) downshift of d-band to preserve its filling due to charge conservation. Reprinted Figure 1 with permission from [114]. Copyright (2021) by the American Physical Society.
Figure 17.
Effect of tensile strain, which lowers coordination on width and position of d-band of an early transition metal: (a) less than half-filled d-band; (b) reduced bandwidth due to decreased overlap or lower coordination; and (c) downshift of d-band to preserve its filling due to charge conservation. Reprinted Figure 1 with permission from [114]. Copyright (2021) by the American Physical Society.

Figure 18.
Implications of strain on CoO catalyst. (a) Adsorption/dissociation of water on a CoO {111} surface with O-vacancies. (b) Hydrogen binding energy (HBE) as a function of applied strain. (c) Band structures of pristine and strained catalyst, where small strain-imposed change has pronounced effects on d- and 2p-orbital [117].
Figure 18.
Implications of strain on CoO catalyst. (a) Adsorption/dissociation of water on a CoO {111} surface with O-vacancies. (b) Hydrogen binding energy (HBE) as a function of applied strain. (c) Band structures of pristine and strained catalyst, where small strain-imposed change has pronounced effects on d- and 2p-orbital [117].

Figure 19.
(a) Illustration of cell setup for in situ XAS. (b) In situ L-edge X-ray adsorption near edge spectroscopy (XANES) of RhO-Rh-NP catalyst at various chronoamperometric test durations at 10 mA cm and (c) under various degrees of polarization. Both reproduced from [119] with permission from Wiley–VCH.
Figure 19.
(a) Illustration of cell setup for in situ XAS. (b) In situ L-edge X-ray adsorption near edge spectroscopy (XANES) of RhO-Rh-NP catalyst at various chronoamperometric test durations at 10 mA cm and (c) under various degrees of polarization. Both reproduced from [119] with permission from Wiley–VCH.

Figure 20.
(a) XRD displaying FeP electrode before and after 12 h immersion in 1.0 M KOH. (b) Regenerating effect of etching the electrode in HCl for 60 s. Reprinted from [122], with permission from Elsevier.
Figure 20.
(a) XRD displaying FeP electrode before and after 12 h immersion in 1.0 M KOH. (b) Regenerating effect of etching the electrode in HCl for 60 s. Reprinted from [122], with permission from Elsevier.

Figure 21.
Triangular plot displaying all three activity descriptors for hydrogen evolution reaction. Utilizing , and as screening parameters for searching for HER catalyst materials.
Figure 21.
Triangular plot displaying all three activity descriptors for hydrogen evolution reaction. Utilizing , and as screening parameters for searching for HER catalyst materials.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ferriday, T.B.; Middleton, P.H.; Kolhe, M.L. Review of the Hydrogen Evolution Reaction—A Basic Approach. Energies 2021, 14, 8535. https://doi.org/10.3390/en14248535
AMA Style
Ferriday TB, Middleton PH, Kolhe ML. Review of the Hydrogen Evolution Reaction—A Basic Approach. Energies. 2021; 14(24):8535. https://doi.org/10.3390/en14248535
Chicago/Turabian StyleFerriday, Thomas B., Peter Hugh Middleton, and Mohan Lal Kolhe. 2021. "Review of the Hydrogen Evolution Reaction—A Basic Approach" Energies 14, no. 24: 8535. https://doi.org/10.3390/en14248535
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.