

Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data

1
School of Biological and Chemical Sciences, University of Galway, University Road, H91 TK33 Galway, Ireland
2
Department of Bioproduct and Biosystems, Aalto University, 02150 Espoo, Finland
3
SSPC—The SFI Research Centre for Pharmaceuticals, University of Galway, University Road, H91 TK33 Galway, Ireland
*
Authors to whom correspondence should be addressed.
Chemistry 2022, 4(3), 983-1009; https://doi.org/10.3390/chemistry4030067
Submission received: 26 July 2022
/
Revised: 26 August 2022
/
Accepted: 31 August 2022
/
Published: 6 September 2022
(This article belongs to the Special Issue Discovery of Bioactive Ingredients from Natural Products)
Abstract
:The human myeloid leukemia cell differentiation protein (hMcl-1) is an anti-apoptotic multi-partner protein, belonging to the B-cell lymphoma-2 (Bcl-2) family of proteins. Studies have linked hMcl-1 alleviated expression with resistance to hemopoietic chemotherapeutics, which makes it a key drug target in blood cancers. However, most of the developed small- to medium-sized hMcl-1 inhibitors have typical off-target activity towards other members of the Bcl-2 family. To improve the hMcl-1 inhibitor design, especially exploring a suitable scaffold with pharmacophoric features, we focused on natural hMcl-1 inhibitors. To date, seven classes of natural compounds have been isolated, which display a low micromolar affinity for hMcl-1 and have limited biophysical studies. We screened hMcl-1 co-crystal structures, and identified nine co-crystal structures of hMcl-1 protein, which were later evaluated by multiple receptor conformations (which indicates that the differences between hMcl-1 in crystal structures are low (RMSD values between 0.52 and 1.13 Å, average RMSD of 0.638–0.888 Å, with a standard deviation of 0.102–0.185Å)), and multiple ligand conformations (which led to the selection of the PDB structure, 3WIX (RMSD value = 0.879 Å, standard deviation 0.116 Å), to accommodate various Mcl-1 ligands from a range of co-crystal PDB files) methods. Later, the three adopted docking methods were assessed for their ability to reproduce the conformation bound to the crystal as well as predict trends in Ki values based on calculated RMSD and docking energies. Iterative docking and clustering of the docked pose within ≤1.0 Å was used to evaluate the reproducibility of the adopted docking methods and compared with their experimentally determined hMcl-1 affinity data.
1. Introduction
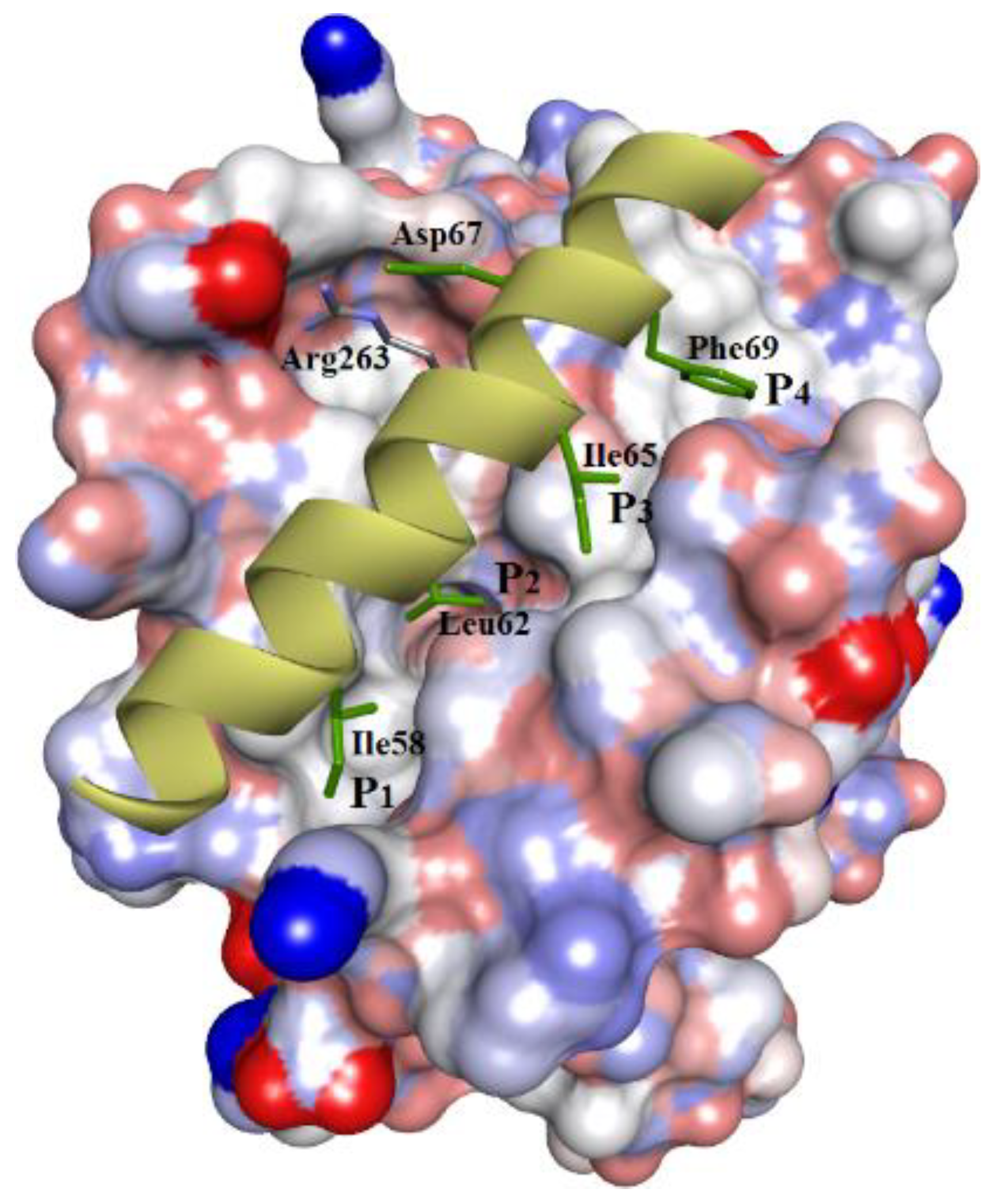
The human myeloid leukemia cell differentiation protein, hMcl-1, is a multi-partner, anti-apoptotic protein, belonging to the B-cell lymphoma-2 family (Bcl-2 family) of proteins [1]. Several reports have established a link between the upregulation of hMcl-1 with the development of resistance to various anticancer chemotherapies [2,3,4,5,6]. Furthermore, amplification of the hMcl-1 gene is one of the most commonly observed genetic aberrations in human blood cancers [7], which makes it a key drug target in hemopoietic malignancies. Additionally, its RNA-mediated knockdown and Mcl-1 gene silencing demonstrated a substantial decrease in cancer cell growth and restored the sensitivity of chemo-resistant cells [2,8,9] to chemotherapy. This Bcl-2 family comprises anti-apoptotic proteins other than hMcl-1 (especially Bcl-2 itself, Bcl-xL [10], and Bcl-w), as well as proapoptotic and apoptotic proteins (Bax, Bak, Bik, Bid, and NOXA). The interplay between their intracellular levels (anti-apoptotic proteins versus apoptotic proteins) and their protein–protein interactions decides the fate of cell survival. Therefore, altering or blocking these protein–protein interactions (as “PPIs”) is one of the strategies used to develop inhibitors. However, targeting PPIs with small molecules can be challenging, as these PPIs mainly occur over relatively large and flat surfaces. Although, there are small pockets between these interfaces (hot spots), which accommodate high-affinity binding interactions of small functional groups and, generally, is the side chain of an amino acid of the other binding partner protein [11]. In this context, the cellular expression of Mcl-1 is tightly regulated by its partner proteins, both proapoptotic and apoptotic, which utilize their alpha-helical peptide regions (more specifically, “BH3-helix domain”) for selective binding to the pockets of hMcl-1 and inhibit its functions. In the case of hMcl-1 (Figure 1), for example, the BH3 α-helix uses hydrophobic amino acid side chains to bind to hMcl-1 via interactions with four hydrophobic pockets, P1–4. Therefore, BH3 helix-based ligand design has been used to develop various inhibitor types, such as oligopeptides, stapled α-helixes, α-/β-peptide foldamers, reverse BH3 (rBH3) peptide, and small molecules, as Mcl-1 inhibitors, but none have attained clinical application so far [12]. Developed hMcl-1 inhibitors based on the BH3-helix have been plagued with affinities/activities for other anti-apoptotic proteins of the Bcl-2 family as hMcl-1 shares high structural homology with other proteins (especially Bcl-2 and Bcl-xL), and therefore most discovered inhibitors against hMcl-1 are either dual- or multi-targeting inhibitors [12].
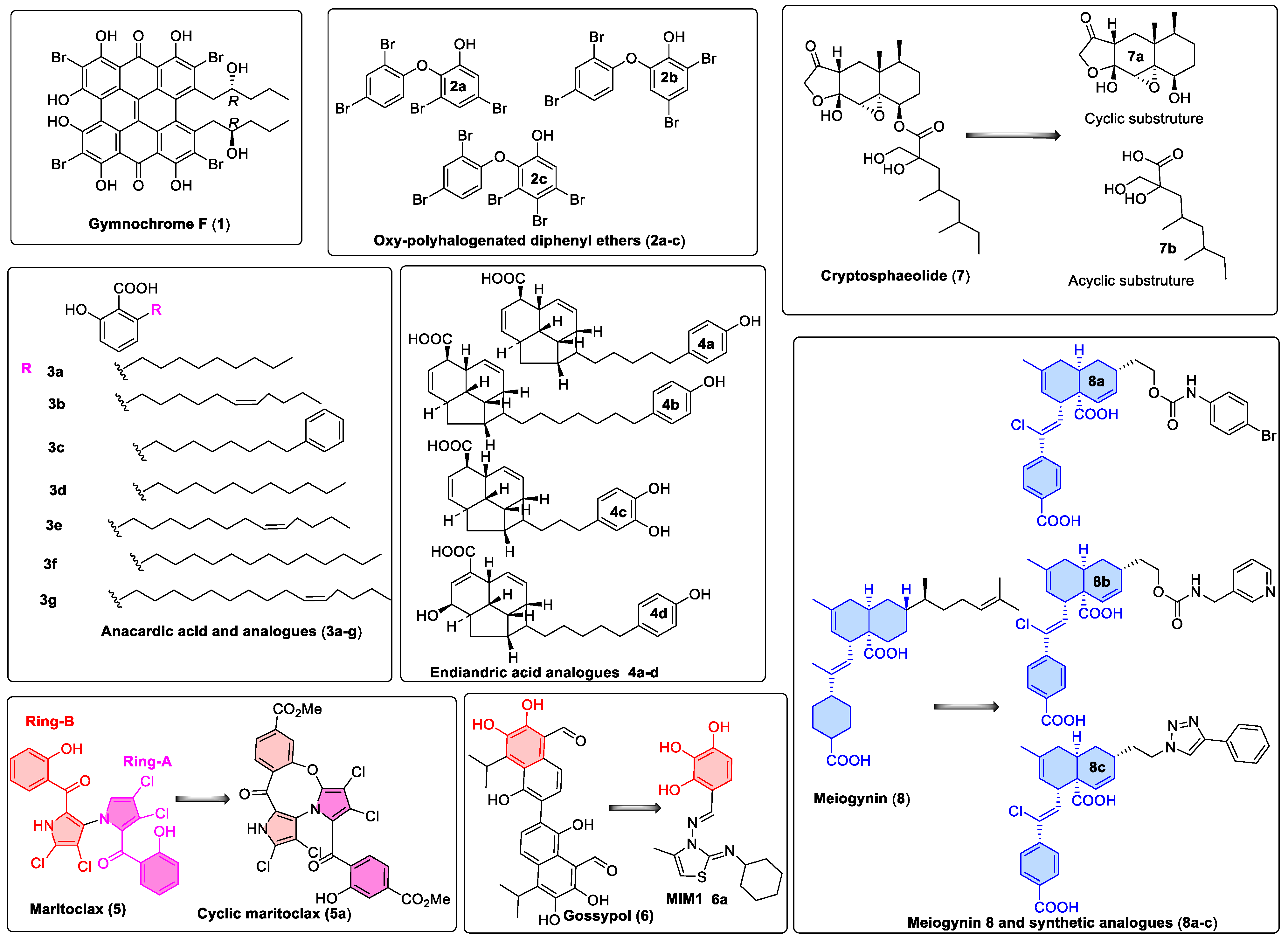
In recent years, seven classes were reported as inhibitors of hMcl-1 (six from marine sources: gymnochrome-F, oxy-polyhalogenated diphenyl ethers, anacardic acids, endiandric acids, maritoclax, and cryptosphaerolide, while one natural compound was from a terrestrial plant source: meiogynin). Their structures are summarized in Figure 2, and their inhibitory data are provided in Table 7. To date, no other biophysical studies were performed on these compounds prior to measuring their binding behavior with hMcl-1. Herein, we provide details of a modeling study to gain insight into their binding modes. This includes validation of the computational protocols and correlation of results obtained with published, experimentally determined affinity data, as well as with X-ray crystal structural data for hMcl-1 bound to small molecules [13].
2. Materials and Methods
2.1. Protein Structure Preparation for Modeling
All protein crystal structural coordinates were downloaded from the protein databank (www.rcsb.org). They were further prepared for modeling in CCG MOE 2019.01 by: (a) removal of solvent molecules from their crystal structures, (b) converting any protein downloaded in a dimer to the monomeric state (Mcl-1 protein inhibition/activation takes place through its BH3 domain, which is a monomer), (c) adding any missing atoms of amino acid residues in the loops, and (d) protonation to ensure appropriate ionization and tautomeric forms for amino acid residues of the hMcl-1 protein.
Our study is limited to ligand-based human Mcl-1 (hMcl-1) inhibitor design; therefore, those hMcl-1 with no co-crystallized ligand or sourced from a different organism other than human, or those with truncated or mutated protein structures, were ignored. In some cases, the NMR-based 15N-HSQC-based mouse Mcl-1 (mMcl-1) perturbation studies were performed, which were taken into account to determine the homologous amino acids in hMcl-1.
2.2. Ligand Structure Preparation
The ligands were collected from various crystal structures, as downloaded from the RCSB website (see in Table 1). In the case of naturally occurring Mcl-1 inhibitors, their structures were built using the MOE builder tool. In the case of cryptosphaerolide, as the absolute configuration of three stereogenic centers remains unknown, all eight possible stereoisomers were constructed. The generated models of the structures were energy-minimized in MOE (MMFF94x forcefield) with a gradient of 0.00001. All energy-minimized structures were compiled into a single library ligand database file, formatted into .mdb format as required for MOE docking. In the case of AutoDock, structures in .mol format were converted to .pdbqt files as required for docking using this software, while .mol format was used directly for docking using VlifeDock.
2.3. Superposition and Docking Protocols and Analysis
The atoms of the protein backbones, of all nine X-ray co-crystal structures of hMcl-1, which had small molecules bound, were superposed in MOE version 2019.01, and RMSD values were calculated. In our previous study, we compared different placement methods of MOE (triangle matcher, alpha triangle, and alpha PMI methods) for their suitability for various types of scaffolds. During our studies, we compared the resulted docking energies of eight inhibitors (whose native X-ray conformation with fucosidase enzyme was known) docked on the fucosidase enzyme using three different placement methods (triangle matcher, alpha triangle, and alpha PMI methods), and we found that the correlation factor (R2 = 0.908) for the triangle matcher was comparatively higher. Therefore, we used the triangle matcher method as a reasonable placement method and GB/VI as a reasonable scoring algorithm [14] for various types of ligands [15]. Here, again, we used the triangle matcher placement method (100,000 poses with retention time 3000 s and 10,000 returned poses) and refinement using rigid structure with poses = 1000 (gradient = 0.0001 and iteration = 5000). The triangle matcher placement method generates the inhibitor-protein poses by aligning the ligand atoms in triplet on triplets of alpha spheres in a more methodical way. Using this method, the docked conformation of each ligand was compared with their crystal conformation to calculate RMSD values. Docked conformations were arranged into clusters of similar binding conformations from the 500 lowest energy poses (RMSD value ≤ 2.0 Å). The data for the lowest energy-minimized pose from the largest cluster are displayed in the tables and shown in the various figures. A similar approach was used to analyze output from docking attained from the AutoDock and VlifeDock methods. Genetic algorithm-based grid docking in AutoDock was used. Thus, hMcl-1 protein crystal and ligand structures were saved in .pdbqt format for AutoDock 1.5.6 [16,17]. The AutoGrid was mapped around the co-crystallized ligand using the following dimensions, 60 Å × 60 Å × 60 Å (representing the three coordinates X, Y, and Z of the grid), with 0.400 Å spacing. The docking parameters were set up as follows: (a) a maximum of 25 conformers were considered for each ligand, (b) population size was kept to 250 with the individual conformers initialized randomly, (c) maximum numbers of generations were 27,000, and (d) the number of evaluations was set as 2,500,000. Docking with VlifeDock [17] involved the use of the in-house placement method (a Genetic Algorithm) and the scoring was calculated by VlifeDock’s GRIP function. A grid was initially generated for docking (60 Å × 60 Å × 60 Å). This docking method generates a population of initial poses, which evolves into the optimal binding mode. After docking, the interactions (H-bonding, hydrophobic, ionic, and aromatic π-π), measurements, and labeling were carried out with the Accelrys discovery studio visualizer. Studying interactions are not only limited to understand the biophysical properties of small molecules with macromolecules but also could be used for choosing better drug delivery materials for natural compounds (which have limited physiochemical properties), such as nanoencapsulation of medicinal essential oils [18].
3. Results
3.1. Validation of Molecular Modeling Protocols
Nineteen structures (see Table 1) of coordinates for hMcl-1 co-crystallized with ligands from the protein databank (www.rcsb.org) were retrieved. Although these crystal structures belong to the same species (Homo sapiens), their quality varied. Validation metrics are shown in Table 1, which are independent of the ligand. There were hMcl-1 crystal structures that contained peptide ligands (Table 1, entries 10–19), and these showed typical interactions with amino acids of the cavity, including those in the pockets: P1, P2, P3, and P4 (Figure 1). We did not proceed with further study for those that had peptide ligands, which interacted with P1–P4. Small-molecule inhibitors (SMIs) co-crystallized (Table 1, entries 1–9) with hMcl-1 have a wider diversity of scaffolds compared to peptides, with a greater variety of binding modes. It was decided that these nine SMI structures would be used to generate binding modes for the natural products.
Figure 2.
Natural product and derivatives identified as hMcl-1 inhibitors: 1. Gymnochrome-F, a phenanthroperylenequinone-containing core compound isolated from a deep-water Crinoid, Halopus rangii [19]. 2. Oxy-polyhalogenated diphenyl ethers from a sponge Dysidea (Lamellodysidea) herbacea [20]. 3. Anacardic acids from Knema hookeriana [21]. 4. Endiandric acids analogues from Beilschmiedia ferruginea [22]. 5. Maritoclax, obtained from Actinomycete strain CNQ-418, which shares 98.1% 16S rRNA gene sequence identity with Streptomyces sannurensis, suggesting that it may be a new Streptomyces species [23,24], and its synthetic analog, the maritoclax structure, can be divided into ring-A (magenta) and ring-B (red) for easy presentation. 6. Gossypol [25,26] and hMcl-1 inhibitor molecule 1 (MIM1) [27], a high-throughput screening compound, is not a natural product but shares features with Gossypol. 7. Cryptosphaerolide from a marine-derived ascomycete related to the genus Cryptosphaeria [28] and derivatives. 8. Meiogynin (obtained from ethyl acetate extract of the bark of Meiogyne cylindrocarpa) and synthetic derivatives (8a–c) [29,30].
Figure 2.
Natural product and derivatives identified as hMcl-1 inhibitors: 1. Gymnochrome-F, a phenanthroperylenequinone-containing core compound isolated from a deep-water Crinoid, Halopus rangii [19]. 2. Oxy-polyhalogenated diphenyl ethers from a sponge Dysidea (Lamellodysidea) herbacea [20]. 3. Anacardic acids from Knema hookeriana [21]. 4. Endiandric acids analogues from Beilschmiedia ferruginea [22]. 5. Maritoclax, obtained from Actinomycete strain CNQ-418, which shares 98.1% 16S rRNA gene sequence identity with Streptomyces sannurensis, suggesting that it may be a new Streptomyces species [23,24], and its synthetic analog, the maritoclax structure, can be divided into ring-A (magenta) and ring-B (red) for easy presentation. 6. Gossypol [25,26] and hMcl-1 inhibitor molecule 1 (MIM1) [27], a high-throughput screening compound, is not a natural product but shares features with Gossypol. 7. Cryptosphaerolide from a marine-derived ascomycete related to the genus Cryptosphaeria [28] and derivatives. 8. Meiogynin (obtained from ethyl acetate extract of the bark of Meiogyne cylindrocarpa) and synthetic derivatives (8a–c) [29,30].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Parameters of co-crystal structures from the protein databank where hMcl-1 binds to a ligand [31,32].
| Entry | PDB Code | Res. (Å) | Co-Crystallized Ligand Type | R-Value | R-Free | Clash Score | Ramachandran Outliers (%) | Side-Chain Outliers (%) | RSRZ Outliers (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4WMR | 1.70 | SMI * | 0.171 | 0.206 | 1 | 0 | 0 | 4.0 |
| 2 | 4ZBF | 2.20 | SMI | 0.184 | 0.233 | 6 | 0.2 | 4.5 | 2.0 |
| 3 | 4ZBI | 2.50 | SMI | 0.183 | 0.242 | 7 | 0.6 | 8 | 1.7 |
| 4 | 4OQ5 | 2.86 | SMI | 0.200 | 0.235 | 9 | 2.1 | 11.3 | 8.1 |
| 5 | 4OQ6 | 1.81 | SMI | 0.203 | 0.205 | 11 | 0 | 7.3 | 1.4 |
| 6 | 3WIX | 1.90 | SMI | 0.246 | 0.291 | 4 | 0 | 3.3 | 3.2 |
| 7 | 3WIY | 2.15 | SMI | 0.214 | 0.283 | 3 | 0.7 | 4.0 | 2.5 |
| 8 | 4HW2 | 2.80 | SMI | 0.217 | 0.251 | 32 | 1.7 | 17.6 | 1.8 |
| 9 | 4HW3 | 2.40 | SMI | 0.216 | 0.269 | 13 | 1 | 11.4 | 3.2 |
| 10 | 4HW4 | 1.53 | Peptide | 0.140 | 0.182 | 2 | 0 | 0 | 2.1 |
| 11 | 3TWU | 1.80 | Peptide | 0.184 | 0.223 | 2 | 0 | 0 | 2.3 |
| 12 | 3PK1 | 2.49 | Peptide | 0.213 | 0.245 | 3 | 0.9 | 8.1 | 0.9 |
| 13 | 3MK8 | 2.32 | Peptide | 0.233 | 0.275 | 9 | 0.7 | 0 | 1.9 |
| 14 | 3KZ0 | 2.35 | Peptide | 0.224 | 0.270 | 10 | 0 | 0 | 9.9 |
| 15 | 3KJ0 | 1.70 | Peptide | 0.187 | 0.223 | 8 | 0 | 0.6 | 4.4 |
| 16 | 3KJ1 | 1.95 | Peptide | 0.188 | 0.213 | 3 | 1.2 | 0.7 | 9.4 |
| 17 | 3KJ2 | 2.35 | Peptide | 0.210 | 0.233 | 2 | 0 | 1.3 | 6.7 |
| 18 | 3IO9 | 2.40 | Peptide | 0.211 | 0.269 | 7 | 0.6 | 4.9 | 4.1 |
| 19 | 2PQK | 2.00 | Peptide | 0.196 | 0.234 | 6 | 0 | 1.4 | 7 |
* SMI = small-molecule inhibitor. PDB = ID code from the protein databank (www.rcsb.org). Res: resolution. R-Value: a measure of how a simulated diffraction pattern matches the experimentally observed diffraction pattern: a perfect fit would have a value of 0, and typical values are about 0.20. R-free: 10% of experimental observations were removed from the dataset and then refinement was performed using the remaining 90%. The R-free value is calculated from how well the model predicts the 10% of observations that were not used in refinement. R-free is typically found to be slightly higher than the R-value and with a value of close to 0.26. Clash score: the number of pairs of atoms clashing sterically per 1000 atoms. Ramachandran outliers: fraction of amino acids with non-favorable dihedral angles. Side-chain outliers: Off-rotamer positions or less-stable conformations of amino acid side chains compared to the on-rotamer position or statistically more favored conformation. The score is calculated as the percentage of residues with unusual side-chain conformation to the total number of amino acid residues. RSRZ outliers: An amino acid residue is said to be an RSRZ outlier if its RSRZ value is more than two. The RSRZ outlier score depends on the fraction of polypeptide residues that do not fit electron density well when compared with other instances of the same residues in structures at a similar resolution [33].
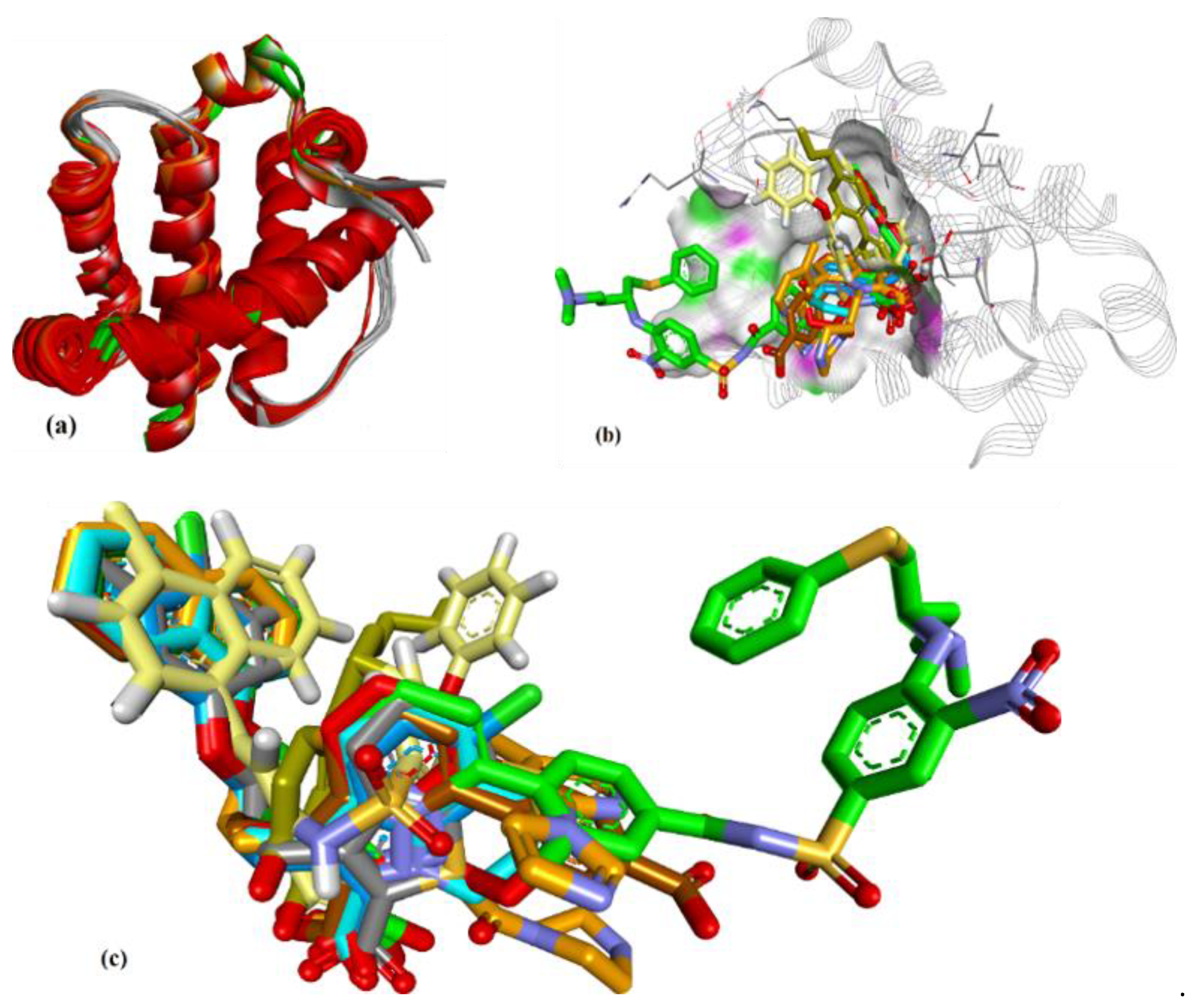
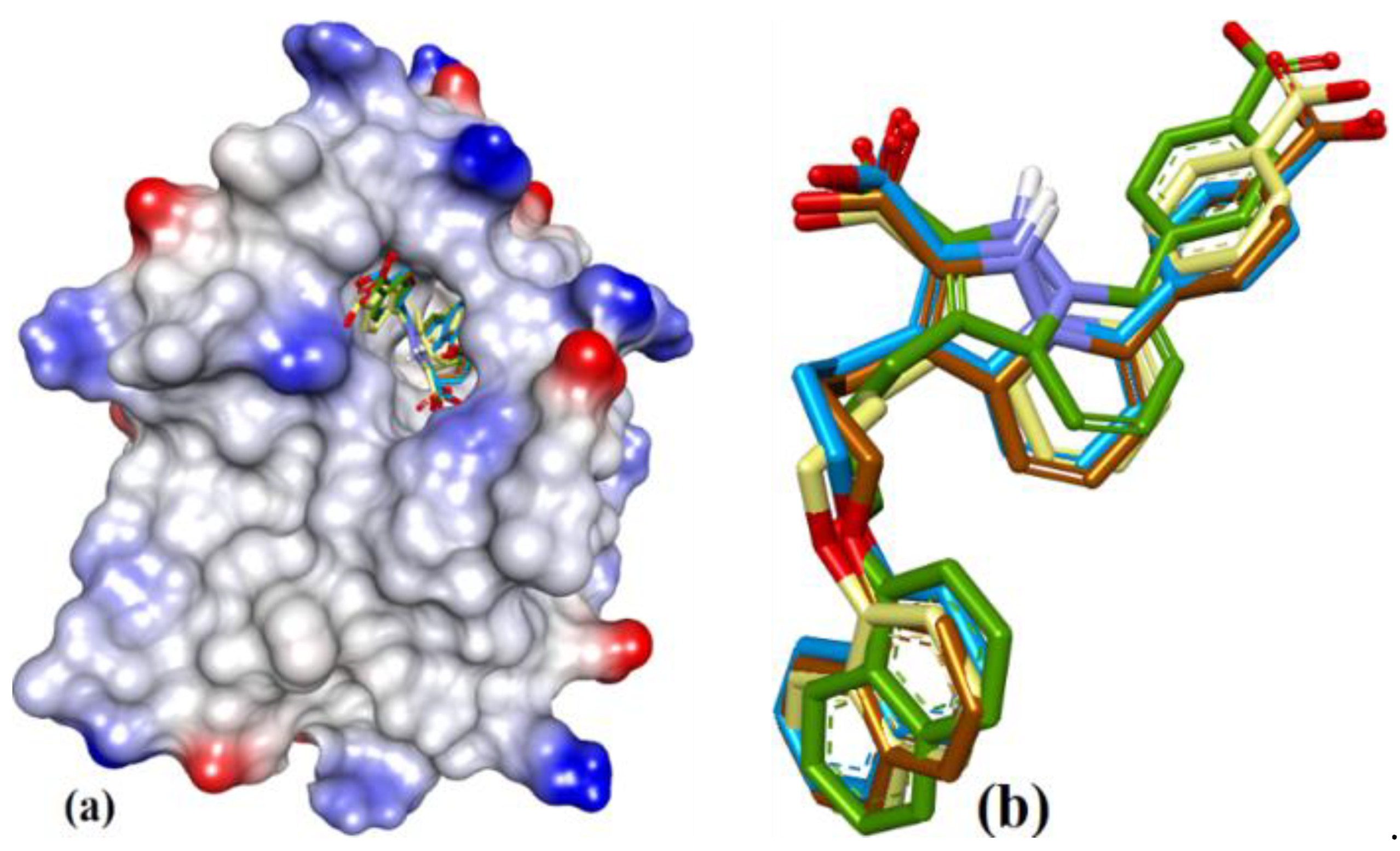
Having selected nine co-crystal structures of hMcl-1 with SMIs, the multiple receptor conformations-based approach (MRC) [34] was implemented, using the ensemble of conformations from the crystal structures (Figure 3a). Thus, the backbone atoms of these conformers were superimposed (entries 1–9, Table 1, excluding the small molecules), with subsequent calculation of the various RMSD values (Table 2). The hMcl-1 backbone conformation was found to be conserved based on RMSD values between 0.52 and 1.13 Å, and these values were considered acceptable given the total number of amino acids (157) in the protein backbones (Table 2). The average RMSD values are also reported. Next, the multiple ligand conformations-based approach (MLC) was applied, which involved docking all SMIs from co-crystals to each of the nine hMcl-1 structures downloaded from the protein databank. The conformations of the docked structures were then compared to that in the X-ray crystal structure and RMSD values calculated (Table 3), and the average RMSD values for the various ligands ranged from 0.879 to 1.390 Å with standard deviations from 0.116 to 0.331 Å. The structure 3WIX was selected as the PDB structure on which to proceed with further docking experiments as it accommodated co-crystal ligands to other PDBs with the lowest RMSD average. Through this approach, we also obtained structural information that helped to identify the active and passive amino acids in the cavity (mapped in Figure 3b) involved in the binding of all nine ligands to the 3WIX PDB (the superimposed ligands are shown in Figure 3c).
3.2. Evaluation of the Precision and Accuracy of Docking Methods
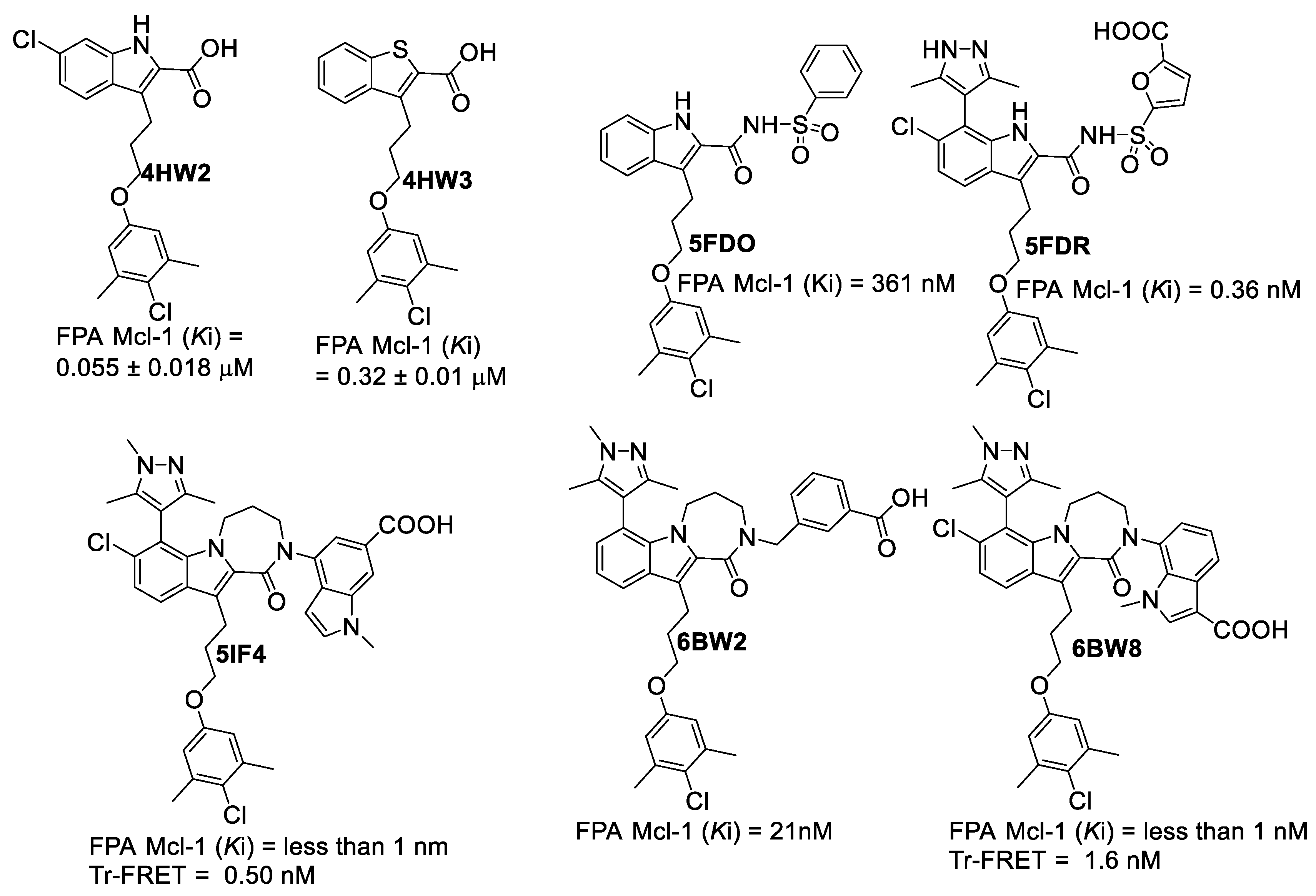
To evaluate the precision and accuracy of docking methods, docking scores of a set of hMcl-1 inhibitors (as shown in Figure 4) were obtained and compared with their corresponding Ki values [35]. These hMcl-1 inhibitors were chosen as they were prepared by the same group (Fesik’s group) and the binding assay conditions were uniform for these inhibitors. Secondly, they have all been reported in co-crystal structures with hMcl-1, providing the experimental basis to validate docking methods. We employed three docking methods, with each ligand being docked into its own crystal structure (self-docking), and the results are summarized in Table 4 [13]. The self-docking [17] generally showed that the use of MOE [36] afforded the lowest RMSD values for its docked poses when compared with the crystal structure-bound ligand conformation in hMcl-1, followed by AutoDock [37] and then VLifeDock [38].
To further assess the accuracy of the docking methods, iterative self-docking was performed using 3WIX [43], where the co-crystal ligand [7-(4-carboxyphenyl)-3-[3-(naphthalen-1-yloxy)propyl]pyrazolo[1,5-a]pyridine-2-carboxylic acid] of 3WIX was self-docked five times into its binding site, and each time it was compared with the orientations of its own conformation in the co-crystal structure by RMSD, as shown in Table 5. The choice of using 3WIX for iterative self-docking was based on its co-crystallized ligand’s higher selectivity (above 60-fold) to Mcl-1 protein compared to Bcl-xL (Mcl-1 IC50 = 0.54 µM; Bcl-xL IC50 > 30 µM). The docking was commenced from random conformations of the ligand, and this, as well as affecting accuracy, can affect precision [44]. The precision of docking is also a concern in drug design, with precision defined by comparing RMSD values of poses obtained after repeated docking of the same ligand. The measurement of reproducibility of accuracy (NRMSE) was determined from the RMSD between the docked pose that is closest to the RMSD to that of the co-crystallized ligand of 3WIX, as shown in Table 5. The “RMSE” value is the difference between the docked pose that has the highest RMSD value and the lowest RMSD value of the docked pose and is thus a measure of the precision of the docking. These values are provided in Table 5, and the poses are shown in Figure 5. The MOE triangle matcher docking method was the most accurate (NRMSE = 86%) compared to AutoDock (genetic algorithm, 55%) and VLifeDock (GRIP method-based docking: 36%). The MOE method was also the most precise as it had the lowest RMSE value. We also observed comparatively fast, accurately bound conformation prediction from VLifeDock when compared with AutoDock. However, AutoDock was more precise in generating docking poses [45] but less accurate in reproducing the co-crystal pose as compared to VLifeDock.
Table 5.
Reproducibility of self-docking experiments, performed five times, on 3WIX and its co-crystal ligand *.
Table 5.
Reproducibility of self-docking experiments, performed five times, on 3WIX and its co-crystal ligand *.
| Docking Phase Trials | RMSD (Å) | ||
|---|---|---|---|
| MOE | AutoDock | VLifeDock | |
| 1 | 0.689 | 1.007 | 0.902 |
| 2 | 0.681 | 0.989 | 0.884 |
| 3 | 0.682 | 0.996 | 0.879 |
| 4 | 0.681 | 0.996 | 0.857 |
| 5 | 0.681 | 0.994 | 0.871 |
| RMDSE (Δ = Å) | 0.008 | 0.018 | 0.025 |
| NRMSE | 85.35 | 55.36 | 36.08 |
* Lower values for RMSE indicate higher precision. Lower values for RMSD indicate higher accuracy. RMSDE: Root-mean-square deviation error = RMSD (maximum value)−RMSD (minimum value), and a lower RMSDE value indicates more reproducibility. Reproducibility was measured in terms of normalized root-mean-square deviation or error (NRMSD or NRMSE: this is a fraction number which measures the degree of reproducibility) [46,47,48]. NRMSE = (average RMSD attained/RMSDE). NRMSE value for any provided method tells the frequency of the accuracy of getting docking conformation, and higher values indicate better accuracy.
3.3. Docking of Naturally Occurring hMcl-1 Inhibitors and Related Compounds
After evaluating the docking methods, screening of seven classes of naturally occurring hMcl-1 inhibitors was carried out. All these ligands had shown affinity in the low micromolar range.
3.3.1. Gymnochrome-F
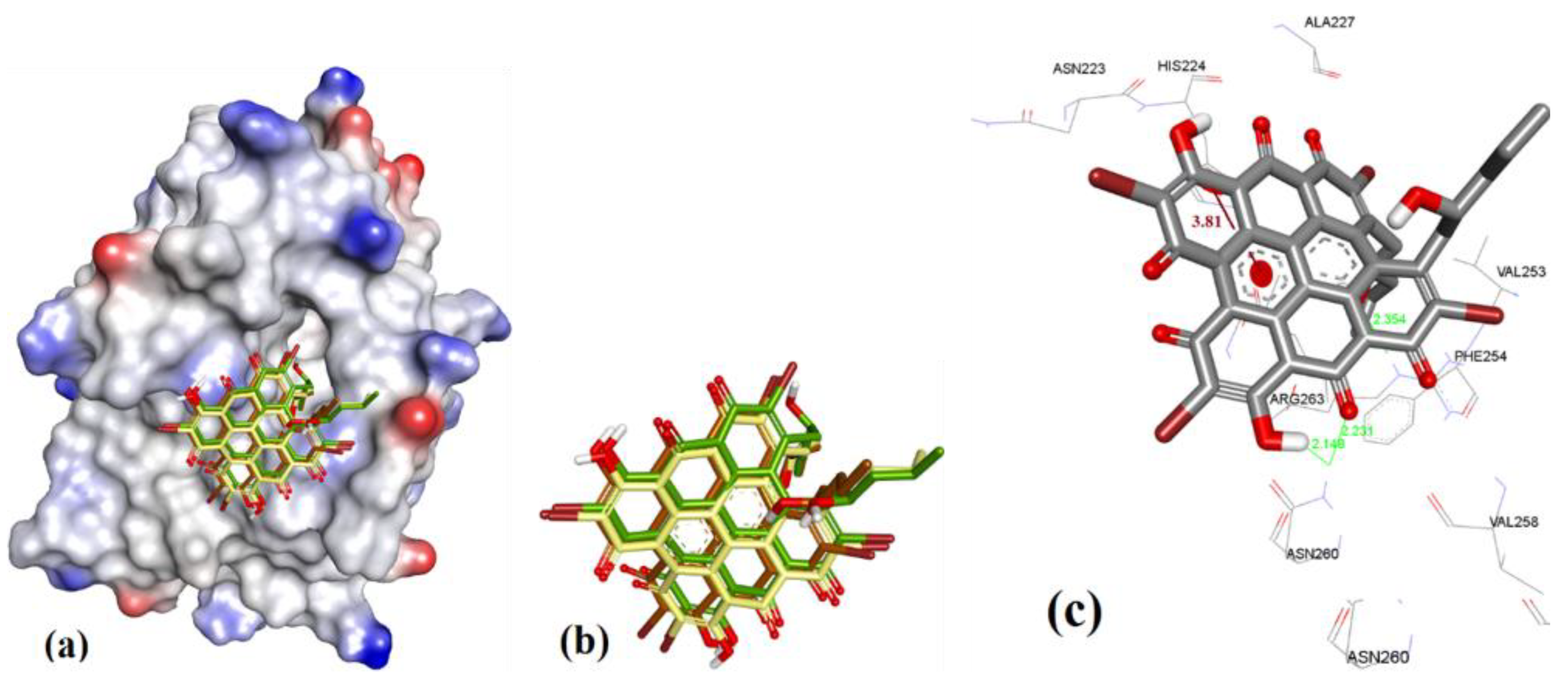
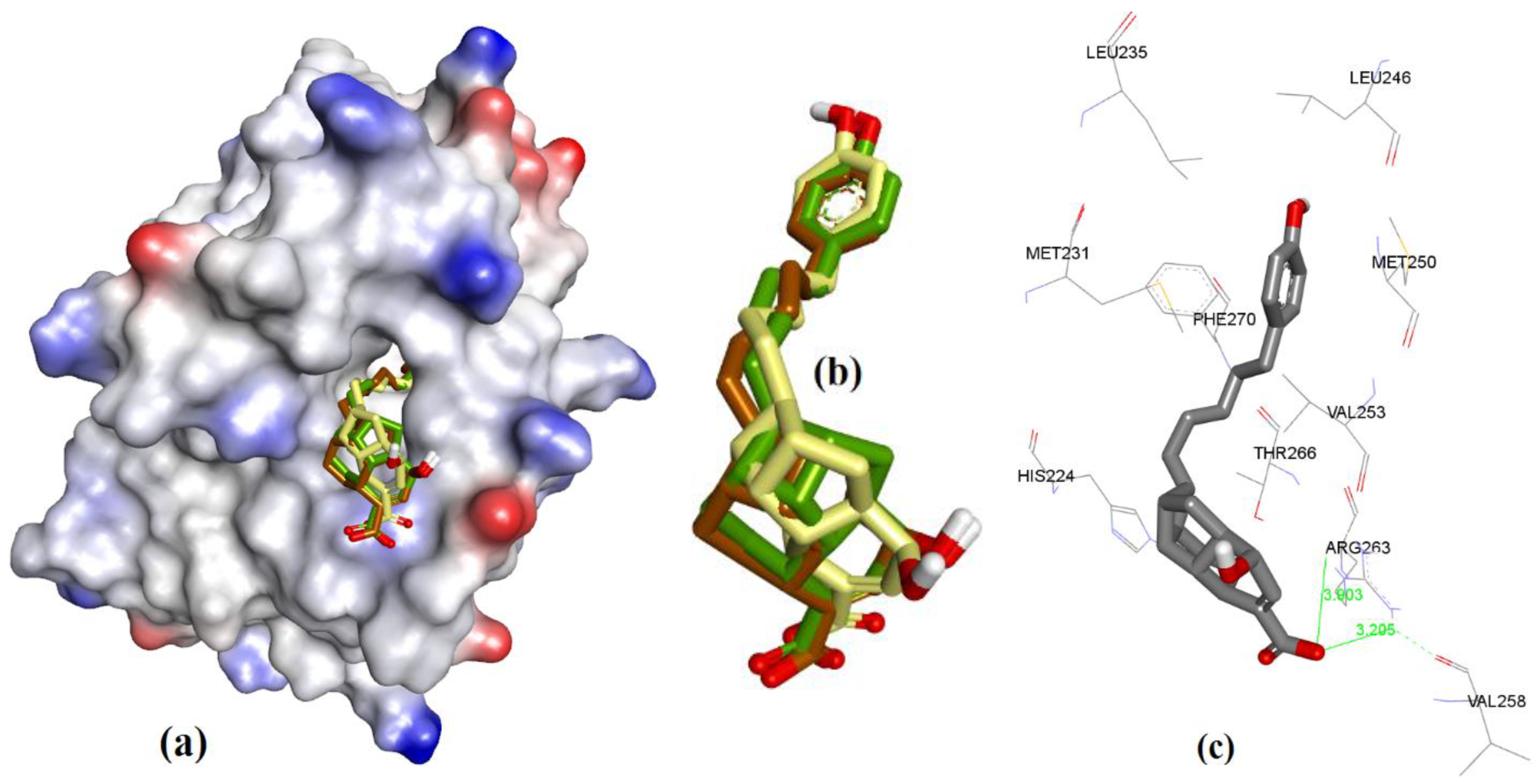
To determine the binding conformation of Gymnochrome-F (1), we employed docking to 3WIX using MOE, AutoDock, and VlifeDock. Similar-pose ensemble clustering [51] confirmed that the attained docking poses from these three different methods were all within RMSD 2 Å. This low RMSD value attained from these methods could be expected due to the high rigidity possessed by the fused heteroaromatic rings in the ligand structure, which limits the number of binding conformations of Gymnochrome F. The docking of Gymnochrome-F showed its main interactions with His224 (π-π interaction, 3.81 Å) and H-bond donor and acceptor interactions with Asn260 (2.14 and2.23 Å), as shown in Figure 6.
3.3.2. Sponge-Derived Oxy-Polyhalogenated Diphenyl Ethers 2
In case of the diphenyl ether derivatives, we observed various bound conformations, all with similar docking energies (within 0.2 kcal/mol). The smaller structure of these natural product derivatives enabled them to adopt a wide number of potential bound conformations in the larger active site of hMcl-1, as shown in Figure 7. The structures were in proximity to Phe270 but with no interaction with, for example, Arg 263, that interacted with anacardic acid’s salicylic acid residue, for example.
3.3.3. Anacardic Acid Derivatives (3a–g)
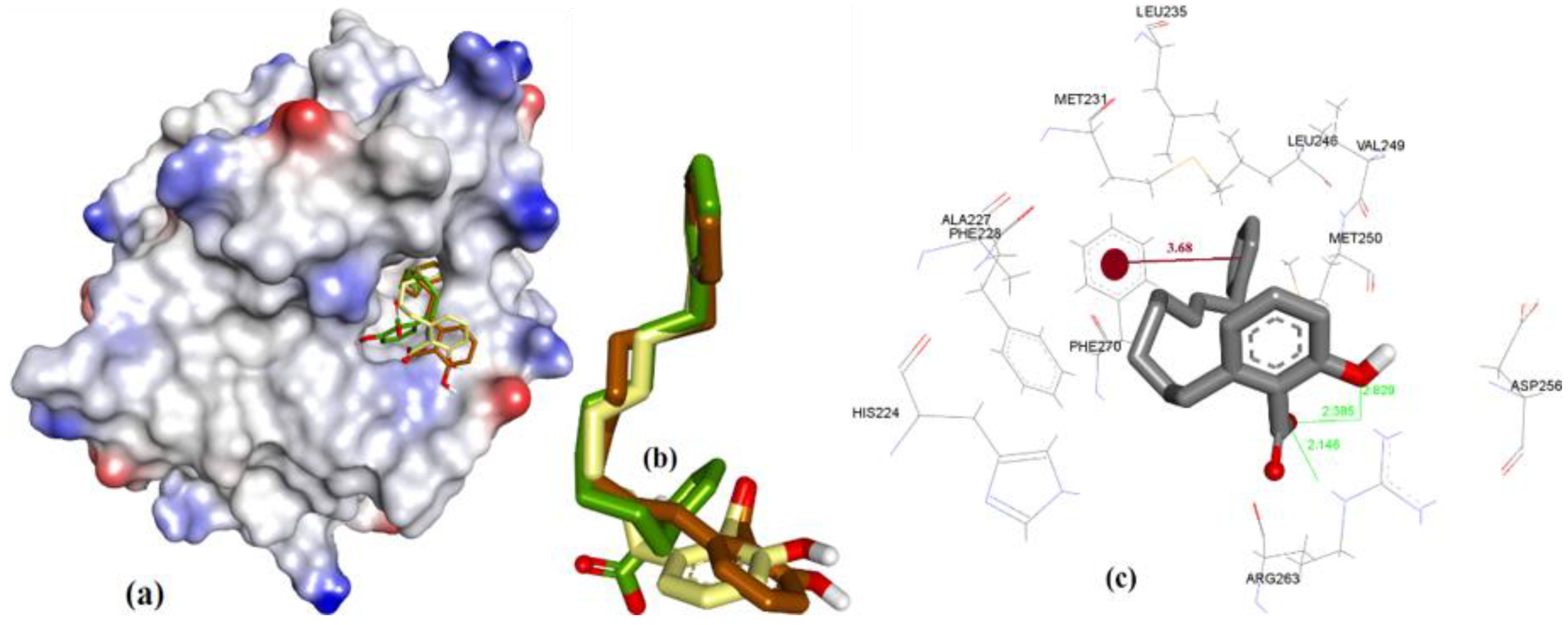
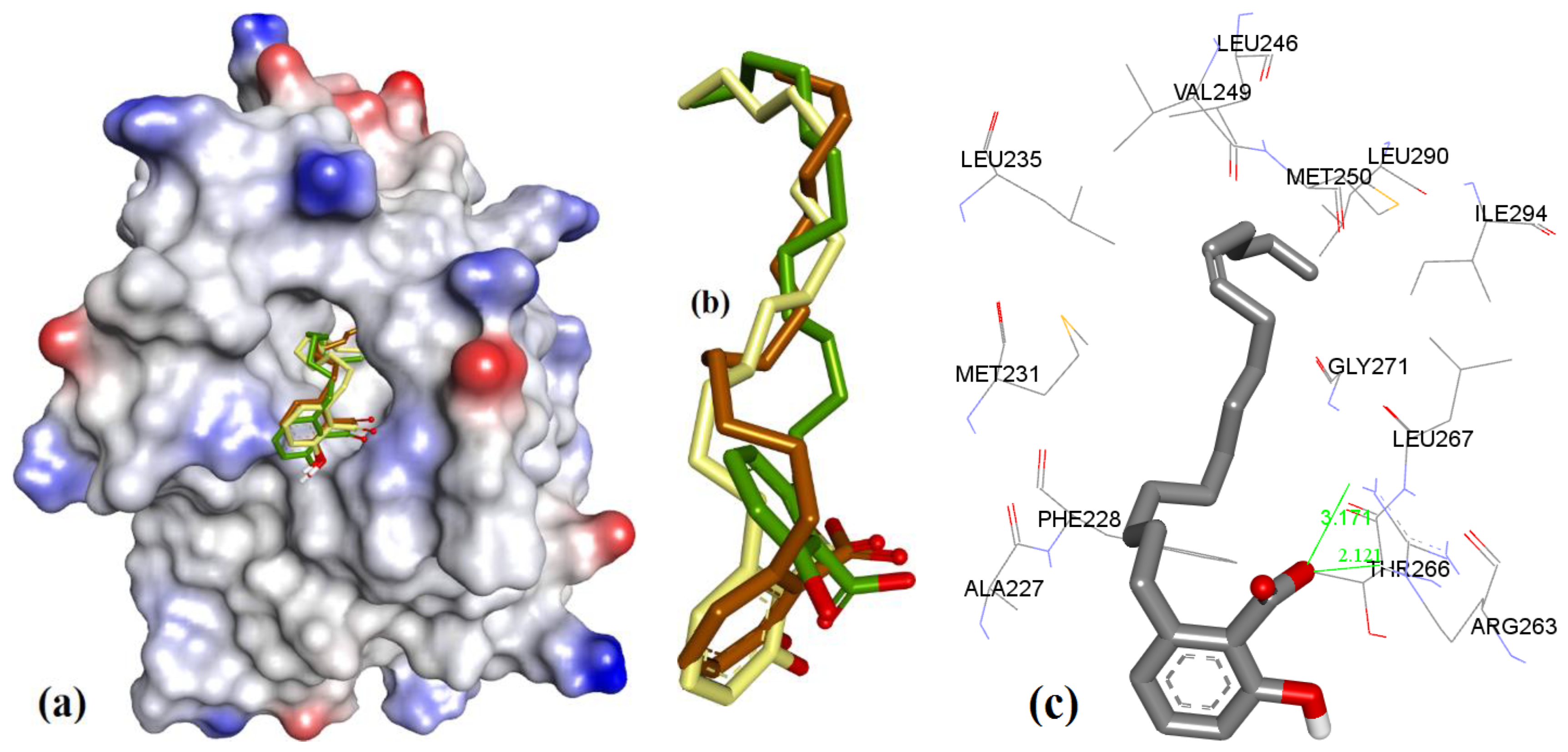
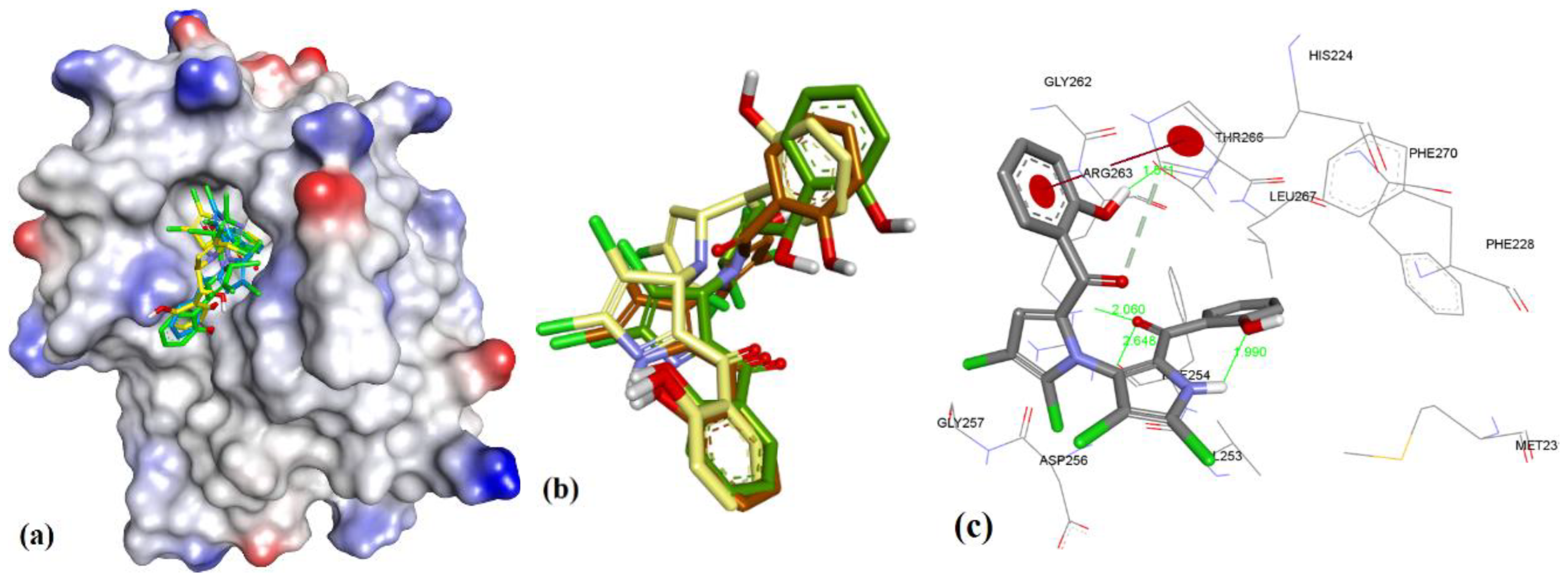
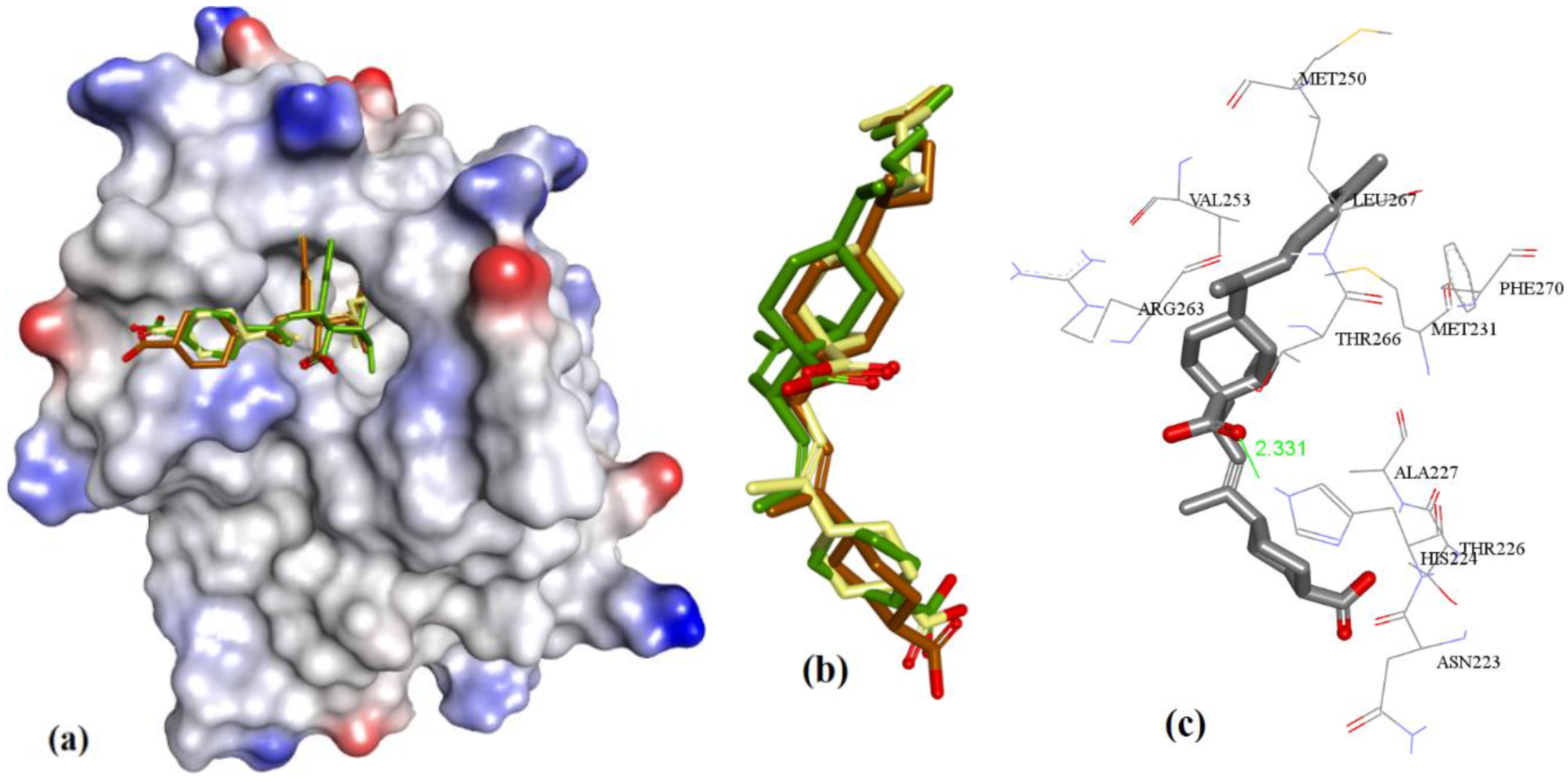
In case of anacardic acids, 3a showed close binding poses from all three docking methods (1.17 Å RMSD). The docking predicted close interactions of polar phenolic head and carboxylic acid with Arg263 (2.36 and 2.21 Å) and an intramolecular interaction (1.81 Å), as shown in Figure 8.
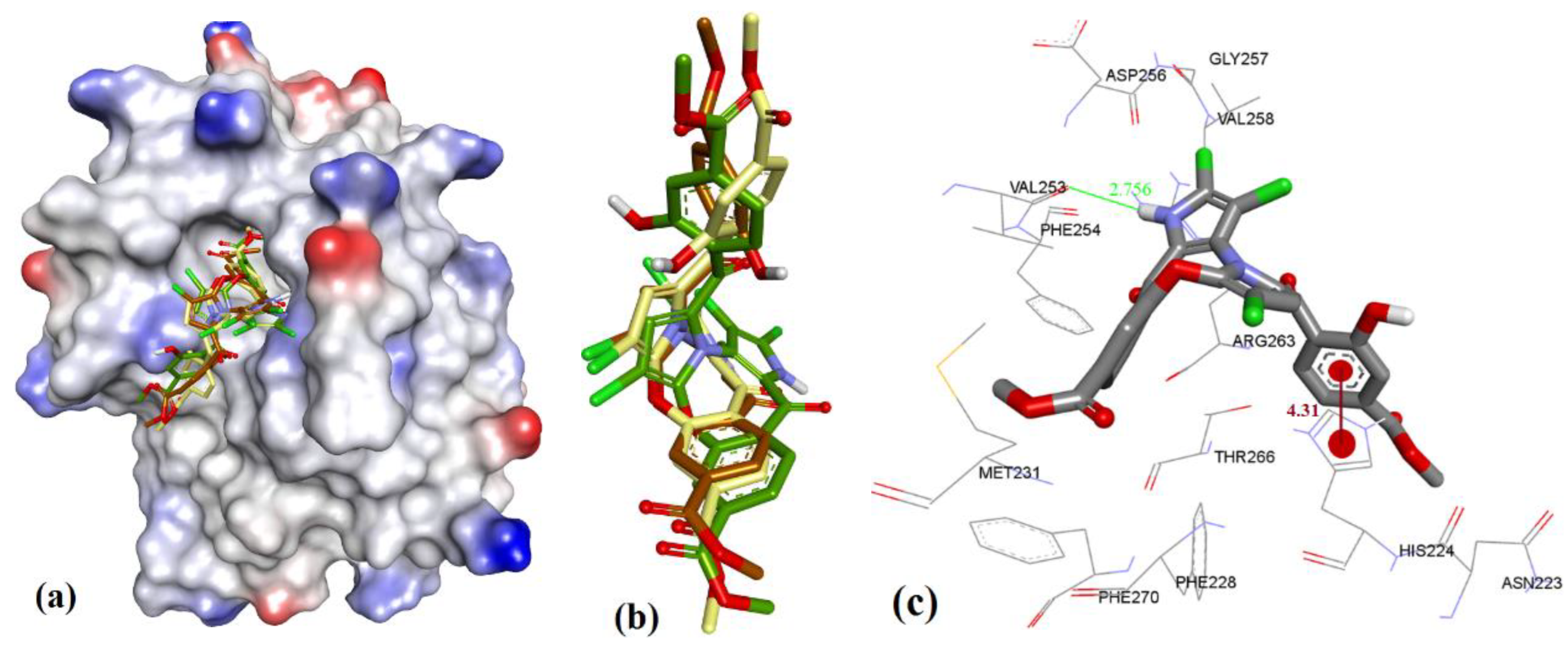
The extra length in the hydrocarbon chain of 3b compared to 3a projected the salicylic acid substructure of 3b more towards the NWGR domain of hMcl-1, where it interacted with Arg263 as a H-bond acceptor (2.0 and 2.14 Å), which also supports its improved hMcl-1 affinity (IC50 = 5.8 µM), compared to that of 3a (IC50 = 17.7 µM), as shown in Figure 9.
In comparison to 3a and 3b, shortening of a hydrocarbon chain and incorporation of a phenyl ring, as in 3c, further improved the affinity for hMcl-1 (IC50 = 0.6 µM), which could be understood with its additional π-π interaction with Phe270, which was missing in previously mentioned anacardic acid derivatives, as shown in Figure 10. Clustering ensemble of docking conformations was found within RMSD of 1.67 Å and showed a π-π interaction with Phe270 (3.68 Å) along with Arg263 (2.14, 2.38, and 2.82 Å).
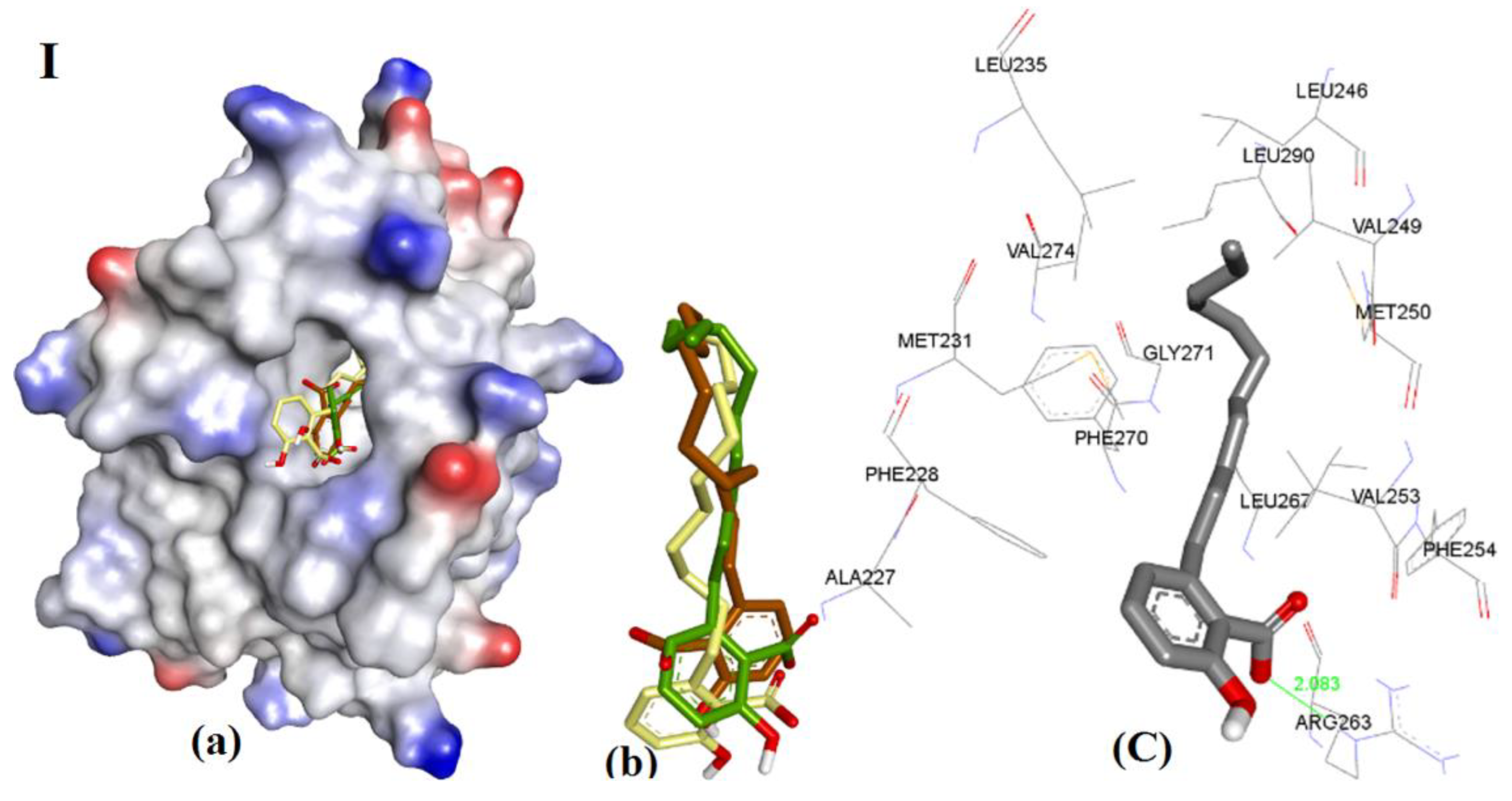
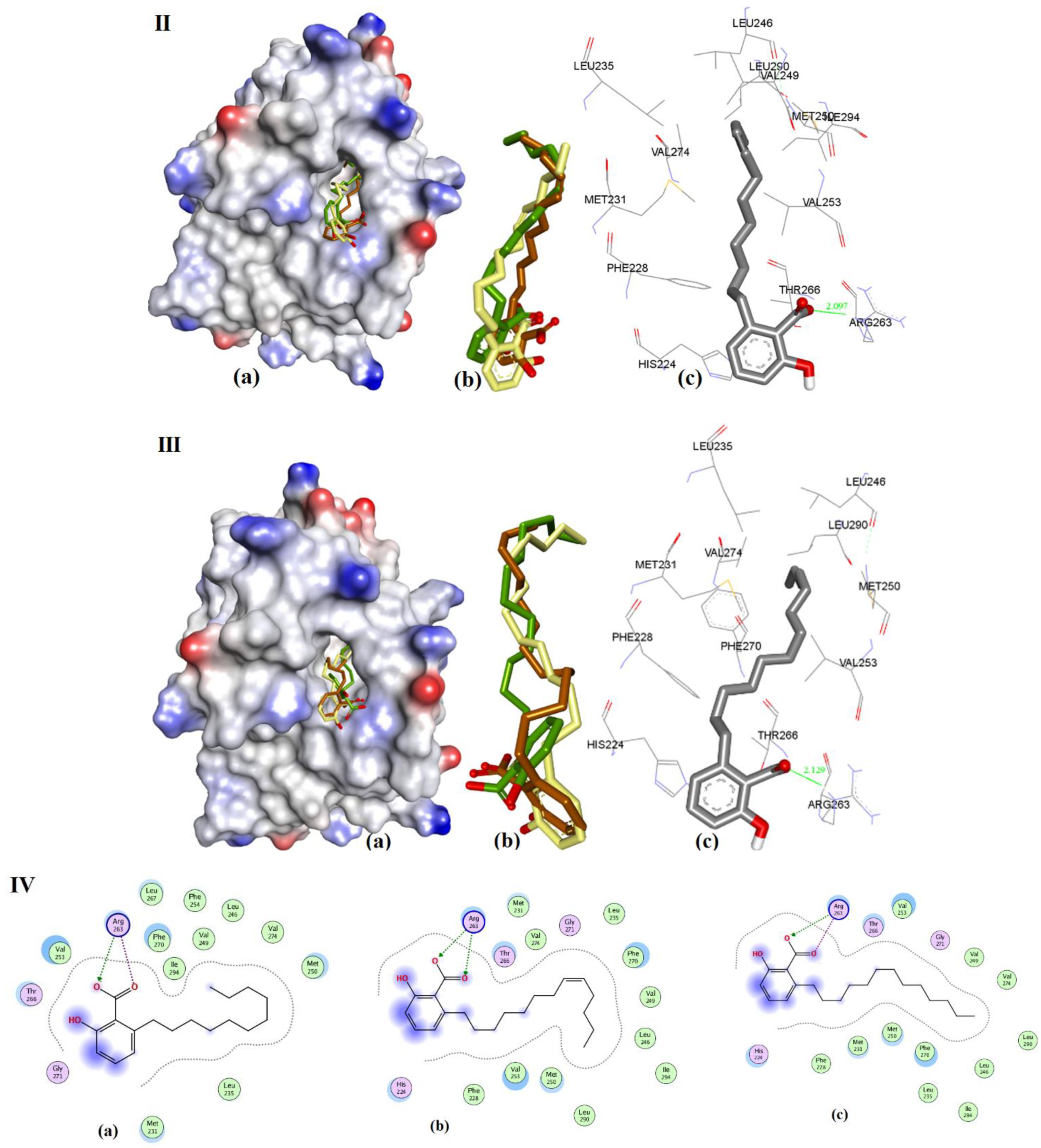
The docking of 3d, 3e, and 3f showed almost identical docking interactions with Arg263, as shown in Figure 11, which is supported by their similar docking energies (−8.34, −8.47, and −8.22 kcal/mol, respectively). These anacardic acids’ binding modes and binding energies are similar, which pointed out that the hydrocarbon chain length of 11–13 carbons plays a less significant role in influencing the binding of these molecules to hMcl-1.
In case of 3g (IC50 = 1.2 µM, ΔG = −8.00 kcal.mol−1), when compared to 3e (IC50 = 0.2 µM, ΔG = −8.47 kcal.mol−1) and 3f (IC50 = 0.2 µM, ΔG = −8.22 kcal.mol−1), it has a longer hydrocarbon chain which is also unsaturated, which might lead to a reduced affinity, which is most likely to be due to its steric effect (as shown in Figure 12).
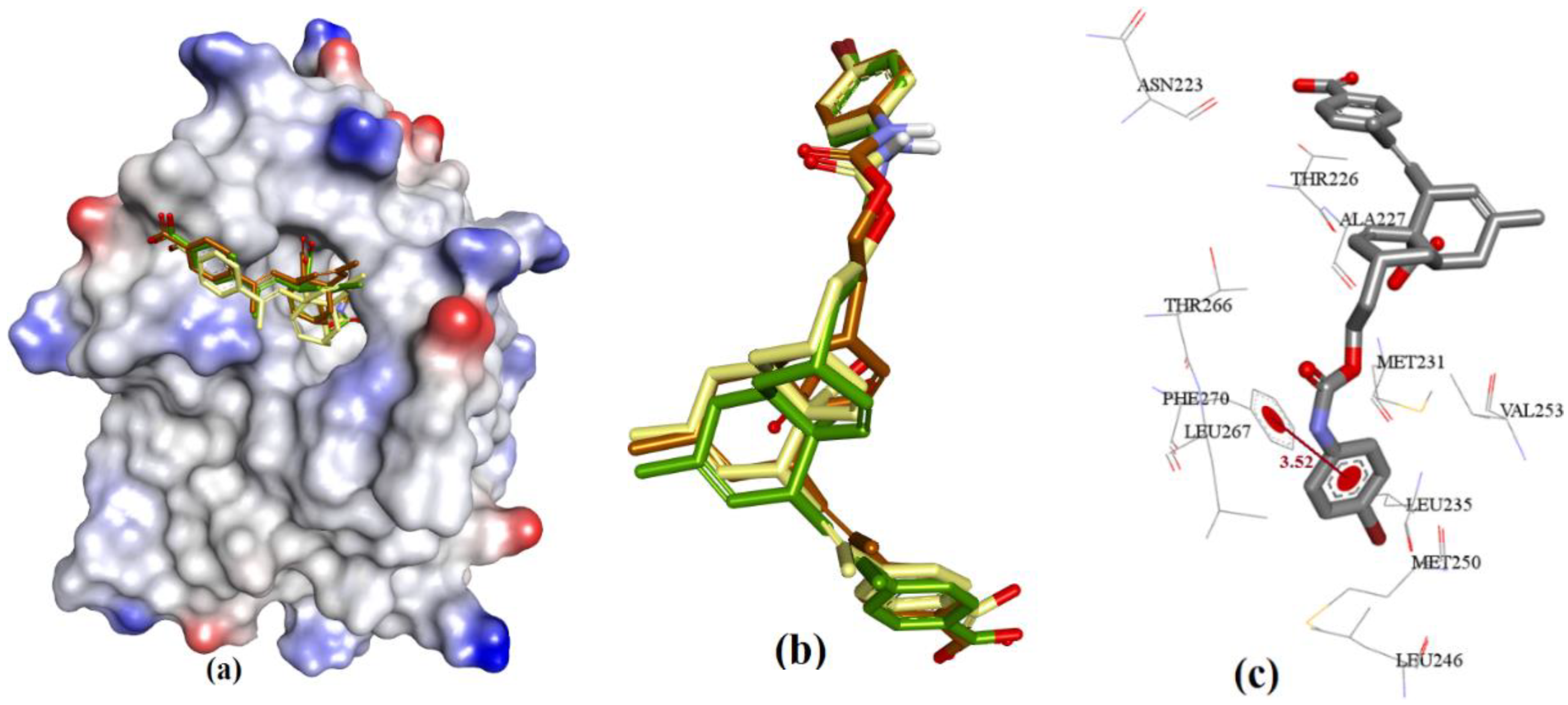
3.3.4. Endiandric Acid Analogues (4a–d)
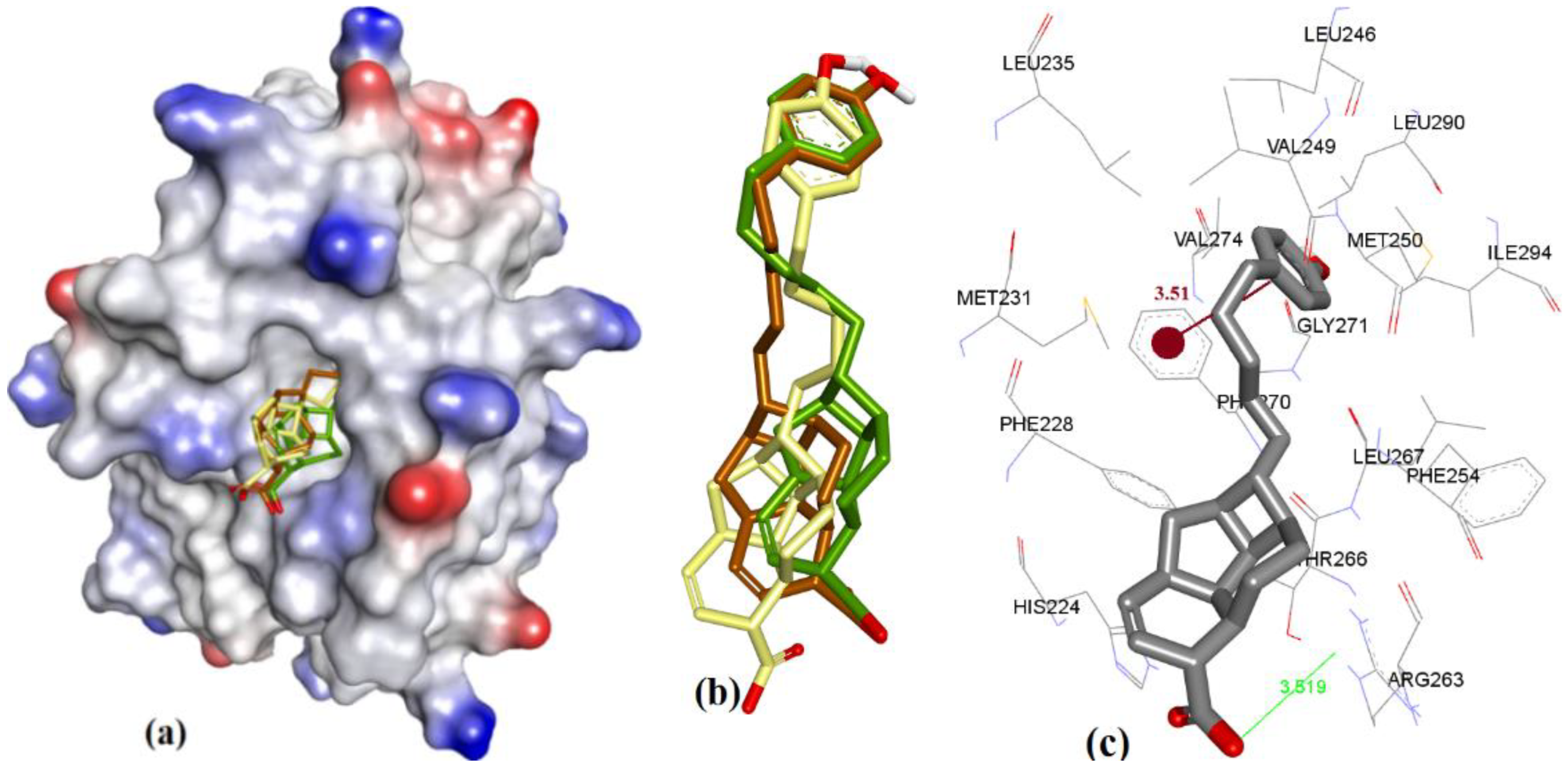
The docking showed that 4a (fused ring with carboxylic acid) binding conformation utilizes similar regions of hMcl-1 as 3c (salicylic acid derivative), which could possibly be based on similarity in their molecular structures (4a and 3c). However, structurally, 4a has only a COOH group in its ring, while 3c has both the acidic phenolic OH and COOH groups, where these polar functional groups were found to consistently interact with Arg263 of hMcl-1. The constrained fused ring in 4a might prevent its adoption of the optimal binding conformation for interaction with Arg263, and therefore could account for the observation of a weak H-bond interaction with Arg263 (3.51 Å) (ΔG = −7.15 kcal.mol−1) when compared to the 3c docking pose (has three H-bond acceptor interactions with Arg263: 2.14, 2.38, and 2.82 Å, ΔG = −7.98 kcal.mol−1) (see Figure 10). Additionally, docking of 4a showed a π-π interaction with Phe270 (3.51 Å), as shown in Figure 13. Similar interactions were also seen with 4b (Ki = 13 µM), where it displayed an improved geometry for H-bonding, as evaluated by its closer proximity to Arg263 (3.02 Å) and its π-π interaction with Phe270 (3.50 Å), as shown in Figure 14. The latter explains the marginal improvement in the in vitro hMcl-1 affinity and docked binding energy, as indicated in Table 7.
The comparatively shorter analog in the endiandric acid class (4c) has an additional phenolic polar head and therefore shows an additional new H-bond donor interaction with the peptide backbone of Leu267, and it has proximity with Phe270 (π-π interaction, 3.85 Å) when compared to longer analogs (4a and 4b), as shown in Figure 15; therefore, this explains why 4c achieved better in vitro hMcl-1 affinity than the latter two, as shown in Table 7.
Ligand 4d has a different stereochemistry in its fused cyclic ring substructure with respect to the other analogs of this series, which leads to its COOH functionality being more inclined towards Arg263 (3.29 and 3.90 Å) as compared to a similar analog (such as 4a), as shown in Figure 16, which may account for its improved binding affinity (5.2 µM) over 4a (14.3 µM).
3.3.5. Marinopyrrole Analogs
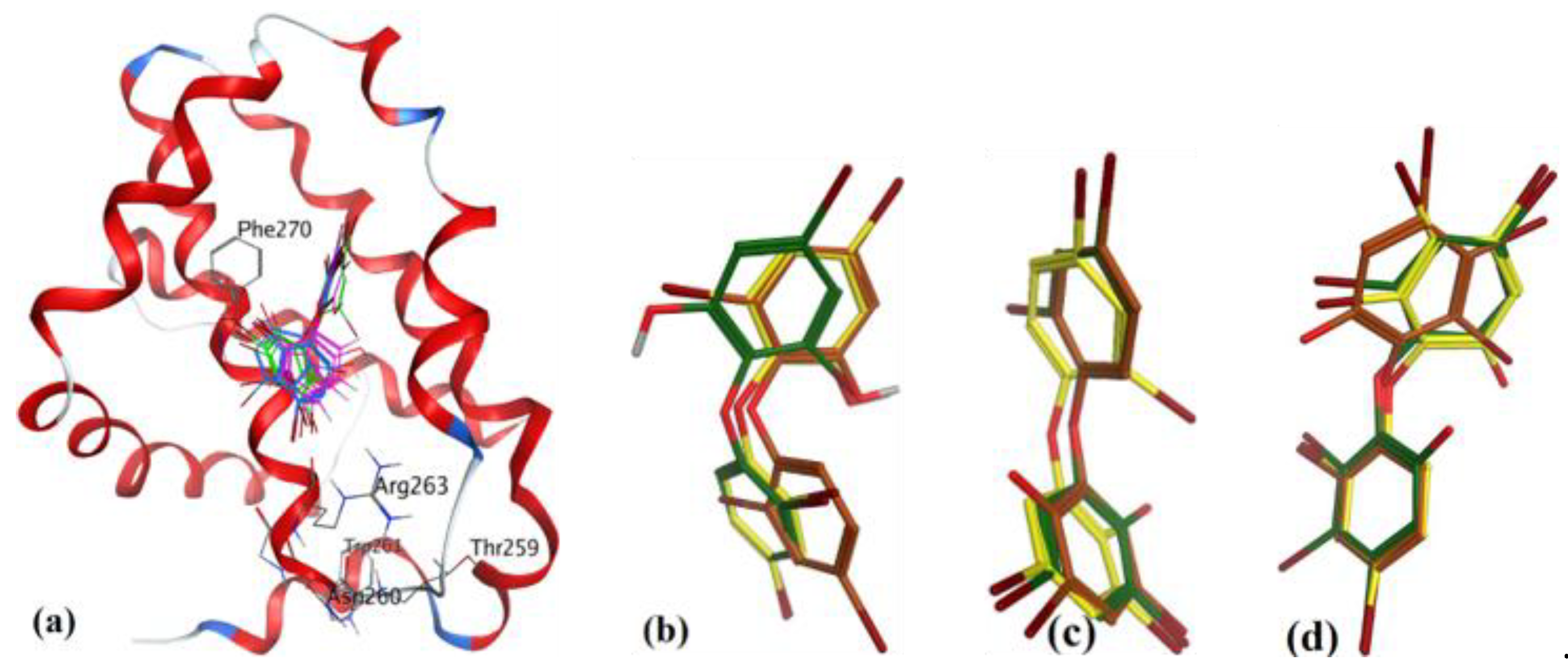
To study the interaction of marinopyrrole-A (maritoclax, 5a), Doi et al. [23] utilized the 1H-15N HSQC spectrum of 15N-labeled mMcl-1 data, and docking with mouse Mcl-1 (mMcl-1). Their study revealed some of the important molecular interactions with mMcl-1 protein: (a) the chloro group in one of the pyrrole rings of maritoclax was facing in towards the binding groove of mMcl-1, (b) the phenol group of the same pyrrole ring had a H-bond with Gly308, (c) the phenol group of another pyrrole group had a H-bond with Thr247, and (d) the carbonyl group had a H-bond with Asn204. Later, we utilized comparative sequence alignment of hMcl-1 with mMcl-1 to identify the corresponding amino acids of hMcl-1 with respect to those of mMcl-1 (as shown Table 6). Furthermore, our study revealed those amino acids of hMcl-1 which had a preference to interact with marinopyrrole-A, with key species-specific differences: (a) identified two point-mutations, G222-D241 and V262-I281, and (b) revealed the hydrophobic character of the cavity of mMcl-1 compared to hMcl-1 (15 out of 25 in mMcl-1 and 13 out of 22 in hMcl-1 are hydrophobic amino acids) (see Table 6).
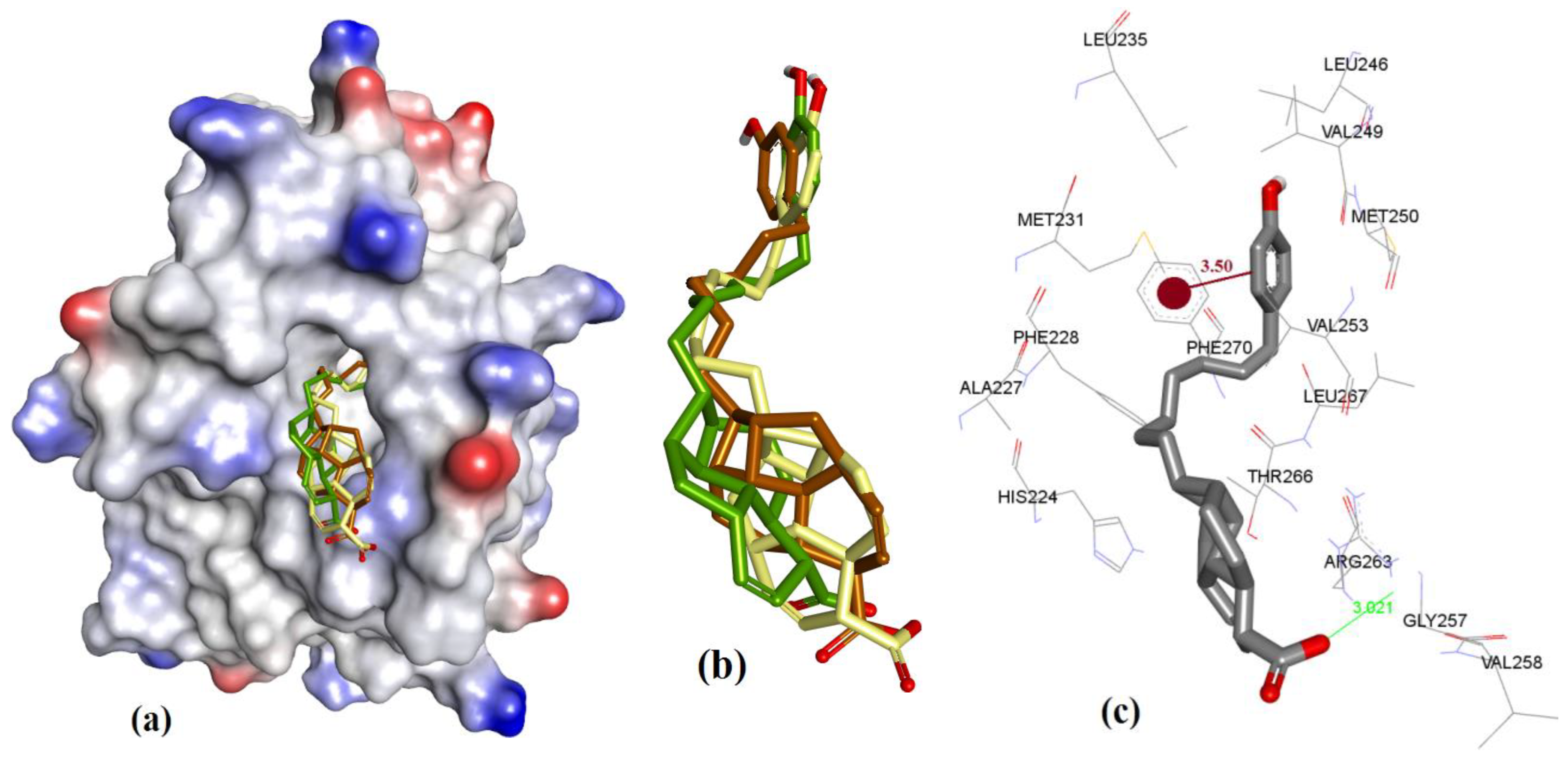
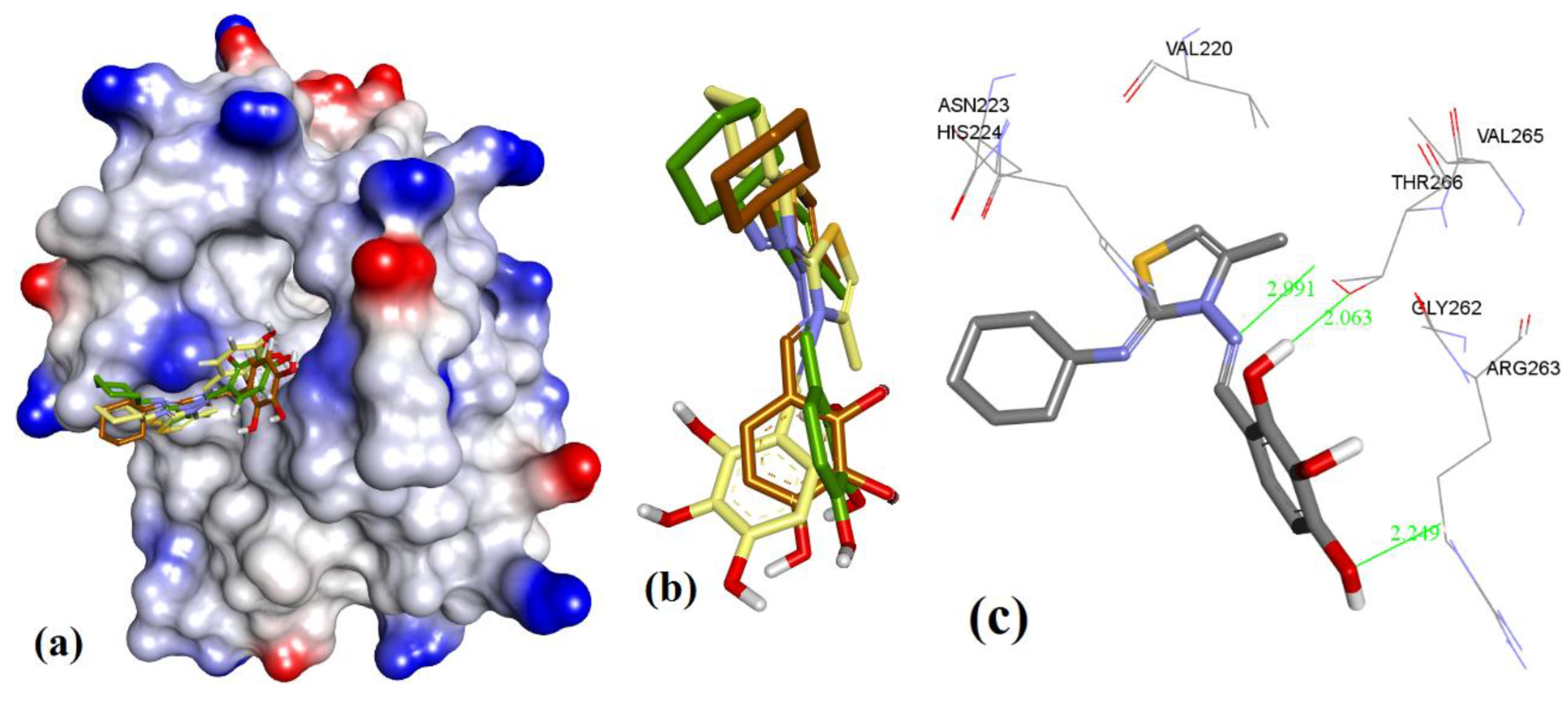
Highly substituted non-symmetric N-heterocycles are difficult to synthesis and, therefore scaffolds derived from nature with such structural specificity are of prime importance [52]. Maritoclax is one such example. Although maritoclax (5a) showed similar bound conformation as that reported for mMcl-1 in our docking studies with hMcl-1, key amino acids residues’ interactions were found missing. To gain a better understanding, the hMcl-1 structure (PDB code: 3WIX) was superimposed with the mMcl-1 (PDB ID: 2JM6), where the RMSD value was found as 6.91 Å. This high deviation provides the basis to explain differences observed in binding conformations of these proteins with maritoclax (5a). In our observation, maritoclax (5a) is a dimer which contains two pyrroles: ring-A and ring-B (Figure 2). The docking studies of maritoclax (5a) with hMcl-1 showed that the phenyl substituent on ring-A displays a π-π interaction with His224 and phenolic OH makes a H-bond donor interaction with Thr266 (1.51 Å) (Figure 17), whereas ring-B showed an intramolecular H-bond interaction between NH-pyrrole and OH-phenyl and the carbonyl functionality showed two H-bond acceptor interactions with the NH-functional group of the guanidine moiety of Arg263 (2.06 and 2.64 Å). It has been observed that the maritoclax (5a) is in close proximity to Asp256, Met231, Phe254, Leu267, His224, and Phe228 found in the cavity, which were also reported by Doi et al. [23].
Later, Cheng et al. chemically modified maritoclax into cyclic derivatives which were evaluated for their Mcl-1 potencies. This work led to cyclic maritoclax (5b) with improved drug-like properties and a more potent hMcl-1 IC50 value (4.3 µM) compared to the parent maritoclax (8.9 µM) [53]. The correlation of improved drug-like properties and selective potency with cyclization of inhibitor is based on the fact that, in drug design, cyclization restricts the conformational flexibility of a ligand to improve its selectivity and potency for a given physiological target. However, docking evaluated herein failed to explain the differences in binding energies between maritoclax and its cyclic derivative (Figure 18), which shows an intrinsic deficiency of docking algorithms to possess physicochemical parameters of ligand metrics.
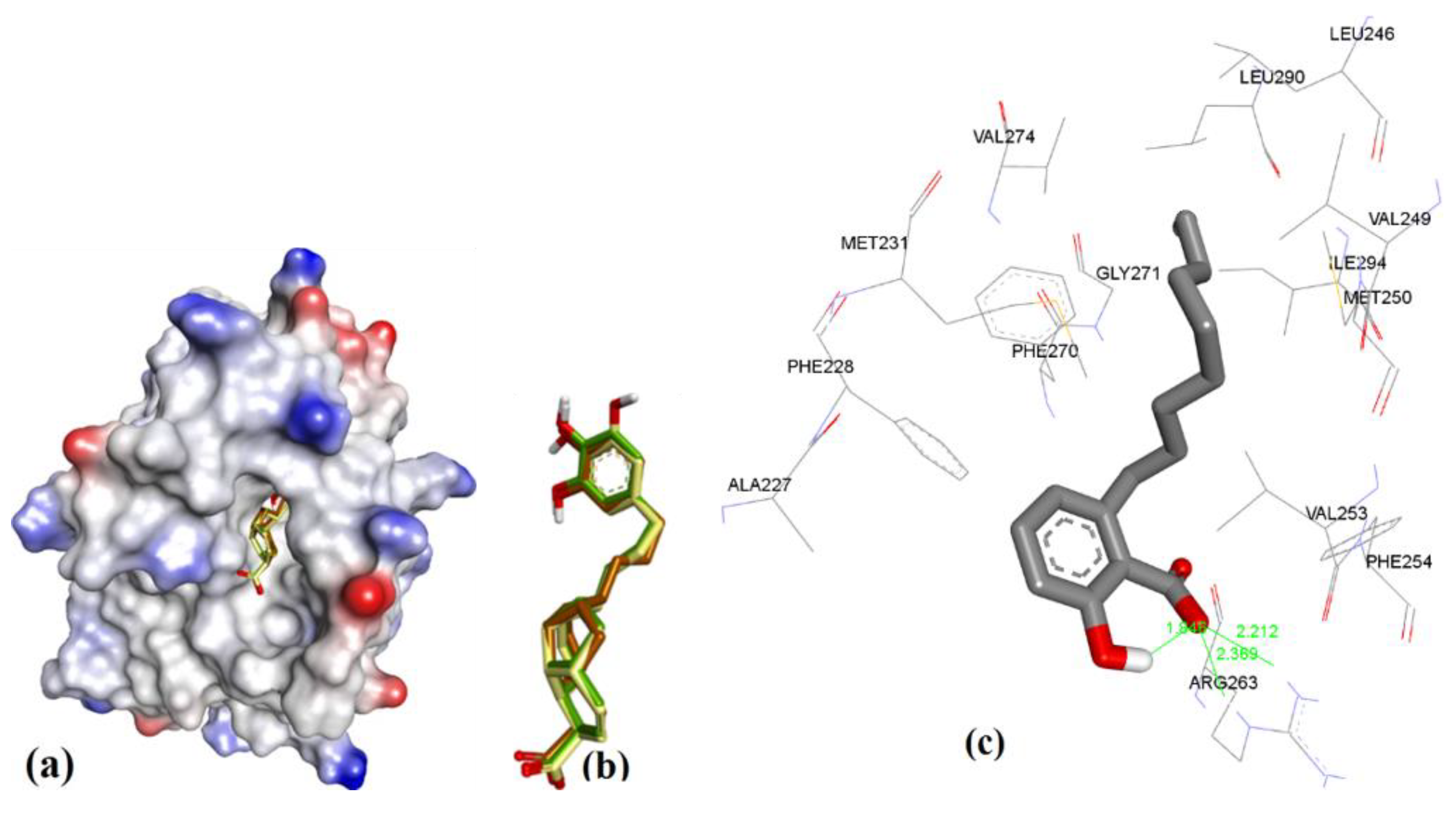
3.3.6. MIM1 (Mcl-1 Inhibitor Molecule 1)
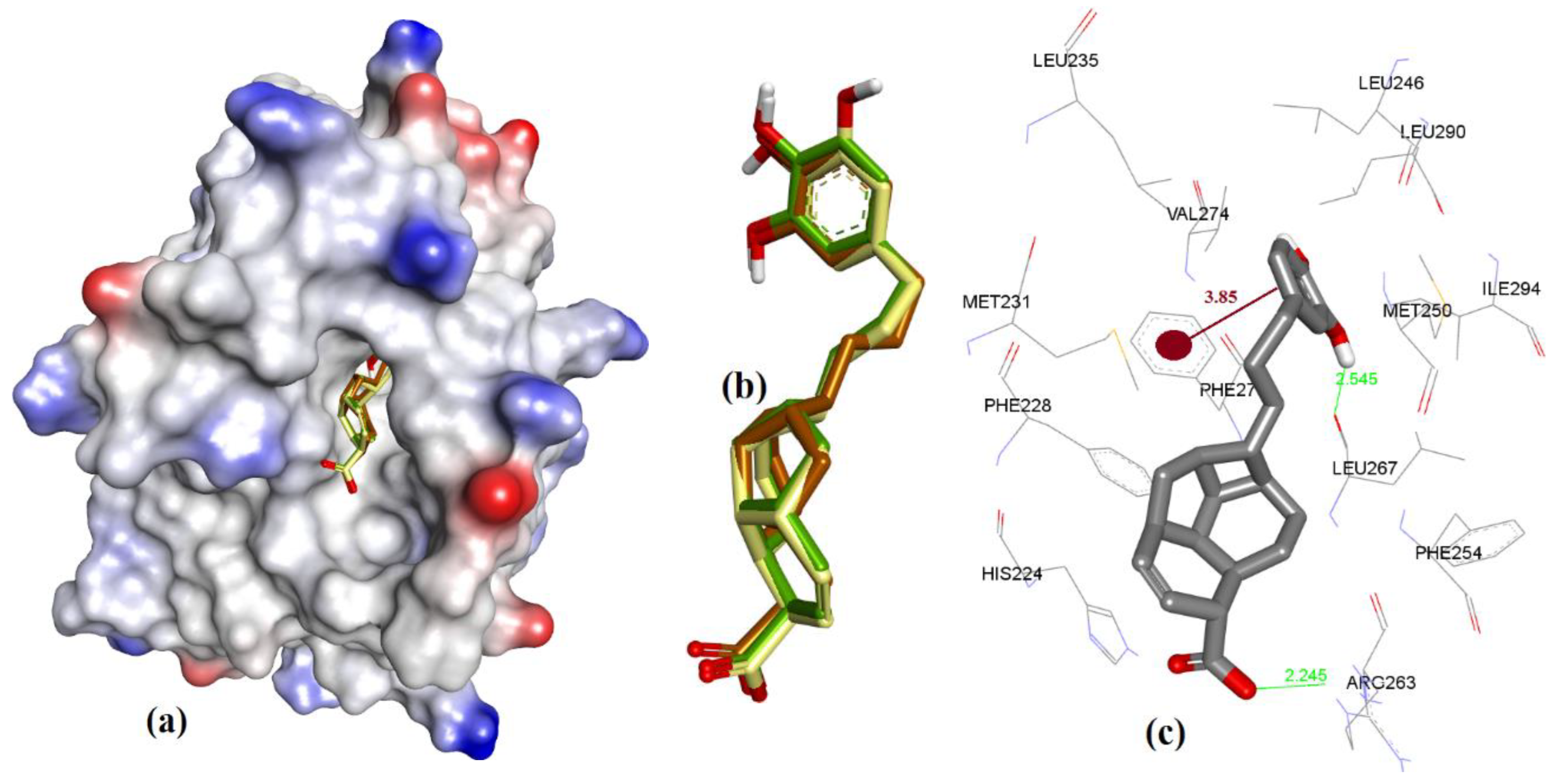
Similar to maritoclax (5), NMR-assisted docking was performed to identify the binding pose of MIM1 (6a) by Cohen et al. [27]. The ensemble clustering of docked poses from MOE, AutoDock, and VlifeDock had RMSD values of 1.07 Å, 1.61 Å, and 2.02 Å, respectively (see Figure 19). The attained docking pose in our study was compared with the previous reported binding of 6a, as was studied with hMcl-1 protein. In a previous report, it had been reported that the 6a had a cyclohexyl group hydrophobic interaction with Val216 and Leu213 [27], which was not seen in our case as both residues were away from cyclohexyl functionality (>5.5 Å), which is too long even for a weak hydrophobic interaction. However, other interactions were found similar to the previous study: (a) imidazole ring of His224 with a CH-π interaction (4.19 Å) with the methylated thiazole, and (b) H-bond donor interaction (2.49 Å) with ortho-substituted –OH of pyrogallol functionality. On the other hand, the guanidine moiety of the side chain of Arg263 and the alcoholic side chain (–OH) of Thr266 showed a H-bond acceptor interaction (2.47 and 2.06 Å) with para-substituted and ortho-substituted –OH of pyrogallol functionality. We also observed that the Asp256 is in close proximity to Arg263 (2.63 Å), and could be better-suited to contribute to binding the pyrogallol group via H-bonding [27].
3.3.7. Cryptosphaerolide
Cryptosphaerolide (7, TR-FRET, Ki = 11.4 µM) contains 2 substructures: cyclic (7a) and acyclic (7b). The absolute stereochemistry was reported for the cyclic portion but not for the acyclic substructure. Therefore, we generated all eight possible stereoisomers and docked them. These stereoisomers were later ranked based on their docking scores (see Table 7). As shown in Figure 20, docking of 7 showed that the cyclic substructure of all eight stereoisomers (coded in different colors in the figure) does not fit into the cavity and is projected out from the binding cavity. This explains why Fenical and co-workers [28] did not observe any hMcl-1 inhibition activity for 7a.
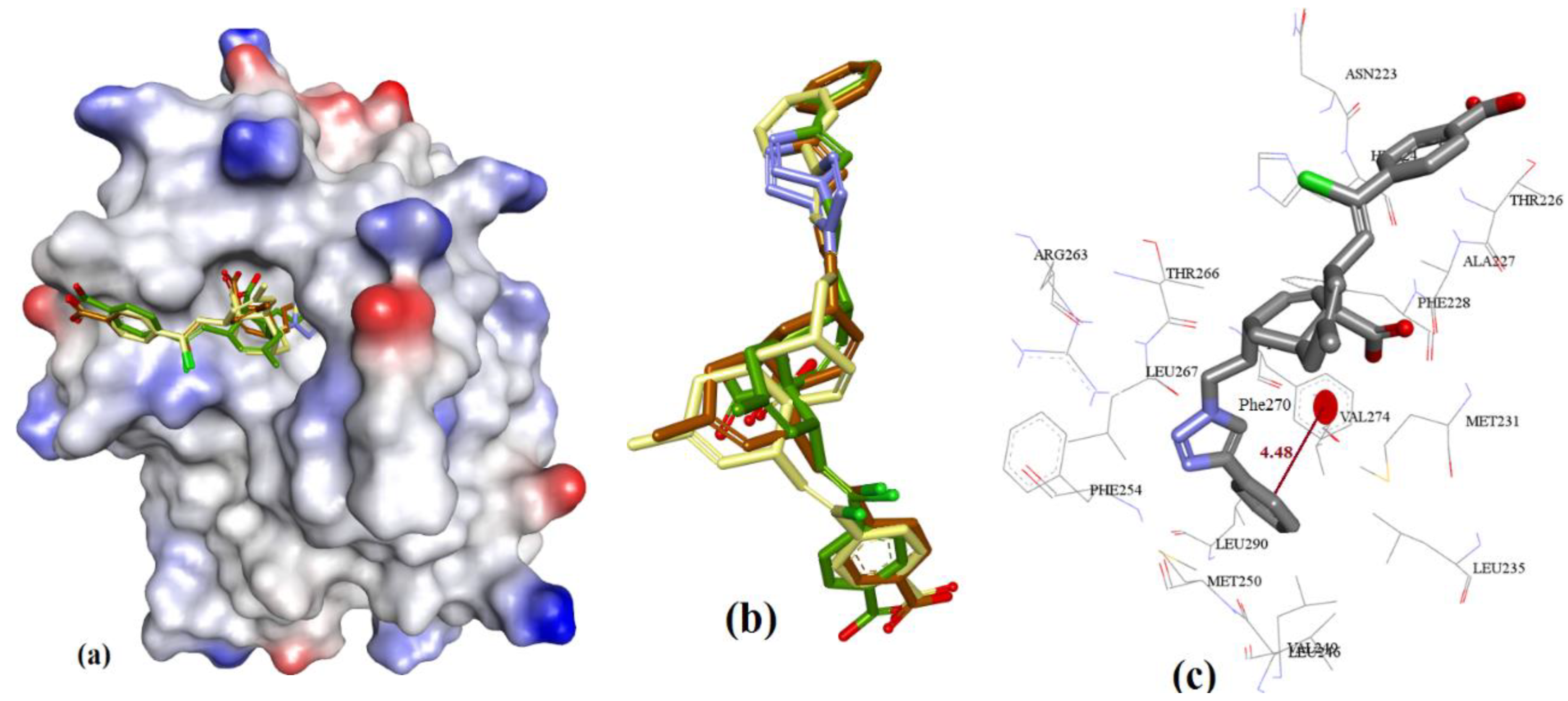
3.3.8. Meiogynin-Derived hMcl-1 Inhibitors (8a–c)
Meiogynin-A (8)-derived compounds have shown a wide range of activity against anti-apoptotic proteins of the Bcl-2 family [29]. Docking predicted similar binding interactions for meiogynins, except in the case of their parent molecule 8 (Ki = 5.2 µM), which showed H-acceptor interactions with the imidazole side chain of His224. Meiogynin derivatives showed π-π interactions with Phe270. Previous docking studies of 8, which showed proximal binding with Arg100 and Tyr195, were also observed in our docking experiments (meiogynin-A was found within 5 Å of these residues). However, the docking generated distinctive poses compared to those previously disclosed [54,55] for meiogynins, as shown in Figure 21, Figure 22, Figure 23 and Figure 24.
4. Conclusions
A number of the small-sized hMcl-1 inhibitors were reported in recent years [12], but the majority of them have failed due to either their off-target binding or therapeutic efficacy, or both, which leads to adverse effects such as drug–drug interactions (e.g., alteration of eicosanoid metabolism [56]) and drug-tissue toxicity (such as an increase in the progression of neurodegeneration of neurons in geriatric patients). As nearly 70% of marketed drugs are derived from natural products, this certainly proves natural products as a good source of exploration of new drug molecules, and a number have been reported as inhibitors of Mcl-1 in recent years. Our studies in this paper focused on gaining insights into the binding mode of the seven natural product class inhibitors of hMcl-1 and a limited number of derivatives that have been synthesized. Selection of a crystal structure from the protein databank was followed by evaluation of three independent scoring functions and three docking methods as to their suitability to study docking to hMcl-1. This was carried out by self-docking using various ligands with experimentally measured hMcl-1 affinities (Ki values) and for which crystal structure data were available showing their binding modes. Based on these three scoring methods, we further disclosed the binding conformations for the most potent naturally derived compounds of these seven classes. However, we also used the clustering of similar docked poses within 2.0 Å and chose the largest cluster to identify the most likely binding conformation. We have also repetitively seen that the NWGR (asparagine-tryptophan-glycine-arginine) domain of hMcl-1 is consistently within 4.5 Å of the conformations of the natural products and closely related derivatives. This domain also has an active role in the binding of BH3 helix of proapoptotic proteins. The attained binding conformations were compared with the experimental hMcl-1 affinity data (in vitro IC50) to validate or evaluate the reliability of the adopted biophysical methodologies [13]. Some of the highlights of this manuscript are regarding how subtle structural changes alter the binding conformation of a molecule as well as respective affinities. The docking with cryptosphaerolide and its isomers, for example, showed that its polycyclic fragment has little if any interaction with the binding groove, whereas docking with maritoclax showed its cyclic framework having more interaction with the groove. Furthermore, various non-covalent interactions, such as hydrogen bonding, Van der Waal’s forces, and electrostatic forces, are commonly observed in the binding of these natural products, which finds key intrinsic molecular features required for a structure to achieve low micromolar Mcl-1 inhibitory activity. Such insights with the methodologies adopted in this manuscript could be helpful and appealing to various types of chemists (such as natural product chemists, chemical biologists, medicinal chemists, or biophysicists) in choosing how to deploy resources when looking to develop natural products in drug discovery programs.
Author Contributions
A.N. carried out all experimental work and data analysis, and contributed to the drafting, revision, and finalizing of the manuscript; P.V.M. supervised the work and contributed to the writing, revision, and finalizing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are grateful to the Irish Research Council (GOIPG/2014/1191 to A.N.) and Science Foundation Ireland (Investigator award 16/IA/4419 to P.V.M.) for the financial support. The APC was covered by A.N.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| hMcl-1 | Human Mcl-1 protein |
| mMcl-1 | Mouse Mcl-1 protein |
| MRC | Multiple receptor conformations |
| MLC | Multiple ligand conformations |
| RMSD | Root-mean-square deviation |
| RMSE | Root-mean-square deviation error |
| NRMSE | Normalized root-mean-square deviation or error |
| MIM1 | Mcl-1 inhibitor molecule 1 |
| SMI | Small-molecule inhibitor |
References
- Denis, C.; Sopková-de Oliveira Santos, J.; Bureau, R.; Voisin-Chiret, A.S. Hot-Spots of Mcl-1 Protein: Miniperspective. J. Med. Chem. 2019, 63, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Michels, J.; Obrist, F.; Vitale, I.; Lissa, D.; Garcia, P.; Behnam-Motlagh, P.; Kohno, K.; Wu, G.S.; Brenner, C.; Castedo, M. MCL-1 dependency of cisplatin-resistant cancer cells. Biochem. Pharmacol. 2014, 92, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.; Al-Harbi, S.; Mazumder, S.; Hill, B.; Smith, M.; Bodo, J.; Hsi, E.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef]
- Williams, M.M.; Cook, R.S. Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015, 6, 3519–3530. [Google Scholar] [CrossRef]
- Wei, A.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef]
- Keuling, A.M.; Felton, K.E.; Parker, A.A.; Akbari, M.; Andrew, S.E.; Tron, V.A. RNA silencing of Mcl-1 enhances ABT-737-mediated apoptosis in melanoma: Role for a caspase-8-dependent pathway. PLoS ONE 2009, 4, e6651. [Google Scholar] [CrossRef]
- Kang, M.H.; Wan, Z.; Kang, Y.H.; Sposto, R.; Reynolds, C.P. Mechanism of synergy of N-(4-hydroxyphenyl) retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J. Natl. Cancer Inst. 2008, 100, 580–595. [Google Scholar] [CrossRef]
- Negi, A.; Voisin-Chiret, A.S. Strategies to Reduce the On-Target Platelet Toxicity of Bcl-xL Inhibitors: PROTACs, SNIPERs and Prodrug-Based Approaches. ChemBioChem 2022, 23, e202100689. [Google Scholar] [CrossRef]
- Negi, A.; Reilly, C.O.; Jarikote, D.V.; Zhou, J.; Murphy, P.V. Multi-targeting protein-protein interaction inhibitors: Evolution of macrocyclic ligands with embedded carbohydrates (MECs) to improve selectivity. Eur. J. Med. Chem. 2019, 176, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.; Murphy, P.V. Development of Mcl-1 inhibitors for cancer therapy. Eur. J. Med. Chem. 2020, 210, 113038. [Google Scholar] [CrossRef] [PubMed]
- Kairys, V.; Baranauskiene, L.; Kazlauskiene, M.; Matulis, D.; Kazlauskas, E. Binding affinity in drug design: Experimental and computational techniques. Expert Opin. Drug Discov. 2019, 14, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Negi, A.; Mirallai, S.I.; Warta, R.; Herold-Mende, C.; Carty, M.P.; Ye, X.-S.; Murphy, P.V. N-Alkyl-1, 5-dideoxy-1, 5-imino-L-fucitols as fucosidase inhibitors: Synthesis, molecular modelling and activity against cancer cell lines. Bioorganic Chem. 2019, 84, 418–433. [Google Scholar] [CrossRef]
- Negi, A.; Zhou, J.; Sweeney, S.; Murphy, P.V. Ligand design for somatostatin receptor isoforms 4 and 5. Eur. J. Med. Chem. 2019, 163, 148–159. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Negi, A.; Bhandari, N.; Shyamlal, B.R.K.; Chaudhary, S. Inverse docking based screening and identification of protein targets for Cassiarin alkaloids against Plasmodium falciparum. Saudi Pharm. J. 2018, 26, 546–567. [Google Scholar] [CrossRef]
- Negi, A.; & Kesari, K.K. Chitosan Nanoparticle Encapsulation of Antibacterial Essential Oils. Micromachines 2022, 13, 1265. [Google Scholar] [CrossRef]
- Kemami Wangun, H.V.; Wood, A.; Fiorilla, C.; Reed, J.K.; McCarthy, P.J.; Wright, A.E. Gymnochromes E and F, cytotoxic phenanthroperylenequinones from a deep-water crinoid, Holopus rangii. J. Nat. Prod. 2010, 73, 712–715. [Google Scholar] [CrossRef]
- Calcul, L.; Chow, R.; Oliver, A.G.; Tenney, K.; White, K.N.; Wood, A.W.; Fiorilla, C.; Crews, P. NMR strategy for unraveling structures of bioactive sponge-derived oxy-polyhalogenated diphenyl ethers. J. Nat. Prod. 2009, 72, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Gény, C.; Rivière, G.; Bignon, J.r.; Birlirakis, N.; Guittet, E.; Awang, K.; Litaudon, M.; Roussi, F.; Dumontet, V. Anacardic acids from Knema hookeriana as modulators of Bcl-xL/Bak and Mcl-1/Bid interactions. J. Nat. Prod. 2016, 79, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Apel, C.c.; Gény, C.; Dumontet, V.; Birlirakis, N.; Roussi, F.; Pham, V.C.; Doan Thi Mai, H.; Nguyen, V.H.; Chau, V.M.; Litaudon, M. Endiandric acid analogues from Beilschmiedia ferruginea as dual inhibitors of Bcl-xL/Bak and Mcl-1/Bid interactions. J. Nat. Prod. 2014, 77, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Li, R.; Sung, S.-S.; Wu, H.; Liu, Y.; Manieri, W.; Krishnegowda, G.; Awwad, A.; Dewey, A.; Liu, X. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Biol. Chem. 2012, 287, 10224–10235. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.C.; Prieto-Davo, A.; Jensen, P.R.; Fenical, W. The marinopyrroles, antibiotics of an unprecedented structure class from a marine Streptomyces sp. Org. Lett. 2008, 10, 629–631. [Google Scholar] [CrossRef]
- Etxebarria, A.; Landeta, O.; Antonsson, B.; Basañez, G. Regulation of antiapoptotic MCL-1 function by gossypol: Mechanistic insights from in vitro reconstituted systems. Biochem. Pharmacol. 2008, 76, 1563–1576. [Google Scholar] [CrossRef]
- Hron, R.; Kuk, M.; Abraham, G.; Wan, P. Ethanol extraction of oil, gossypol and aflatoxin from cottonseed. J. Am. Oil Chem. Soc. 1994, 71, 417–421. [Google Scholar] [CrossRef]
- Cohen, N.A.; Stewart, M.L.; Gavathiotis, E.; Tepper, J.L.; Bruekner, S.R.; Koss, B.; Opferman, J.T.; Walensky, L.D. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem. Biol. 2012, 19, 1175–1186. [Google Scholar] [CrossRef]
- Oh, H.; Jensen, P.R.; Murphy, B.T.; Fiorilla, C.; Sullivan, J.F.; Ramsey, T.; Fenical, W. Cryptosphaerolide, a cytotoxic Mcl-1 inhibitor from a marine-derived ascomycete related to the genus Cryptosphaeria. J. Nat. Prod. 2010, 73, 998–1001. [Google Scholar] [CrossRef]
- Samra, A.A.; Robert, A.; Gov, C.; Favre, L.; Eloy, L.; Jacquet, E.; Bignon, J.; Wiels, J.; Desrat, S.; Roussi, F. Dual inhibitors of the pro-survival proteins Bcl-2 and Mcl-1 derived from natural compound meiogynin A. Eur. J. Med. Chem. 2018, 148, 26–38. [Google Scholar] [CrossRef]
- Litaudon, M.; Bousserouel, H.; Awang, K.; Nosjean, O.; Martin, M.-T.; Dau, M.E.T.H.; Hadi, H.A.; Boutin, J.A.; Sevenet, T.; Gueritte, F. A dimeric sesquiterpenoid from a Malaysian Meiogyne as a new inhibitor of Bcl-xL/BakBH3 domain peptide interaction. J. Nat. Prod. 2009, 72, 480–483. [Google Scholar] [CrossRef]
- Gore, S.; García, E.S.; Hendrickx, P.M.; Gutmanas, A.; Westbrook, J.D.; Yang, H.; Feng, Z.; Baskaran, K.; Berrisford, J.M.; Hudson, B.P. Validation of structures in the Protein Data Bank. Structure 2017, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Joosten, R.P.; Vriend, G. New biological insights from better structure models. J. Mol. Biol. 2016, 428, 1375–1393. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Harris, M.R.; Zou, J.y.; Taylor, T.C.; Wählby, A.; Jones, T.A. The Uppsala electron-density server. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2240–2249. [Google Scholar] [CrossRef]
- Rueda, M.; Bottegoni, G.; Abagyan, R. Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes. J. Chem. Inf. Modeling 2009, 49, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. -Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef]
- Molecular Operating Environment; Software; Chemical Computing Group: Montreal, QC, Canada, 2014.
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905. [Google Scholar] [CrossRef] [PubMed]
- Molecular Design Suite; Software; VLife Sciences Technologies Pvt. Ltd.: Pune, India, 2008.
- Friberg, A.; Vigil, D.; Zhao, B.; Daniels, R.N.; Burke, J.P.; Garcia-Barrantes, P.M.; Camper, D.; Chauder, B.A.; Lee, T.; Olejniczak, E.T. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013, 56, 15–30. [Google Scholar] [CrossRef]
- Pelz, N.F.; Bian, Z.; Zhao, B.; Shaw, S.; Tarr, J.C.; Belmar, J.; Gregg, C.; Camper, D.V.; Goodwin, C.M.; Arnold, A.L. Discovery of 2-indole-acylsulfonamide myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods. J. Med. Chem. 2016, 59, 2054–2066. [Google Scholar] [CrossRef]
- Lee, T.; Bian, Z.; Zhao, B.; Hogdal, L.J.; Sensintaffar, J.L.; Goodwin, C.M.; Belmar, J.; Shaw, S.; Tarr, J.C.; Veerasamy, N. Discovery and biological characterization of potent myeloid cell leukemia-1 inhibitors. FEBS Lett. 2017, 591, 240–251. [Google Scholar] [CrossRef]
- Shaw, S.; Bian, Z.; Zhao, B.; Tarr, J.C.; Veerasamy, N.; Jeon, K.O.; Belmar, J.; Arnold, A.L.; Fogarty, S.A.; Perry, E. Optimization of Potent and Selective Tricyclic Indole Diazepinone Myeloid Cell Leukemia-1 Inhibitors Using Structure-Based Design. J. Med. Chem. 2018, 61, 2410–2421. [Google Scholar] [CrossRef]
- Svensson, F.; Norinder, U.; Bender, A. Improving screening efficiency through iterative screening using docking and conformal prediction. J. Chem. Inf. Modeling 2017, 57, 439–444. [Google Scholar] [CrossRef]
- Ferreira, L.; dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Conformational adaptation in drug-target interactions and residence time. Future Med. Chem. 2011, 3, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Ashtawy, H.M.; Mahapatra, N.R. Task-specific scoring functions for predicting ligand binding poses and affinity and for screening enrichment. J. Chem. Inf. Modeling 2017, 58, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Pons, J.-L.; Bourguet, W.; Labesse, G. Towards accurate high-throughput ligand affinity prediction by exploiting structural ensembles, docking metrics and ligand similarity. bioRxiv 2019. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Yamashita, Y.; Mashimo, T.; Nakamura, H. Prediction of Protein−compound Binding Energies from Known Activity Data: Docking-score-based Method and its Applications. Mol. Inform. 2018, 37, 1700120. [Google Scholar] [CrossRef]
- Greenidge, P.; Kramer, C.; Mozziconacci, J.-C.; Sherman, W. Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. J. Chem. Inf. Modeling 2014, 54, 2697–2717. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- Makeneni, S.; Thieker, D.F.; Woods, R.J. Applying pose clustering and MD simulations to eliminate false positives in molecular docking. J. Chem. Inf. Modeling 2018, 58, 605–614. [Google Scholar] [CrossRef]
- Negi, A.; Mirallai, S.I.; Konda, S.; & Murphy, P.V. An improved method for synthesis of non-symmetric triarylpyridines. Tetrahedron 2022, 121, 132930. [Google Scholar] [CrossRef]
- Cheng, C.; Liu, Y.; Balasis, M.E.; Simmons, N.L.; Li, J.; Song, H.; Pan, L.; Qin, Y.; Nicolaou, K.; Sebti, S.M. Cyclic marinopyrrole derivatives as disruptors of Mcl-1 and Bcl-xL binding to Bim. Mar. Drugs 2014, 12, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Desrat, S.; Remeur, C.; Geny, C.; Riviere, G.; Colas, C.; Dumontet, V.; Birlirakis, N.; Iorga, B.; Roussi, F. From meiogynin A to the synthesis of dual inhibitors of Bcl-xL and Mcl-1 anti-apoptotic proteins. Chem. Commun. 2014, 50, 8593–8596. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Roussi, F.; Iorga, B. Focused ligand libraries as tools for in silico design of anti-apoptotic proteins inhibitors. Chem. Life Sci. 2011, 41–46. [Google Scholar]
- Zhang, T.; Walensky, L.D.; Saghatelian, A. A nonapoptotic role for BAX and BAK in eicosanoid metabolism. ACS Chem. Biol. 2015, 10, 1398–1403. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The crystal structure of hMcl-1 protein bound with a BH3 α-helix peptide provides information on the binding event (PDB id: 2PQK). The structure shows 4 pockets (P1, P2, P3, P4) in the active site, with each hydrophobic amino acid side chain of the BH3 helix (H1, H2, H3, H4) utilizing these pockets.
Figure 1.
The crystal structure of hMcl-1 protein bound with a BH3 α-helix peptide provides information on the binding event (PDB id: 2PQK). The structure shows 4 pockets (P1, P2, P3, P4) in the active site, with each hydrophobic amino acid side chain of the BH3 helix (H1, H2, H3, H4) utilizing these pockets.

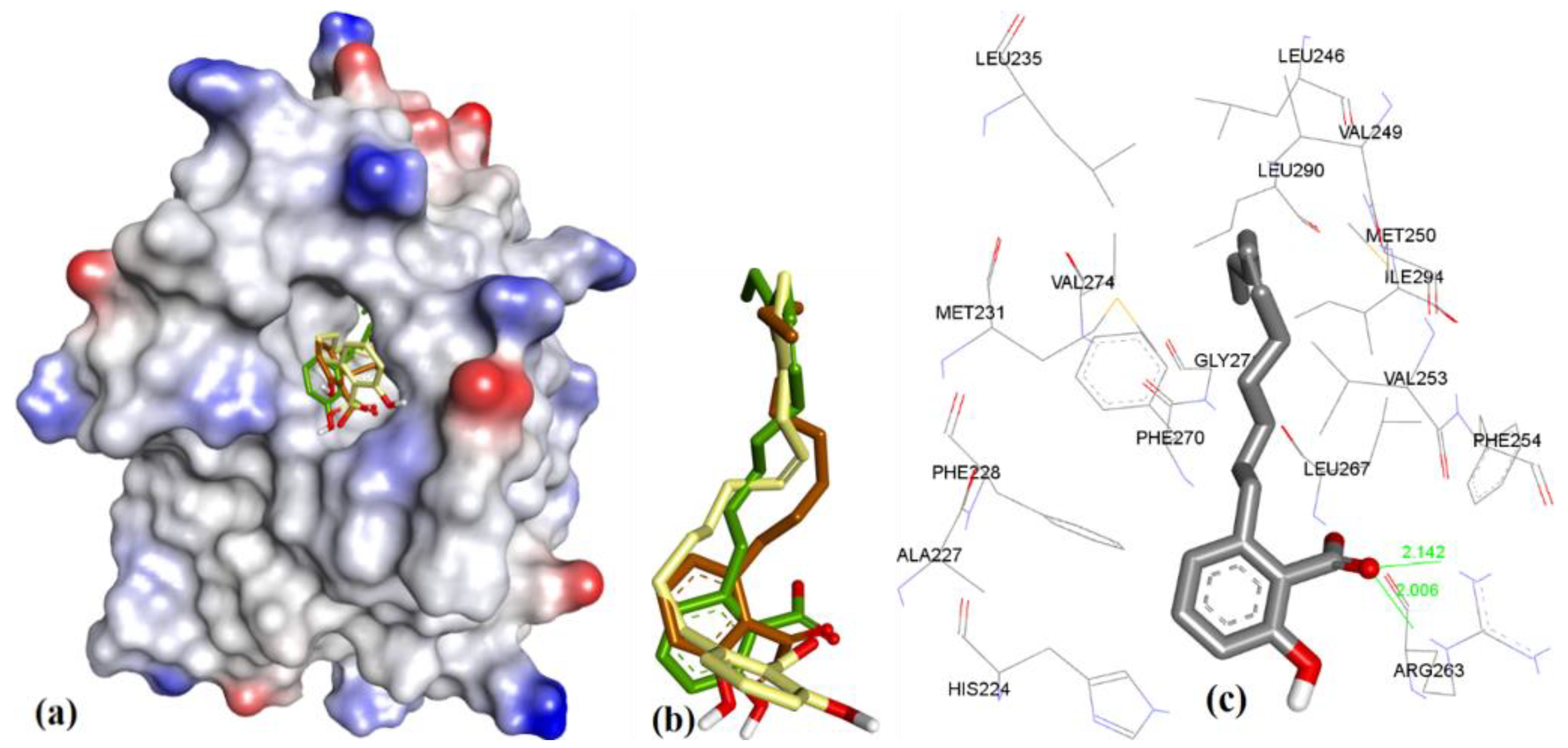
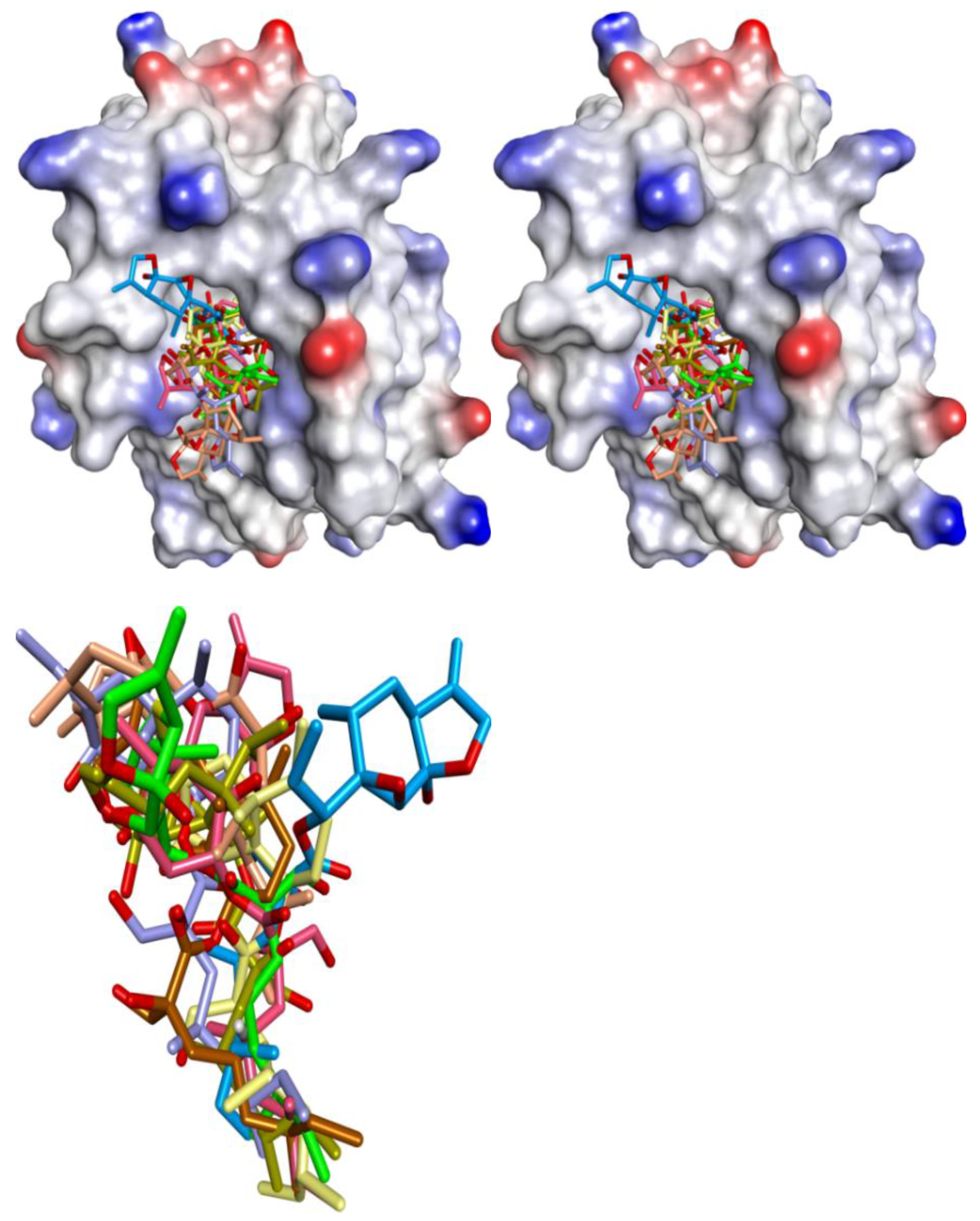
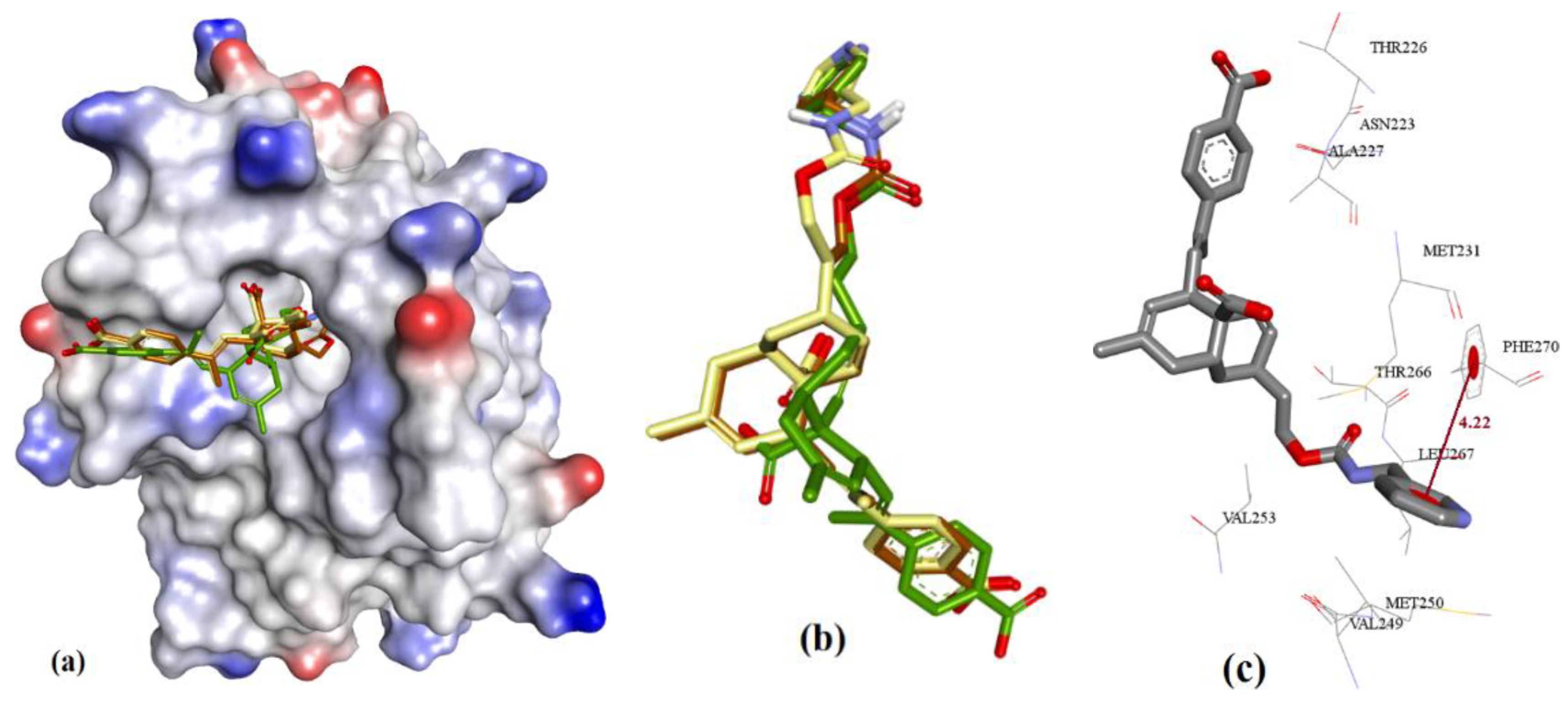
Figure 3.
(a) Superposition of conformers of hMcl-1 found in crystal structures with small-molecule inhibitors. (b) H-donor and acceptor cavity in hMcl-1 identified by MLC (docking of ligands to 3WIX). (c) Superimposed binding poses of ligands obtained from co-crystal structures docked to 3WIX.
Figure 3.
(a) Superposition of conformers of hMcl-1 found in crystal structures with small-molecule inhibitors. (b) H-donor and acceptor cavity in hMcl-1 identified by MLC (docking of ligands to 3WIX). (c) Superimposed binding poses of ligands obtained from co-crystal structures docked to 3WIX.

Figure 4.
Fesik’s group’s ligands, which were obtained from co-crystal structures with hMcl-1 (PDB: 4HW2 and 4HW3 [39], PDB: 5FDO and 5FDR [40], PDB: 5IF4 [41], PDB: 6BW2 and 6BW8 [42]).

Figure 5.
Self-docking of 3WIX PDB to its ligand using three methods (together), along with similar-pose ensemble clustering [49]. (a) The left-hand side (LHS) figure shows the ionized solvent-exposed surface mapping of hMcl-1, where ligands were fitted to the binding site. (b) The right-hand side (RHS) shows the similar-pose clustering [50,51]: showing the following overlayed structures—co-crystal-bound conformation of the ligand (in blue), MOE-based docking conformation (in brown), AutoDock-based docking conformation (in green), and GRIP-based docking conformation (in yellow).
Figure 5.
Self-docking of 3WIX PDB to its ligand using three methods (together), along with similar-pose ensemble clustering [49]. (a) The left-hand side (LHS) figure shows the ionized solvent-exposed surface mapping of hMcl-1, where ligands were fitted to the binding site. (b) The right-hand side (RHS) shows the similar-pose clustering [50,51]: showing the following overlayed structures—co-crystal-bound conformation of the ligand (in blue), MOE-based docking conformation (in brown), AutoDock-based docking conformation (in green), and GRIP-based docking conformation (in yellow).

Figure 6.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of Gymnochrome-F from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity, and all are within 1.08 Å RMSD. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of Gymnochrome-F with hMcl-1.
Figure 6.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of Gymnochrome-F from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity, and all are within 1.08 Å RMSD. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of Gymnochrome-F with hMcl-1.

Figure 7.
(a) Binding modes of diphenyl derivatives: 2a (magenta), 2b (Green), and 2c (Blue). (b) Docked poses of 2a from MOE (in brown), AutoDock (in Green), and VlifeDock (in yellow). (c) Docked poses of 2b from MOE (in brown), AutoDock (in Green), and VlifeDock (in yellow). (d) Docked poses of 2c from MOE (in brown), AutoDock (in Green), and VlifeDock (in yellow).
Figure 7.
(a) Binding modes of diphenyl derivatives: 2a (magenta), 2b (Green), and 2c (Blue). (b) Docked poses of 2a from MOE (in brown), AutoDock (in Green), and VlifeDock (in yellow). (c) Docked poses of 2b from MOE (in brown), AutoDock (in Green), and VlifeDock (in yellow). (d) Docked poses of 2c from MOE (in brown), AutoDock (in Green), and VlifeDock (in yellow).

Figure 8.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 3a from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3a with hMcl-1.
Figure 8.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 3a from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3a with hMcl-1.

Figure 9.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 3b from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3b.
Figure 9.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 3b from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3b.

Figure 10.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 3c from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3c.
Figure 10.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 3c from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3c.

Figure 11.
Representation of modeling for (I) 3d, (II) 3e, and (III) 3f: (a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3d/e/f. (IV) 2D interaction of 3d (LHS), 3e (Middle), and 3f (RHS).
Figure 11.
Representation of modeling for (I) 3d, (II) 3e, and (III) 3f: (a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3d/e/f. (IV) 2D interaction of 3d (LHS), 3e (Middle), and 3f (RHS).


Figure 12.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 3g from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3g.
Figure 12.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 3g from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 3g.

Figure 13.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 4a from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 4a.
Figure 13.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 4a from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 4a.

Figure 14.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 4b from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 4b.
Figure 14.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 4b from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 4b.

Figure 15.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 4c from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 4c.
Figure 15.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 4c from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 4c.

Figure 16.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 4d from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 4d.
Figure 16.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 4d from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 4d.

Figure 17.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of marinopyrrole from MOE (in blue), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods, MOE (in brown), AutoDock (in green), and VlifeDock (in yellow). (c) Binding mode of marinopyrrole.
Figure 17.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of marinopyrrole from MOE (in blue), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods, MOE (in brown), AutoDock (in green), and VlifeDock (in yellow). (c) Binding mode of marinopyrrole.

Figure 18.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of cyclic marinopyrrole (5b) from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of cyclic marinopyrrole (5b).
Figure 18.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of cyclic marinopyrrole (5b) from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of cyclic marinopyrrole (5b).

Figure 19.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of MIM1 from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses attained from the three different docking methods. (c) Binding mode of MIM1.
Figure 19.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of MIM1 from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses attained from the three different docking methods. (c) Binding mode of MIM1.

Figure 20.
Eight stereoisomeric analogs of cryptosphaerolide can be possible based on specific stereochemistry at three chiral centers, therefore all eight isomers were docked. The cyclic substructures of all 8 isomers were not involved in binding (as seen in the binding mode in the figure for any of the eight stereoisomers (b: on the left-hand side) in the figure with hMcl-1).
Figure 20.
Eight stereoisomeric analogs of cryptosphaerolide can be possible based on specific stereochemistry at three chiral centers, therefore all eight isomers were docked. The cyclic substructures of all 8 isomers were not involved in binding (as seen in the binding mode in the figure for any of the eight stereoisomers (b: on the left-hand side) in the figure with hMcl-1).

Figure 21.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 8 from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 8.
Figure 21.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 8 from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 8.

Figure 22.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 8a from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 8a.
Figure 22.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 8a from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 8a.

Figure 23.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 8b from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 8b.
Figure 23.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 8b from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 8b.

Figure 24.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 8c from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 8c.
Figure 24.
(a) The ionized solvent-exposed surface mapping of hMcl-1 along with the docked poses of 8c from MOE (in brown), AutoDock (in green), and VlifeDock (in yellow) show their fitting to the cavity. (b) Similar-pose ensemble clustering of all three poses was attained from the three different docking methods. (c) Binding mode of 8c.

Table 2.
MRC approach: superposition of backbone of multiple hMcl-1 from various crystal structures and their calculated RMSD values (Å).
Table 2.
MRC approach: superposition of backbone of multiple hMcl-1 from various crystal structures and their calculated RMSD values (Å).
| Proteins | 4WMR | 4ZBF | 4ZBI | 4OQ5 | 4OQ6 | 3WIX | 3WIY | 4HW2 | Average | SD |
|---|---|---|---|---|---|---|---|---|---|---|
| Proteins | ||||||||||
| 4WMR | 0.888 | 0.174 | ||||||||
| 4ZBF | 0.86 | 0.644 | 0.156 | |||||||
| 4ZBI | 0.68 | 0.43 | 0.638 | 0.134 | ||||||
| 4OQ5 | 0.66 | 0.83 | 0.73 | 0.858 | 0.143 | |||||
| 4OQ6 | 1.06 | 0.78 | 0.85 | 0.90 | 0.873 | 0.102 | ||||
| 3WIX | 1.05 | 0.65 | 0.67 | 1.07 | 0.96 | 0.829 | 0.161 | |||
| 3WIY | 0.72 | 0.46 | 0.46 | 0.71 | 0.70 | 0.74 | 0.658 | 0.117 | ||
| 4HW2 | 1.13 | 0.62 | 0.72 | 1.05 | 0.88 | 0.70 | 0.78 | 0.808 | 0.185 | |
| 4HW3 | 0.94 | 0.52 | 0.56 | 0.91 | 0.85 | 0.79 | 0.69 | 0.58 | 0.730 | 0.155 |
Table 3.
MLC approach: docking of ligands and their respective RMSD values (Å).
| Proteins | 4WMR | 4ZBF | 4ZBI | 4OQ5 | 4OQ6 | 3WIX | 3WIY | 4HW2 | 4HW3 |
|---|---|---|---|---|---|---|---|---|---|
| Ligands | |||||||||
| 4WMR | 0.73 | 1.12 | 1.23 | 1.61 | 1.45 | 0.81 | 0.89 | 1.26 | 1.32 |
| 4ZBF | 1.27 | 0.75 | 1.19 | 1.32 | 1.09 | 0.90 | 1.03 | 1.31 | 1.42 |
| 4ZBI | 1.19 | 1.12 | 0.96 | 1.12 | 1.11 | 0.86 | 1.12 | 1.21 | 1.36 |
| 4OQ5 | 1.70 | 0.95 | 1.09 | 0.81 | 1.18 | 0.93 | 0.82 | 1.44 | 1.24 |
| 4OQ6 | 1.57 | 0.83 | 1.12 | 0.95 | 1.02 | 1.09 | 1.11 | 1.38 | 1.42 |
| 3WIX | 1.20 | 0.98 | 1.03 | 0.99 | 1.08 | 0.69 | 0.78 | 1.25 | 1.07 |
| 3WIY | 1.33 | 1.26 | 0.92 | 1.04 | 0.80 | 0.74 | 0.79 | 1.18 | 1.04 |
| 4HW2 | 1.87 | 1.14 | 1.30 | 1.61 | 1.49 | 0.98 | 0.86 | 1.12 | 1.13 |
| 4HW3 | 1.71 | 1.32 | 1.21 | 1.53 | 1.34 | 0.82 | 0.78 | 1.20 | 0.97 |
| Average | 1.397 | 1.052 | 1.117 | 1.220 | 1.173 | 0.869 | 0.909 | 1.261 | 1.219 |
| SD | 0.331 | 0.179 | 0.121 | 0.288 | 0.207 | 0.116 | 0.132 | 0.095 | 0.162 |
Table 4.
Comparison of scores from self-docking using various methods with hMcl-1 inhibitor Ki values.
Table 4.
Comparison of scores from self-docking using various methods with hMcl-1 inhibitor Ki values.
| PDB File | Ki Value (nM) | MOE | AutoDock | VLifeDock | |||
|---|---|---|---|---|---|---|---|
| Score a | RMSD b | Score a | RMSD b | Score a | RMSD b | ||
| 5FDO | 361 | −7.29 | 1.81 | −8.30 | 1.94 | −7.02 | 3.49 |
| 5FDR | 0.94 | −8.59 | 2.82 | −7.22 | 3.07 | −7.48 | 3.14 |
| 6BW2 | 21.0 | −10.52 | 1.86 | −9.94 | 2.30 | −9.11 | 2.33 |
| 6BW8 | <1.00 | −10.34 | 0.81 | −9.84 | 1.36 | −9.17 | 1.45 |
| 5IF4 | <1.00 | −10.76 | 1.24 | −10.14 | 1.36 | −10.87 | 1.51 |
| 4HW2 | 55 ± 18 | −8.41 | 1.12 | −7.99 | 1.27 | −8.02 | 1.30 |
| 4HW3 | 320 ± 10 | −6.46 | 0.97 | −6.47 | 1.19 | −6.10 | 1.43 |
a Scores in kcal/mol. b RMSD in Å.
Table 6.
Comparison between mMcl-1 and hMcl-1 active site residues *.
| Corresponding Amino Acids | |||||
|---|---|---|---|---|---|
| mMcl-1 | hMcl-1 | mMcl-1 | hMcl-1 | mMcl-1 | hMcl-1 |
| G150 | - | N241 | N260 | R229 | R248 |
| I163 | I182 | G243 | G262 | F235 | F254 |
| T172 | T191 | I245 | I264 | D237 | D256 |
| G184 | G203 | S250 | S269 | R244 | R263 |
| G200 | G219 | F251 | F270 | V246 | V265 |
| Q202 | Q221 | V255 | V274 | ||
| R203 | R222 | V262 | I281 | ||
| N204 | N223 | V278 | V297 | ||
| R214 | R233 | L279 | L298 | ||
| L216 | L235 | F300 | F319 | ||
| N220 | N239 | Q306 | |||
| G222 | D241 | G307 | |||
| S228 | S247 | ||||
* The enlisted amino acids of mMcl-1 showed chemical shift perturbation (1H-15N HSQC spectrum) during the addition of maritoclax (5a). The amino acids in black color were detected through changes in the 1H-15N HSQC spectrum of 15N-labeled mMcl-1. The un-bolded and bolded amino acids of mMcl-1 in black color showed a 0.05 to 0.08 ppm chemical shift (δ) change, respectively. However, the amino acids shown in green were undetected due to a large signal intensity change. Furthermore, we used sequence alignment to identify the corresponding amino acids of hMcl-1. Red colored amino acids in hMcl-1 represent the species-based point mutations among rat and human Mcl-1 proteins.
Table 7.
Comparison of Ki’s on hMcl-1 inhibition with their respective cumulative docking energies of naturally derived molecules.
Table 7.
Comparison of Ki’s on hMcl-1 inhibition with their respective cumulative docking energies of naturally derived molecules.
| Entry | Compound | hMcl-1 Ki/IC50 in µM | Bcl-2 a/Bcl-xL b Ki/IC50 = µM | Ref. | Triangle Matcher f | AutoDock f | GRIP Docking f | RMSD (Å) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 3.3 d | NR | [19] | −7.36 | −7.71 | −6.17 | 1.08 |
| 2 | 2a | 2.4 ± 0.1 | NR | [20] | −8.33 | −8.29 | −8.76 | 0.81 |
| 3 | 2b | 8.9 | NR | [20] | −8.42 | −8.22 | −8.67 | 0.72 |
| 4 | 2c | 7.3 | NR | [20] | −8.27 | −8.16 | −8.64 | 1.04 |
| 5 | 3a | 17.7 ± 3.1 d | >23 b,d | [21] | −7.14 | −6.82 | −7.17 | 1.22 |
| 6 | 3b | 5.8 ± 0.3 d | 3.2 ± 0.1 b,d | [21] | −7.83 | −7.14 | −7.67 | 2.09 |
| 7 | 3c | 3.7 ± 2.0 d | 16.3 ± 0.5 b,d | [21] | −7.98 | −7.61 | −8.13 | 1.58 |
| 8 | 3d | 0.7 ± 0.1 d | 1.2 ± 0.1 b,d | [21] | −8.34 | −7.42 | −8.91 | 2.18 |
| 9 | 3e | 0.2 ± 0.1 d | 0.3 ± 0.1 b,d | [21] | −8.47 | −8.22 | −9.09 | 1.97 |
| 10 | 3f | 0.2 ± 0.1 d | 0.2 ± 0.1 b,d | [21] | −8.22 | −7.53 | −8.76 | 2.05 |
| 11 | 3g | 1.2 ± 0.9 d | 5.7 ± 0.6 b,d | [21] | −8.00 | −7.68 | −8.27 | 2.31 |
| 12 | 4aa | 14 ± 3.3 e | 19.2 ± 1.6 b,e | [22] | −7.15 | −6.71 | −7.19 | 1.86 |
| 13 | 4b | 13 ± 5.0 e | 12.6 ± 0.2 b,e | [22] | −7.42 | −6.32 | −7.53 | 1.59 |
| 14 | 4c | 5.2 ± 0.2 e | >100 b,e | [22] | −8.02 | −7.60 | −8.11 | 0.88 |
| 15 | 4d | 5.9 ± 0.5 e | 19.4 ± 3.0 b,e | [22] | −7.89 | −7.14 | −8.02 | 1.03 |
| 17 | 5 | 8.9 ± 1.0 d | 16.4 ± 3.3 b,d | [53] | −7.51 | −6.67 | −7.92 | 1.23 |
| 18 | 5a | 4.3 ± 1.5 d | 3.4 ± 0.9 b,d | [53] | −7.06 | −6.39 | −7.84 | 1.70 |
| 19 | 6a | 4.72 d | NR | [27] | −8.77 | −7.88 | −8.90 | 2.13 |
| 20 | 7 (R, R, R) | NAc | NR | [28] | −6.88 | −5.29 | −6.16 | 2.64 |
| 21 | 7 (R, R, S) | NAc | NR | [28] | −7.17 | −6.02 | −6.31 | 2.37 |
| 22 | 7 (R, S, S) | NAc | NR | [28] | −7.04 | −5.89 | −6.20 | 3.14 |
| 23 | 7 (S, S, S) | NAc | NR | [28] | −7.22 | −6.17 | −6.47 | 1.76 |
| 24 | 7 (S, R, R) | NAc | NR | [28] | −6.53 | −5.56 | −5.90 | 2.22 |
| 25 | 7 (S, S, R) | NAc | NR | [28] | −6.70 | −5.82 | −6.13 | 2.90 |
| 26 | 7 (R, S, R) | NAc | NR | [28] | −7.34 | −6.31 | −6.41 | 2.46 |
| 27 | 7 (S, R, S) | NAc | NR | [28] | −7.12 | −6.12 | −6.28 | 1.94 |
| 29 | 8 | 5.2 ± 1.2 e | 1.46 ± 0.12 a,e/8.30 ± 1.20 b,e | [29] | −6.66 | −6.42 | −6.24 | 1.48 |
| 30 | 8a | 0.46 ± 0.06 e | 0.83 ± 0.16 a,e/2.19 ± 0.09 b,e | [29] | −7.22 | −7.14 | −6.94 | 1.14 |
| 31 | 8b | 5.92 ± 0.47 e | >23 a,e/8.48 ± 0.40 b,e | [29] | −7.33 | −7.25 | −7.22 | 1.85 |
| 32 | 8c | 0.56 ± 0.04 e | 1.54 ± 0.44 a,e/2.44 ± 0.02 b,e | [29] | −6.97 | −6.89 | −6.77 | 1.57 |
a Bcl-2 protein inhibition, b Bcl-xL protein inhibition, NA c Not Applicable for isolated cryptosphaerolide (7) had a Ki of 11.4 µM, so all isomers were listed here, d IC50 values, e Ki values, NR not reported, f kcal/mol.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Negi, A.; Murphy, P.V. Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data. Chemistry 2022, 4, 983-1009. https://doi.org/10.3390/chemistry4030067
AMA Style
Negi A, Murphy PV. Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data. Chemistry. 2022; 4(3):983-1009. https://doi.org/10.3390/chemistry4030067
Chicago/Turabian StyleNegi, Arvind, and Paul V. Murphy. 2022. "Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data" Chemistry 4, no. 3: 983-1009. https://doi.org/10.3390/chemistry4030067