Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death


1
Graduate College of Biomedical Sciences, Western University of Health Sciences, Pomona, CA 91766, USA
2
College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
*
Author to whom correspondence should be addressed.
Cells 2020, 9(12), 2698; https://doi.org/10.3390/cells9122698
Submission received: 18 August 2020
/
Revised: 8 December 2020
/
Accepted: 14 December 2020
/
Published: 16 December 2020
(This article belongs to the Special Issue Calpains in Health and Diseases)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Calpains are a family of soluble calcium-dependent proteases that are involved in multiple regulatory pathways. Our laboratory has focused on the understanding of the functions of two ubiquitous calpain isoforms, calpain-1 and calpain-2, in the brain. Results obtained over the last 30 years led to the remarkable conclusion that these two calpain isoforms exhibit opposite functions in the brain. Calpain-1 activation is required for certain forms of synaptic plasticity and corresponding types of learning and memory, while calpain-2 activation limits the extent of plasticity and learning. Calpain-1 is neuroprotective both during postnatal development and in adulthood, while calpain-2 is neurodegenerative. Several key protein targets participating in these opposite functions have been identified and linked to known pathways involved in synaptic plasticity and neuroprotection/neurodegeneration. We have proposed the hypothesis that the existence of different PDZ (PSD-95, DLG and ZO-1) binding domains in the C-terminal of calpain-1 and calpain-2 is responsible for their association with different signaling pathways and thereby their different functions. Results with calpain-2 knock-out mice or with mice treated with a selective calpain-2 inhibitor indicate that calpain-2 is a potential therapeutic target in various forms of neurodegeneration, including traumatic brain injury and repeated concussions.
1. Introduction
Calcium-activated neutral proteases (CANPs) were discovered in 1964 by Guroff [1], but in 1980 Murachi changed their name to calpain, as a contraction of calpain and papain, and also coined the name calpastatin for their endogenous inhibitor [2]. Since then, many studies have been directed at understanding the physiological as well as the pathological function(s) of this family of proteases in the brain and other organs. We initially proposed in 1984 that calpain played a critical role in long-term potentiation (LTP) and learning and memory [3]. This hypothesis was recently validated by studies performed first in hippocampal slices from calpain-4 knock-out (KO) mice [4] and later in slices from calpain-1 KO mice [5,6]. Following our initial studies on the potential role of calpain in LTP and learning and memory, a large number of studies focused on the potential role of calpain in neuronal death and neurodegeneration [7,8,9,10,11,12,13,14]. During the same period, a plethora of calpain isoforms were identified and we now know that calpains constitute a family of enzymes with at least 15 members [15,16], with calpain-1 (aka μ-calpain) and calpain-2 (aka m-calpain) being the most ubiquitous isoforms in all tissues and organs, including the brain. While there is strong evidence that calpain plays a role in neurodegeneration, there are only a handful of studies addressing the question of which calpain isoform(s) is (are) involved and of which are the signaling pathways leading to neurodegeneration. Genetic studies have provided some information regarding the potential contributions of various calpain isoforms in human diseases [17]. In particular, defects in the gene encoding the muscle-specific calpain-3 lead to a particular type of dystrophy, limb-girdle muscular dystrophy 2A (LGMD-2A) [18,19]. There is also good evidence for a link between calpain-10 and diabetes mellitus, based on genetic studies [20]. More recently, calpain-14 has been linked to eosinophilic esophagitis, due to its abundance in the upper gastro-intestinal tract [21]. Mutations in calpain-5 have recently been associated with autoimmune uveitis and photoreceptor degeneration [22]. Very recent studies have also linked mutations in calpain-15 with various types of developmental eye disorders in humans [23]. Our laboratory has focused mostly on the study of the roles of calpain-1 (aka, µ-calpain) and calpain-2 (aka m-calpain) in the brain [14]. Calpain-1 and calpain-2 exhibit a high degree (>70%) of homology. Early in vitro biochemical studies suggested the major difference consisted in their calcium requirement for activation, with calpain-1 requiring micromolar calcium concentrations and calpain-2 millimolar calcium concentrations [2]. This presented a significant challenge to study the potential role of calpain-2 in the brain, as such a high calcium requirement for calpain-2 activation made it unlikely that cytoplasmic calpain-2 could be activated under physiological and most pathological conditions. We were thus left with the paradox of explaining how the same enzyme, calpain-1, could be involved in both synaptic plasticity and neurodegeneration. This review will summarize our work over the last 20–25 years directed at resolving this paradox. What we found turned out to be quite remarkable: in short, our studies revealed that calpain-1 and calpain-2 play opposite functions in the brain, with calpain-1 activation being required for triggering certain forms of synaptic plasticity and thereby in various forms of learning and memory; in addition, calpain-1 is neuroprotective both during postnatal development and in adulthood. In contrast, calpain-2 activation limits the magnitude of synaptic plasticity and the extent of learning and is neurodegenerative [14]. These different functions of calpain-1 and calpain-2 and the signaling pathways they regulate to perform these functions will be discussed in greater details in this review.
2. Calpain-1 Role in Synaptic Plasticity
As mentioned above, the hypothesis that calpain plays a significant role in LTP and learning and memory was proposed in 1984 [3]. Over the following 10 years, several findings indicated that calpain-1 and calpain-2, by cleaving several key proteins, participated in the regulation of dendritic structure and local protein synthesis. The first hint that calpain was linked to the regulation of local protein translation was provided by a report that calpain cleaved dicer and released dicer and eIF2c from postsynaptic densities, thereby facilitating the processing of miRNA and regulating protein translation [24]. It was later reported that calpain could cleave and inactivate the suprachiasmatic nucleus circadian oscillatory protein (SCOP, also known as PH domain and leucine-rich repeat protein phosphatase 1 (PHLPP1β)) [25], a negative regulator of the extracellular signal-regulated kinase (ERK), a kinase with numerous links to LTP [26], thus linking calpain activation to activation of the ERK pathway. Calpain was also shown to cleave β-catenin, generating an active fragment, which regulates gene transcription, thereby providing a mechanism by which NMDA (N-Methyl-D-Aspartate) receptor stimulation, which has been repeatedly linked to calpain activation [27,28], could modify gene expression [29]. We also found that several scaffolding proteins were calpain substrates, including PSD95 [30], GRIP [31], SAP97 [32], and more recently ankyrin repeat-rich membrane spanning protein (ARMS) or kinase D-interacting substrate of 220 kDa (Kidins220) was shown to be regulated by calpain-mediated truncation [33]. By degrading the translational repressor poly(A)-binding protein (PABP)-interacting protein 2A (PAIP2A), an inhibitor of PABP, calpain could also relieve translational inhibition of proteins, a mechanism that has been involved in synaptic plasticity and learning and memory [34].
More recently, the role of calpain in long-term potentiation (LTP) was supported by the study using hippocampal slices from mice with a conditional downregulation of calpain-4, the small subunit required by both calpain-1 and calpain-2 for functional activity. Theta burst stimulation (TBS)-induced LTP induction was impaired in hippocampal slices prepared from these mice [4].
Interestingly, other targets of calpain also link calpain activation to some of the molecular/cellular mechanisms known to participate in LTP; in particular, we identified a pathway linking actin polymerization in dendritic spines to the synaptic structural modifications associated with LTP [35]. Three actin-signaling pathways involving the Rho family of small GTPases, RhoA, Rac, and Cdc42, are ubiquitously implicated in the mechanisms of actin filament assembly, disassembly, or stabilization in most cells [36], and have been shown to be critically involved in LTP consolidation [37,38]. We discovered that, like PHLPP1β, RhoA is rapidly degraded and then resynthesized after LTP through the sequential activation of calpain-1 and calpain-2 [39].
The results obtained with calpain inhibitors or the calpain-4 KO mice were further confirmed by experiments showing that calpain-1 KO mice were impaired in TBS-LTP and in hippocampus-dependent learning [6], clearly indicating that calpain-1 activation following synaptic NMDA receptor stimulation is essential for theta burst stimulation-induced LTP in field CA1 of the hippocampus. More recently, we also found that calpain-1 activation was required for mGluR-dependent long-term depression (LTD) in hippocampus [40]. The molecular pathway involved in this case is the inactivation of PP2A due to the cleavage of B56α, a regulatory subunit of PP2A. This leads to mTOR stimulation and a local increase in the synthesis of activity-regulated cytoskeleton-associated protein (Arc) and reduced levels of GluA1-containing a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. In support of this finding, LTD impairment in calpain-1 KO mice was rescued by PP2A inhibitors [40]. We also found that calpain-1 was involved in various forms of LTD in the cerebellum, and the molecular pathways involved were quite similar to those we found in the hippocampus. Thus, dihydroxyphenylglycine (DHPG)-LTD was enhanced by the application of okadaic acid, a PP2A inhibitor, in cerebellar slices from both WT and calpain-1 KO mice. These results indicated that the same signaling pathway, calpain-1-→PP2A-→Arc-→AMPA receptor internalization, is involved in both hippocampal and cerebellar LTD [41]. Previous studies had reported that cerebellar LTD required the dephosphorylation of transmembrane AMPAR regulatory protein γ-2 (TARP γ-2), a process known to result in AMPA receptor internalization [42]. As we also found that TARP γ-2 is a calpain substrate [43], we proposed that calpain-mediated truncation of TARP γ-2 contributes to cerebellar LTD.
Over the last 20 years, LTP and LTD at the parallel fiber to Purkinje cell synapses have been proposed to participate in the learning of various cerebellum-dependent tasks, including classical conditioning of motor responses [44,45,46], although some evidence suggests that LTD at the parallel fiber to Purkinje cell synapses is not required for cerebellar motor learning [47]. In agreement with the role of calpain-1 in LTD in the cerebellum, we found that calpain-1 KO mice were impaired in the initial acquisition of classically conditioned eye-blink responses, although they were able to reach the same level of performance as the WT mice with continued training [41]. In addition, calpain-1 KO mice did not show any impairment during extinction of classical conditioning eye-blink responses.
Overall, all the studies reported above clearly indicate that calpain-1 plays an important role in various forms of synaptic plasticity and related types of learning and memory. Some of the molecular pathways regulated by calpain-1 activation in these mechanisms have been identified and these findings might lead to future potential treatments for diseases associated with learning and memory impairment.
3. Calpain-1 Role in Neuroprotection
The first hint that calpain-1 activation was neuroprotective came from studies showing that calpain-1 could cleave PHLPP1, resulting in Akt activation, a known pathway leading to neuronal survival [48]. This idea was further confirmed by demonstrating that calpain-1-mediated truncation of PHLPP1 and Akt activation was involved in NMDAR-dependent survival of cerebellar granule cells (CGCs) [49]. Later studies indicated that calpain-1 KO mice exhibited abnormal cerebellar development, including enhanced apoptosis of CGCs during the early postnatal period, reduced granule cell density and impaired synaptic transmission from the parallel fibers to Purkinje cells, resulting in cerebellar ataxia [49]. All these defects were due to deficits in the calpain-1/PHLPP1/Akt pro-survival pathway in developing granule cells, since treatment with an Akt activator during the postnatal period or crossing calpain-1 KO mice with PHLPP1 KO mice restored most of the observed alterations in cerebellar structure and function in calpain-1 KO mice [49]. Interestingly, several human families carrying CAPN1 mutations were found to exhibit cerebellar ataxia [49,50] and Russell terrier dogs carrying a missense mutation in calpain-1 also exhibit spinocerebellar ataxia [51].
To further demonstrate the role of calpain-1 in neuroprotection, we determined the extent of neuronal damage elicited by acute insults in wild-type mice and calpain-1 KO mice. We postulated that the extent of damage would be greater in the KO mice than in the WT littermates. Thus, we found that, following traumatic brain injury (TBI), cell death and lesion volume were larger in calpain-1 KO mice than in WT mice [52]. In addition, both retinal ganglion cell death following acute increased intraocular pressure and hippocampal neuronal death following kainate-induced seizure were enhanced in calpain-1 KO mice, as compared to WT mice [53,54]. Calpain deletion also results in neuronal degeneration across different species including human, dog, mice, zebrafish, fruit fly, and Caenorhabditis elegans [49,51,55]. All these results suggest a neuroprotective role for calpain-1 both during the postnatal period and in the adult.
4. Calpain-2 Role in Synaptic Plasticity
While the studies discussed above clearly supported a necessary role for calpain-1 activation in the induction phase of LTP, very little information was available concerning the potential role of calpain-2 activation in synaptic plasticity and in LTP. As mentioned, a key issue was the high calcium concentration required for calpain-2 activation, and there was no evidence that such a concentration could be reached intracellularly under physiological conditions. Two major findings completely changed our understanding of the roles of calpain-1 and calpain-2 in LTP. First, we discovered that brain-derived neurotrophic factor (BDNF), a neurotrophic factor critically involved in synaptic plasticity and in LTP [56], could activate calpain-2 though ERK-mediated phosphorylation at serine 50 [57]. Moreover, we later found that BDNF-induced stimulation of local protein synthesis, another critical process in LTP formation [58], was mediated by calpain-2 activation of mTOR [59]. Detailed analysis of this mechanism indicated that calpain-2, but not calpain-1, cleaved phosphatase and tensin homolog (PTEN), a negative mTOR regulator, thus suggesting that calpain-2 activation following LTP induction could lead to increased local protein synthesis and participate in LTP consolidation [59]. The other major finding that changed our understanding of the role of calpain in LTP was provided by results from experiments performed with small modifications of the original protocol that was used to demonstrate the role of calpain in theta burst stimulation (TBS)-induced LTP in acute hippocampal slices [60,61]. In these experiments, a non-selective calpain inhibitor, calpain inhibitor III, was applied before delivering the theta burst. However, when calpain inhibitor III was applied immediately after TBS, the magnitude of LTP was dramatically enhanced; treatment with calpain inhibitor III 1 h after TBS had no effect [5]. These surprising results led us to propose a completely new model for LTP induction and consolidation, at least in field CA1 of the hippocampus. In our model, calpain-1 activation following synaptic NMDA receptor stimulation results in PHLPP1β degradation and ERK activation. However, calpain-2 activation following TBS, possibly resulting from BDNF-mediated ERK activation extrasynaptically, leads to PTEN truncation and mTOR activation followed by the stimulation of local protein synthesis and, in particular, of PHLPP1β synthesis, which would limit the duration of ERK activation [5]. To validate the model, we tested the effects of a selective calpain-2 inhibitor, the dipeptide ketoamide, Z-Leu-Abu-CONH-CH2-C6H3[3,5-(OMe)2] (C2I), which exhibits a more than 20-fold selectivity for calpain-2 over calpain-1, on LTP induction when applied before or after TBS [5]. The results were unambiguous, as C2I application either before or 10 min after TBS produced the same enhancement in LTP magnitude as when calpain inhibitor III was applied after TBS.
The differential effects of calpain-1 and calpain-2 on SCOP/PHLPP1β could be due to a different subcellular localization of the proteases. In particular, a fraction of PHLPP1β has been shown to be present in membrane rafts, and this fraction could be degraded following NMDA receptor channel opening and the resulting calpain-1 activation. On the other hand, calpain-2 might be present in a different subcellular compartment, and in particular, at the base of dendritic spines where local dendritic protein synthesis is taking place. Thus, the biphasic effect of TBS on ERK activation, i.e., initial activation followed by delayed prevention of activation, could reflect the sequential activation of calpain-1 and calpain-2 following TBS, as well as their opposite effects on PHLPP1β levels and ERK activation. An alternative, although not exclusive, hypothesis is suggested by the role of BDNF on calpain-2 activation through ERK-mediated phosphorylation. The release of BDNF during TBS, as evidenced by the activation of its TrkB receptor at synapses, is known to facilitate the cytoskeletal changes required for LTP consolidation [62,63]. In addition, the effect of BDNF on calpain-2 activation indicates that, by stimulating local protein synthesis and in particular the synthesis of SCOP/PHLPP1β, it triggers ERK inhibition, thereby limiting the magnitude of the LTP. Further studies using mice with a deletion of calpain-2 in hippocampal pyramidal neurons will verify the critical role of calpain-2 in limiting LTP magnitude following TBS. Further studies will also be directed at evaluating the potential role of calpain-2 in other forms of synaptic plasticity at various synapses. Likewise, further studies will determine whether calpain-2 activation also participates in different types of learning and memory.
5. Calpain-2 Role in Neuronal Death
While an extensive literature links calpain activation with neurodegeneration, very few studies have explored the specific contributions of calpain-1 and calpain-2 in neurodegeneration. As discussed above, we demonstrated that calpain-1 activation is neuroprotective both during the postnatal period and in the adult. This conclusion would therefore lead to the hypothesis that it is calpain-2 that is responsible for the reported role of calpain in neurodegeneration, unless some other calpain isoform would perform this function. Using primary neuronal cultures, we first showed that calpain-2, but not calpain-1 activation was responsible for NMDA-induced excitotoxicity through the activation of Striatal-Enriched Protein Tyrosine Phosphatase (STEP) [48]. A similar study indicated that the downregulation of calpain-2 but not calpain-1 increased neuronal survival following NMDA treatment of cultured hippocampal neurons [64]. It has been proposed that the activation of synaptic and extrasynaptic NMDA receptors have opposite effects on neuronal survival and degeneration [65]. Our hypothesis is that calpain-1 activation is directly downstream of synaptic NMDA receptors, while calpain-2 activation results from stimulation of extrasynaptic NMDA receptors. These receptors are enriched in NR2B subunits [66], which bind RasGRF1, thereby providing a link between NMDAR activation and ERK activation [67]. This pathway could therefore be responsible for the prolonged activation of calpain-2 following the stimulation of extrasynaptic NMDA receptors. In addition, calpain cleaves striatal-enriched tyrosine phosphatase (STEP), resulting in the activation of p38 and downstream cell death signaling pathways [68,69].
Other pathways could link calpain-2 activation to cell death. A recent study demonstrated that calpain-2 inhibition or knock-down enhanced autophagy and reduced cell death after ischemia/reperfusion in liver [70]. Similarly, calpain inhibitors promoted mTOR-independent autophagy and rescued Huntington’s disease phenotypes in zebrafish [71]. In Alzheimer’s disease (AD), calpain-2 was found to be hyper-activated in synaptosomes in presymptomatic AD, and the activation was correlated with a decline in cognitive function and an increase in levels of β-amyloid deposits [72]. Calpain activation has also been shown to switch cellular programs from autophagy to apoptosis in various preparations [73,74,75]. Likewise, calpain activation has been repeatedly shown to stimulate apoptosis pathways through multiple mechanisms [76]. More recently, we discovered that calpain-2 could cleave and inactivate the protein tyrosine phosphatase, PTPN13, aka Fas-associated protein-1 (FAP1) [77]. This phosphatase is an inhibitor of apoptosis, and therefore, by inactivating it, calpain-2 activation would stimulate apoptosis. Calpains also cleave several members of the Bcl-2 family of proteins, including Bax, Bid, and Bcl-xL, leading to cytochrome c release [78,79,80] and caspase-3 activation [81]. Calpain also converts pro-caspase-7 to caspase-7 [82]. Therefore, many studies indicate that calpain-2 activation prevents autophagy and stimulates apoptosis. Consequently, it is highly likely that calpain-2 activation represents a critical step towards cell death.
We previously identified another mechanism linking calpain activation to neuronal death through the truncation of mGluR1α, following NMDA receptor stimulation-induced calpain activation [83]. Under control conditions, mGluR1α receptors are coupled to PI3K-Akt signaling and their activation is neuroprotective. Brief mGluR1α activation leads to calcium release from internal stores, but the extent of calcium release does not produce significant toxic effects. Following NMDA receptor stimulation or ischemia onset, calpain activation leads to mGluR1α truncation, disrupting the neuroprotective effect of the mGluR1α-PI3K-Akt signaling cascade. Importantly, truncated mGluR1α receptors maintain their stimulation of calcium release from intracellular stores, which further contributes to calcium overload through NMDA receptors and enhances neurotoxicity [83].
Calpain activation has long been shown to be involved in the pathology of traumatic brain injury (TBI) [84,85,86]. Calpain activation results in the truncation of brain spectrin and several spectrin breakdown products (SBDPs) have been used as biomarkers for TBI in both cerebro-spinal fluid (CSF) and blood [87,88,89,90,91,92,93,94]. Surprisingly, none of these studies has addressed the respective roles of calpain-1 and calpain-2 either in brain pathology or in the generation of the blood biomarkers. This is likely due to the lack of isoform-selective calpain inhibitors, the lack of animals with selective deletion of calpain-1 or calpain-2, and the lack of markers for calpain-1 and calpain-2 activation. These limitations could account for several conflicting results. In particular, in some studies, the calpain inhibitors AK295 and ALLM were reported to protect the cytoskeletal structure of injured neurons and to attenuate motor and cognitive deficits after TBI [95,96]. In other studies, two other calpain inhibitors, SNJ-1945 and MDL-28170, which were shown to cross the blood–brain barrier, did not exhibit significant efficacy in a model of controlled cortical impact [97,98].
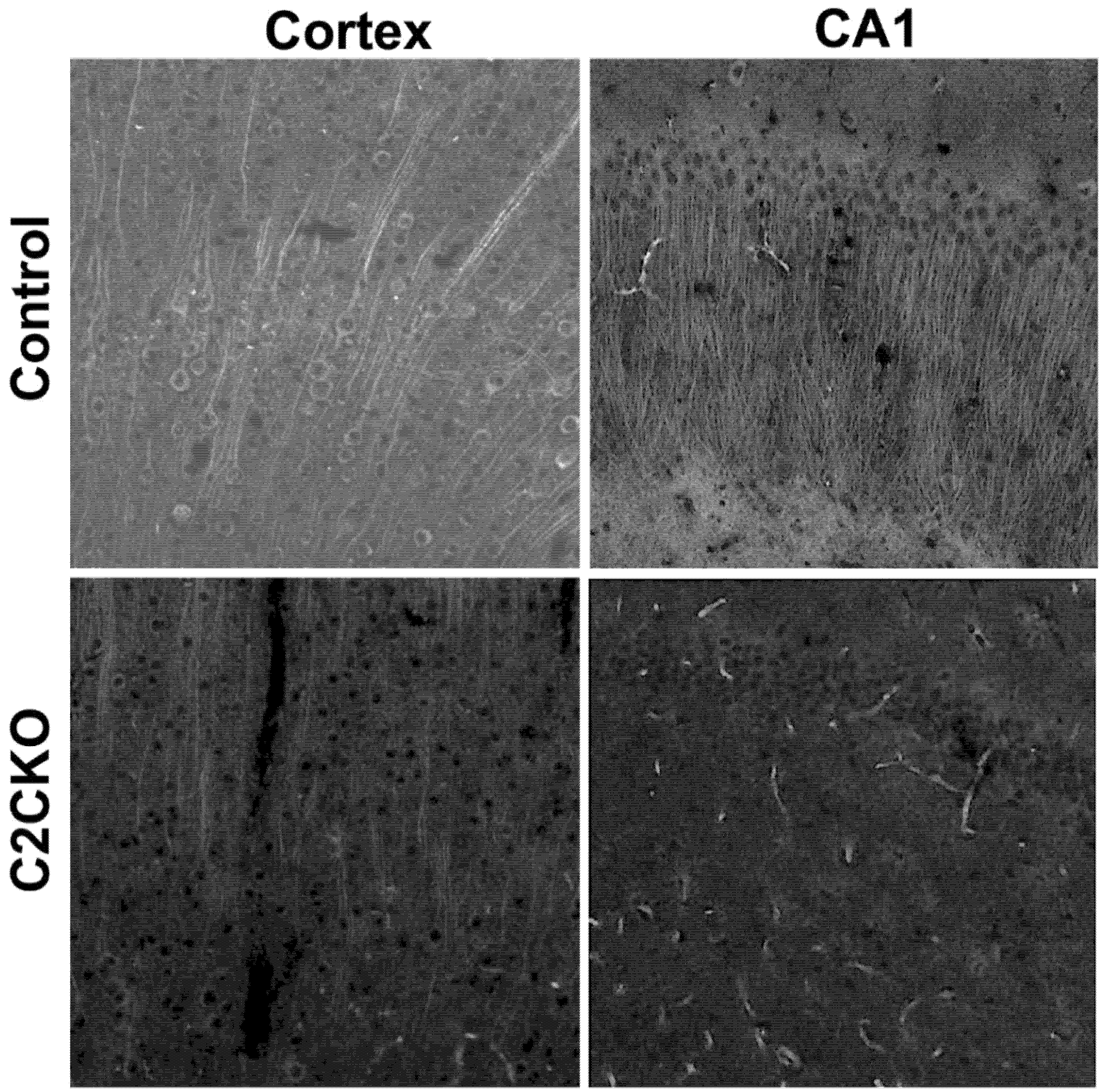
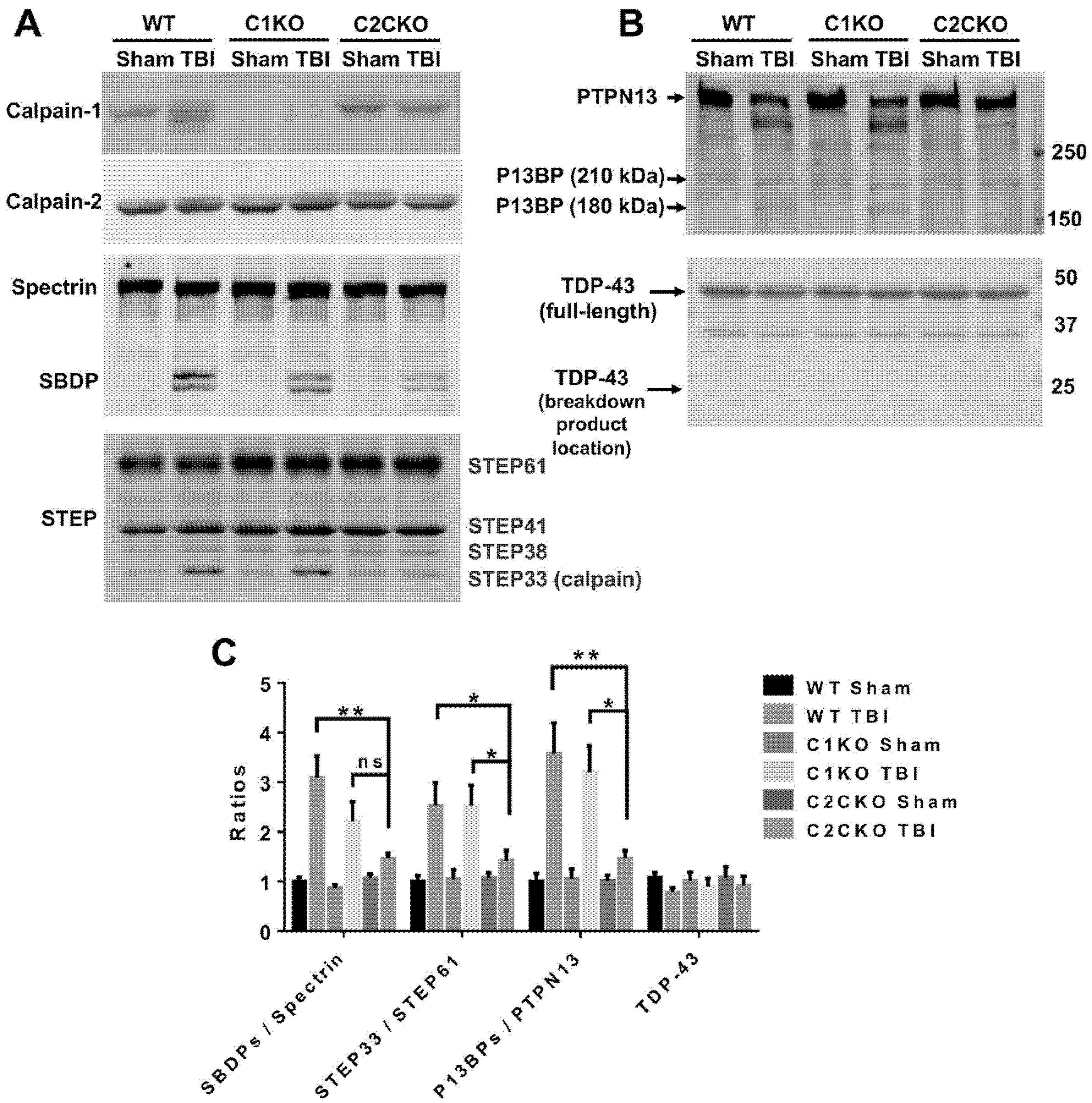
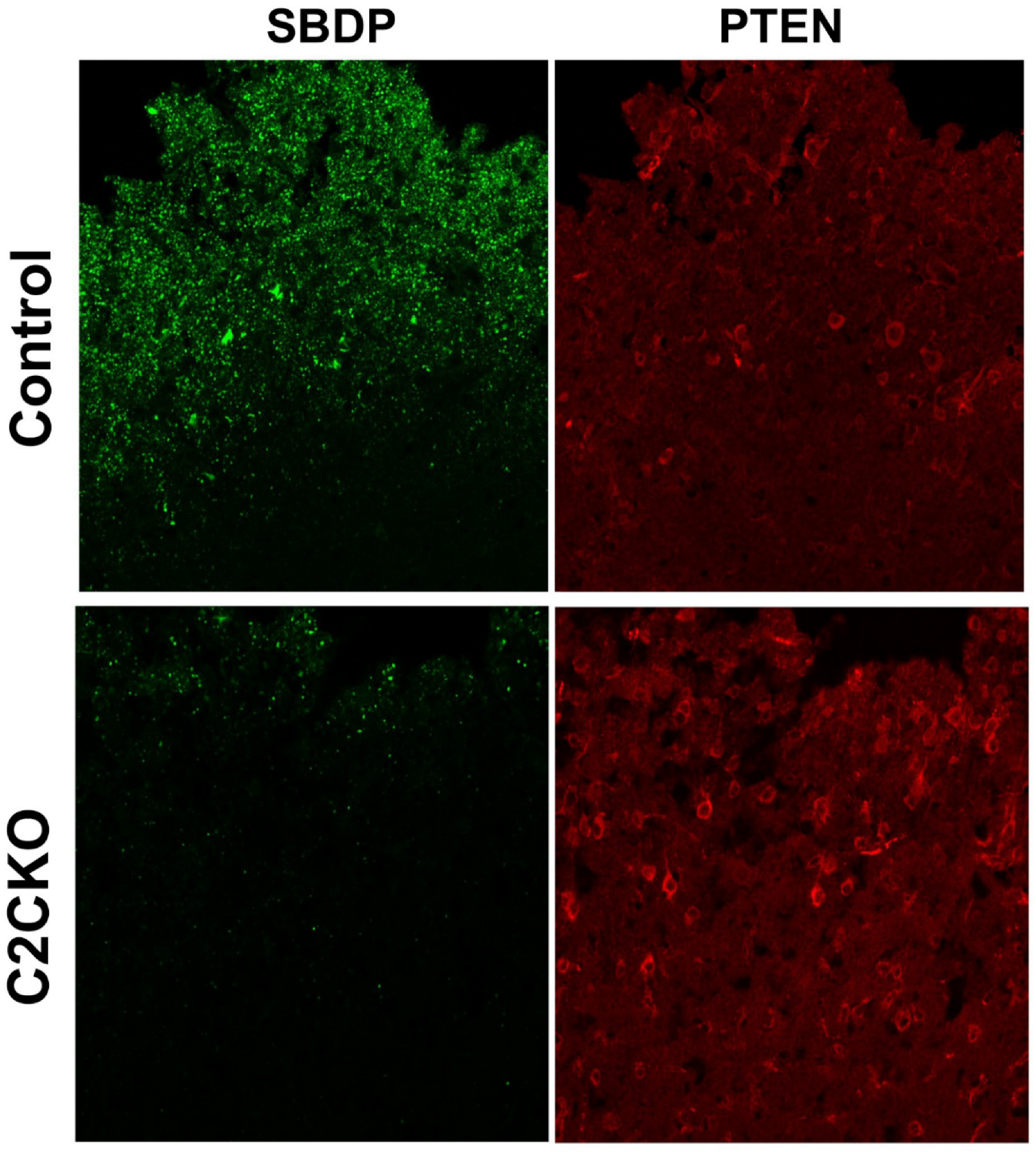
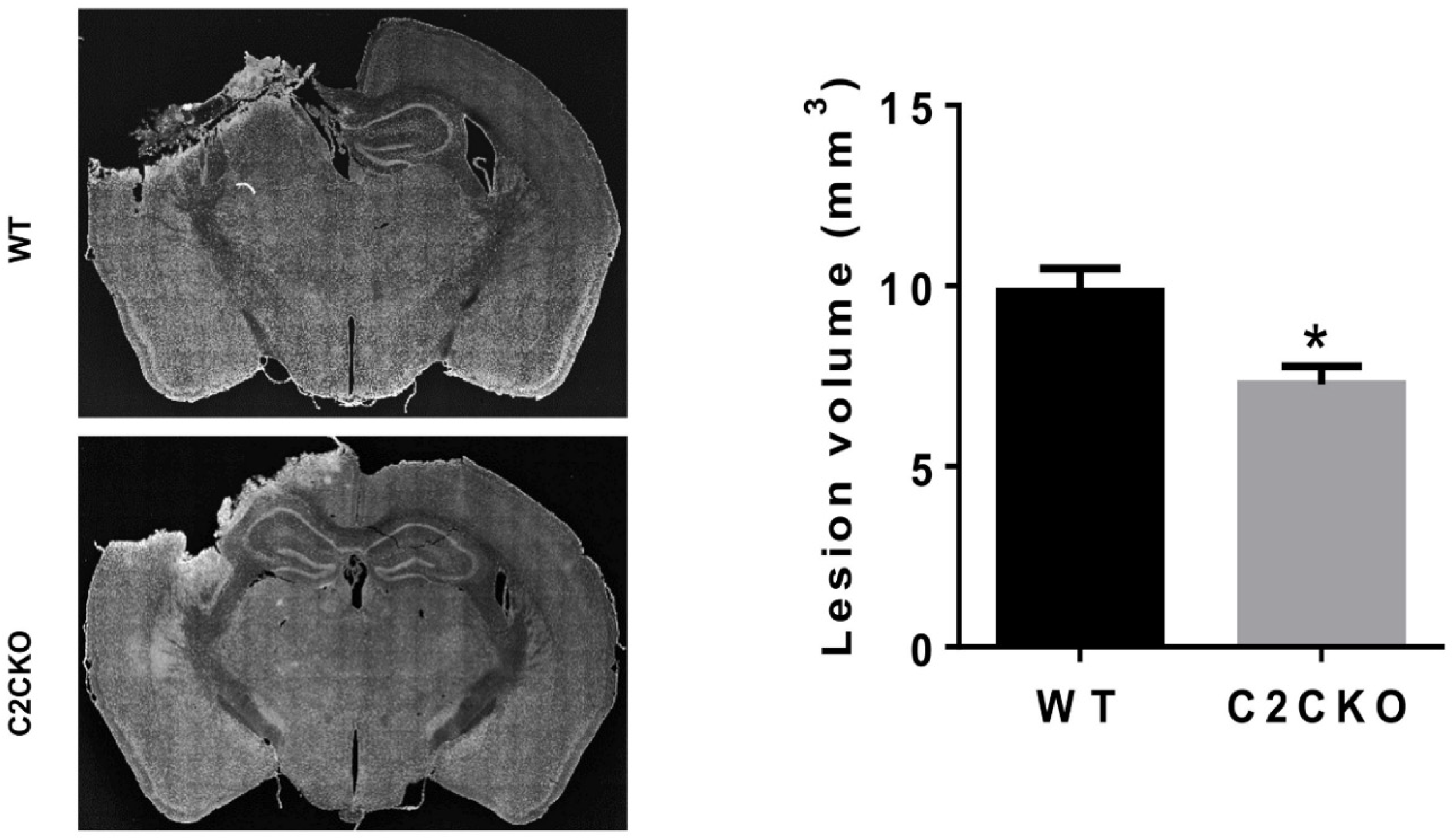
As mentioned above, using global calpain-1 KO mice, we established that calpain-1 is neuroprotective in a mouse model of TBI, the controlled cortical impact (CCI). To address the question of the roles of calpain-2 in both the pathology and the production of the blood biomarker, we used mice with selective deletion of calpain-2 in excitatory neurons of the forebrain, or selective calpain-2 inhibition provided by injection of C2I. Selective deletion of calpain-2 in excitatory neurons of the forebrain was obtained by crossing loxP-calpain-2 mice with CmKII-Cre mice, producing calpain-2 conditional KO (C2CKO) mice; immunohistochemistry confirmed that calpain-2 was significantly deleted in the cerebral cortex and in hippocampus, including field CA1 (Figure 1). Changes in SBDP levels in the cortex after TBI represent both calpain-1 and calpain-2 activation, as spectrin can be cleaved by both calpain-1 and calpain-2 [99]. However, by comparing the changes in SBDP at various times after TBI in wild-type (WT) mice and calpain-1 KO mice, we could estimate the respective contributions of calpain-1 and calpain-2 activation in SBDP generation [52]. Moreover, since PTEN is selectively cleaved by calpain-2 but not calpain-1 [59], analyzing changes in PTEN at various times after TBI also reflects the temporal activation of calpain-2 after TBI. Our results indicated that calpain-1 is rapidly and transiently activated in the cortex surrounding the impact site after TBI, with a maximal activation 6 h after TBI, but was no longer activated 24 h after TBI. In contrast, calpain-2 activation was delayed, starting between 4 and 8 h after TBI and was still present 3 days after TBI [52]. Western blots from cortical homogenates confirmed that 24 h after TBI, the increase in the levels of the 145–150 Kd SBDP fragments was similar in WT and calpain-1 KO mice but almost completely absent in C2CKO mice (Figure 2). Additionally shown in Figure 2 are western blots showing that the truncation of STEP61 resulting in the formation of the neurodegenerative fragment STE33 is also completely mediated by calpain-2. Similarly, the truncation of PTPN13 and the formation of breakdown products, labeled P13BPs, is entirely mediated by calpain-2 activation. Interestingly, in this model, we did not detect the truncation of TDP43, a protein known to be involved in Amyotrophic Lateral Sclerosis (ALS), fronto-temporal dementia (FTD), and repeated concussions [100,101]. Results from immunohistochemistry confirmed that the large increase in SBDP in the cortex surrounding the lesion site 24 h after TBI was entirely due to calpain-2 activation, as it was absent in C2CKO mice (Figure 3). Moreover, the large decrease in PTEN levels was also absent in C2CKO mice (Figure 3). Using Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and Fluoro-Jade staining to identify dying cells, we found that the extent of calpain-2 activation was positively correlated with the extent of cell death [52]. Systemic administration of C2I, 1 or 4 h after TBI, significantly reduced calpain-2 activation and the number of degenerating cells in the cortex surrounding the impact site, further demonstrating the neurodegenerative role of calpain-2. In good agreement with this result, the lesion volume 7 days after TBI was significantly smaller in C2CKO mice than in control mice (Figure 4). Finally, we also determined the levels of calpain-cleaved αII-spectrin N-terminal fragment (SNTF), a blood biomarker shown to be present in human subjects after concussion [92,93,94], in the blood of mice subjected to TBI with or without treatment with a selective calpain-2 inhibitor (Figure 5). First of all, we found that SNTF blood levels were very well correlated with the levels of calpain-2 activation in the brain; in addition, treatment of the mice with C2I 1 h after TBI prevented the increase in blood SNTF elicited by TBI. In a recent study, we also observed that calpain-2 deletion or semi-chronic treatment with a selective calpain-2 inhibitor prevented all the pathological manifestations resulting from repeated concussions in mice [101].
The same pattern of a brief activation of calpain-1 and delayed but chronic activation of calpain-2 was also found in retinal ganglion cells after retinal ischemia/reperfusion injury, a mouse model of primary angle-closure glaucoma (PACG) [53]. All these results are consistent with the idea that calpain-2 activation is delayed and prolonged following acute insults or repeated concussions. These results suggest that calpain-2 activation is responsible for the neurodegeneration as well as the brain inflammation resulting from brain insults. Calpain-2 activation may also be responsible for the cognitive impairment associated with the neuropathology observed weeks and months after the insults.
6. Conclusions
The long journey that started 40 years ago when we were trying to understand the role of calpain in synaptic plasticity and learning and memory has led us to explore a vast array of pathways and mechanisms. It has also uncovered the remarkable finding that the two major isoforms of calpain in the brain, calpain-1 and calpain-2, play opposite functions in many of these mechanisms. In particular, our studies revealed that calpain-1 activation is critical to trigger various types of synaptic plasticity in both hippocampus and cerebellum, which are engaged in various types of learning. Furthermore, calpain-1 activation is neuroprotective both during the postnatal development and in adulthood. Interestingly, several human families with null mutations in CANP1 have been identified and shown to exhibit profound neurological disorders, and in some cases cognitive impairment [49]. In contrast, calpain-2 activation limits the extent of synaptic plasticity and of learning during a brief consolidation period lasting about 1 h. The signaling pathways underlying these opposite functions of calpain-1 and calpain-2 have started to be identified and we have postulated that they are due to the association of calpain-1 and calpain-2 with different PDZ binding domains, resulting in their linkages to different signaling cascades [14]. These opposite functions are schematically represented in Figure 6, in which we stress the point that calpain-1 is preferentially linked to synaptic NMDA receptors while calpain-2 is proposed to be downstream of extra-synaptic NMDA receptors. Even more importantly, prolonged calpain-2 activation following a variety of acute brain insults is responsible for the neuronal damage resulting from these insults. It is also responsible for the brain inflammation associated with such insults as well as the long-term cognitive impairments produced by these insults. The identification of selective calpain-2 inhibitors that can prevent the neuropathological manifestations of these acute insults provide the hope that it will be possible to translate the animal results into clinical applications.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4409/9/12/2698/s1.
Author Contributions
Y.W. performed experiments, prepared figures and worked on the manuscript. Y.L. performed some of the experiments. X.B. analyzed data, worked on the figures and on the manuscript. M.B. oversaw the project, directed the experiments, and worked on figures and manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Defense Medical Research and Development Program under Award No. W81XWH-19-1-0329. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. Grant #BA170606. “Optimization of a selective calpain-2 inhibitor for prolonged field care in Traumatic Brain Injury”. X.B. is supported in part by funds from the Daljit and Elaine Sarkaria Chair.
Acknowledgments
The authors want to thank Robert Siman from the University of Pennsylvania for performing the SNTF assays in plasma 24 h after TBI. They also want to thank Anthony McCloud who helped with several experiments.
Conflicts of Interest
M.B., X.B. and Y.W. are cofounders of NeurAegis, a startup company focusing on developing selective calpain-2 inhibitors for the treatment of acute neurodegeneration. M.B. is an inventor on a Provisional Patent “New selective calpain-2 inhibitors for the treatment of neurodegeneration”. The other authors declare no competing interests.
References
- Guroff, G. A neutral, calcium-activated proteinase from the soluble fraction of rat brain. J. Biol. Chem. 1964, 239, 149–155. [Google Scholar] [PubMed]
- Murachi, T.; Tanaka, K.; Hatanaka, M.; Murakami, T. Intracellular Ca2+-dependent protease (calpain) and its high-molecular-weight endogenous inhibitor (calpastatin). Adv. Enzym. Regul. 1981, 19, 407–424. [Google Scholar] [CrossRef]
- Lynch, G.; Baudry, M. The biochemistry of memory—A new and specific hypothesis. Science 1984, 224, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Amini, M.; Ma, C.-L.; Farazifard, R.; Zhu, G.; Zhang, Y.; Vanderluit, J.; Zoltewicz, J.S.; Hage, F.; Savitt, J.M.; Lagace, D.C. Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. J. Neurosci. 2013, 33, 5773–5784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhu, G.; Briz, V.; Hsu, Y.T.; Bi, X.; Baudry, M. A molecular brake controls the magnitude of long-term potentiation. Nat. Commun. 2014, 5, 3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Liu, Y.; Wang, Y.; Bi, X.; Baudry, M. Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J. Neurosci. 2015, 35, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Saito, K.I.; Grynspan, F.; Griffin, W.R.; Katayama, S.; Honda, T.; Mohan, P.S.; Shea, T.B.; Beermann, M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1994, 747, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Vanderklish, P.W.; Bahr, B.A. The pathogenic activation of calpain: A marker and mediator of cellular toxicity and disease states. Int. J. Exp. Pathol. 2000, 81, 323–339. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef]
- Camins, A.; Verdaguer, E.; Folch, J.; Pallàs, M. Involvement of calpain activation in neurodegenerative processes. CNS Drug Rev. 2006, 12, 135–148. [Google Scholar] [CrossRef]
- Bevers, M.B.; Neumar, R.W. Mechanistic role of calpains in postischemic neurodegeneration. J. Cereb. Blood Flow Metab. 2008, 28, 655–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosler, P.; Brennan, C.; Chen, J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 2008, 38, 78–100. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, M.C.; Wang, K. Calpain in the CNS: From synaptic function to neurotoxicity. Sci. Signal. 2008, 1, re1. [Google Scholar] [CrossRef]
- Baudry, M.; Bi, X. Calpain-1 and Calpain-2: The Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci. 2016, 39, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Sorimachi, H.; Ono, Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc. Res. 2012, 96, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macqueen, D.J.; Wilcox, A.H. Characterization of the definitive classical calpain family of vertebrates using phylogenetic, evolutionary and expression analyses. Open Biol. 2014, 4, 130219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Wang, K.K. The calpain family and human disease. Trends Mol. Med. 2001, 7, 355–362. [Google Scholar] [CrossRef]
- Beckmann, J.S.; Bushby, K.M. Advances in the molecular genetics of the limb-girdle type of autosomal recessive progressive muscular dystrophy. Curr. Opin. Neurol. 1996, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, E.; de Luna, N.; Diaz-Manera, J.; Rojas-García, R.; Gonzalez-Quereda, L.; Flix, B.; de Morrée, A.; van der Maarel, S.; Illa, I. Comparison of dysferlin expression in human skeletal muscle with that in monocytes for the diagnosis of dysferlin myopathy. PLoS ONE 2011, 6, e29061. [Google Scholar] [CrossRef]
- Harris, F.; Biswas, S.; Singh, J.; Dennison, S.; Phoenix, D.A. Calpains and their multiple roles in diabetes mellitus. Ann. N. Y. Acad. Sci. 2006, 1084, 452–480. [Google Scholar] [CrossRef]
- Litosh, V.A.; Rochman, M.; Rymer, J.K.; Porollo, A.; Kottyan, L.C.; Rothenberg, M.E. Calpain-14 and its association with eosinophilic esophagitis. J. Allergy Clin. Immunol. 2017, 139, 1762–1771. [Google Scholar] [CrossRef]
- Mahajan, V.B.; Skeie, J.M.; Bassuk, A.G.; Fingert, J.H.; Braun, T.A.; Daggett, H.T.; Folk, J.C.; Sheffield, V.C.; Stone, E.M. Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet. 2012, 8, e1003001. [Google Scholar] [CrossRef]
- Zha, C.; Abi Farah, C.; Fonov, V.; Holt, R.; Ceroni, F.; Ragges, N.; Rudko, D.; Sossin, W.S. Disruption of Capn15 in mice leads to brain and eye deficits. bioRxiv 2019, 763888. [Google Scholar] [CrossRef] [Green Version]
- Lugli, G.; Larson, J.; Martone, M.E.; Jones, Y.; Smalheiser, N.R. Dicer and eIF2c are enriched at postsynaptic densities in adult mouse brain and are modified by neuronal activity in a calpain-dependent manner. J. Neurochem. 2005, 94, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Phan, T.; Mansuy, I.M.; Storm, D.R. Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell 2007, 128, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, M.G. The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev. Neurosci. 2006, 17, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Pieri, M.; Ciotti, M.T.; Carunchio, I.; Canu, N.; Calissano, P.; Zona, C.; Severini, C. Substance P provides neuroprotection in cerebellar granule cells through Akt and MAPK/Erk activation: Evidence for the involvement of the delayed rectifier potassium current. Neuropharmacology 2007, 52, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- del Cerro, S.; Arai, A.; Kessler, M.; Bahr, B.A.; Vanderklish, P.; Rivera, S.; Lynch, G. Stimulation of NMDA receptors activates calpain in cultured hippocampal slices. Neurosci. Lett. 1994, 167, 149–152. [Google Scholar] [CrossRef]
- Abe, K.; Takeichi, M. NMDA-receptor activation induces calpain-mediated β-catenin cleavages for triggering gene expression. Neuron 2007, 53, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Rong, Y.; Baudry, M. Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neurosci. Lett. 2000, 286, 149–153. [Google Scholar] [CrossRef]
- Lu, X.; Wyszynski, M.; Sheng, M.; Baudry, M. Proteolysis of glutamate receptor-interacting protein by calpain in rat brain: Implications for synaptic plasticity. J. Neurochem. 2001, 77, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Jourdi, H.; Lu, X.; Yanagihara, T.; Lauterborn, J.C.; Bi, X.; Gall, C.M.; Baudry, M. Prolonged positive modulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors induces calpain-mediated PSD-95/Dlg/ZO-1 protein degradation and AMPA receptor down-regulation in cultured hippocampal slices. J. Pharmacol. Exp. Ther. 2005, 314, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-Y.; Lynch, D.R. Calpain and synaptic function. Mol. Neurobiol. 2006, 33, 215–236. [Google Scholar] [CrossRef]
- Khoutorsky, A.; Yanagiya, A.; Gkogkas, C.G.; Fabian, M.R.; Prager-Khoutorsky, M.; Cao, R.; Gamache, K.; Bouthiette, F.; Parsyan, A.; Sorge, R.E. Control of synaptic plasticity and memory via suppression of poly (A)-binding protein. Neuron 2013, 78, 298–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudry, M.; Bi, X.; Gall, C.; Lynch, G. The biochemistry of memory: The 26 year journey of a ‘new and specific hypothesis’. Neurobiol. Learn. Mem. 2011, 95, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Rex, C.S.; Casale, M.S.; Gall, C.M.; Lynch, G. Changes in synaptic morphology accompany actin signaling during LTP. J. Neurosci. 2007, 27, 5363–5372. [Google Scholar] [CrossRef] [PubMed]
- Rex, C.S.; Chen, L.Y.; Sharma, A.; Liu, J.; Babayan, A.H.; Gall, C.M.; Lynch, G. Different Rho GTPase–dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J. Cell Biol. 2009, 186, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Briz, V.; Zhu, G.; Wang, Y.; Liu, Y.; Avetisyan, M.; Bi, X.; Baudry, M. Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J. Neurosci. 2015, 35, 2269–2282. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Briz, V.; Seinfeld, J.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 deletion impairs mGluR-dependent LTD and fear memory extinction. Sci. Rep. 2017, 7, 42788. [Google Scholar] [CrossRef]
- Heysieattalab, S.; Lee, K.-H.; Liu, Y.; Wang, Y.; Foy, M.R.; Bi, X.; Baudry, M. Impaired cerebellar plasticity and eye-blink conditioning in calpain-1 knock-out mice. Neurobiol. Learn. Mem. 2019, 170, 106995. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Kakegawa, W.; Matsuda, S.; Kohda, K.; Nishiyama, J.; Takahashi, T.; Yuzaki, M. Cerebellar long-term depression requires dephosphorylation of TARP in Purkinje cells. Eur. J. Neurosci. 2012, 35, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Rostamiani, K.; Hsu, Y.-T.; Wang, Y.; Bi, X.; Baudry, M. Calpain-mediated regulation of stargazin in adult rat brain. Neuroscience 2011, 178, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, J.H. Cerebellar learning mechanisms. Brain Res. 2015, 1621, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Van Beugen, B.J.; De Zeeuw, C.I. Distributed synergistic plasticity and cerebellar learning. Nat. Rev. Neurosci. 2012, 13, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Hansel, C. Cerebellar long-term potentiation: Cellular mechanisms and role in learning. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 117, pp. 39–51. [Google Scholar]
- Schonewille, M.; Gao, Z.; Boele, H.-J.; Veloz, M.F.V.; Amerika, W.E.; Šimek, A.A.; De Jeu, M.T.; Steinberg, J.P.; Takamiya, K.; Hoebeek, F.E. Reevaluating the role of LTD in cerebellar motor learning. Neuron 2011, 70, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Briz, V.; Chishti, A.; Bi, X.; Baudry, M. Distinct roles for mu-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration. J. Neurosci. 2013, 33, 18880–18892. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hersheson, J.; Lopez, D.; Hammer, M.; Liu, Y.; Lee, K.H.; Pinto, V.; Seinfeld, J.; Wiethoff, S.; Sun, J.; et al. Defects in the CAPN1 Gene Result in Alterations in Cerebellar Development and Cerebellar Ataxia in Mice and Humans. Cell Rep. 2016, 16, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Shetty, A.; Gan-Or, Z.; Ashtiani, S.; Ruskey, J.A.; van de Warrenburg, B.; Wassenberg, T.; Kamsteeg, E.-J.; Rouleau, G.A.; Suchowersky, O. CAPN1 mutations: Expanding the CAPN1-related phenotype: From hereditary spastic paraparesis to spastic ataxia. Eur. J. Med Genet. 2019, 62, 103605. [Google Scholar] [CrossRef]
- Forman, O.P.; De Risio, L.; Mellersh, C.S. Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the Parson Russell Terrier dog breed. PLoS ONE 2013, 8, e64627. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Y.; Lopez, D.; Lee, M.; Dayal, S.; Hurtado, A.; Bi, X.; Baudry, M. Protection against TBI-induced neuronal death with post-treatment with a selective calpain-2 inhibitor in mice. J. Neurotrauma 2017, 35, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lopez, D.; Davey, P.G.; Cameron, D.J.; Nguyen, K.; Tran, J.; Marquez, E.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and calpain-2 play opposite roles in retinal ganglion cell degeneration induced by retinal ischemia/reperfusion injury. Neurobiol. Dis. 2016, 93, 121–128. [Google Scholar] [CrossRef]
- Seinfeld, J.; Baudry, N.; Xu, X.; Bi, X.; Baudry, M. Differential activation of calpain-1 and calpain-2 following kainate-induced seizure activity in rats and mice. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Bouslam, N.; Birouk, N.; Lissouba, A.; Chambers, D.B.; Vérièpe, J.; Androschuk, A.; Laurent, S.B.; Rochefort, D.; Spiegelman, D. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am. J. Hum. Genet. 2016, 98, 1038–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, G.; Afonso, P.M.; Salazar, I.L.; Duarte, C.B. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 2015, 1621, 82–101. [Google Scholar] [CrossRef] [PubMed]
- Zadran, S.; Jourdi, H.; Rostamiani, K.; Qin, Q.; Bi, X.; Baudry, M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J. Neurosci. 2010, 30, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.; Comprido, D.; Duarte, C.B. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology 2014, 76, 639–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briz, V.; Hsu, Y.-T.; Li, Y.; Lee, E.; Bi, X.; Baudry, M. Calpain-2-mediated PTEN degradation contributes to BDNF-induced stimulation of dendritic protein synthesis. J. Neurosci. 2013, 33, 4317–4328. [Google Scholar] [CrossRef] [Green Version]
- Oliver, M.W.; Baudry, M.; Lynch, G. The protease inhibitor leupeptin interferes with the development of LTP in hippocampal slices. Brain Res. 1989, 505, 233–238. [Google Scholar] [CrossRef]
- Denny, J.B.; Polan-Curtain, J.; Rodriguez, S.; Wayner, M.J.; Armstrong, D.L. Evidence that protein kinase M does not maintain long-term potentiation. Brain Res. 1990, 534, 201–208. [Google Scholar] [CrossRef]
- Panja, D.; Bramham, C.R. BDNF mechanisms in late LTP formation: A synthesis and breakdown. Neuropharmacology 2014, 76, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Rex, C.S.; Lin, C.-Y.; Kramár, E.A.; Chen, L.Y.; Gall, C.M.; Lynch, G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J. Neurosci. 2007, 27, 3017–3029. [Google Scholar] [CrossRef] [PubMed]
- Bevers, M.B.; Lawrence, E.; Maronski, M.; Starr, N.; Amesquita, M.; Neumar, R.W. Knockdown of m-calpain increases survival of primary hippocampal neurons following NMDA excitotoxicity. J. Neurochem. 2009, 108, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papouin, T.; Oliet, S.H. Organization, control and function of extrasynaptic NMDA receptors. Philos. Trans. R. Soc. B 2014, 369, 20130601. [Google Scholar] [CrossRef] [PubMed]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Ivanov, A.; Tyzio, R.; Pellegrino, C.; Ben-Ari, Y.; Clapham, D.E.; Medina, I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 2003, 40, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kurup, P.; Zhang, Y.; Goebel-Goody, S.M.; Wu, P.H.; Hawasli, A.H.; Baum, M.L.; Bibb, J.A.; Lombroso, P.J. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 2009, 29, 9330–9343. [Google Scholar] [CrossRef]
- Gladding, C.M.; Sepers, M.D.; Xu, J.; Zhang, L.Y.; Milnerwood, A.J.; Lombroso, P.J.; Raymond, L.A. Calpain and STriatal-Enriched protein tyrosine phosphatase (STEP) activation contribute to extrasynaptic NMDA receptor localization in a Huntington’s disease mouse model. Hum. Mol. Genet. 2012, 21, 3739–3752. [Google Scholar] [CrossRef]
- Zhao, Q.; Guo, Z.; Deng, W.; Fu, S.; Zhang, C.; Chen, M.; Ju, W.; Wang, D.; He, X. Calpain 2-mediated autophagy defect increases susceptibility of fatty livers to ischemia–reperfusion injury. Cell Death Dis. 2016, 7, e2186. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Das, D.; Kommaddi, R.P.; Diwakar, L.; Gowaikar, R.; Rupanagudi, K.V.; Bennett, D.A.; Ravindranath, V. Isoform-specific hyperactivation of calpain-2 occurs presymptomatically at the synapse in Alzheimer’s disease mice and correlates with memory deficits in human subjects. Sci. Rep. 2018, 8, 13119. [Google Scholar] [CrossRef] [PubMed]
- Gordy, C.; He, Y.-W. The crosstalk between autophagy and apoptosis: Where does this lead? Protein Cell 2012, 3, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Fimia, G.M.; Piacentini, M. Dismantling the autophagic arsenal when it is time to die: Concerted AMBRA1 degradation by caspases and calpains. Autophagy 2012, 8, 1255–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwood, S.M.; Yaqoob, M.M.; Allen, D.A. Caspase and calpain function in cell death: Bridging the gap between apoptosis and necrosis. Ann. Clin. Biochem. 2005, 42, 415–431. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hall, R.A.; Lee, M.; Kamgar-parsi, A.; Bi, X.; Baudry, M. The tyrosine phosphatase PTPN13/FAP-1 links calpain-2, TBI and tau tyrosine phosphorylation. Sci. Rep. 2017, 7, 11771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.S.; Lee, E.H.; Chung, C.W.; Jung, Y.K.; Jin, B.K.; Kim, S.U.; Oh, T.H.; Saido, T.C.; Oh, Y.J. Cleavage of Bax is mediated by caspase-dependent or-independent calpain activation in dopaminergic neuronal cells: Protective role of Bcl-2. J. Neurochem. 2001, 77, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Takano, J.; Tomioka, M.; Tsubuki, S.; Higuchi, M.; Iwata, N.; Itohara, S.; Maki, M.; Saido, T.C. Calpain Mediates Excitotoxic DNA Fragmentation via Mitochondrial Pathways in Adult Brains evidence from calpastatin mutant mice. J. Biol. Chem. 2005, 280, 16175–16184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.K. Calpain and caspase: Can you tell the difference? Trends Neurosci. 2000, 23, 20–26. [Google Scholar] [CrossRef]
- Ruiz-Vela, A.; de Buitrago, G.G.; Martínez-A, C. Implication of calpain in caspase activation during B cell clonal deletion. EMBO J. 1999, 18, 4988–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Wong, T.P.; Chery, N.; Gaertner, T.; Wang, Y.T.; Baudry, M. Calpain-mediated mGluR1α truncation: A key step in excitotoxicity. Neuron 2007, 53, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampfl, A.; Posmantur, R.; Zhao, X.; Schmutzhard, E.; Clifton, G.; Hayes, R. Mechanisms of calpain proteolysis following traumatic brain injury: Implications for pathology and therapy: A review and update. J. Neurotrauma 1997, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.; Larner, S.F.; Robinson, G.; Hayes, R.L. Neuroprotection targets after traumatic brain injury. Curr. Opin. Neurol. 2006, 19, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Yin, F.; Zhang, J.; Qian, Y. The role of calpains in traumatic brain injury. Brain Inj. 2014, 28, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Pike, B.R.; Zhao, X.; Newcomb, J.K.; Posmantur, R.M.; Wang, K.K.; Hayes, R.L. Regional calpain and caspase-3 proteolysis of α-spectrin after traumatic brain injury. Neuroreport 1998, 9, 2437–2442. [Google Scholar] [CrossRef]
- Pike, B.R.; Flint, J.; Dutta, S.; Johnson, E.; Wang, K.K.; Hayes, R.L. Accumulation of non-erythroid αII-spectrin and calpain-cleaved αII-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. J. Neurochem. 2001, 78, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Pineda, J.A.; Lewis, S.B.; Valadka, A.B.; Papa, L.; Hannay, H.J.; Heaton, S.C.; Demery, J.A.; Liu, M.C.; Aikman, J.M.; Akle, V. Clinical significance of α II-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J. Neurotrauma 2007, 24, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Brophy, G.M.; Pineda, J.A.; Papa, L.; Lewis, S.B.; Valadka, A.B.; Hannay, H.J.; Heaton, S.C.; Demery, J.A.; Liu, M.C.; Tepas, J.J., III. αII-Spectrin breakdown product cerebrospinal fluid exposure metrics suggest differences in cellular injury mechanisms after severe traumatic brain injury. J. Neurotrauma 2009, 26, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Mondello, S.; Robicsek, S.A.; Gabrielli, A.; Brophy, G.M.; Papa, L.; Tepas, J., III; Robertson, C.; Buki, A.; Scharf, D.; Jixiang, M. αII-spectrin breakdown products (SBDPs): Diagnosis and outcome in severe traumatic brain injury patients. J. Neurotrauma 2010, 27, 1203–1213. [Google Scholar] [CrossRef] [Green Version]
- Siman, R.; Giovannone, N.; Hanten, G.; Wilde, E.A.; McCauley, S.R.; Hunter, J.V.; Li, X.; Levin, H.S.; Smith, D.H. Evidence That the Blood Biomarker SNTF Predicts Brain Imaging Changes and Persistent Cognitive Dysfunction in Mild TBI Patients. Front. Neurol. 2013, 4, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siman, R.; Shahim, P.; Tegner, Y.; Blennow, K.; Zetterberg, H.; Smith, D.H. Serum SNTF increases in concussed professional ice hockey players and relates to the severity of postconcussion symptoms. J. Neurotrauma 2015, 32, 1294–1300. [Google Scholar] [CrossRef] [Green Version]
- Siman, R.; Cui, H.; Wewerka, S.; Hamel, L.; Smith, D.H.; Zwank, M. Serum SNTF, a Surrogate Marker of Axonal Injury, Is Prognostic for Lasting Brain Dysfunction in Mild TBI Treated in the Emergency Department. Front. Neurol. 2020, 11, 249. [Google Scholar] [CrossRef] [PubMed]
- Saatman, K.E.; Murai, H.; Bartus, R.T.; Smith, D.H.; Hayward, N.J.; Perri, B.R.; McIntosh, T.K. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc. Natl. Acad. Sci. USA 1996, 93, 3428–3433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posmantur, R.; Kampfl, A.; Siman, R.; Liu, S.; Zhao, X.; Clifton, G.; Hayes, R. A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neuroscience 1997, 77, 875–888. [Google Scholar] [CrossRef]
- Thompson, S.N.; Carrico, K.M.; Mustafa, A.G.; Bains, M.; Hall, E.D. A pharmacological analysis of the neuroprotective efficacy of the brain- and cell-permeable calpain inhibitor MDL-28170 in the mouse controlled cortical impact traumatic brain injury model. J. Neurotrauma 2010, 27, 2233–2243. [Google Scholar] [CrossRef] [Green Version]
- Bains, M.; Cebak, J.E.; Gilmer, L.K.; Barnes, C.C.; Thompson, S.N.; Geddes, J.W.; Hall, E.D. Pharmacological analysis of the cortical neuronal cytoskeletal protective efficacy of the calpain inhibitor SNJ-1945 in a mouse traumatic brain injury model. J. Neurochem. 2013, 125, 125–132. [Google Scholar] [CrossRef]
- Seubert, P.; Baudry, M.; Dudek, S.; Lynch, G. Calmodulin stimulates the degradation of brain spectrin by calpain. Synapse 1987, 1, 20–24. [Google Scholar] [CrossRef]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Y.; Nham, A.; Sherbaf, A.; Quach, D.; Yahya, E.; Ranburger, D.; Bi, X.; Baudry, M. Calpain-2 as a therapeutic target in repeated concussion–induced neuropathy and behavioral impairment. Sci. Adv. 2020, 6, eaba5547. [Google Scholar] [CrossRef]
Figure 1.
Calpain-2 deletion in cortex and hippocampal field CA1 in calpain-2 conditional knock-out (C2CKO) mice. Immunohistochemistry (IHC) with calpain-2 antibody (1:300; LS-C337641, LSBio) in cortex and CA1 of control (calpain-2 loxP) and C2CKO mice. Note the very large decrease in calpain-2 immunoreactivity in cortex and field CA1 of hippocampus. Scale bar: 50 µm. Details for the methods can be found in the Supplementary Materials.
Figure 1.
Calpain-2 deletion in cortex and hippocampal field CA1 in calpain-2 conditional knock-out (C2CKO) mice. Immunohistochemistry (IHC) with calpain-2 antibody (1:300; LS-C337641, LSBio) in cortex and CA1 of control (calpain-2 loxP) and C2CKO mice. Note the very large decrease in calpain-2 immunoreactivity in cortex and field CA1 of hippocampus. Scale bar: 50 µm. Details for the methods can be found in the Supplementary Materials.

Figure 2.
Changes in spectrin, striatal-enriched tyrosine phosphatase (STEP), PTPN13 breakdown product (P13BP), and TAR DNA-binding protein of about 43 kDa (TDP-43) in brain of WT, calpain-1 knock out (C1KO), and C2CKO mice 24 h after traumatic brain injury (TBI). (A,B) Representative western blots (WBs) of ipsilateral cortical tissue (P2 fraction) from WT, C1KO, and C2CKO mice collected 24 h after TBI or sham surgery. Note that calpain-1 is absent in C1KO mice and calpain-2 levels were reduced in C2CKO mice. Several calpain substrates including spectrin (1:500; MAB1622, EMD Millipore, Burlington, MA, USA), STEP (1:1000, NB300-202, Novus Biologicals, Colorado, CO, USA), PTPN13 (1:1000, PA5-72906, Thermo Fisher Scientific, Waltham, MA, USA), and TDP-43 (1:1000, 10782-2-AP, Proteintech, Chicago, IL, USA) were probed, and their fragments generated by calpain cleavage including SBDP, STEP33, and P13BPs were detected after TBI. (C) Quantification of WB. Ratios of SBDPs to spectrin, STEP33 to STEP61, P13BPs to PTPN13 were significantly increased in WT and C1KO but not in C2CKO mice after TBI. Levels of TDP-43 were not changed after TBI in all genotypes. * p < 0.05, ** p < 0.01. ns, no significant difference. N = 4. One way ANOVA followed by Bonferroni’s test. Full immunoblots for the blots shown here are presented in the Supplementary Materials.
Figure 2.
Changes in spectrin, striatal-enriched tyrosine phosphatase (STEP), PTPN13 breakdown product (P13BP), and TAR DNA-binding protein of about 43 kDa (TDP-43) in brain of WT, calpain-1 knock out (C1KO), and C2CKO mice 24 h after traumatic brain injury (TBI). (A,B) Representative western blots (WBs) of ipsilateral cortical tissue (P2 fraction) from WT, C1KO, and C2CKO mice collected 24 h after TBI or sham surgery. Note that calpain-1 is absent in C1KO mice and calpain-2 levels were reduced in C2CKO mice. Several calpain substrates including spectrin (1:500; MAB1622, EMD Millipore, Burlington, MA, USA), STEP (1:1000, NB300-202, Novus Biologicals, Colorado, CO, USA), PTPN13 (1:1000, PA5-72906, Thermo Fisher Scientific, Waltham, MA, USA), and TDP-43 (1:1000, 10782-2-AP, Proteintech, Chicago, IL, USA) were probed, and their fragments generated by calpain cleavage including SBDP, STEP33, and P13BPs were detected after TBI. (C) Quantification of WB. Ratios of SBDPs to spectrin, STEP33 to STEP61, P13BPs to PTPN13 were significantly increased in WT and C1KO but not in C2CKO mice after TBI. Levels of TDP-43 were not changed after TBI in all genotypes. * p < 0.05, ** p < 0.01. ns, no significant difference. N = 4. One way ANOVA followed by Bonferroni’s test. Full immunoblots for the blots shown here are presented in the Supplementary Materials.

Figure 3.
Changes in spectrin breakdown product (SBDP) and Phosphatase and Tensin Homolog (PTEN) around the lesion site in control and C2CKO mice after TBI. IHC with antibodies targeting SBDP (1:500, a gift from Dr. Saido, Riken, Japan) and full-length PTEN (1:600, 9556, Cell Signaling) around the lesion site was performed in control and C2CKO mice 24 h after TBI. Note that the levels of SBDP were much lower while PTEN levels were much higher in C2CKO mice as compared to control mice. Scale bar: 50 µm.
Figure 3.
Changes in spectrin breakdown product (SBDP) and Phosphatase and Tensin Homolog (PTEN) around the lesion site in control and C2CKO mice after TBI. IHC with antibodies targeting SBDP (1:500, a gift from Dr. Saido, Riken, Japan) and full-length PTEN (1:600, 9556, Cell Signaling) around the lesion site was performed in control and C2CKO mice 24 h after TBI. Note that the levels of SBDP were much lower while PTEN levels were much higher in C2CKO mice as compared to control mice. Scale bar: 50 µm.

Figure 4.
Reduced lesion volume after TBI in C2CKO mice. (Left): Representative images of Nissl-stained brain sections (Bregma 1.58 mm) in WT and C2CKO mice collected 3 days after TBI. Scale bar: 1 mm. (Right): Lesion volume in WT and C2CKO mice measured 3 days after TBI. Lesion areas were measured in eight sections (Bregma 1.54, 0.50, −0.58, −1.58, −1.94, −2.30, −2.70, and −3.40 mm) from each brain. Total lesion volume in each brain was calculated based on the lesion area in each section and the distance between sections. N = 5 for WT, N = 4 for C2CKO. * p < 0.05, t-test.
Figure 4.
Reduced lesion volume after TBI in C2CKO mice. (Left): Representative images of Nissl-stained brain sections (Bregma 1.58 mm) in WT and C2CKO mice collected 3 days after TBI. Scale bar: 1 mm. (Right): Lesion volume in WT and C2CKO mice measured 3 days after TBI. Lesion areas were measured in eight sections (Bregma 1.54, 0.50, −0.58, −1.58, −1.94, −2.30, −2.70, and −3.40 mm) from each brain. Total lesion volume in each brain was calculated based on the lesion area in each section and the distance between sections. N = 5 for WT, N = 4 for C2CKO. * p < 0.05, t-test.

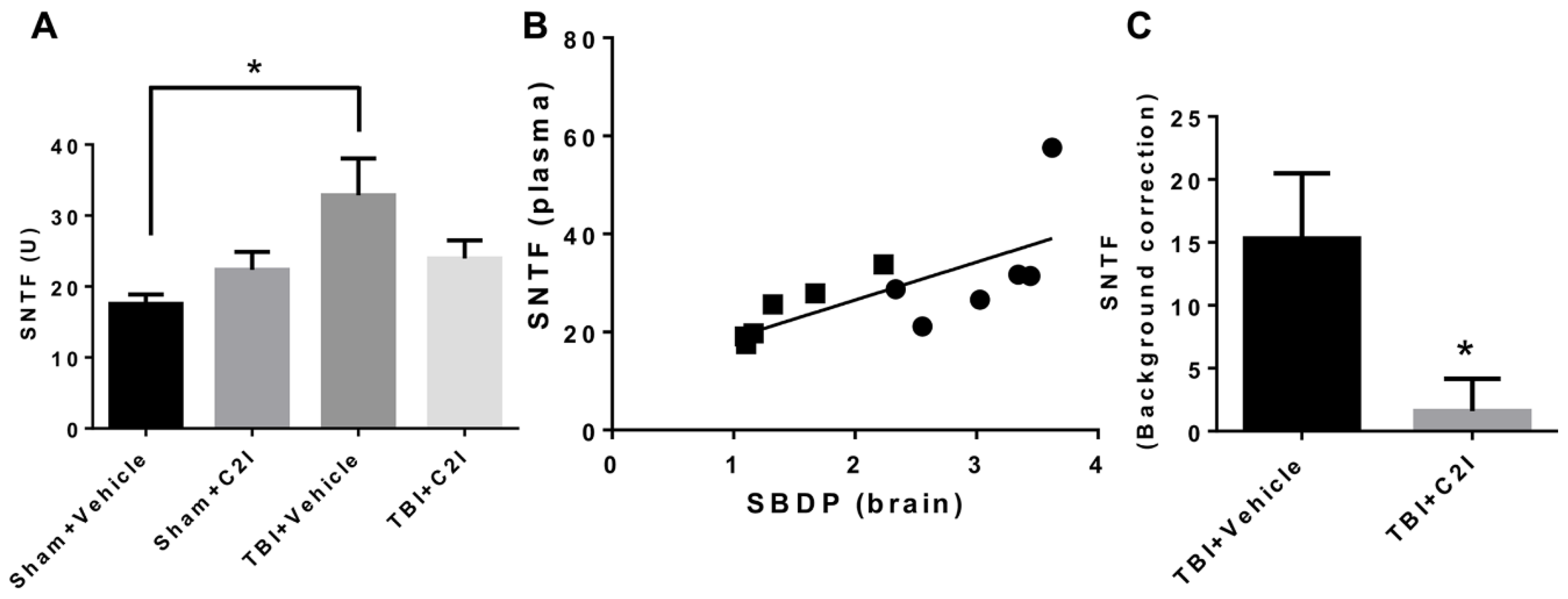
Figure 5.
Levels of Spectrin N-Terminal Fragment (SNTF) in mouse blood 24 h after TBI with or without treatment with calpain-2 inhibitor (C2I). (A) Levels of SNTF in plasma of CD-1 mice collected 24 h after TBI. C2I (0.3 mg/kg) or vehicle was injected intravenously 1 h after TBI. * p < 0.05. N = 4 for Sham + Vehicle and Sham + C2I. N = 6 for TBI + Vehicle and TBI + C2I. One way ANOVA followed by Bonferroni’s test. (B) Correlation between SNTF levels in plasma and SBDP levels in brain of CD-1 mice 24 h after TBI. Black circles: TBI + Veh; black rectangles: TBI + C2I. R = 0.70. (C) Increase in SNTF levels after TBI in plasma of CD-1 mice collected 24 h after TBI. SNTF levels after sham surgery (background SNTF level) were subtracted from SNTF levels after TBI. * p < 0.05. N = 6, t-test.
Figure 5.
Levels of Spectrin N-Terminal Fragment (SNTF) in mouse blood 24 h after TBI with or without treatment with calpain-2 inhibitor (C2I). (A) Levels of SNTF in plasma of CD-1 mice collected 24 h after TBI. C2I (0.3 mg/kg) or vehicle was injected intravenously 1 h after TBI. * p < 0.05. N = 4 for Sham + Vehicle and Sham + C2I. N = 6 for TBI + Vehicle and TBI + C2I. One way ANOVA followed by Bonferroni’s test. (B) Correlation between SNTF levels in plasma and SBDP levels in brain of CD-1 mice 24 h after TBI. Black circles: TBI + Veh; black rectangles: TBI + C2I. R = 0.70. (C) Increase in SNTF levels after TBI in plasma of CD-1 mice collected 24 h after TBI. SNTF levels after sham surgery (background SNTF level) were subtracted from SNTF levels after TBI. * p < 0.05. N = 6, t-test.

Figure 6.
Different subcellular locations and functions of calpain-1 and calpain-2 in excitatory neurons. Calpain-1 is preferentially linked to synaptic N-Methyl-D-Aspartate (NMDA) receptors while calpain-2 is downstream of extra-synaptic NMDA receptors in neurons. Synaptic NMDAR activation activates calpain-1, which cleaves and inhibits PHLPP1α/β. PHLPP1α/β inhibits AKT and ERK. Thus, calpain-1-mediated cleavage of PHLPP1 activates AKT and ERK, which trigger long-term potentiation (LTP) and promote neuronal survival. On the other hand, extra-synaptic NMDAR activation induces phosphorylation and prolonged activation of calpain-2. Calpain-2 cleaves its substrates such as PTEN and STEP, leading to reduced LTP magnitude and neurodegeneration.
Figure 6.
Different subcellular locations and functions of calpain-1 and calpain-2 in excitatory neurons. Calpain-1 is preferentially linked to synaptic N-Methyl-D-Aspartate (NMDA) receptors while calpain-2 is downstream of extra-synaptic NMDA receptors in neurons. Synaptic NMDAR activation activates calpain-1, which cleaves and inhibits PHLPP1α/β. PHLPP1α/β inhibits AKT and ERK. Thus, calpain-1-mediated cleavage of PHLPP1 activates AKT and ERK, which trigger long-term potentiation (LTP) and promote neuronal survival. On the other hand, extra-synaptic NMDAR activation induces phosphorylation and prolonged activation of calpain-2. Calpain-2 cleaves its substrates such as PTEN and STEP, leading to reduced LTP magnitude and neurodegeneration.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, Y.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells 2020, 9, 2698. https://doi.org/10.3390/cells9122698
AMA Style
Wang Y, Liu Y, Bi X, Baudry M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells. 2020; 9(12):2698. https://doi.org/10.3390/cells9122698
Chicago/Turabian StyleWang, Yubin, Yan Liu, Xiaoning Bi, and Michel Baudry. 2020. "Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death" Cells 9, no. 12: 2698. https://doi.org/10.3390/cells9122698
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.