Mating System in a Native Norway Spruce (Picea abies [L.] KARST.) Stand-Relatedness and Effective Pollen Population Size Show an Association with the Germination Percentage of Single Tree Progenies

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
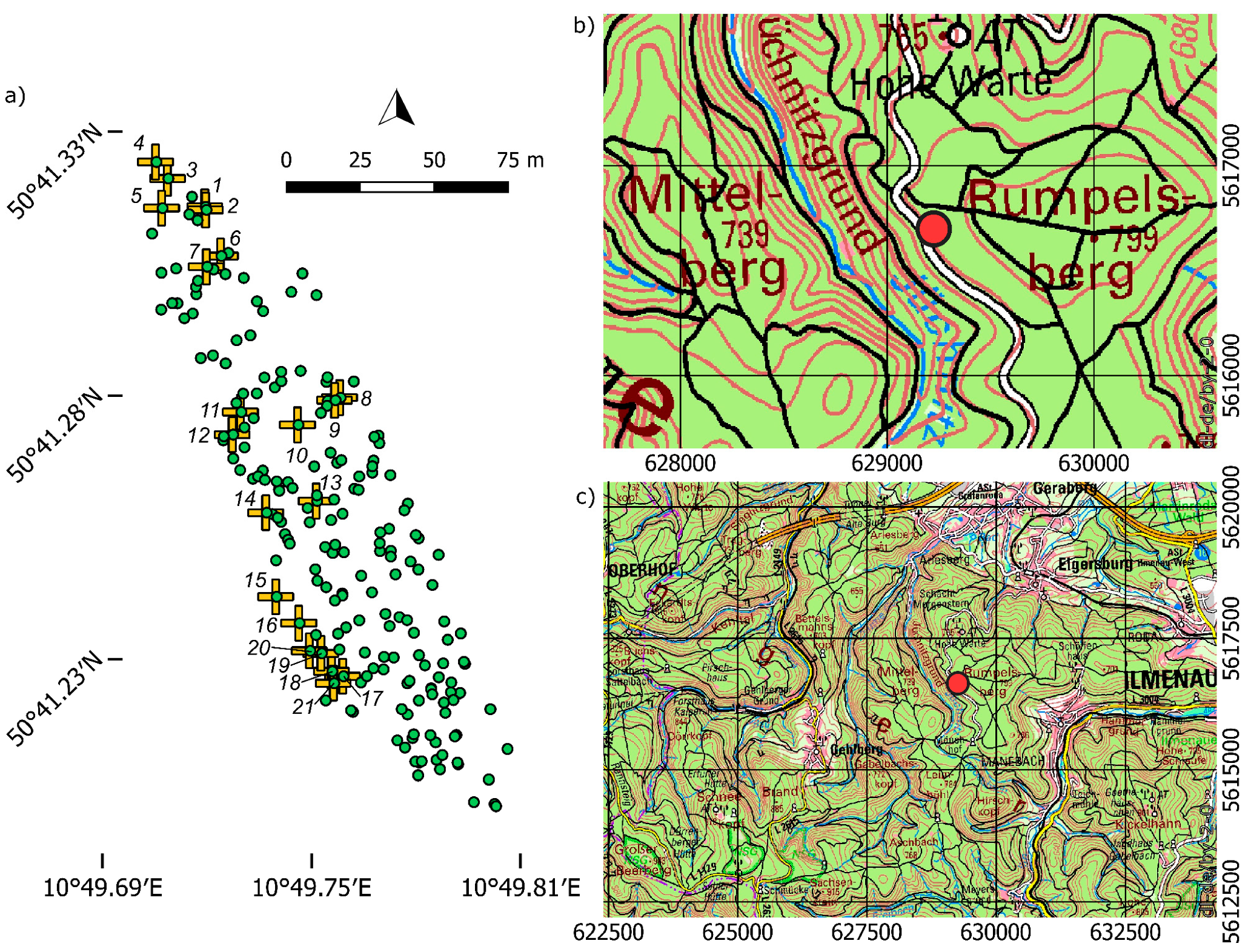
2.1. Plant Material
2.2. Observation of Germination
2.3. DNA Extraction and Genotyping
2.4. Statistical Analysis
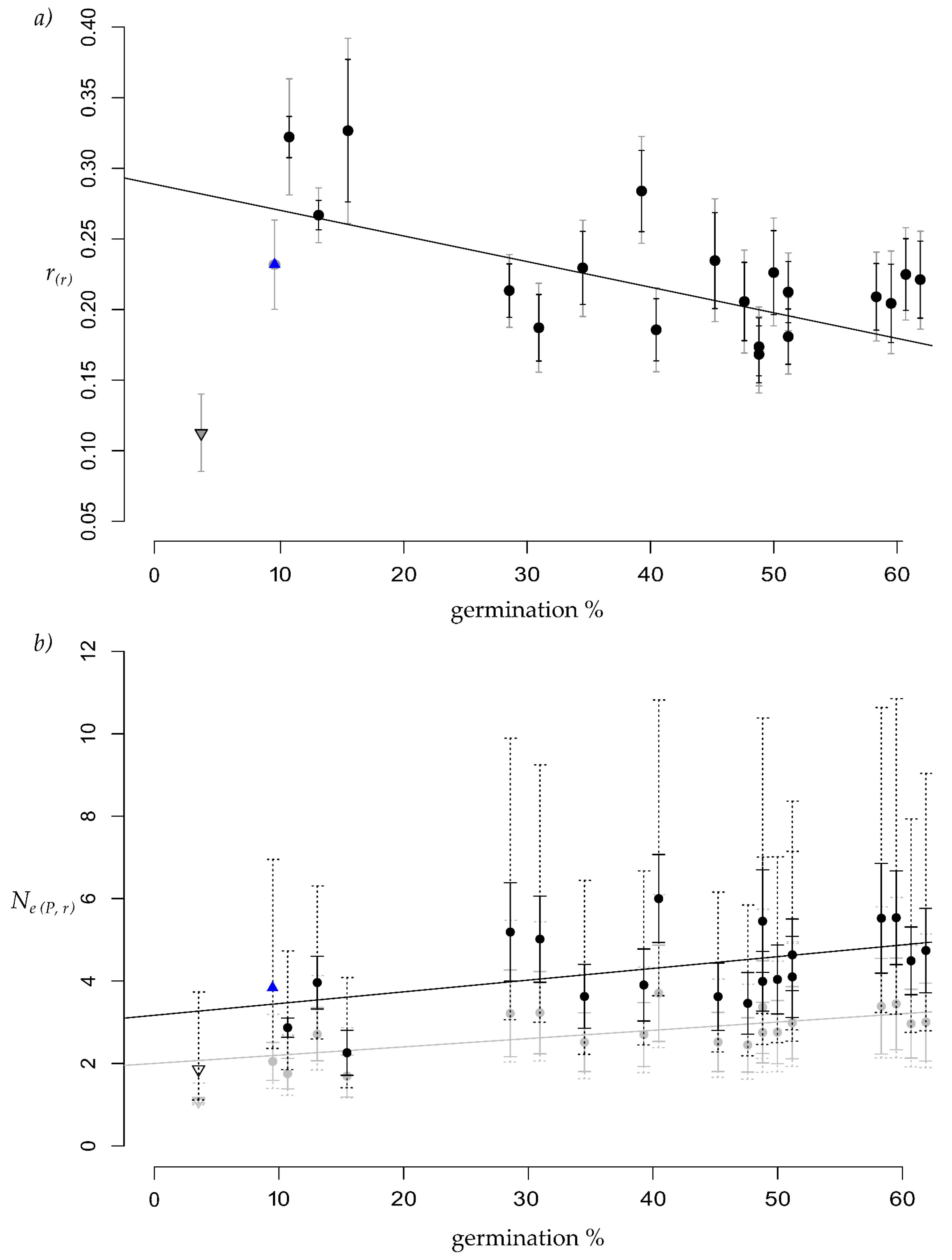
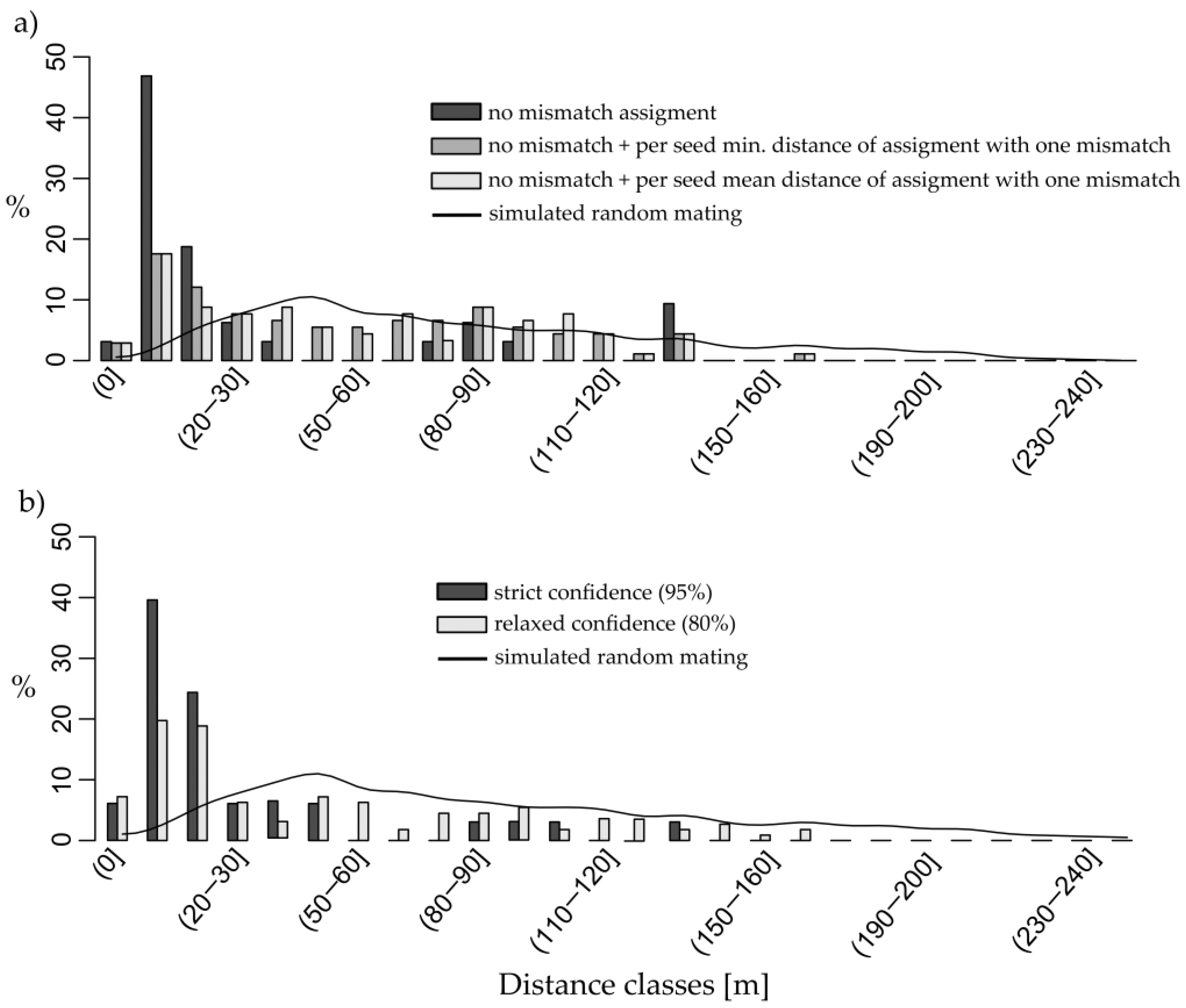
3. Results
4. Discussion
4.1. Germination and Relatedness
4.2. Distances and Distribution
4.3. Selfing and Outcrossing
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paul, M.; Hinrichs, T.; Janßen, A.; Schmitt, H.-P.; Soppa, B.; Stephan, R.; Dörflinger, H. Forest Genetic Resources in Germany—Concept for the Conservation and Sustainable Utilization of Forest Genetic Resources in the Federal Republic of Germany; Federal Ministry of Food Agriculture and Consumer Protection (BMELV) in Cooperation with the Federal Government/Länder Working Group “Forest Genetic Resources and Legislation on Forest Reproductive Material” (BLAG-FGR), Ed.; BMELV: Bonn, Germany, 2010. [Google Scholar]
- Schmidt-Vogt, H. Taxonomie, Verbreitung, Morphologie, Ökologie, Waldgesellschaften. In Die Fichte—Ein Handbuch in zwei Bänden; Bd. 1.; Parey: Hamburg/Berlin, Germany, 1977; p. 647. [Google Scholar]
- Geburek, T.; Robitschek, K.; Milasowszky, N. A tree of many faces: Why are there different crown types in Norway spruce (Picea abies [L.] Karst.)? Flora Morphol. Distrib. Funct. Ecol. Plants 2008, 203, 126–133. [Google Scholar] [CrossRef]
- Caré, O.; Gailing, O.; Müller, M.; Krutovsky, K.V.; Leinemann, L. Crown morphology in Norway spruce (Picea abies [Karst.] L.) as adaptation to mountainous environments is associated with single nucleotide polymorphisms (SNPs) in genes regulating seasonal growth rhythm. Tree Genet. Genomes 2020, 16. [Google Scholar] [CrossRef]
- Xie, C.Y.; Knowles, P. Mating system and effective pollen immigration in a Norway spruce plantation (Picea abies). Silvae Genet. 1994, 43, 48–51. [Google Scholar]
- Burczyk, J.; Lewandowski, A.; Chalupka, W. Local pollen dispersal and distant gene flow in Norway spruce (Picea abies [L.] Karst.). For. Ecol. Manag. 2004, 197, 39–48. [Google Scholar] [CrossRef]
- Williams, C.G. Long-distance pine pollen still germinates after meso-scale dispersal. Am. J. Bot. 2010, 97, 846–855. [Google Scholar] [CrossRef]
- Mitton, J.B.; Williams, C.G. Gene flow in conifers. In Landscapes, Genomics and Transgenic Conifers. Managing Forest Ecosystems; Williams, C., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 147–168. [Google Scholar] [CrossRef]
- Hagman, M. Incompatibility in forest trees. Proc. R. Soc. London. Ser. B. Biol. Sci. 1975, 188, 313–326. [Google Scholar] [CrossRef]
- Mergen, F.; Burley, J.; Furnival, G.M. Embryo and seedling development in Picea glauca (Moench) Voss after self-, cross-, and wind-pollination. Silvae Genet. 1965, 14, 188–194. [Google Scholar] [CrossRef]
- Skrøppa, T. Diallel crosses in Picea abies II. Perdormance and inbreeding depression of selfed families. For. Genet. 1996, 3, 69–79. [Google Scholar]
- Eriksson, G.; Schelander, B.; Åkerbrand, V. Inbreeding depression in an old experimental plantation of Picea abies. Hereditas 1973, 73, 185–193. [Google Scholar] [CrossRef]
- Di-Giovanni, F.; Kevan, P.G. Factors affecting pollen dynamics and its importance to pollen contamination: A review. Can. J. For. Res. 1991, 21, 1155–1170. [Google Scholar] [CrossRef]
- Di-Giovanni, F.; Kevan, P.G.; Arnold, J. Lower planetary boundary layer profiles of atmospheric conifer pollen above a seed orchard in northern Ontario, Canada. For. Ecol. Manag. 1996, 83, 87–97. [Google Scholar] [CrossRef]
- Tollefsrud, M.M.; Sønstebø, J.H.; Brochmann, C.; Johnsen, Ø.; Skrøppa, T.; Vendramin, G.G. Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European Picea abies. Heredity 2009, 102, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutovsky, K.; Burczyk, J.; Chybicki, I. Gene Flow, Spatial Structure, Local Adaptation, and Assisted Migration in Trees. In Genomics of Tree Crops; Schnell, R.J., Priyadarshan, P.M., Eds.; Springer: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2012; pp. 71–116. ISBN 9781461409199. [Google Scholar]
- Caré, O.; Müller, M.; Vornam, B.; Höltken, A.; Kahlert, K.; Krutovsky, K.; Gailing, O.; Leinemann, L. High morphological differentiation in crown architecture contrasts with low population genetic structure of German Norway spruce stands. Forests 2018, 9, 752. [Google Scholar] [CrossRef] [Green Version]
- Thüringer Landesamt für Bodenmanagement und Geoinformation (Ed.) Geoportal-Th. Available online: https://www.geoportal-th.de (accessed on 4 May 2020).
- Scotti, I.; Magni, F.; Paglia, G.P.; Morgante, M. Trinucleotide microsatellites in Norway spruce (Picea abies): Their features and the development of molecular markers. Theor. Appl. Genet. 2002, 106, 40–50. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Olivieri, A.M.; Morgante, M. Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 1997, 40, 411–419. [Google Scholar] [CrossRef]
- Besnard, G.; Acheré, V.; Rampant, P.F.; Favre, J.M.; Jeandroz, S. A set of cross-species amplifying microsatellite markers developed from DNA sequence databanks in Picea (Pinaceae). Mol. Ecol. Notes 2003, 3, 380–383. [Google Scholar] [CrossRef]
- Rungis, D.; Bérubé, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Hamming, R.W. Error Detecting and error correcting codes. Bell Syst. Tech. J. 1950, 29, 147–160. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamvar, Z.N.; Brooks, J.C.; Grünwald, N.J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, T.-Y. NB: Maximum Likelihood Method in Estimating Effective Population Size from Genetic Data. R Package Version 0.9. 2014. Available online: https://rdrr.io/cran/NB/ (accessed on 2 May 2020).
- Hui, T.-Y.J.; Burt, A. Estimating Effective Population Size from Temporally Spaced Samples with a Novel, Efficient Maximum-Likelihood Algorithm. Genetics 2015, 200, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J. Triadic IBD coefficients and applications to estimating pairwise relatedness. Genet. Res. 2007, 89, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Pew, J.; Muir, P.H.; Wang, J.; Frasier, T.R. related: An R package for analyzing pairwise relatedness from codominant molecular markers. Mol. Ecol. Resour. 2015, 15, 557–561. [Google Scholar] [CrossRef]
- Ritland, K. Estimators for pairwise relatedness and individual inbreeding coefficients. Genet. Res. 1996, 67, 175–185. [Google Scholar] [CrossRef]
- Milligan, B.G. Maximum-likelihood estimation of relatedness. Genetics 2003, 163, 1153–1167. [Google Scholar]
- Kalinowski, S.; Taper, M.; Marshall, T. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Bivand, R.; Keitt, T.; Rowlingson, B. rgdal: Bindings for the “Geospatial” Data Abstraction Library. R Package Version 1.3-6. Available online: https://CRAN.R-project.org/package=rgdal (accessed on 26 June 2020).
- Hijmans, R.J. Geosphere: Spherical Trigonometry. R Package Version 1.5-7. Available online: https://CRAN.R-project.org/package=geosphere (accessed on 26 May 2020).
- Lemon, J. Plotrix: A package in the red light district of R. R-News 2006, 6, 8–12. [Google Scholar]
- Breusch, T.S.; Pagan, A.R. A Simple test for heteroscedasticity and random coefficient variation. Econometrica 1979, 47, 1287–1294. [Google Scholar] [CrossRef]
- Andersson, E. Cone and seed studies in Norway spruce (Pices abies (L.) Karst). Studia For. Suec. 1965, 23, 214. [Google Scholar]
- Fowler, D.P.; Park, Y.S. Population studies of white spruce. I. Effects of self-pollination. Can. J. For. Res. 1983, 13, 1133–1138. [Google Scholar] [CrossRef]
- Coles, J.F.; Fowler, D.P. Inbreeding in neighboring trees in two white spruce populations. Silvae Genet. 1976, 25, 29–34. [Google Scholar]
- Kuittinen, H.; Savolainen, O. Picea omorika is a self-fertile but outcrossing conifer. Heredity 1992, 68, 183–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullin, T.J.; Persson, T.; Abrahamsson, S.; Andersson Gull, B. Effects of inbreeding depression on seed production in scots pine (Pinus sylvestris). Can. J. For. Res. 2019, 49, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Berrill, J.; Libby, W.J. Comparing growth and form of coast redwood selfs and outcrosses. In Coast. Redwood Science Symposium—2016: Past Successes and Future Direction; Standiford, R.B., Valachovic, Y., Eds.; Gen. Tech. Rep. PSW-GTR-258; U.S. Department of Agriculture, Forest Service, Pacific Southwest Research Station: Albany, CA, USA, 2016; pp. 229–240. [Google Scholar]
- Woods, J.H.; Heaman, J.C. Effect of different inbreeding levels on filled seed production in Douglas-fir. Can. J. For. Res. 1989, 19, 54–59. [Google Scholar] [CrossRef]
- Müller-Starck, G.; Ziehe, M.; Schubert, R. Genetic Diversity Parameters Associated with Viability Selection, Reproductive Efficiency, and Growth in Forest Tree Species. In Forest Diversity and Function; Scherer-Lorenzen, M., Körner, C., Schulze, E.D., Eds.; Springer: Heidelberg, Germany, 2005; Volume 176, pp. 87–108. [Google Scholar]
- Wojacki, J.; Eusemann, P.; Ahnert, D.; Pakull, B.; Liesebach, H. Genetic diversity in seeds produced in artificial Douglas-fir (Pseudotsuga menziesii) stands of different size. For. Ecol. Manag. 2019, 438, 18–24. [Google Scholar] [CrossRef]
- Del Castillo, R.F.; Trujillo-Argueta, S.; Sánchez-Vargas, N.; Newton, A.C. Genetic factors associated with population size may increase extinction risks and decrease colonization potential in a keystone tropical pine. Evol. Appl. 2011, 4, 574–588. [Google Scholar] [CrossRef]
- Lynch, M. The genetic interpretation of inbreeding depression and outbreeding depression. Evolution 1991, 45, 622–629. [Google Scholar] [CrossRef]
- Kärkkäinen, K.; Savolainen, O.; Koski, V. Why do plants abort so many developing seeds: Bad offspring or bad maternal genotypes? Evol. Ecol. 1999, 13, 305–317. [Google Scholar] [CrossRef]
- Goto, S.; Iijima, H.; Ogawa, H.; Ohya, K. Outbreeding depression caused by intraspecific hybridization between local and nonlocal genotypes in Abies sachalinensis. Restor. Ecol. 2011, 19, 243–250. [Google Scholar] [CrossRef]
- Sønstebø, J.H.; Tollefsrud, M.M.; Myking, T.; Steffenrem, A.; Nilsen, A.E.; Edvardsen, M.; Johnskås, O.R.; El-Kassaby, Y.A. Genetic diversity of Norway spruce (Picea abies (L.) Karst.) seed orchard crops: Effects of number of parents, seed year, and pollen contamination. For. Ecol. Manag. 2018, 411, 132–141. [Google Scholar] [CrossRef]
- O’Connell, L.M.; Mosseler, A.; Rajora, O.P. Impacts of forest fragmentation on the mating system and genetic diversity of white spruce (Picea glauca) at the landscape level. Heredity 2006, 97, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimono, A.; Wang, X.R.; Torimaru, T.; Lindgren, D.; Karlsson, B. Spatial variation in local pollen flow and mating success in a Picea abies clone archive and their implications for a novel “breeding without breeding” strategy. Tree Genet. Genomes 2011, 7, 499–509. [Google Scholar] [CrossRef]
- Wright, J.W. Pollen Dispersion of Some Forest Trees; Station Paper NE-46; United States Department of Agriculture, Forest Service, Northeastern Forest Experiment Station Upper Darby: Pennsylvania, PA, USA, 1952; p. 42.
- Finkeldey, R. Homogeneity of pollen allele frequencies of single seed trees in Picea abies (L.) Karst plantations. Heredity 1995, 74, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Pakkanen, A.; Nikkanen, T.; Pulkkinen, P. Annual variation in pollen contamination and outcrossing in a Picea abies seed orchard. Scand. J. For. Res. 2000, 15, 399–404. [Google Scholar] [CrossRef]
- Dering, M.; Misiorny, A.; Chałupka, W. Inter-year variation in selfing, background pollination, and paternal contribution in a Norway spruce clonal seed orchard. Can. J. For. Res. 2014, 44, 760–767. [Google Scholar] [CrossRef]
- Muona, O.; Paule, L.; Szmidt, A.E.; Kärkkäinen, K. Mating system analysis in a central and northern European population of Picea abies. Scand. J. For. Res. 1990, 5, 97–102. [Google Scholar] [CrossRef]
- Rajora, O.P.; Mosseler, A.; Major, J.E. Indicators of population viability in red spruce, Picea rubens. II. Genetic diversity, population structure, and mating behavior. Can. J. Bot. 2000, 78, 941–956. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Halb-sib Family | Germination 1 (%) | Number of Seeds | Ne (P; r) [CI] | Ho [SD] | He | FIS | r(r) [SD] | ||
|---|---|---|---|---|---|---|---|---|---|
| Genotyped | Assigned by Comparison 2 | Assigned by Cervus 3 | |||||||
| 1 | 3.6 | 4 | 0 | 1 | 1.1 [1, 1.5] | 0.533 [0.076] | 0.616 | −0.034 | 0.11 [0.03] |
| 2 | 40.5 | 20 | 6 | 7 | 3.7 [2.4, 6.1] | 0.700 [0.040] | 0.657 | −0.068 * | 0.19 [0.03] |
| 3 | 13.1 | 13 | 2 | 2 | 2.7 [1.8, 4.1] | 0.679 [0.035] | 0.628 | −0.106 * | 0.27 [0.02] |
| 4 | 15.5 | 13 | 3 | 0 | 1.7 [1.2, 2.9] | 0.554 [0.067] | 0.536 | −0.081 | 0.33 [0.07] |
| 5 | 48.8 | 20 | 5 | 2 | 2.7 [1.8, 4.5] | 0.571 [0.040] | 0.587 | −0.006 | 0.17 [0.03] |
| 6 | 50.0 | 20 | 8 | 8 | 2.8 [1.8, 4.5] | 0.667 [0.065] | 0.639 | −0.122** | 0.23 [0.04] |
| 7 | 61.9 | 20 | 4 | 7 | 3.0 [1.9, 5.1] | 0.558 [0.043] | 0.513 | −0.112** | 0.22 [0.03] |
| 8 | 39.3 | 20 | 7 | 5 | 2.7 [1.8, 4.3] | 0.552 [0.053] | 0.532 | −0.048 | 0.28 [0.04] |
| 9 | 58.3 | 20 | 6 | 6 | 3.4 [2.1, 5.8] | 0.655 [0.032] | 0.581 | −0.131 ** | 0.21 [0.03] |
| 10 | 45.2 | 20 | 8 | 10 | 2.5 [1.7, 4] | 0.582 [0.055] | 0.647 | −0.024 | 0.23 [0.04] |
| 11 | 28.6 | 20 | 8 | 10 | 3.2 [2, 5.5] | 0.616 [0.045] | 0.584 | −0.065 * | 0.21 [0.03] |
| 12 | 47.6 | 20 | 7 | 10 | 2.4 [1.6, 3.9] | 0.571 [0.057] | 0.563 | −0.048 | 0.21 [0.04] |
| 13 | 9.5 | 9 | 2 | 4 | 2.0 [1.4, 3.2] | 0.632 [0.051] | 0.636 | −0.023 | 0.23 [0.03] |
| 14 | 48.8 | 20 | 1 | 3 | 3.4 [2.1, 5.7] | 0.632 [0.029] | 0.616 | −0.079 * | 0.17 [0.03] |
| 15 | 51.2 | 20 | 1 | 5 | 3.0 [1.9, 5] | 0.526 [0.054] | 0.563 | 0.024 | 0.18 [0.03] |
| 16 | 51.2 | 24 | 4 | 8 | 3.0 [1.9, 4.9] | 0.607 [0.049] | 0.583 | −0.047 | 0.21 [0.03] |
| 17 | 10.7 | 11 | 4 | 3 | 1.8 [1.2, 2.7] | 0.555 [0.048] | 0.525 | −0.096 | 0.32 [0.04] |
| 18 | 59.5 | 21 | 3 | 2 | 3.4 [2.1, 6] | 0.717 [0.054] | 0.660 | −0.105 * | 0.20 [0.04] |
| 19 | 31.0 | 20 | 5 | 8 | 3.2 [2.1, 5.4] | 0.624 [0.045] | 0.628 | −0.086 * | 0.19 [0.03] |
| 20 | 34.5 | 20 | 6 | 5 | 2.5 [1.6, 4.2] | 0.606 [0.049] | 0.614 | −0.109 * | 0.23 [0.03] |
| 21 | 60.7 | 20 | 2 | 6 | 3.0 [1.9, 4.9] | 0.598 [0.032] | 0.562 | −0.089 * | 0.22 [0.03] |
| pooled | 38.5 | 375 | 92 | 112 | 85.3 [52.6, 159.2] | 0.607 [0.011] | 0.696 | −0.071 *** | 0.22 [0.03] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caré, O.; Gailing, O.; Müller, M.; Krutovsky, K.V.; Leinemann, L. Mating System in a Native Norway Spruce (Picea abies [L.] KARST.) Stand-Relatedness and Effective Pollen Population Size Show an Association with the Germination Percentage of Single Tree Progenies. Diversity 2020, 12, 266. https://doi.org/10.3390/d12070266
Caré O, Gailing O, Müller M, Krutovsky KV, Leinemann L. Mating System in a Native Norway Spruce (Picea abies [L.] KARST.) Stand-Relatedness and Effective Pollen Population Size Show an Association with the Germination Percentage of Single Tree Progenies. Diversity. 2020; 12(7):266. https://doi.org/10.3390/d12070266
Chicago/Turabian StyleCaré, Oliver, Oliver Gailing, Markus Müller, Konstantin V. Krutovsky, and Ludger Leinemann. 2020. "Mating System in a Native Norway Spruce (Picea abies [L.] KARST.) Stand-Relatedness and Effective Pollen Population Size Show an Association with the Germination Percentage of Single Tree Progenies" Diversity 12, no. 7: 266. https://doi.org/10.3390/d12070266