
Prenatal Diagnosis of True Fetal Mosaicism with Small Supernumerary Marker Chromosome Derived from Chromosome 16 by Funipuncture and Molecular Cytogenetics Including Chromosome Microarray

 , and
, and 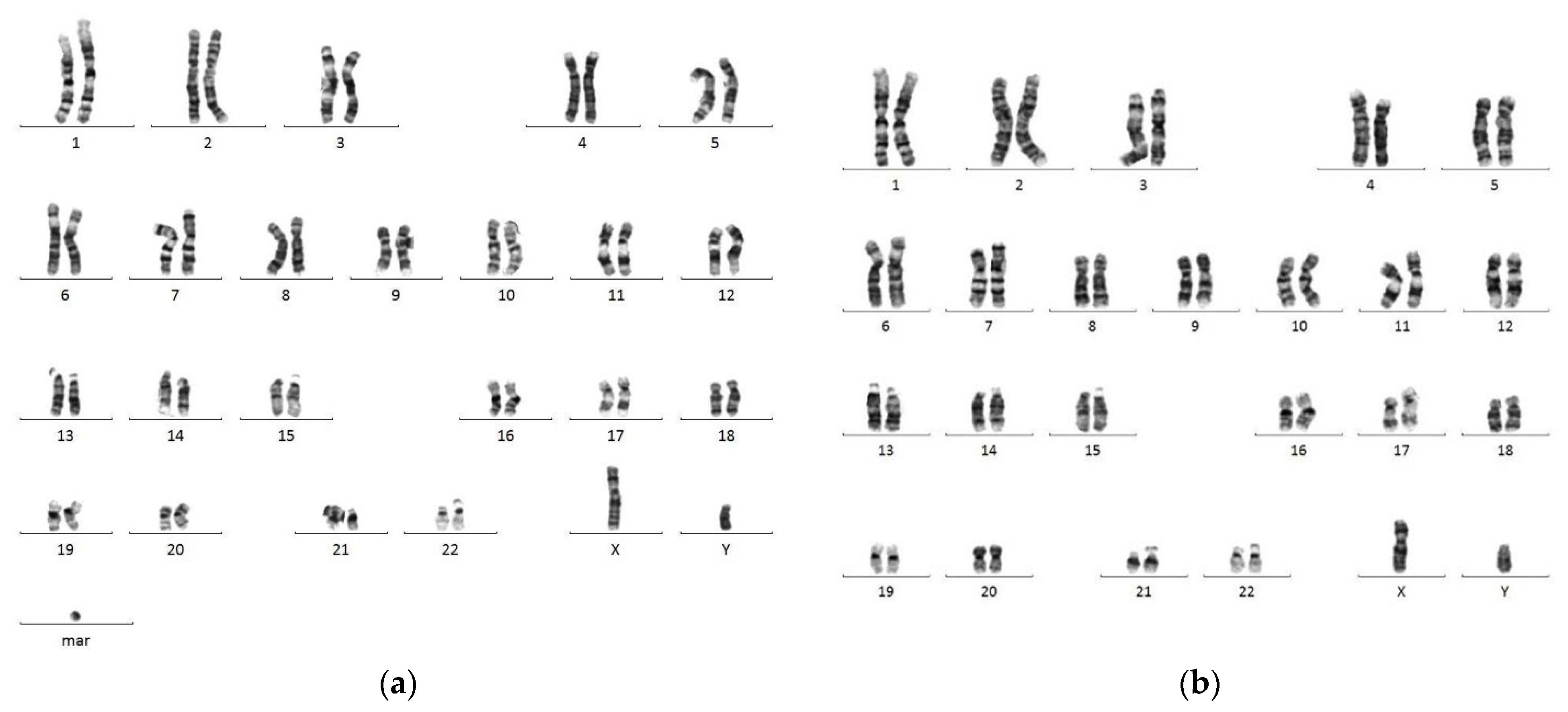{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Description
2.2. Cytogenetic and Molecular Cytogenetic Analysis of Amniocytes
2.3. Molecular Cytogenetic and Chromosome Microarray Analysis of Cord Blood
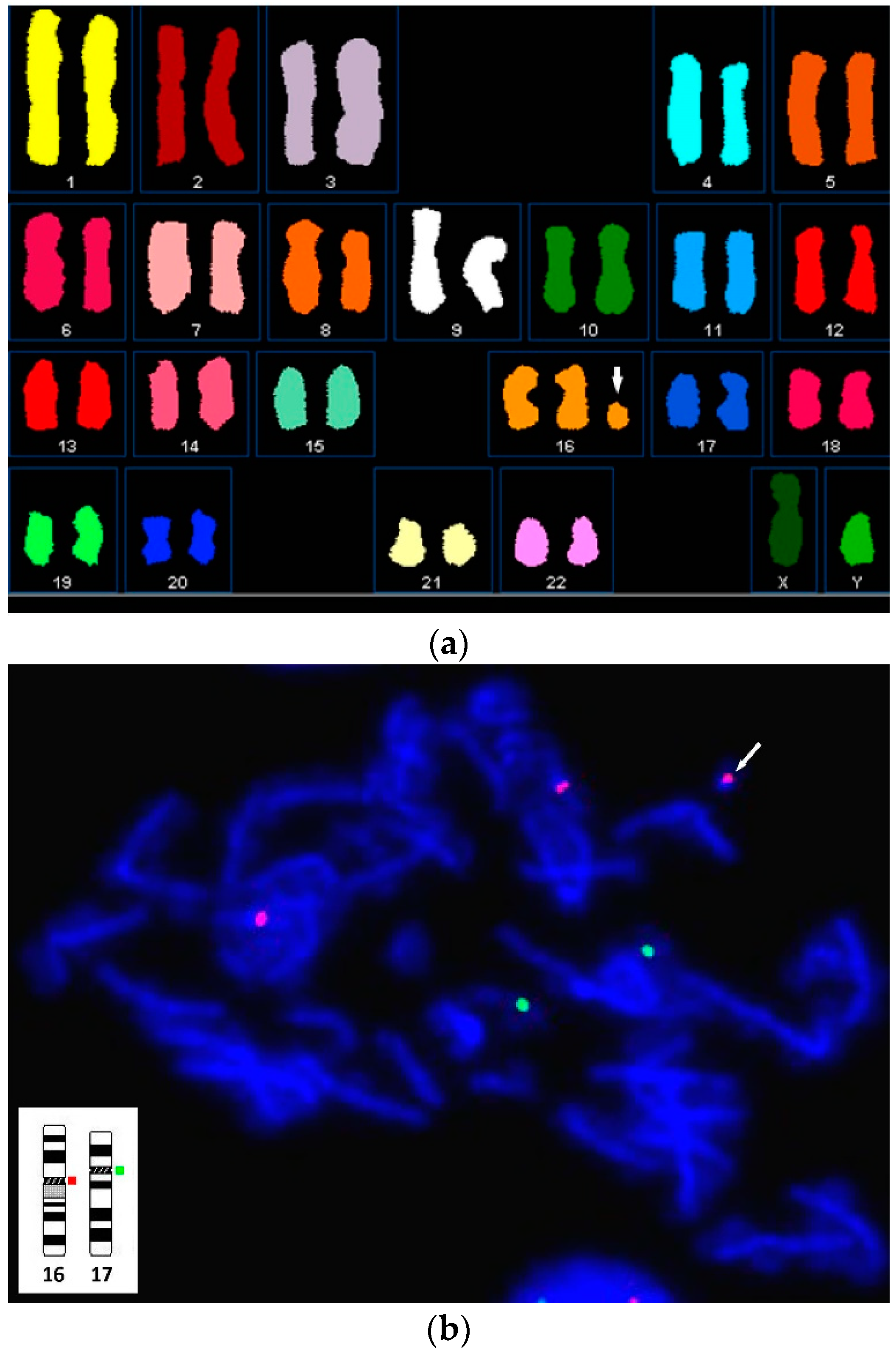
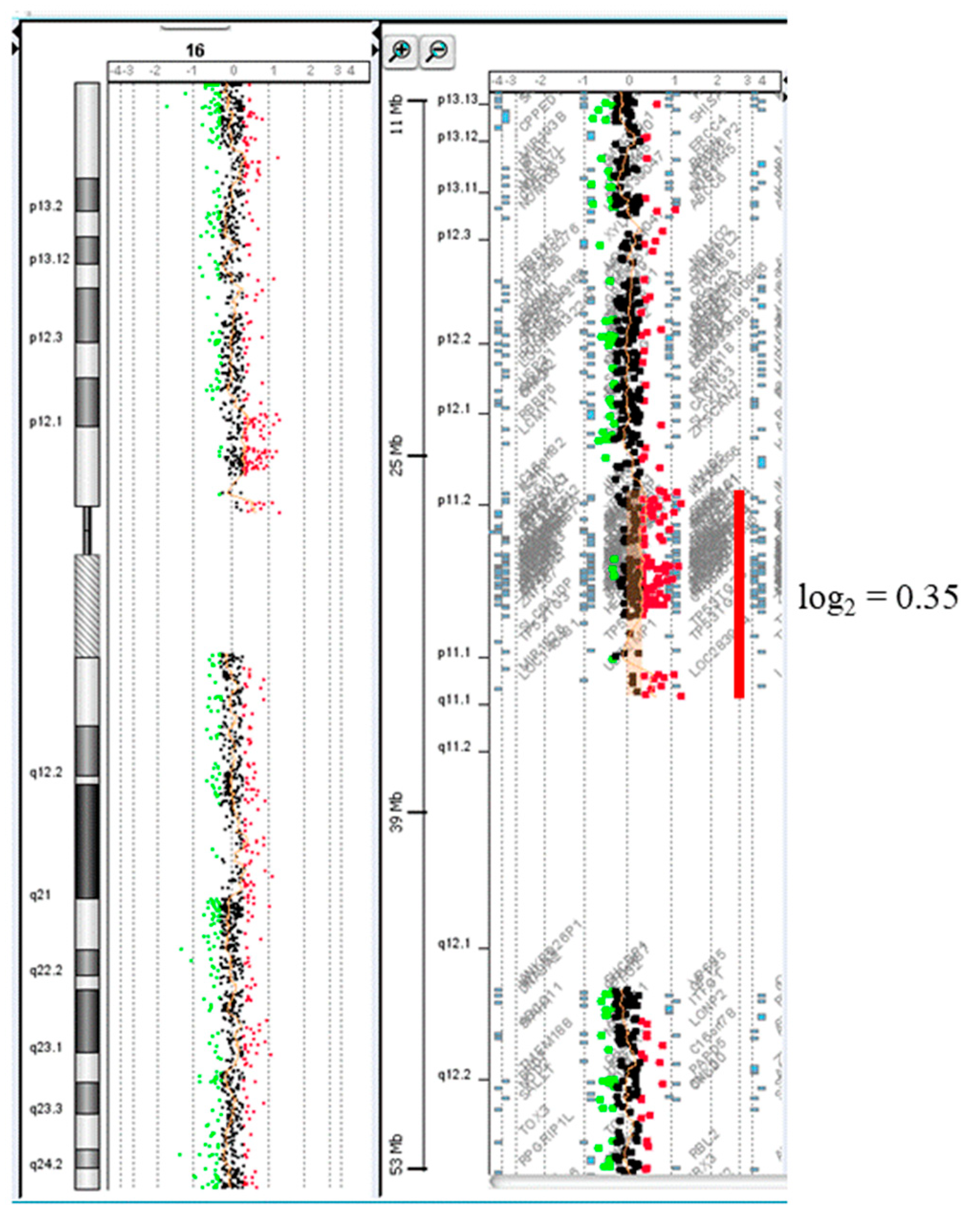
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liehr, T.; Claussen, U.; Starke, H. Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet. Genome Res. 2004, 107, 55–67. [Google Scholar] [CrossRef]
- Liehr, T.; Weise, A. Frequency of small supernumerary marker chromosomes in prenatal, newborn, developmentally retarded and infertility diagnostics. Int. J. Mol. Med. 2007, 19, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Chang, S.P.; Yin, P.L.; Sapeta, M.; Barringer, S.; Kuo, S.J.; Yu, H.T.; Wang, B.B. Prenatal identification of small supernumerary marker chromosomes by FISH in an infant born with mild congenital anomalies. Prenat. Diagn. 2006, 26, 383–387. [Google Scholar] [CrossRef]
- Chen, M.; Yeh, G.P.; Shih, J.C.; Wang, B.T. Trisomy 13 mosaicism: Study of serial cytogenetic changes in a case from early pregnancy to infancy. Prenat. Diagn. 2004, 24, 137–143. [Google Scholar] [CrossRef]
- Lin, C.C.; Hsieh, Y.Y.; Wang, C.H.; Li, Y.C.; Hsieh, L.J.; Lee, C.C.; Tsai, C.H.; Tsai, F.J. Prenatal detection and characterization of a small supernumerary marker. chromosome (sSMC) derived from chromosome 22 with apparently normal phynoetype. Prenat. Diagn. 2006, 26, 898–902. [Google Scholar] [CrossRef]
- Sung, P.L.; Chang, S.P.; Wen, K.C.; Chang, C.M.; Yang, M.J.; Chen, L.C.; Chao, K.C.; Huang, C.Y.F.; Li, Y.C.; Lin, C.C. Small supernumerary marker chromosome originating from chromosome 10 associated with an apparently normal phenotype. Am. J. Med. Genet. A 2009, 149, 2768–2774. [Google Scholar] [CrossRef]
- Chen, C.P.; Chen, M.; Wu, C.H.; Lin, C.J.; Chern, S.R.; Wu, P.S.; Chen, Y.N.; Chen, S.W.; Chang, S.P.; Chen, L.F.; et al. Prenatal diagnosis and molecular cytogenetic. characterization of mosaicism for a small supernumerary marker chromosome derived from chromosome 21q11.2q21.1 and a literature review. Taiwan. J. Obstet. Gynecol. 2017, 56, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Huang, H.; Wang, Y.; An, G.; Zhang, M.; Xu, L.; Lin, Y. Molecular cytogenetic identification of small supernumerary marker chromosome using chromosome microarray analysis. Mol. Cytogenet. 2019, 12, 13. [Google Scholar] [CrossRef]
- Chen, C.P.; Chen, M.; Wang, L.K.; Chern, S.R.; Wu, P.S.; Ma, G.C.; Chang, S.P.; Chen, S.W.; Wu, F.T.; Lee, C.C.; et al. Low-level mosaicism for trisomy 16 at amniocentesis in a pregnancy associated with intrauterine growth restriction and a favorable outcome. Taiwan. J. Obstet. Gynecol. 2021, 60, 345–349. [Google Scholar] [CrossRef]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.; Green, T.; et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.; Han, C.; Gordon, L.A.; Terry, A.; Prabhakar, S.; She, X.; Xie, G.; Hellsten, U.; Chan, Y.M.; Altherr, M.; et al. The sequence and analysis of duplication-rich human chromosome 16. Nature 2004, 432, 988–994. [Google Scholar] [CrossRef]
- Pu, L.; Lin, Y.; Pevzner, P.A. Detection and analysis of ancient segmental duplications in mammalian genomes. Genome Res. 2018, 28, 901–909. [Google Scholar] [CrossRef]
- Nuttle, X.; Giannuzzi, G.; Duyzend, M.H.; Schraiber, J.G.; Narvaiza, I.; Sudmant, P.H.; Penn, O.; Chiatante, G.; Malig, M.; Huddleston, J.; et al. Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature 2016, 536, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.H. Trisomy 16, cause of first trimester abortion. J. Pak. Med. Assoc. 2001, 51, 378–379. [Google Scholar] [PubMed]
- Yakut, S.; Toru, H.S.; Çetin, Z.; Özel, D.; Şimşek, M.; Mendilcioğlu, İ.; Lüleci, G. Chromosome abnormalities identified in 457 spontaneous abortions and their histopathological findings. Turk Patoloji Derg. 2015, 31, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, A.; Pinti, É.; Pikó, H.; Jávorszky, E.; David, D.; Tihanyi, M.; Gönczi, É.; Kiss, E.; Tóth, Z.; Tory, K.; et al. Clinical and genetic findings in Hungarian pediatric patients carrying chromosome 16p copy number variants and a review of the literature. Eur. J. Med. Genet. 2020, 63, 104027. [Google Scholar] [CrossRef]
- Fernandez, B.A.; Roberts, W.; Chung, B.; Weksberg, R.; Meyn, S.; Szatmari, P.; Joseph-George, A.M.; Mackay, S.; Whitten, K.; Noble, B.; et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J. Med. Genet. 2010, 47, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Shinawi, M.; Liu, P.; Kang, S.H.; Shen, J.; Belmont, J.W.; Scott, D.A.; Probst, F.J.; Craigen, W.J.; Graham, B.H.; Pursley, A.; et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2010, 47, 332–341. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.E.; Makarov, V.; Kirov, G.; Addington, A.M.; McClellan, J.; Yoon, S.; Perkins, D.O.; Dickel, D.E.; Kusenda, M.; Krastoshevsky, O.; et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009, 41, 1223–1227. [Google Scholar] [CrossRef]
- Loviglio, M.N.; Leleu, M.; Männik, K.; Passeggeri, M.; Giannuzzi, G.; van der Werf, I.; Waszak, S.M.; Zazhytska, M.; Roberts-Caldeira, I.; Gheldof, N.; et al. Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Mol. Psychiatry 2017, 22, 836–849. [Google Scholar] [CrossRef]
- Haslinger, D.; Waltes, R.; Yousaf, A.; Lindlar, S.; Schneider, I.; Lim, C.K.; Tsai, M.M.; Garvalov, B.K.; Acker-Palmer, A.; Krezdorn, N.; et al. Loss of the Chr16p11.2 ASD candidate gene QPRT leads to aberrant neuronal differentiation in the SH-SY5Y neuronal cell model. Mol. Autism. 2018, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Golzio, C.; Willer, J.; Talkowski, M.E.; Oh, E.C.; Taniguchi, Y.; Jacquemont, S.; Reymond, A.; Sun, M.; Sawa, A.; Gusella, J.F.; et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012, 485, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Escamilla, C.O.; Filonova, I.; Walker, A.K.; Xuan, Z.X.; Holehonnur, R.; Espinosa, F.; Liu, S.; Thyme, S.B.; López-García, I.A.; Mendoza, D.B.; et al. Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 2017, 551, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tang, J.; Lan, X.; Mi, X. Increased expression of DOC2A in human and rat temporal lobe epilepsy. Epilepsy Res. 2019, 151, 78–84. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, T.-Y.; Wu, W.-J.; Law, K.-S.; Lee, M.-H.; Chang, S.-P.; Lee, D.-J.; Lin, W.-H.; Chen, M.; Ma, G.-C. Prenatal Diagnosis of True Fetal Mosaicism with Small Supernumerary Marker Chromosome Derived from Chromosome 16 by Funipuncture and Molecular Cytogenetics Including Chromosome Microarray. Diagnostics 2021, 11, 1457. https://doi.org/10.3390/diagnostics11081457
Yao T-Y, Wu W-J, Law K-S, Lee M-H, Chang S-P, Lee D-J, Lin W-H, Chen M, Ma G-C. Prenatal Diagnosis of True Fetal Mosaicism with Small Supernumerary Marker Chromosome Derived from Chromosome 16 by Funipuncture and Molecular Cytogenetics Including Chromosome Microarray. Diagnostics. 2021; 11(8):1457. https://doi.org/10.3390/diagnostics11081457
Chicago/Turabian StyleYao, Tien-Yu, Wan-Ju Wu, Kim-Seng Law, Mei-Hui Lee, Shun-Ping Chang, Dong-Jay Lee, Wen-Hsiang Lin, Ming Chen, and Gwo-Chin Ma. 2021. "Prenatal Diagnosis of True Fetal Mosaicism with Small Supernumerary Marker Chromosome Derived from Chromosome 16 by Funipuncture and Molecular Cytogenetics Including Chromosome Microarray" Diagnostics 11, no. 8: 1457. https://doi.org/10.3390/diagnostics11081457