

BSR and Full-Length Transcriptome Approaches Identified Candidate Genes for High Seed Ratio in Camellia vietnamensis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Trait Evaluation
2.3. Full-Length Transcriptome Sequencing
2.4. Prediction and Functional Annotation of Full-Length Transcripts and Their Open Reading Frames
2.5. BSR-Seq Analysis
2.6. Analysis of BSR-Seq Data
2.7. SNP and InDel Detection
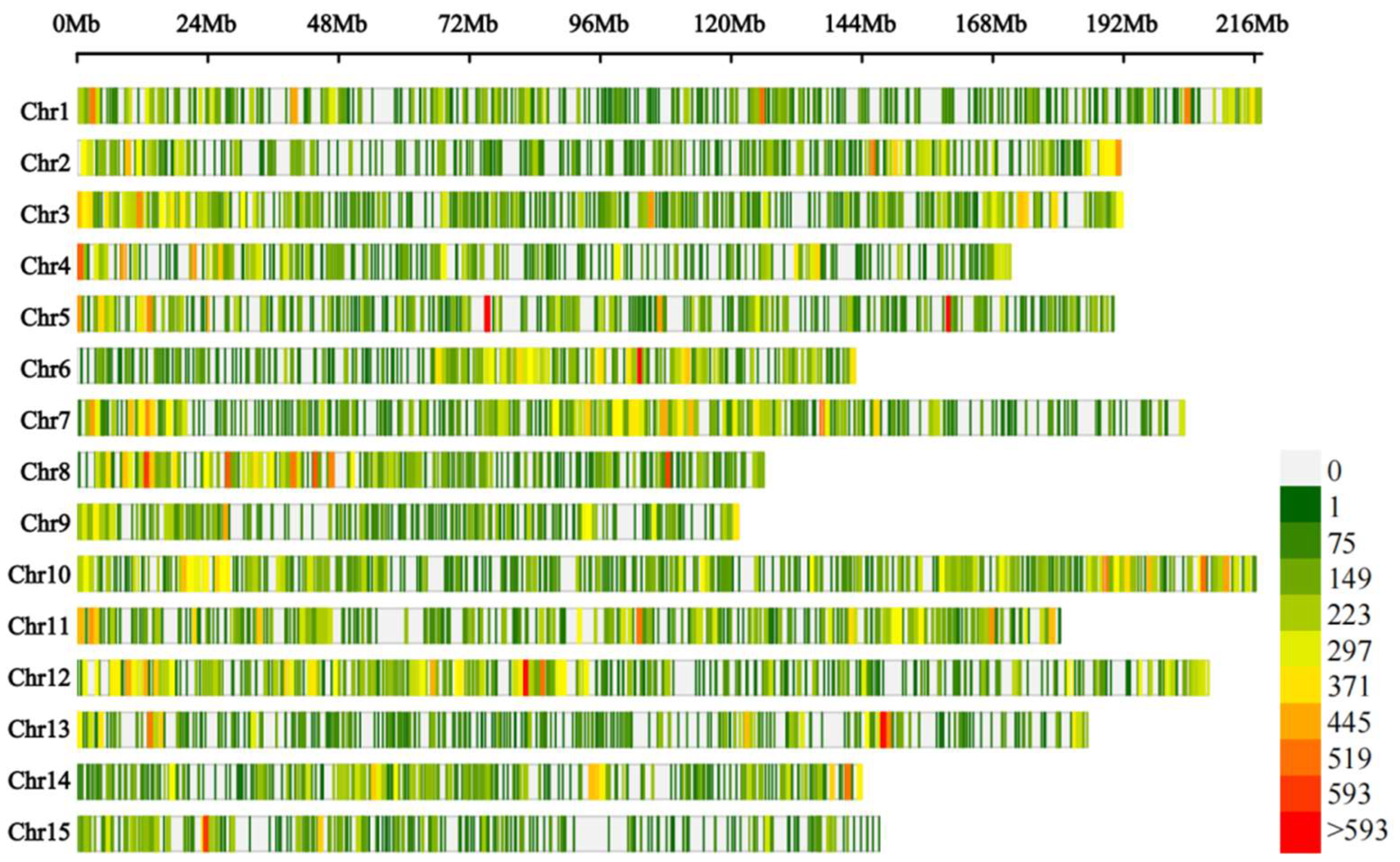
2.8. Association Analysis
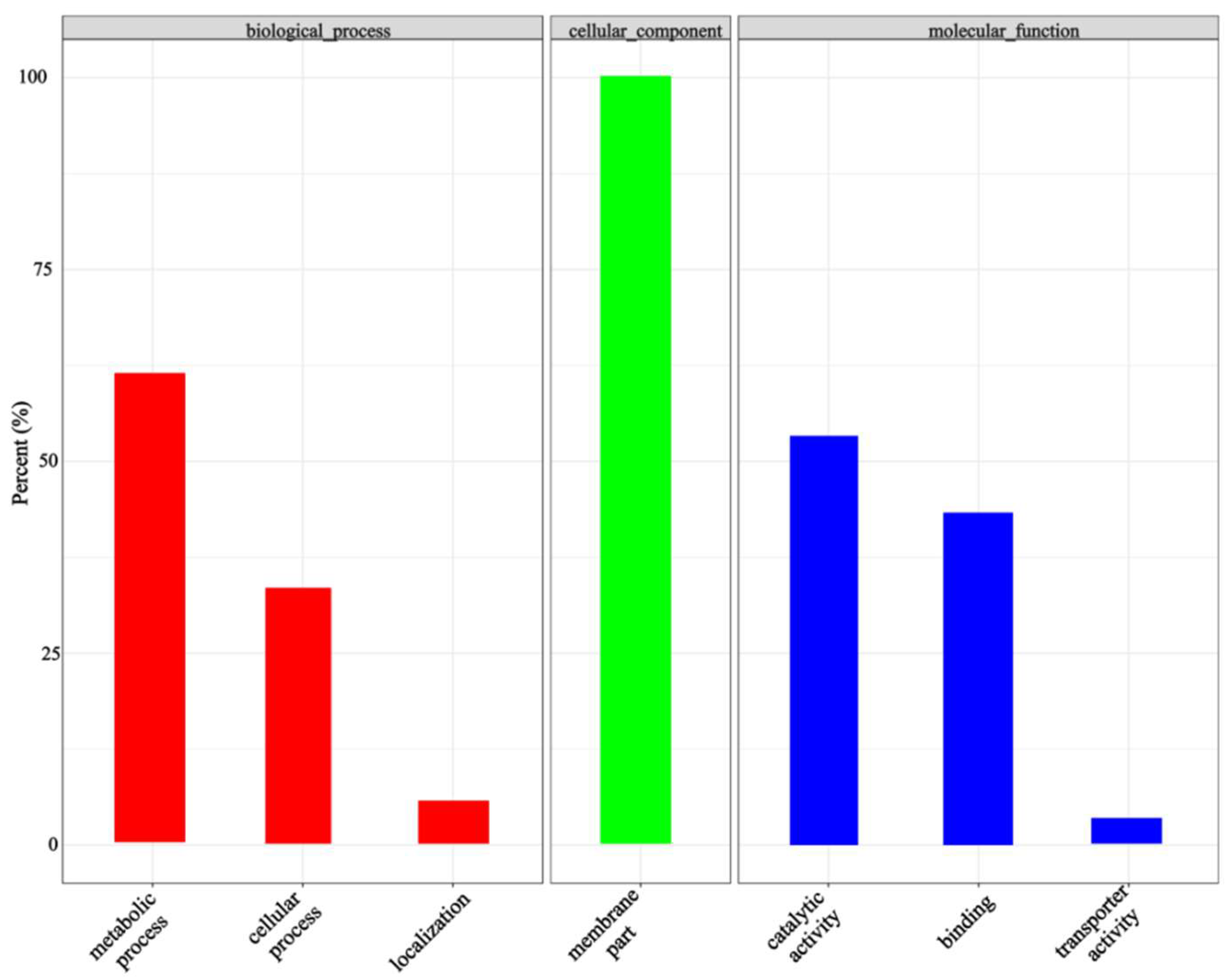
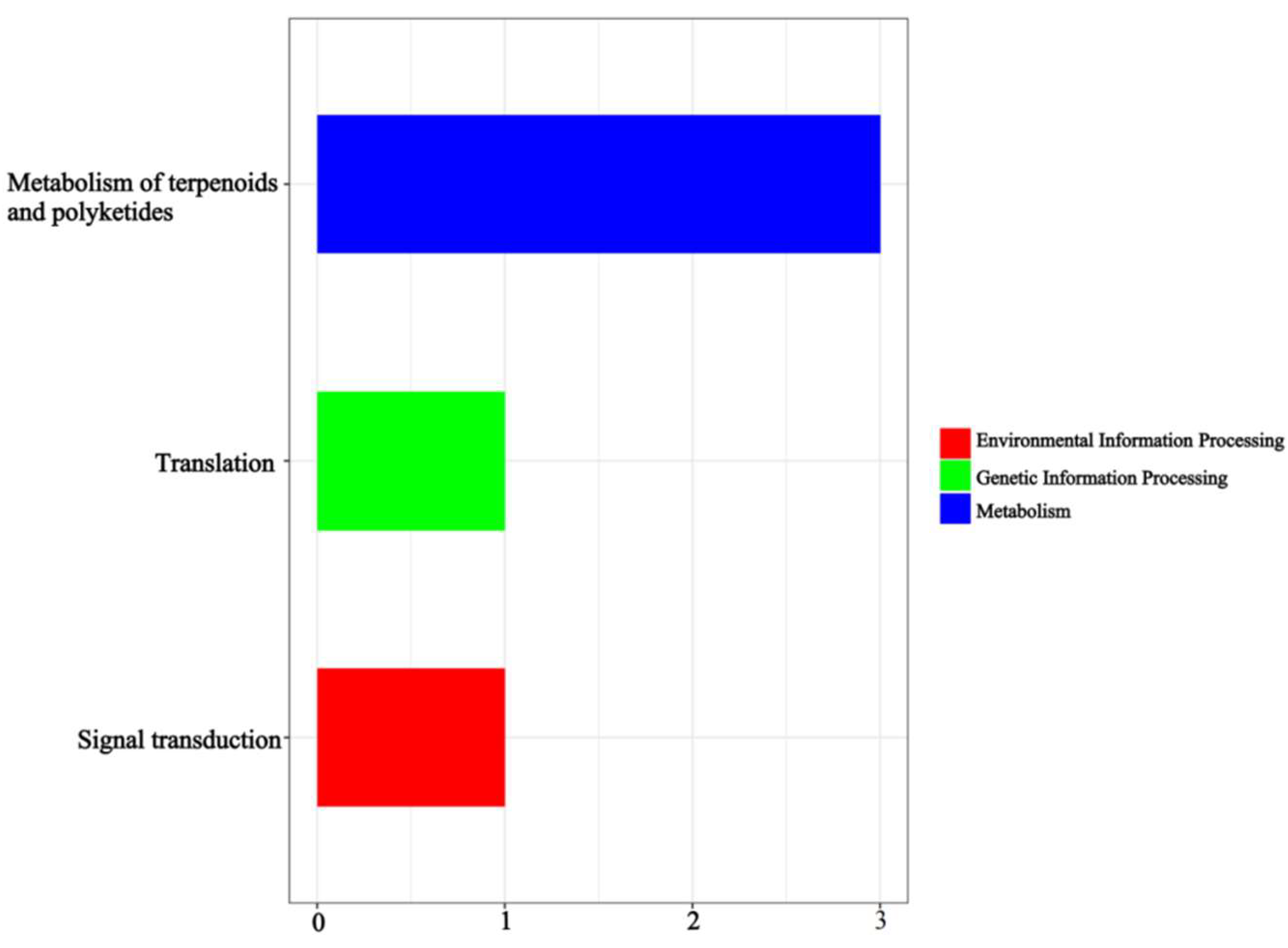
2.9. Functional Annotation and Expression Analysis of Associated Genes or Transcripts
3. Results
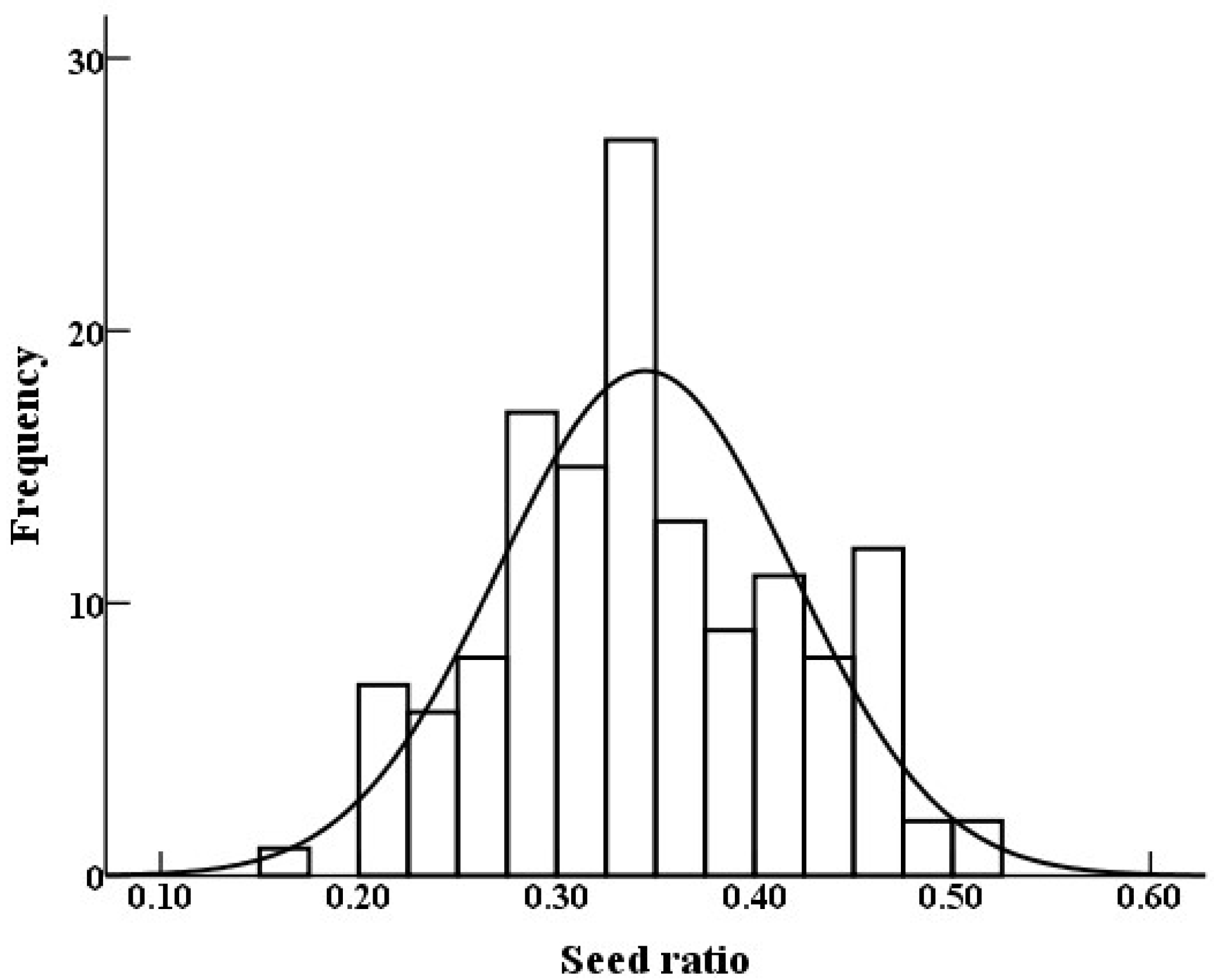
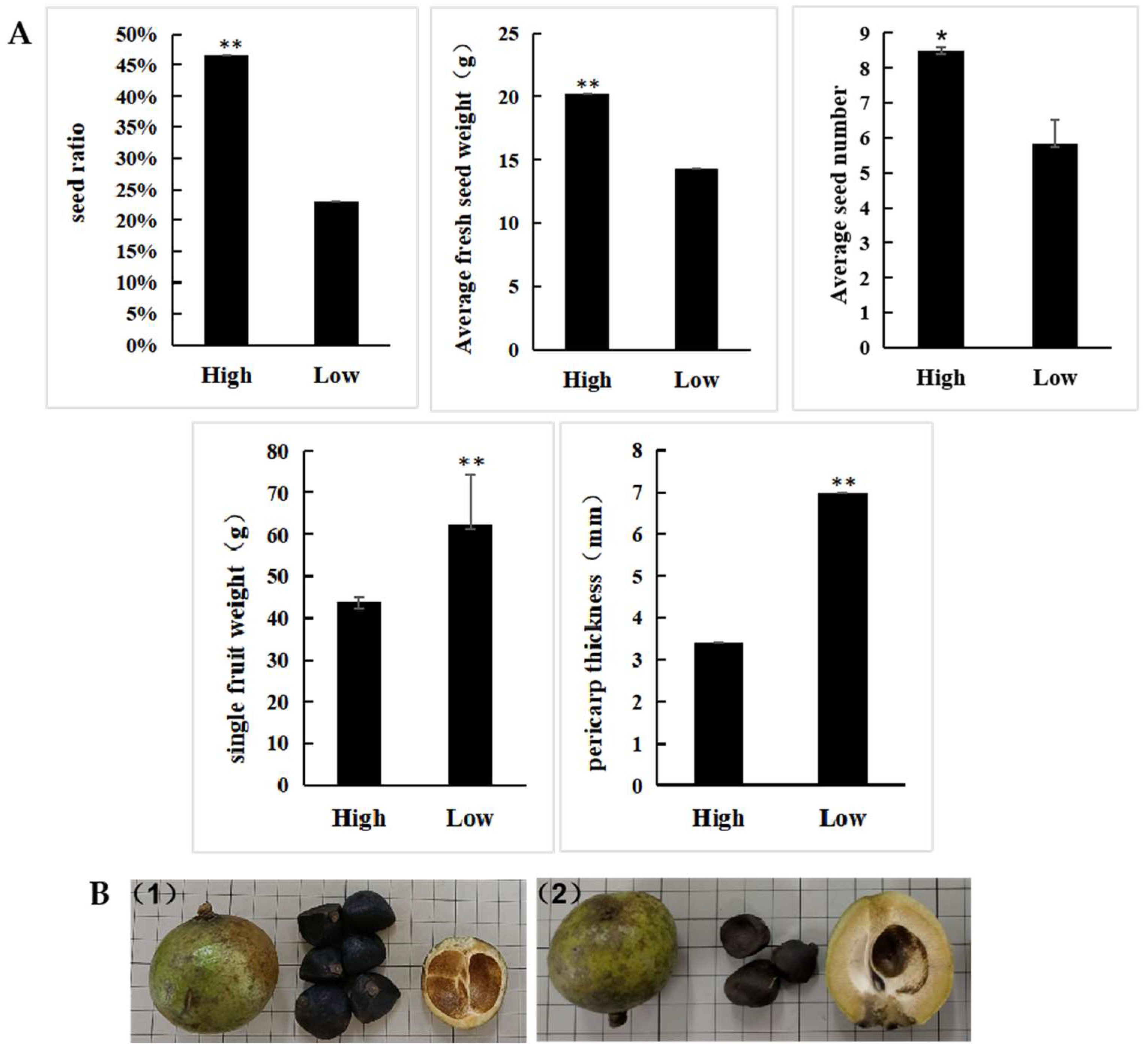
3.1. Phenotypic Data
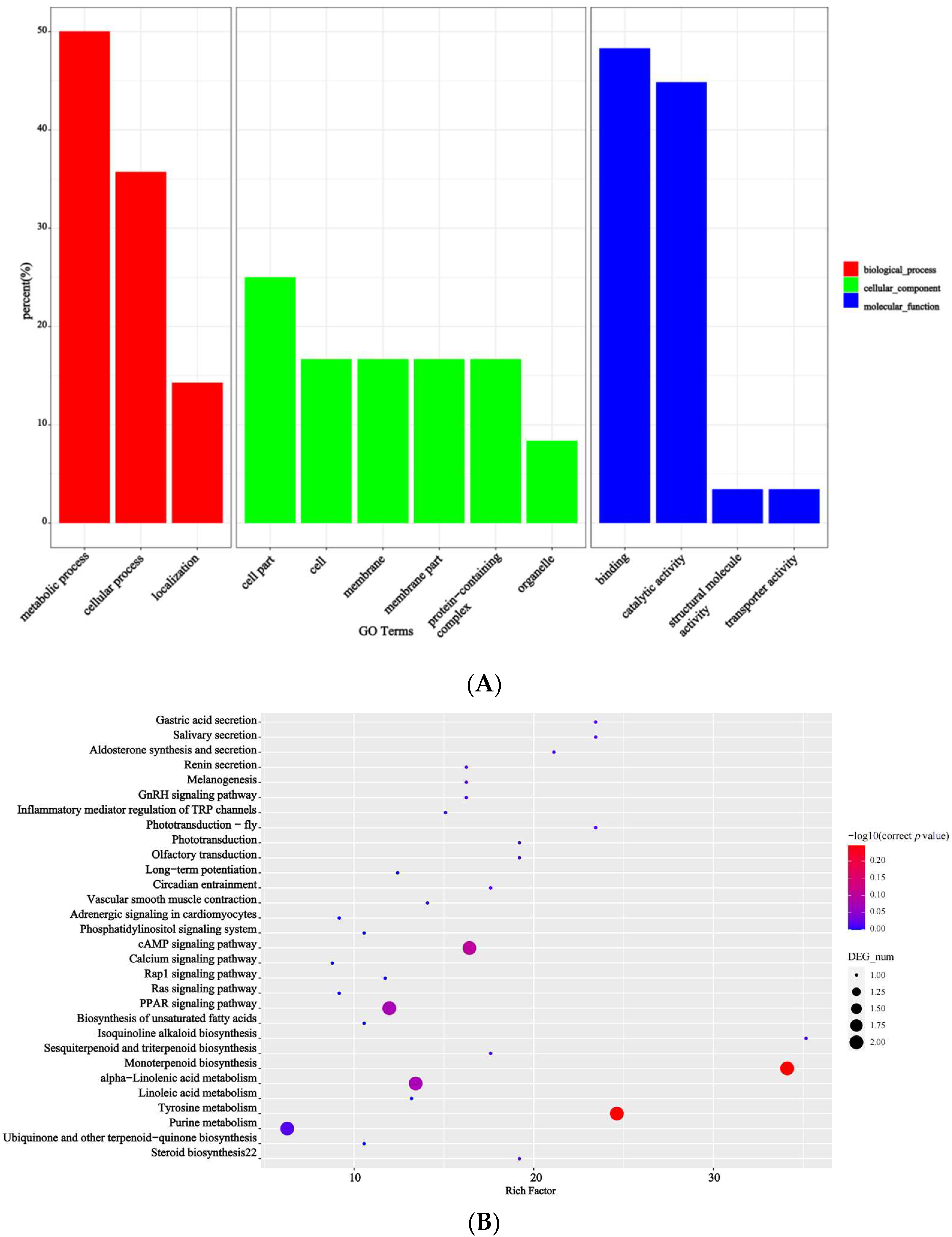
3.2. Transcriptomic Analysis via SMRT Sequencing
3.3. Differential Expression of Transcripts in Relation to Seed Ratio
3.4. Differential Expression of Genes in Relation to Seed Ratios
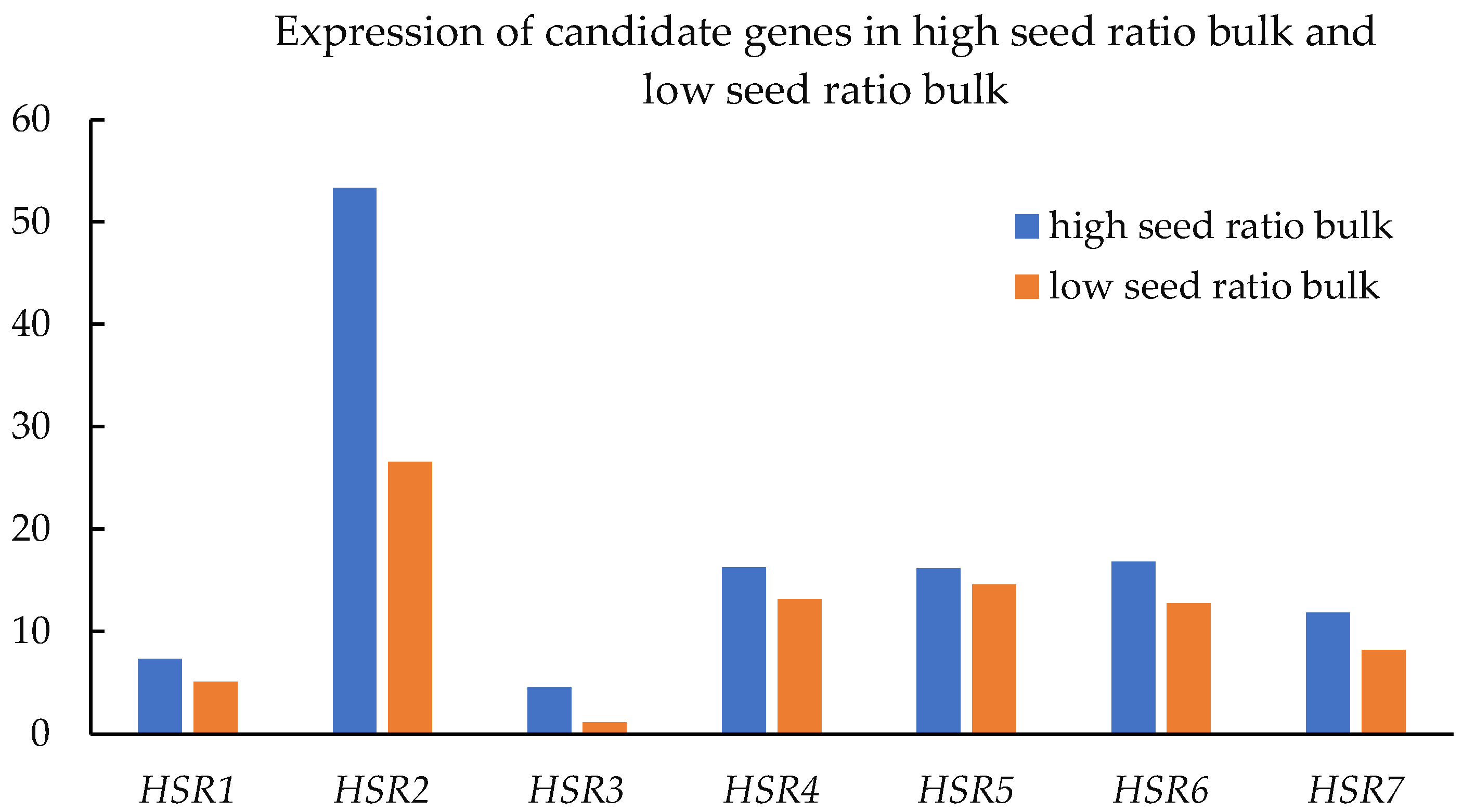
3.5. Expression of Associated Transcripts/Genes
4. Discussion
4.1. Cytochrome p450 Regulation in C. vietnamensis Seed Ratio
4.2. Phytohormone Regulation in C. vietnamensis Seed Ratio
4.3. Ubiquitin-Specific Protease Regulation in C. vietnamensis Seed Ratio
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, C.; Chen, L.; Tang, W.; Peng, S.; Li, M.; Deng, N.; Chen, Y. Predicting Potential Distribution and Evaluating Suitable Soil Condition of Oil Tea Camellia in China. Forests 2018, 9, 487. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Su, S.; Ma, L.; Yang, S.; Wang, Y.; Wang, X. Effects of canopy microclimate on fruit yield and quality of Camellia oleifera. Sci. Hortic. 2018, 235, 132–141. [Google Scholar]
- Wu, L.; Li, J.; Li, Z.; Zhang, F.; Tan, X. Transcriptomic Analyses of Camellia oleifera ’Huaxin’ Leaf Reveal Candidate Genes Related to Long-Term Cold Stress. Int. J. Mol. Sci. 2020, 21, 846. [Google Scholar] [CrossRef] [Green Version]
- Snyder, J.M.; Mounts, T.L.; Holloway, R.K. An analysis scheme for estimation of crude oil quality. J. Am. Oil Chem. Soc. 1991, 68, 285–288. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.P.; Yen, G.C. Antioxidant activity and bioactive compounds of tea seed (Camellia oleifera Abel.) oil. J. Agric. Food Chem. 2006, 3, 54. [Google Scholar] [CrossRef]
- Gao, C.; Yuan, D. Pollen Tube Growth and Double Fertilization in Camellia oleifera. J. Am. Soc. Hortic. Sci. 2015, 140, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Zhou, J.; Masabni, J.; Yuan, J. Phosphorus relieves aluminum toxicity in oil tea seedlings by regulating the metabolic profiling in the roots. Plant Physiol. Biochem. 2020, 152, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.C.; Stone, E.A.; Ayroles, J.F. The genetics of quantitative traits: Challenges and prospects. Nat. Rev. Genet. 2009, 10, 565–577. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Xu, F.; Sun, X.; Chen, Y.; Huang, Y.; Tong, C.; Bao, J. Rapid identification of major QTLs associated with rice grain weight and their utilization. PLoS ONE 2015, 10, e0122206. [Google Scholar] [CrossRef]
- Livaja, M.; Wang, Y.; Wieckhorst, S.; Haseneyer, G.; Seidel, M.; Hahn, V.; Knapp, S.J.; Taudien, S.; Schön, C.C.; Bauer, E. BSTA: A targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genom. 2013, 14, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illa-Berenguer, E.; Van Houten, J.; Huang, Z.; van der Knaap, E. Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 2015, 128, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Khan, A.W.; Jaganathan, D.; Thudi, M.; Roorkiwal, M.; Takagi, H.; Garg, V.; Kumar, V.; Chitikineni, A.; Gaur, P.M.; et al. QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotechnol. J. 2016, 14, 2110–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Chen, X.; Wang, Z.; Liu, Q.; Li, H.; Zhang, Y.; Ma, J.; Yang, J.; Zhang, X. Genetic mapping of the LOBED LEAF 1 (ClLL1) gene to a 127.6-kb region in watermelon (Citrullus lanatus L.). PLoS ONE 2017, 12, e0180741. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Khan, A.W.; Singh, V.K.; Vishwakarma, M.K.; Shasidhar, Y.; Kumar, V.; Garg, V.; Bhat, R.S.; Chitikineni, A.; Janila, P.; et al. QTL-seq approach identified genomic regions and diagnostic markers for rust and late leaf spot resistance in groundnut (Arachis hypogaea L.). Plant Biotechnol. J. 2017, 15, 927–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, C.; Sun, S.; Li, Y.; Duan, C.; Zhu, Z. Next-generation sequencing to identify candidate genes and develop diagnostic markers for a novel Phytophthora resistance gene, RpsHC18, in soybean. Theor. Appl. Genet. 2018, 131, 525–538. [Google Scholar] [CrossRef]
- Sun, J.; Yang, L.; Wang, J.; Liu, H.; Zheng, H.; Xie, D.; Zhang, M.; Feng, M.; Jia, Y.; Zhao, H.; et al. Identification of a cold-tolerant locus in rice (Oryza sativa L.) using bulked segregant analysis with a next-generation sequencing strategy. Rice 2018, 11, 24. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, X.; Zhu, W.; Qin, X.; Xu, J.; Cheng, C.; Lou, Q.; Li, J.; Chen, J. Complete resistance to powdery mildew and partial resistance to downy mildew in a Cucumis hystrix introgression line of cucumber were controlled by a co-localized locus. Theor. Appl. Genet. 2018, 131, 2229–2243. [Google Scholar] [CrossRef]
- Morandin, C.; Pulliainen, U.; Bos, N.; Schultner, E. De novo transcriptome assembly and its annotation for the black ant Formica fusca at the larval stage. Sci. Data 2018, 5, 180282. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.; Tosatto, S.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. The KEGG database. Novartis Found Symp. 2002, 247, 91–101, discussion 101–103, 119–128, 244–252. [Google Scholar]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Wang, K.; Wang, Y.; Hu, Z.; Yan, C.; Huang, H.; Ma, X.; Cao, Y.; Long, W.; Liu, W.; et al. The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication. Genome Biol. 2022, 23, 14. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Li, J.; Xing, S.; Du, S.; Wang, Z.; Sun, L.; Liu, X. RNA-Seq and Bulked Segregant Analysis of a Gene Related to High Growth in Ginkgo Biloba Half-Siblings. bioRxiv 2015, preprint. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Han, K. Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coord. Chem. Rev. 2012, 256, 1137–1150. [Google Scholar]
- Jun, X.U.; Wang, X.Y.; Guo, W.Z. The cytochrome P450 superfamily: Key players in plant development and defense. J. Integr. Agric. 2015, 14, 1673–1686. [Google Scholar]
- Sotelo-Silveira, M.; Cucinotta, M.; Chauvin, A.L.; Chávez, M.R.; Colombo, L.; Marsch-Martínez, N.; de Folter, S. Cytochrome P450 CYP78A9 is involved in Arabidopsis reproductive development. Plant Physiol. 2013, 162, 779–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Wang, Y.; McGregor, C.; Liu, S.; Luan, F.; Gao, M.; Weng, Y. Genetic architecture of fruit size and shape variation in cucurbits: A comparative perspective. Theor. Appl. Genet. 2020, 133, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Zhang, N.; Sauvage, C.; Muños, S.; Blanca, J.; Cañizares, J.; Diez, M.J.; Schneider, R.; Mazourek, M.; McClead, J.; et al. A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc. Natl. Acad. Sci. USA 2013, 110, 17125–17130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Liu, C.; Song, L.; Li, Y.; Li, M. PaCYP78A9, a Cytochrome P450, Regulates Fruit Size in Sweet Cherry (Prunus avium L.). Front. Plant Sci. 2017, 8, 2076. [Google Scholar] [CrossRef] [Green Version]
- Gunupuru, L.R.; Arunachalam, C.; Malla, K.B.; Kahla, A.; Perochon, A.; Jia, J.; Thapa, G.; Doohan, F.M. A wheat cytochrome P450 enhances both resistance to deoxynivalenol and grain yield. PLoS ONE 2018, 13, e0204992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignolli, F.; Vidoz, M.L.; Picciarelli, P.; Mariotti, L. Gibberellins modulate auxin responses during tomato (Solanum lycopersicum L.) fruit development. Physiol. Plant. 2019, 165, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Hussain, Q.; Shi, J.; Scheben, A.; Zhan, J.; Wang, X.; Liu, G.; Yan, G.; King, G.J.; Edwards, D.; Wang, H. Genetic and signalling pathways of dry fruit size: Targets for genome editing-based crop improvement. Plant Biotechnol. J. 2020, 18, 1124–1140. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, F.; Zhang, H.; He, H.; Ma, L.; Deng, X.W. Functional characterization of the Arabidopsis ubiquitin-specific protease gene family reveals specific role and redundancy of individual members in development. Plant J. 2008, 55, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, L.; Wang, H. Arabidopsis ubiquitin-conjugating enzyme UBC22 is required for female gametophyte development and likely involved in Lys11-linked ubiquitination. J. Exp. Bot. 2016, 67, 3277–3288. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Reads (bp) | Pair Reads | Reference Sequence | Aligned Concordantly 0 Times a | Aligned Concordantly Exactly 1 Time b | Aligned Concordantly > 1 Times c | Total Alignment Ratio d |
|---|---|---|---|---|---|---|---|
| pool 1 | 31,123,912 | 31,123,912 (100.00%) | full-length transcript | 15,282,712 (49.10%) | 6,617,832 (21.26%) | 9,223,368 (29.63%) | 60.41% |
| pool 2 | 37,549,983 | 37,549,983 (100.00%) | 18,946,996 (50.46%) | 8,326,225 (22.17%) | 10,276,762 (27.37%) | 62.06% |
| Sample | Total Reads (bp) | Pair Reads | Aligned Concordantly 0 Times a | Aligned Concordantly Exactly 1 Time b | Aligned Concordantly > 1 Times c | Total Alignment Ratio d | |
|---|---|---|---|---|---|---|---|
| pool 1 (High) | 31,123,912 | 31,123,912 (100.00%) | Camellia oleifera (eudicots) genome | 18,080,834 (58.09%) | 5,743,157 (18.45%) | 7,299,921 (23.45%) | 62.54% |
| pool 2 (Low) | 37,549,983 | 37,549,983 (100.00%) | 22,110,840 (58.88%) | 6,036,660 (16.08%) | 9,402,483 (25.04%) | 65.82% |
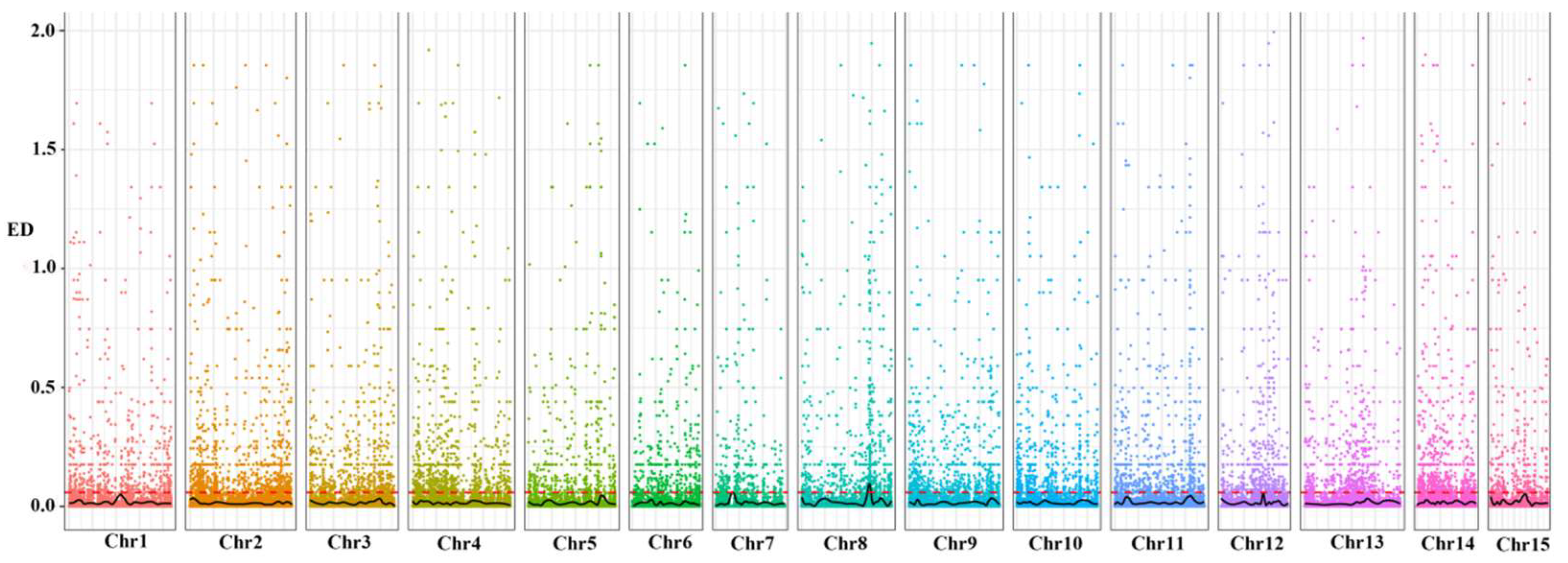
| Chromosome | Start (Mb) | End (Mb) | Length (Mb) | Candidate Gene Number | SNP Number | InDel Number |
|---|---|---|---|---|---|---|
| Chr2 | 140.8 | 148.4 | 7.6 | 97 | 865 | 42 |
| Chr15 | 35.3 | 37.3 | 2.0 | 38 | 24 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, B.-Q.; Liao, H.-Z.; Xia, Y.-Y.; Wang, D.-X.; Ye, H. BSR and Full-Length Transcriptome Approaches Identified Candidate Genes for High Seed Ratio in Camellia vietnamensis. Curr. Issues Mol. Biol. 2023, 45, 311-326. https://doi.org/10.3390/cimb45010022
Hao B-Q, Liao H-Z, Xia Y-Y, Wang D-X, Ye H. BSR and Full-Length Transcriptome Approaches Identified Candidate Genes for High Seed Ratio in Camellia vietnamensis. Current Issues in Molecular Biology. 2023; 45(1):311-326. https://doi.org/10.3390/cimb45010022
Chicago/Turabian StyleHao, Bing-Qing, Hong-Ze Liao, Ying-Ying Xia, Dong-Xue Wang, and Hang Ye. 2023. "BSR and Full-Length Transcriptome Approaches Identified Candidate Genes for High Seed Ratio in Camellia vietnamensis" Current Issues in Molecular Biology 45, no. 1: 311-326. https://doi.org/10.3390/cimb45010022