Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide

Department of Chemistry, Zhejiang University, Hangzhou 310028, China
*
Authors to whom correspondence should be addressed.
Crystals 2018, 8(9), 336; https://doi.org/10.3390/cryst8090336
Submission received: 18 July 2018
/
Revised: 8 August 2018
/
Accepted: 15 August 2018
/
Published: 21 August 2018
Abstract
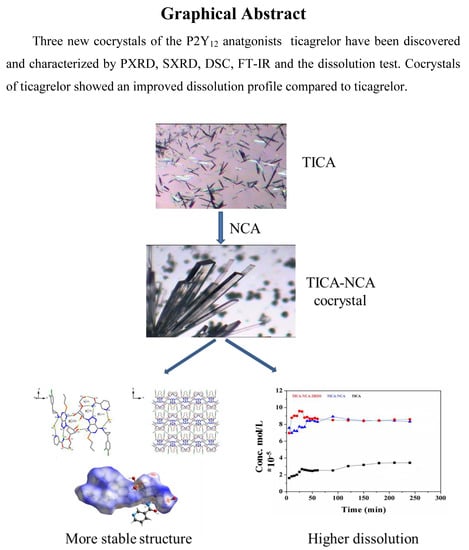
:Two new co-crystals, Ticagrelor with Nicotinamide, have been prepared with improved solubility. Because Ticalegor has a poor solubility and dissolution rate, a novel co-crystallization method with structurally homogenous crystalline material, an active pharmaceutical ingredient (API), and co-former indefinite stoichiometric amount has been made to improve Ticagrelor’s solubility. The co-crystal of Ticagrelor (TICA) with Nicotinamide (NCA) was prepared in ratio (1:1) and confirmed by FTIR, DSC, and XRD characterization. Furthermore, the single crystal structure of TICA-NCA hydrate was analyzed. The solubility of co-crystals was investigated in pH 2 acidic medium, which was a significant improvement as compared to the solubility of a free drug. The in vitro dissolution rate of co-crystal was larger than that of the commercial product.
1. Introduction
Ticagrelor (TICA) is an oral anti-platelet drug used with low dose aspirin to decrease the risk of myocardial infarction and stroke in patients with acute coronary syndromes [1,2,3]. TICA is a non-thienopyridine, reversible inhibitor of adenosine diphosphate (ADP) receptors (P2Y 12) on platelets and is used to decrease the risk of recurrent coronary thromboses. In clinical trials, TICA therapy during acute coronary events decreases the frequency of recurrence of myocardial infarction and stent thrombosis. TICA was approved for use in the United States in 2011. Current indications for potential use are the reduction of recurrent cardiovascular events in patients with acute coronary syndromes. Side effects are not common, but can include bleeding (12%), dyspnea (14%), headaches, nausea, diarrhea, hypotension, and hypersensitivity reactions [4,5,6,7,8,9].
The improvement of the physicochemical properties of active pharmaceutical ingredients (APIs) with solubility-limited bioavailability is of paramount importance in the pharmaceutical development of drug molecules [10,11]. In their ever-increasing efforts to produce materials with improved properties, notably solubility, bioavailability, and stability, pharmaceutical scientists have delved into exploring the scope of solid-state structural variations that can be obtained using salts, polymorphs, and co-crystals of molecules assembled in a single lattice structure [12,13]. Early detection of alternative solid forms, such as polymorphs, hydrate, solvates, salts, co-crystals, and amorphous can lead to significant benefits throughout various stages of drug development [14,15,16,17].
Pharmaceutical co-crystals are a multi-component solid form containing APIs and benign non-toxic conformers [11]. The properties of the drug can be modulated through forming the co-crystal to improve its solubility, dissolution rate [18], stability [19,20,21], hygroscopicity [22], compressibility [23], and thus affecting bioavailability [24]. From a scientific point of view, solvates and hydrates can be considered a subgroup of co-crystals. The solvent, or the water, acts as a co-former, which is the same as other co-formers. Based on the functional groups present in TICA, triazole moiety, pyrimide moiety, hydroxyl, and amino moiety are the most likely groups to form a supramolecular synthon (O–H…O, O–H…N, N–H…O, N–H…N) with other generally regarded as safe (GRAS) co-former molecules. Along these lines, a series of multi-component solid phases with the co-formers involving acetylsalicylic acid, glycolic acid, salicylic acid, capric acid, gentisic acid, glutaric acid, vanillic acid, succinic acid, and malonic acid appeared [25]. Five anhydrous polymorph crystals of TICA—Forms I, II, III, IV and V were reported [3], but their solubility is poor. The form of TICA is the solid form currently available on the market with the solubility of only 16 μg/mL, which is obtained most easily by a common re-crystallization method. Thus far, no crystal structure of the above TICA co-crystals with nicotinamide has been covered.
2. Experimental Section
2.1. Materials
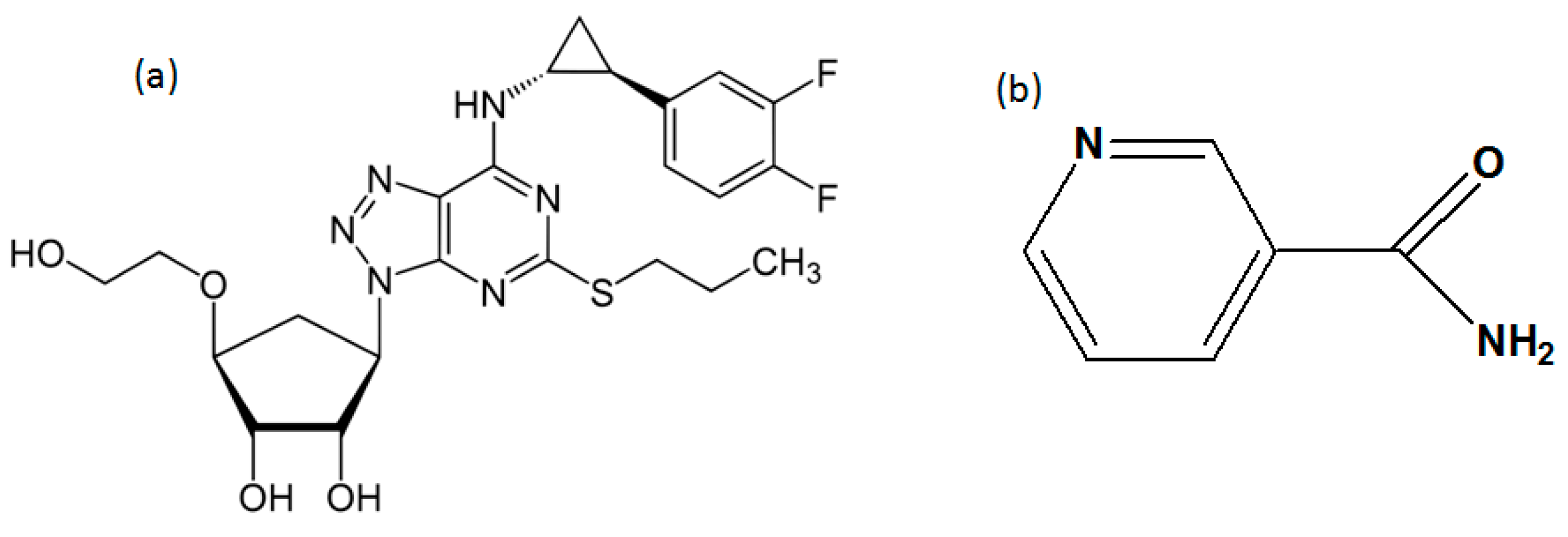
Ticagrelor was provided by Zhejiang Jingxin pharmaceutical Co., Zhejiang, China. Nicotinamide was purchased from Aladdin (Shanghai, China). The molecular structure of TICA and NCA are shown in Scheme 1. All other solvents and chemicals were of analytical grade.
2.2. Preparation of New Solid State Forms
The co-crystals of TICA were prepared using a solution crystallization method. Solution crystallization can yield large, well-formed crystals, from which one may easily evaluate crystal habit and surface features.
TICA-NCA hydrate: Equimolar quantities of TICA (0.57 mol) and NCA (0.57 mol) were added to 5–8 mL ethyl acetate solvent in a 50 mL round-bottomed flask. The slurry was stirred and refluxed at 70 °C for 3 h. The obtained clear solution was cooled down slowly to the room temperature to induce the crystallization. Upon formation of solid crystal were filtered and dried.
TICA-NCA: Equimolar quantities of TICA (2.3 mol) and NCA (2.3 mol) were dissolved in 2–5 mL ethyl at 70 °C. The resulting solution was cooled down slowly to 50 °C and sustained at this temperature for 2 h. Solid crystals were obtained, filtered, dried at 60 °C for 2 h.
Growing single crystals: Single crystal of TICA-NCA hydrate was formed by slow evaporation of saturated solution of co-crystals in ethyl acetate at room temperature.
2.3. Powder X-ray Diffraction (PXRD) Study
The diffraction patterns were measured on Rigaku D/Max-2550PC diffractometer (Rigaku, Tokyo, Japan) using a rotating anode Cu-target X-ray (λ = 1.5406 Å) generator operated at 40 kV and 250 mA. The incidence Cu Ka X-ray beam was monochromatized by a vertical graded multiplayer mirror to remove the Kβ radiation and to obtain a paralleled beam with a width of 0.8 mm. The scans ran from 3.0 to 40.0° 2θ, with an increasing step size of 0.02° and count time of 0.5–1 s.
2.4. Differential Scanning Calorimeter (DSC) Study
DSC analysis was performed on a TA DSCQ100 differential scanning calorimeter (TA Instruments, New Castle, Germany) at a heating rate of 10 °C/min under a nitrogen flow of 50 cm3/min. Approximately 4–7 mg powder samples were used for differential scanning calorimeter (DSC) analyses. A temperature range of 35–220 °C was scanned. The calibrated temperature accuracy was ±0.02 °C.
2.5. Fourier Transform-Infrared Spectroscopy (FT-IR)
FT-IR spectra were recorded using a VECTOR-22 Fourier infrared spectrometer (Bruker Instruments, Karlsruhe, Germany) in the spectral range of 4000–400 cm−1 in the KBr diffuse reflectance mode. Powder samples (about 2 mg) were manually mixed with 100 mg of dry KBr in an agate mortar and pressed into thin pellets. Data were analyzed using Spectrum software.
2.6. Single Crystal X-ray Diffraction (SCXRD)
A single crystal of suitable size and good quality was measured using an area detector on a Rigaku R-AXIS-RAPID diffractometer (Rigaku, Tokyo, Japan) with graphite monochromatic Mo-Kα radiation (λ = 0.71069 Å). Data reduction was performed with Crystal Structure [26]. The crystal structure was solved with direct methods using SHELX-S [27] program and refined on F2′sanisotropic ally by a full-matrix, least-squares method using the SHELX-L program. Anisotropic displacement parameters for no n-hydrogen atoms were applied. Hydrogen atoms were placed at the metrically calculated positions and were refined isotropic ally using a riding mode. ORTEP [28] and Diamond [29] were used for creation of figures.
2.7. Transformation Study
The transformation experiment was done by storing both the TICA-NCA anhydrate and TICA-NCA hydrate in an airtight desiccator saturated with KNO3 solution under the condition of 25 °C/75% RH. The sample was measured in an interval of five days to observe the change on the PXRD patterns. The dehydration experiment was performed by putting the hydrate form in vacuum drying oven (Labcompare, South San Francisco, CA, USA) at 45 °C; the sample was fetched for measurements every hour to observe the changes in the PXRD patterns.
2.8. Solubility and Dissolution Measurement
The solubility studies of TICA, TICA-NCA, and TICA-NCA hydrates were measured by a Thermo Scientific Evolution 300 Ultraviolet–visible (UV–Vis) spectrometer (Thermo Scientific, Waltham, MA, USA) at pH 2 at 37 °C. The concentrations of TICA, TICA-NCA, and TICA-NCA hydrate were calculated by means of a standard graph, which was made by measuring the absorbance of varied concentrations at their respective λmax. From the slope of the calibration curves, molar extinction coefficients for each co-crystal/drug were calculated.
The solubility of TICA, TICA-NCA, and TICA-NCA hydrates were done using excess amounts of samples in acid medium. An excess amount of the sample was added to 5 mL of medium pH 2 acid solution, respectively. The supersaturated solution was stirred at 100 rpm using magnetic stirrer at 37 °C. After 24 h, the suspension was filtered through Whitman’s 0.45 syringe filter paper. The filtered aliquots were diluted sufficiently, and the absorbance was measured at their λmax.
The intrinsic dissolution studies of pure drug and co-crystals were done using equal molar samples. The samples were directly poured into 500 mL pH 2 acidic solutions and stirred with 100 rpm for 240 min at 37 °C. At regular intervals, 5 mL of the dissolution medium was withdrawn and replaced by an equal volume of fresh medium to maintain a constant volume. Each solution removed was measured the absorbance at λmax.
3. Results and Discussion
Preliminary co-crystallization experiments of TICA with NCA revealed the existence of dimorphs of TICA-NCA co-crystals, a novel anhydrate and dihydrate of co-crystal. The hydrates of single-component solids are a common phenomenon [30]. However, the hydrates of multi-component solids are small in size and possess the ability to form multiple hydrogen bond patterns, and furthermore appear during the process to prepare co-crystals [31,32,33] for water. The co-crystal hydrate has some unique and apparent characteristics that are found in the properties of drugs. Due to its unique features, it has a prominent role and important value for diverse and various co-crystal forms. Especially, when the situation is such that the polymorphs of co-crystals are much less than the single compound of APIs, for the possibility of the limited method is applied to screen co-crystal polymorphs [34,35]. Furthermore, co-crystal hydrates bring an alternative route to expand the new solid forms involving APIs as well as to create intellectual property protection. The study about the transformation between co-crystal anhydrate and co-crystal hydrate was also conducted to enrich the dehydration mechanism between co-crystal anhydrate and co-crystal hydrate. The TICA-NCA hydrate can convert to TICA-NCA anhydrate when heating—e.g., in a vacuum drying oven at 45 °C for four hours. Furthermore, the co-crystal dihydrate can be obtained after depositing the co-crystal anhydrate at high humidity condition. There was no crystal transfer found for the co-crystal hydrate at 25 °C and 75% RH, even for 35 days, indicating that the TICA-NCA dihydrate was resistant to the high humidity environment.
All of the newly discovered solid-state forms were further characterized using various analytical techniques.
3.1. Crystal Structure Analyses
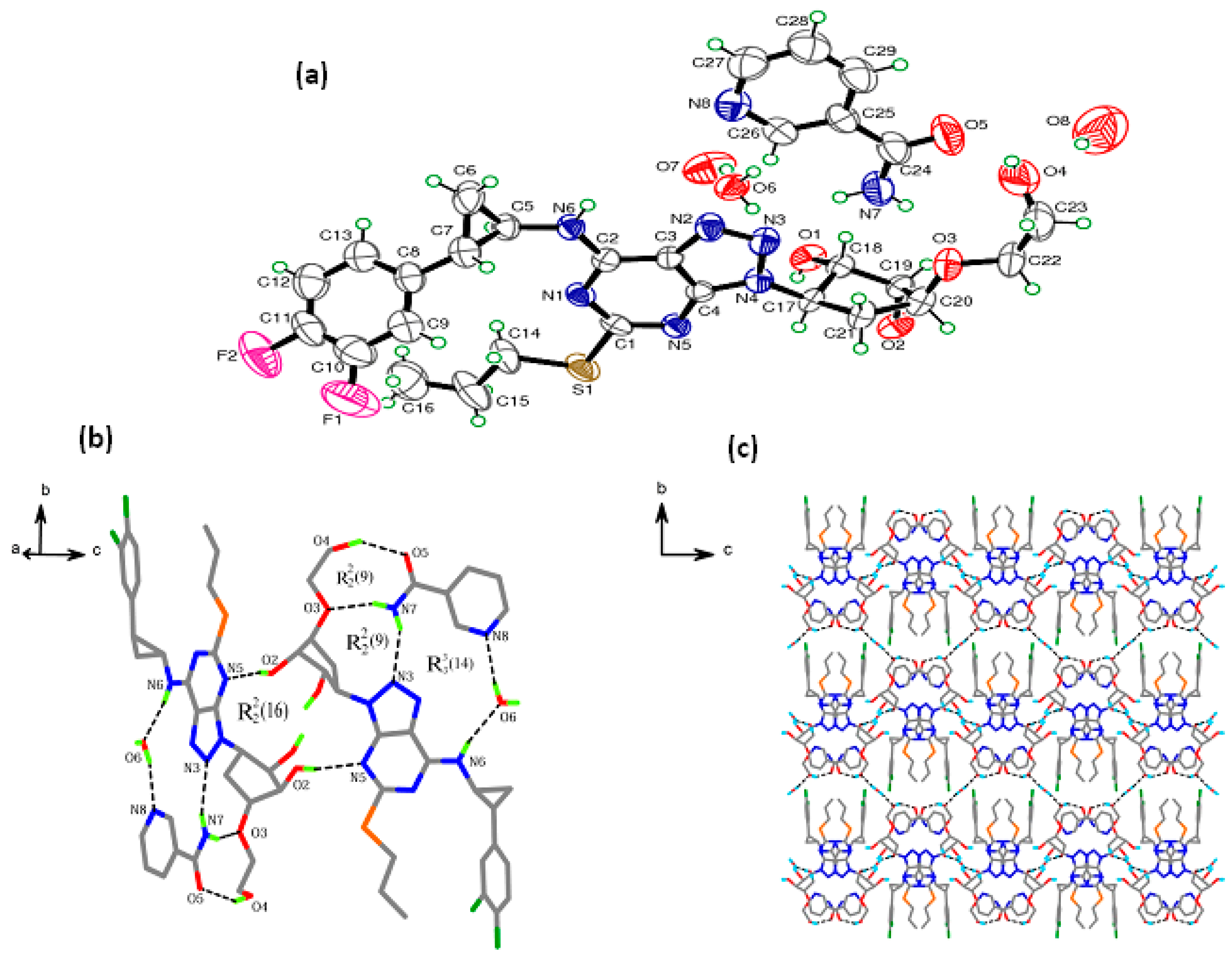
A single crystal of TICA-NCA hydrate was obtained by slow evaporation from ethyl acetate at room temperature. However, we were not successful in growing single crystals of TICA-NCA despite intense efforts following rational approaches. Hence, this section deals with the crystal structure analysis of TICA-NCA hydrate, which provides insights on the crystal packing and hydrogen bond interactions. TICA-NCA hydrate crystallizes in orthorhombic space group P2221 with the asymmetric unit, as shown in Table 1, consisting of one TICA molecule, one NCA molecule, and two molecules of water at a ratio of 1:1:2. There is no proton transfer involved among the TICA and NCA molecules, while two molecules of water trapped in the crystal lattice as shown in Figure 1. Thus, TICA-NCA hydrate is a co-crystal dihydrate. The pyridine ring of NCA is almost coplanar to the triazole ring in TICA. Rich hydrogen bond interactions (Table 2) are built in the crystal packing. The amide group of NCA forms a hetero synthon with the ether group oxygen, hydroxyl group, and amino group of trizole from the Ticagrelor feature, forming O4–H4…O5, N7–H7A…O3, N7–H7B…N3 hydrogen bonds. These interactions lead to the formation of two-ring motifs that can be described by the [] graph set notations [36]. Meanwhile, water molecules play an important role in maintaining the crystal integrity. The O6 atom in a water molecule serves as both the hydrogen bond receptor and donor to form N6–H6…O6ii, N8–H8…O6ii [symmetric code: (ii) 1 − x, y, 0.5 − z], O6–H71…O7 hydrogen bonds with the amino group of TICA, the pyridyl N atom of NCA, and another water molecule respectively forming and [] hydrogen bond motif, as shown in Figure 1b. The complex interactions make the crystal structure extend along the c-axis with the corrugated like ribbon. Furthermore, the paralleled layers are connected by short connect hydrogen bonds stacking along the a-axis, as shown in Figure 1c.
3.2. Powder X-ray Diffraction (PXRD)
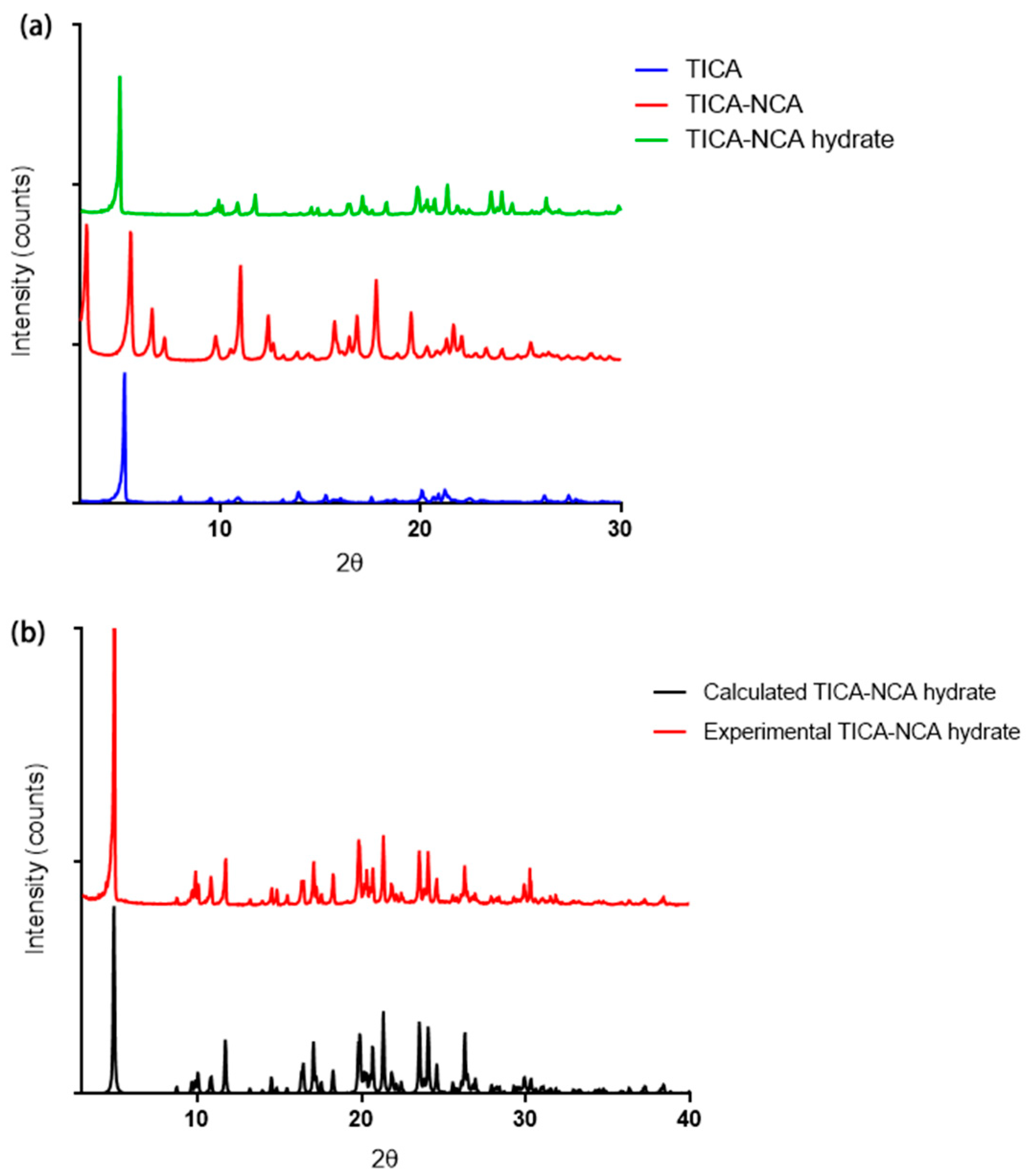
The PXRD pattern of a crystalline sample is considered as the fingerprint of its crystal structure. Every new crystalline material exhibit unique peaks indicative of diffractive from specific atomic planes [37]. The PXRD patterns of pure drug and TICA-NCA co-crystals (anhydrate and hydrate) summarized in Figure 2. TICA-NCA co-crystals (anhydrate and hydrate) exhibits distinguishable diffraction peaks different from those of TICA. The PXRD pattern of co-crystals showed new peaks at 2θ values of 4.9°, 8.7°, 10.8°, 11.7°, 17.0°, 18.2°, 19.8°, 21.3°, and 23.5° for TICA-NCA hydrate and 3.2°, 5.5°, 7.1°, 10.9°, 12.3°, and 15.6° for TICA-NCA. Change in the position and intensity of the peak indicated the formation of new co-crystals. Alternatively, powder diffraction patterns expected for homogenous bulk sample can be simulated when single crystal structures are known. Such simulated patterns can be used as the ‘gold standard’ for unambiguous phase identification (Figure 2b) representing the comparison of the experimental PXRD pattern with the simulated pattern of TICA-NCA hydrate, indicating the crystallographic purity of co-crystal hydrate.
3.3. Differential Scanning Calorimeter (DSC) Analysis
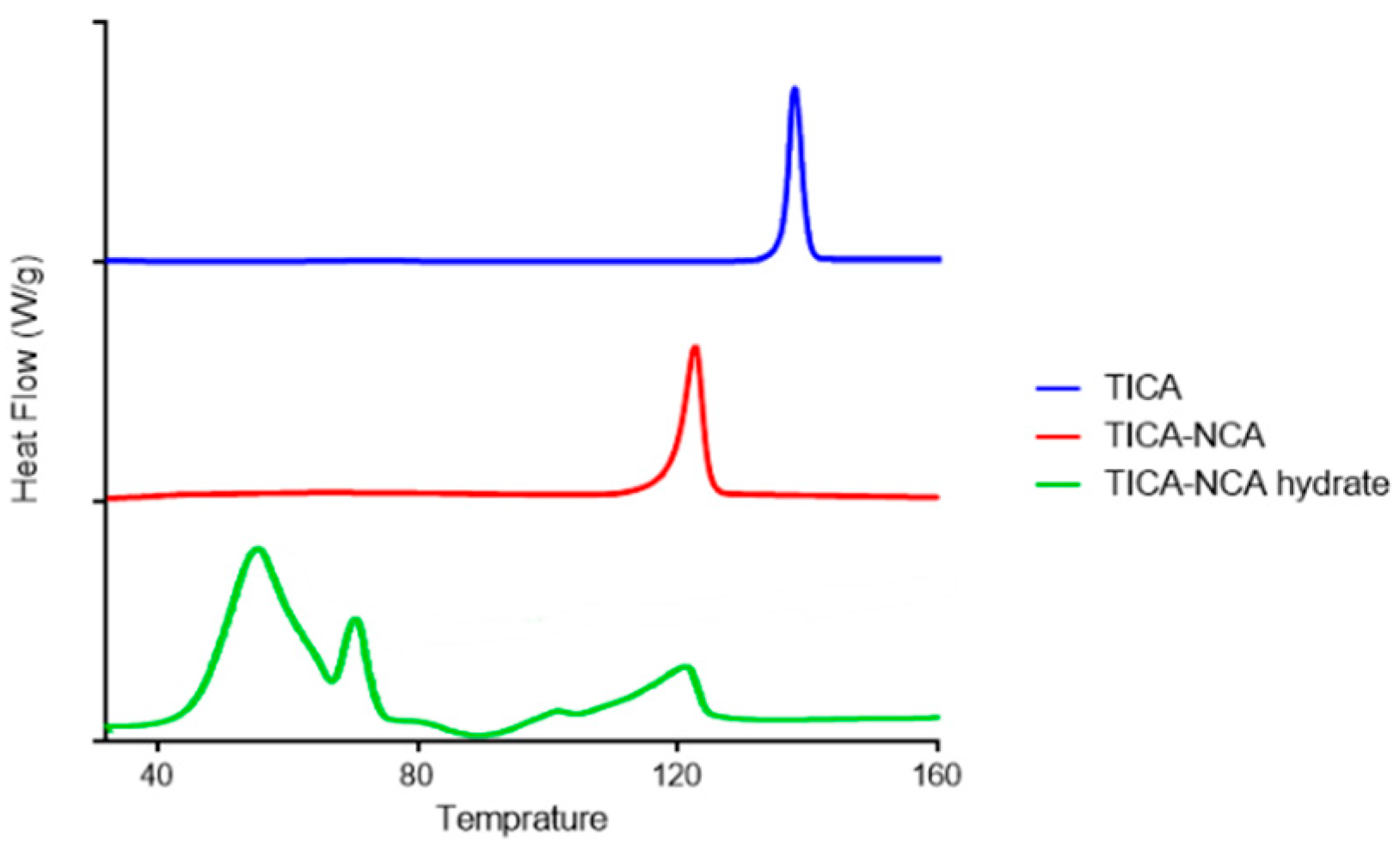
DSC analyses are widely used to characterize co-crystals. In DSC analysis, the thermal curve of pure TICA and the newly prepared co-crystals reported in Figure 3. The thermal curve of the pure drug indicated its crystalline anhydrous state, exhibiting a sharp endothermal effect at 137.5 °C. The two new co-crystals showed a different melting compared to the active pharmaceutical free drug of TICA, thus confirming the co-crystal identity. The DSC scan of TICA-NCA showed single melting endothermic peak at 122.4 °C, which is different than its individual component (TICA form II 137.5 °C). The only existing endothermic peak, due to the melting of co-crystal, confirms the TICA-NCA co-crystal to be anhydrous form. The DSC curve of TICA-NCA hydrate showed two broad endothermic peaks in the temperature range of 50–80 °C at which temperature the dehydration process occurred, followed by melting at 120.5 °C, as shown in Figure 3.
3.4. FT-IR Spectroscopy Analysis
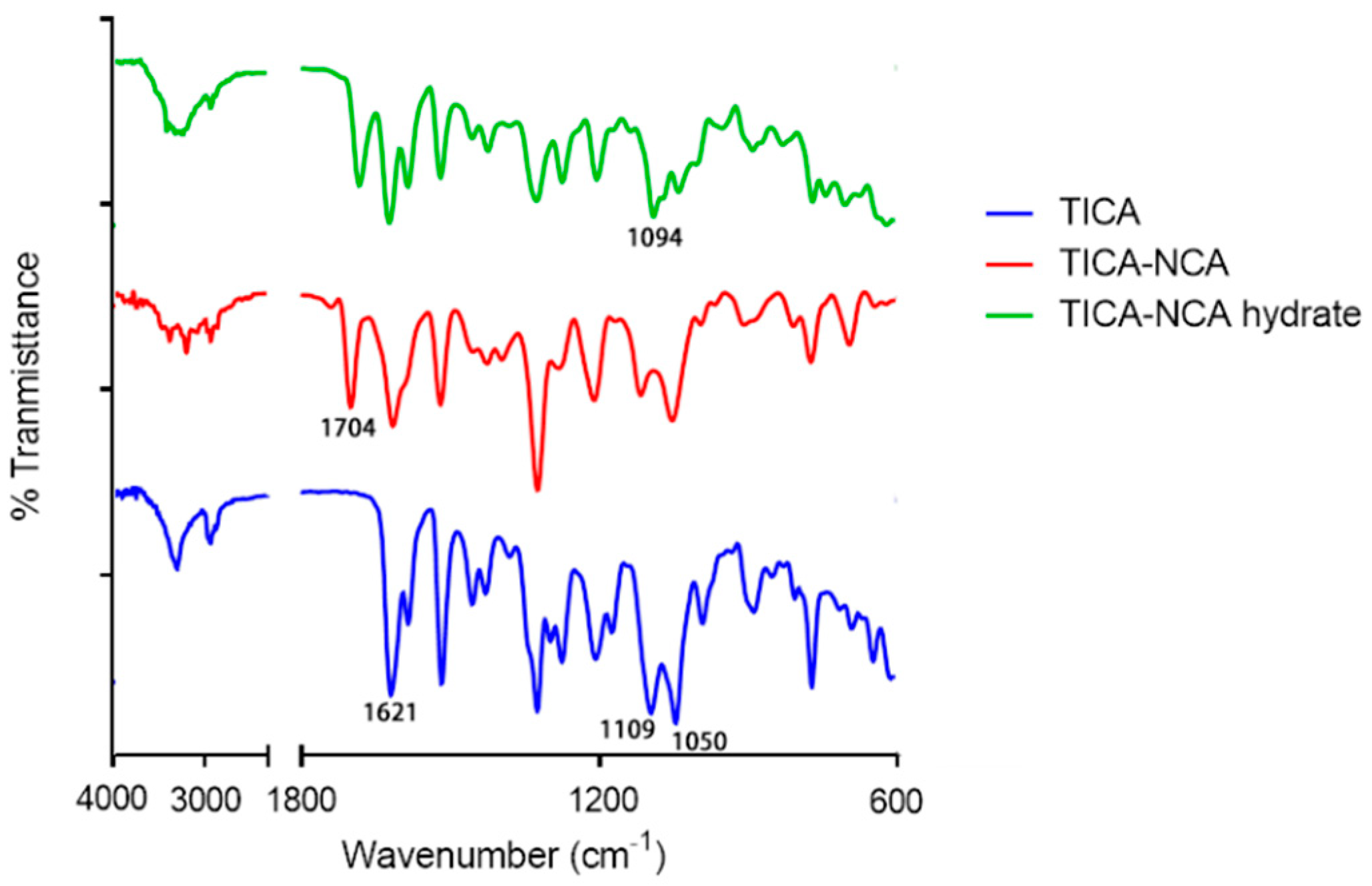
Changes in the vibrational frequencies due to co-crystal formation can be easily monitored by FT-IR spectroscopy. In the case of polymorphic structures, differences in the hydrogen bonding lead to changes in the vibrational frequencies of the functional groups that participated in hydrogen bonds. As shown in Figure 4, TICA exhibited two C–O stretching vibrations at 1109 cm−1 and 1050 cm−1, which merged into a single stretching vibration in the TICA-NCA hydrate at 1094 cm−1, due to a shift caused by the hydrogen bond interaction N–H…O, involving ether group of TICA. When compared to the parent substances, the dominant difference existed in the carbonyl group stretching frequency of TICA, which shifted from 1621 cm−1 to 1704 cm−1. This implies that hydrogen bonds build the carbonyl group of TICA within the TICA-NCA co-crystals. Furthermore, all the shifts in the infrared spectrum confirm the co-crystal identity.
3.5. Hirshfeld Surface (HS) Analysis
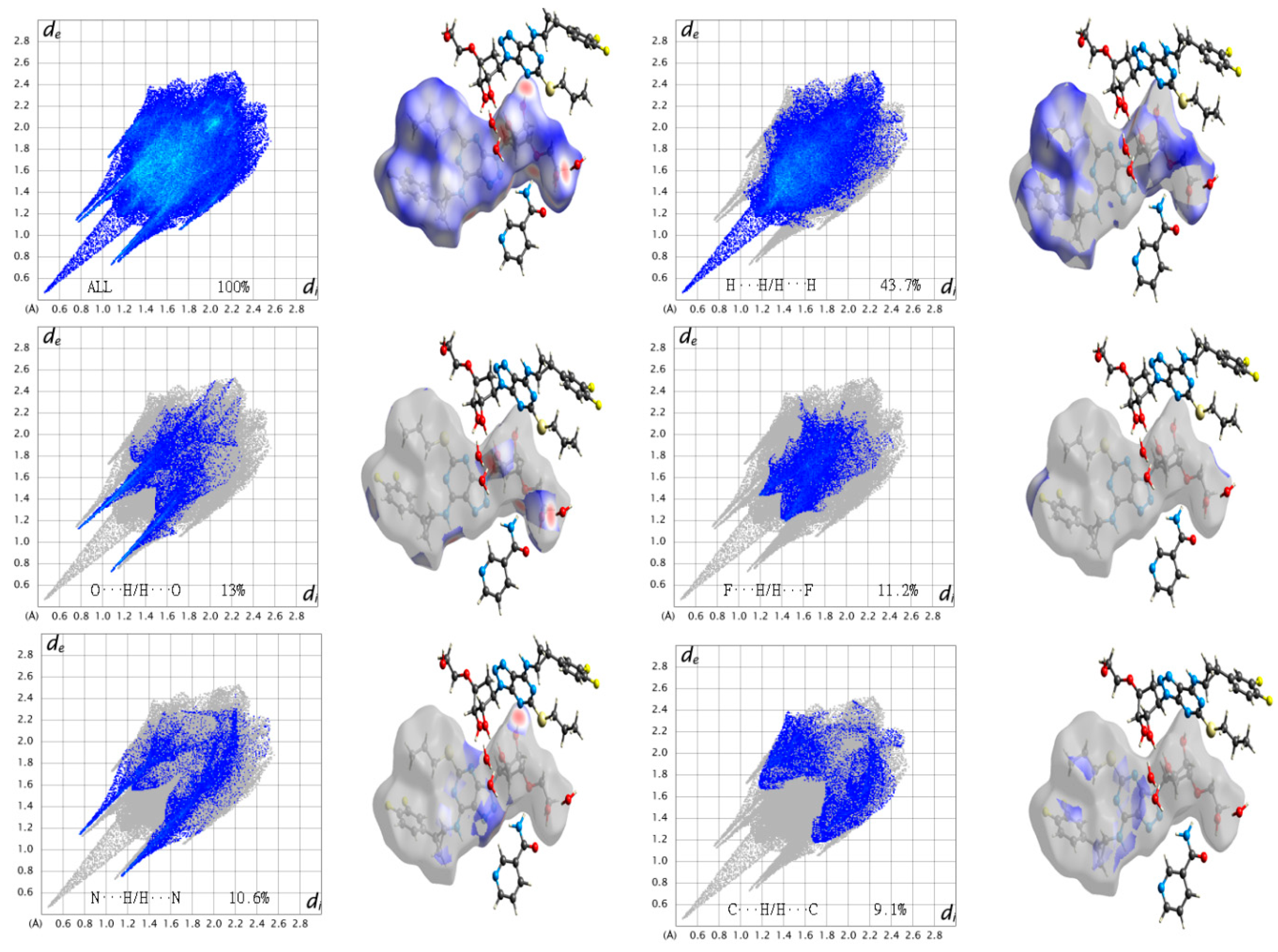
The Hirshfeld surface (HS) emerged from an attempt to define the space occupied by a molecule in a crystal for the purpose of partitioning the crystal electron density into molecular fragments [37]. HS analysis is regarded as a useful tool to assess the intermolecular interaction in molecular crystals (co-former and drug). In order to evaluate the close interaction between the TICA and NCA molecules, the HS analysis were generated using Crystal Explore [38]. The interaction between the amide group of NCA and the ether group oxygen, hydroxyl group, and amino group trizole of TICA, can be seen in the HS as the bright red areas; the HS of TICA and co-former NCA are shown in Figure 5. The red spots on the surface reveal the shortest strong O4–H4…O5, N7–H7A…O3, N7–H7B…N3 hydrogen bonds interacting. The other visible spots on the surface correspond to the suitable H…H contacts. The intermolecular interactions H…H/H…H, O…H/H…O, F…H/H…F, N…H/H…N, and C…H/H…C appear as distinct spikes (Figure 6) in the 2D fingerprint plot.
Fingerprint plots of the Hirshfeld surface of the crystal structure are key tools for the calculation of the density of intermolecular contacts present in a crystal. The combination of (de and di) in the form of a 2D fingerprint plot [39] provides a summary of the intermolecular contacts in the crystal [40]. The fingerprint plots presented here were generated using Crystal Explore. The intermolecular interactions H…H/H…H, O…H/H…O, F…H/H…F, N…H/H…N, and C…H/H…C appear as distinct spikes in the 2D fingerprint plot with 43.7%, 13%, 11.2%, 10.6%, and 9.1% densities, respectively, as shown in Figure 6. Complementary regions are visible in the fingerprint plots, where one molecule acts as a donor (de > di) and the other as an acceptor (de < di). By using a breakdown of the fingerprint plots based on the Hirshfeld surface, we can decompose the fingerprint plots of the crystal structure of TICA-NCA hydrate to highlight particularly close contacts. This decomposition enables separate contributions from different interaction types, which commonly overlap in the full fingerprint, illustrating the decomposition of the fingerprint plots for H…H/H…H, O…H/H…O, F…H/H…F, N…H/H…N, and C…H/H…C in crystal of TICA-NCA hydrate, highlighting the separate intermolecular contacts. To provide context, the outline of the full fingerprint is shown in grey; the blue area shows the separate contact. Through this we can demonstrate that TICA-NCA hydrate is of high stability.
3.6. Solubility and Dissolution Rate
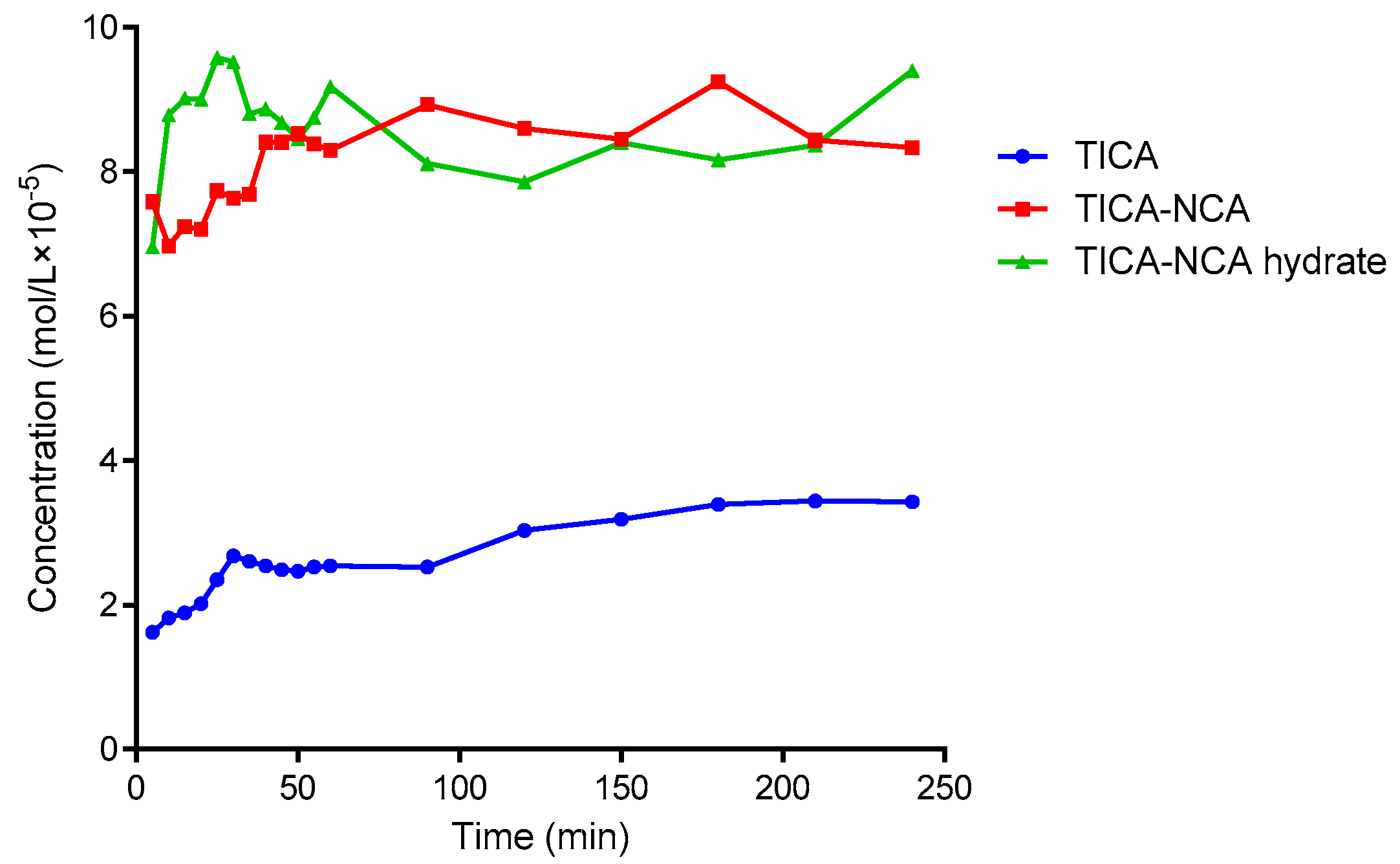
It is well known that the solid form of APIs have a substantial impact on the solubility and dissolution profiles of drug [41]. Therefore, it is important to select an appropriate API solid state forms for successful drug development. According to the Biopharmaceutical Classification System (BCS), TICA was classified as a low solubility and low permeability drug (BCS class IV). The absolute bioavailability of TICA is in range 30–42% [40]. Super saturation API solid forms such as salts and solvates would be effective to increase the bioavailability of a drug [42]. Both TICA-NCA and TICA-NCA hydrate showed about four and four and a half times higher solubility than the commercial, hence demonstrating a significant improvement in the solubility and an advantage in the formulation of co-crystallization, as shown in Figure 7. Furthermore, both co-crystals reached the same equilibrium concentration after four hours though the TICA-NCA hydrate had, and a faster dissolution rate than the TICA-NCA at an early time.
4. Conclusions
Co-crystals represent a promising new approach for improving the crystallinity, physicochemical, solubility, and bioavailability properties of pharmaceutical compounds. The present study focused on the preparation of TICA and NCA co-crystals to enhance the solubility of TICA, as well as obtain further insights into the structure of these solid state forms. Formation of TICA co-crystal with NCA was studied by PXRD, DSC, and FT-IR. With regard to the results, it can be confirmed that the co-crystal was able to overcome the solubility drawback. Consequently, co-crystals of TICA-NCA and TICA-NCA hydrate were presumed as potential APIs possessing enhanced absorption property compared to the free drug TICA.
Supplementary Materials
Supplementary File 1Author Contributions
M.I. was the primary contributor. J.W. contributed to experimental work. J.S. helped with the crystal structure diagrams. C.U.P. revised the manuscript. G.T. and X.H. designed and supervised this work.
Acknowledgments
The authors thank Zhejiang Jingxin Pharmaceutical Co., Ltd. (China) for kindly providing Ticagrelor and Zhejiang University for providing an experimental facility.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Marzio, H.D.; Navarro, V.J. Hepatotoxicity of cardiovascular and antidiabetic drugs. In Drug-Induced Liver Disease, 3rd ed.; Elsevier: New York, NY, USA, 2013; Chapter 29; pp. 519–540. [Google Scholar]
- Bojarska, J.; Remko, M.; Fruziński, A.; Maniukiewicz, W. The experimental and theoretical landscape of a new antiplatelet drug ticagrelor: Insight into supramolecular architecture directed by C-H…F, π-π and C-H…π interactions. J. Mol. Struct. 2018, 1154, 290–300. [Google Scholar] [CrossRef]
- Weitz, J.I. Blood coagulation and anticoagulant, fibrinolytic, and antiplatelet drugs. In Goodman and Gilman’s The Pharmacological Basic of Therapeutics, 12th ed.; McGraw Hill: New York, NY, USA, 2011; Chapter 30; pp. 849–877. [Google Scholar]
- Husted, S.; Emanuelsson, H.; Heptinstall, S.; Sandset, P.M.; Wickens, M.; Peters, G. Pharmacodynamics, pharmacokinetics, and safety of the oral reversible p2y (12) antagonist azd6140 with aspirin in patients with atherosclerosis: A double-blind comparison to clopidogrel with aspirin. Eur. Heart J. 2006, 27, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Emanuelsson, H. Safety, tolerability, and initial efficacy of azd6140, the first reversible oral adenosine diphosphate receptor antagonist, compared with clopidogrel, in patients with non–ST-segment elevation acute coronary syndrome: primary results of the DISPERSE-2 trial. J. Am. Coll. Cardiol. 2007, 50, 1844–1851. [Google Scholar]
- Chalasani, N.; Fontana, R.J.; Bonkovsky, H.L.; Watkins, P.B.; Davern, T.; Serrano, J.; Yang, H.; Rochon, J. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008, 135, 1924. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.A.; Goldberg, T.; Dorsch, M.P.; Cheng, J.W.M. Antiplatelet therapy after placement of a drug-eluting stent: A review of efficacy and safety studies. Clin. Ther. 2010, 32, 2265–2281. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Koch, D.G.; Lee, W.M. Drug-induced acute liver failure: Results of a US. Multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Salmon, D.J. Building co-crystals with molecular sense and supramolecular sensibility. CrystEngComm 2005, 7, 439–448. [Google Scholar] [CrossRef]
- Vishweshwar, P.; Mcmahon, J.A.; Bis, J.A.; Zaworotko, M.J. Pharmaceutical co-crystals. J. Pharm. Sci. 2006, 95, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Zaworotko, M.J. The role of cocrystals in pharmaceutical science. Drug Discovery Today 2008, 13, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Supramolecular synthons in crystal engineering & mdash; a new organic synthesis. Angew. Chem. Int. Ed. 2010, 34, 2311–2327. [Google Scholar]
- Brittain, H.G. Cocrystal systems of pharmaceutical interest: 2010. Cryst. Growth Des. 2012, 12, 1046–1054. [Google Scholar] [CrossRef]
- Editor, N.R.G. Cocrystals: Molecular design of pharmaceutical materials. Mol. Pharm. 2007, 4, 299–300. [Google Scholar]
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Aitipamula, S.; Chow, P.S.; Tan, R.B.H. Dimorphs of a 1:1 cocrystal of ethenzamide and saccharin: Solid-state grinding methods result in metastable polymorph. CrystEngComm 2009, 11, 889–895. [Google Scholar] [CrossRef]
- Childs, S.L.; Chyall, L.J.; Dunlap, J.T.; Smolenskaya, V.N.; Stahly, B.C.; Stahly, G.P. Crystal engineering approach to forming cocrystals of amine hydrochlorides with organic acids. Molecular complexes of fluoxetine hydrochloride with benzoic, succinic, and fumaric acids. J. Am. Chem. Soc. 2004, 126, 13335–13342. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zu, H.; Zhang, J. Enhanced dissolution and stability of adefovir dipivoxil by cocrystal formation. J. Pharm. Pharmacol. 2011, 63, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Maddileti, D.; Jayabun, S.K.; Nangia, A. Soluble cocrystals of the xanthine oxidase inhibitor febuxostat. Cryst. Growth Des. 2013, 13, 3188–3196. [Google Scholar] [CrossRef]
- Aitipamula, S.; Wong, A.B.H.; Chow, P.S.; Tan, R.B.H. Novel solid forms of the anti-tuberculosis drug, isoniazid: Ternary and polymorphic cocrystals. CrystEngComm 2013, 15, 5877–5887. [Google Scholar] [CrossRef]
- Tao, Q.; Chen, J.-M.; Ma, L.; Lu, T.-B. Phenazopyridine cocrystal and salts that exhibit enhanced solubility and stability. Cryst. Growth Des. 2012, 12, 3144–3152. [Google Scholar] [CrossRef]
- Sun, C.C.; Hou, H. Improving mechanical properties of caffeine and methyl gallate crystals by cocrystallization. Cryst. Growth Des. 2008, 8, 1575–1579. [Google Scholar] [CrossRef]
- Hickey, M.B.; Peterson, M.L.; Scoppettuolo, L.A.; Morrisette, S.L.; Vetter, A.; Guzman, H.; Remenar, J.F.; Zhang, Z.; Tawa, M.D.; Haley, S.; et al. Performance comparison of a co-crystal of carbamazepine with marketed product. Eur. J. Pharm. Biopharm. 2007, 67, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, S.D.; Jonaitis, D.T.; Sutch, J.C.D. Novel Ticagrelor Co-Crystal. U.S. Patent 20,160,176,912A1, 23 June 2016. [Google Scholar]
- Rigaku. Process-Auto and Crystalstructure; Rigaku Corporation: Tokyo, Japan, 2006. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A-Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. Wingx and ortep for windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND, version 3.1; Crystal Impact GbR: Bonn, Germany, 2005. [Google Scholar]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs, 2nd ed.; Academic Press: Washington, DC, USA, 1982. [Google Scholar]
- Vangala, V.R.; Chow, P.S.; Tan, R.B.H. Co-crystals and co-crystal hydrates of the antibiotic nitrofurantoin: Structural studies and physicochemical properties. Cryst. Growth Des. 2012, 12, 5925–5938. [Google Scholar] [CrossRef]
- Aitipamula, S.; Vangala, V.R.; Chow, P.S.; Tan, R.B.H. Cocrystal hydrate of an antifungal drug, griseofulvin, with promising physicochemical properties. Cryst. Growth Des. 2012, 12, 5858–5863. [Google Scholar] [CrossRef]
- Ueto, T.; Takata, N.; Muroyama, N.; Nedu, A.; Sasaki, A.; Tanida, S.; Terada, K. Polymorphs and a hydrate of furosemide-nicotinamide 1:1 cocrystal. Cryst. Growth Des. 2012, 12, 485–494. [Google Scholar] [CrossRef]
- Eddleston, M.D.; Sivachelvam, S.; Jones, W. Screening for polymorphs of cocrystals: A case study. CrystEngComm 2012, 15, 175–181. [Google Scholar] [CrossRef]
- Abourahma, H.; Cocuzza, D.S.; Melendez, J.; Urban, J.M. Pyrazinamide cocrystals and the search for polymorphs. CrystEngComm 2011, 13, 6442–6450. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic-compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Newman, A.W.; Byrn, S.R. Solid-state analysis of the active pharmaceutical ingredient in drug products. Drug Discovery Today 2003, 8, 898–905. [Google Scholar] [CrossRef]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Watson, D.G.; Taylor, R. International Tables for Crystallography; D. Reidel: Dordrecht, The Netherlands, 1983. [Google Scholar]
- Chen, P.; Zhang, H.B.; Lin, G.D.; Hong, Q.; Tsai, K.R.; Alvarado, V.; Davis, H.T.; Scriven, L.E.; Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, K.; Shekar, B.C.; Khadgapathi, P.; Bhikshapathi, D.; Renuka, K. Development, characterization and in vivo evaluation of tolvaptan solid dispersions via solvent evaporation technique. Int. J. Drug Deliv. 2015, 7, 32–43. [Google Scholar]
- Sugano, K.; Kataoka, M.; Mathews, C.C.S.; Yamashita, S. Prediction of food effect by bile micelles on oral drug absorption considering free fraction in intestinal fluid. Eur. J. Pharm. Sci. 2010, 40, 118–124. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Molecular structure of (a) Ticagrelor and (b) Nicotinamide.

Figure 1.
The crystal structure of TICA-NCA hydrate (a) ORTEP diagram with 50% probability, (b) Hydrogen bonding connection diagram, and (c) crystal packing along bc direction.
Figure 1.
The crystal structure of TICA-NCA hydrate (a) ORTEP diagram with 50% probability, (b) Hydrogen bonding connection diagram, and (c) crystal packing along bc direction.

Figure 2.
(a) The PXRD patterns of TICA, TICA-NCA, and TICA-NCA hydrate and (b) experimental and simulated XRD patterns of TICA-NCA hydrate.
Figure 2.
(a) The PXRD patterns of TICA, TICA-NCA, and TICA-NCA hydrate and (b) experimental and simulated XRD patterns of TICA-NCA hydrate.

Figure 3.
DSC profile of TICA, TICA-NCA, and TICA-NCA hydrate.

Figure 4.
The FT-IR spectra of TICA, TICA-NCA, and TICA-NCA hydrate.

Figure 5.
Representative Hirshfeld surface of TICA-NCA hydrate.

Figure 6.
Fingerprint plot of TICA-NCA hydrate full and resolved into H…H/H…H, O…H/H…O, F…H/H…F, N…H/H…N, and C…H/H…C contacts showing the percentages of contacts contributed to the total crystal structure.
Figure 6.
Fingerprint plot of TICA-NCA hydrate full and resolved into H…H/H…H, O…H/H…O, F…H/H…F, N…H/H…N, and C…H/H…C contacts showing the percentages of contacts contributed to the total crystal structure.

Figure 7.
The dissolution rate of TICA, TICA-NCA, and TICA-NCA hydrate.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Correspond to Crystallographic data of Ticagrelor (TICA)-Nicotinamide (NCA) hydrate.
| Compound Reference | TICA-NCA Hydrate |
|---|---|
| CCDC No | 1497021 |
| Chemical formula | C29H38F2N8O7S |
| Mr | 608.73 |
| Crystal system | Orthorhombic |
| Space group | P2221 |
| a/Å | 9.1347(5) |
| b/Å | 17.9018(10) |
| c/Å | 20.1929(11) |
| α/° | 90 |
| B/° | 90 |
| γ/° | 90 |
| Unit cell volume/Å | 3302.1(3) |
| No of formula units per unit cell, Z | 4 |
| Radiation type | MoKα |
| μ (mm−1) | 0.167 |
| Temperature/K | 296(2) |
| Crystal size (mm) | 0.43 × 0.38 × 0.07 |
| Dcalc/g cm−3 | 1.369 |
| Rint | 0.1262 |
| Reflections collected | 32621 |
| Unique reflections | 7530 |
| Observed reflections | 3600 |
| Final R1 values (I > 2δ(I)) | 0.0794 |
| Final WR(F2) values (all date) | 0.1609 |
| Goodness-of-fit | 1.006 |
Table 2.
Geometrical parameters for the intermolecular interactions of TICA-NCA hydrate.
| Atom D,H,A | D–H…Aa | H…A/Å | D…A/Å | D–H…A/° |
|---|---|---|---|---|
| N6–H6–O6i | 0.8600 | 2.1400 | 2.982(4) | 168.000 |
| O6–H62–N8i | 0.8200 | 2.0300 | 2.839(5) | 167.000 |
| O1–H1–O1ii | 0.8200 | 1.9400 | 2.714(6) | 155.000 |
| O2–H2–N5ii | 0.8200 | 2.0600 | 2.868(4) | 170.000 |
| N7–H7A–O3 | 0.8600 | 2.1200 | 2.946(5) | 160.000 |
| N7–H7B–N3 | 0.8600 | 2.2100 | 3.005(5) | 154.000 |
| O4–H4–O5 | 0.8200 | 2.0400 | 2.770(6) | 148.000 |
| O6–H61–O1 | 0.8200 | 1.9600 | 2.768(4) | 165.000 |
| O7–H71–O6 | 0.8300 | 2.1000 | 2.859(5) | 152.000 |
| O8–H81–O4 | 0.8200 | 2.1000 | 2.906(6) | 169.000 |
Symmetry Codes (i) −x + 1, y, −z + 1/2 (ii) x,−y + 1, −z + 1.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Inam, M.; Wu, J.; Shen, J.; Phan, C.U.; Tang, G.; Hu, X. Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide. Crystals 2018, 8, 336. https://doi.org/10.3390/cryst8090336
AMA Style
Inam M, Wu J, Shen J, Phan CU, Tang G, Hu X. Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide. Crystals. 2018; 8(9):336. https://doi.org/10.3390/cryst8090336
Chicago/Turabian StyleInam, Muhammad, Jiajia Wu, Jie Shen, Chi Uyen Phan, Guping Tang, and Xiurong Hu. 2018. "Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide" Crystals 8, no. 9: 336. https://doi.org/10.3390/cryst8090336
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.