Comprehensive Exonic Sequencing of Hemiplegic Migraine-Related Genes in a Cohort of Suspected Probands Identifies Known and Potential Pathogenic Variants

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Molecular Analysis of the PRRT2 Mutation Hotspot
2.3. Whole Exome Sequencing of the HM Cohort
2.4. Molecular Analysis of Genes Previously Implicated in HM in WES Data of Cohort
3. Results
3.1. Screening of HM Cases for Variants around the PRRT2 Mutation Hotspot by Sanger Sequencing
3.2. Examination of Whole Exome Sequencing (WES) Data for Pathogenic Variants in HM-Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Russell, M.B.; Ducros, A. Sporadic and familial hemiplegic migraine: Pathophysiological mechanisms, clinical characteristics, diagnosis, and management. Lancet Neurol. 2011, 10, 457–470. [Google Scholar] [CrossRef]
- Chen, S.P.; Tolner, E.A.; Eikermann-Haerter, K. Animal models of monogenic migraine. Cephalalgia 2016, 36, 704–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiekkala, M.E.; Vuola, P.; Artto, V.; Happola, P.; Happola, E.; Vepsalainen, S.; Cuenca-Leon, E.; Lal, D.; Gormley, P.; Hamalainen, E.; et al. The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families. Cephalalgia 2018, 38, 1849–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksemous, N.; Smith, R.A.; Sutherland, H.G.; Maher, B.H.; Ibrahim, O.; Nicholson, G.A.; Carpenter, E.P.; Lea, R.A.; Cader, M.Z.; Griffiths, L.R. Targeted next generation sequencing identifies a genetic spectrum of DNA variants in patients with hemiplegic migraine. Cephalalgia Rep. 2019, 2, 2515816319881630. [Google Scholar] [CrossRef] [Green Version]
- Riant, F.; Roze, E.; Barbance, C.; Meneret, A.; Guyant-Marechal, L.; Lucas, C.; Sabouraud, P.; Trebuchon, A.; Depienne, C.; Tournier-Lasserve, E. PRRT2 mutations cause hemiplegic migraine. Neurology 2012, 79, 2122–2124. [Google Scholar] [CrossRef]
- Marini, C.; Conti, V.; Mei, D.; Battaglia, D.; Lettori, D.; Losito, E.; Bruccini, G.; Tortorella, G.; Guerrini, R. PRRT2 mutations in familial infantile seizures, paroxysmal dyskinesia, and hemiplegic migraine. Neurology 2012, 79, 2109–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi-Fakhari, D.; Saffari, A.; Westenberger, A.; Klein, C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain 2015, 138, 3476–3495. [Google Scholar] [CrossRef] [Green Version]
- Dale, R.C.; Gardiner, A.; Antony, J.; Houlden, H. Familial PRRT2 mutation with heterogeneous paroxysmal disorders including paroxysmal torticollis and hemiplegic migraine. Dev. Med. Child. Neurol. 2012, 54, 958–960. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-J.; Lin, Y.; Xiong, Z.-Q.; Wei, W.; Ni, W.; Tan, G.-H.; Guo, S.-L.; He, J.; Chen, Y.-F.; Zhang, Q.-J.; et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat. Genet. 2011, 43, 1252. [Google Scholar] [CrossRef]
- Wang, J.-L.; Cao, L.; Li, X.-H.; Hu, Z.-M.; Li, J.-D.; Zhang, J.-G.; Liang, Y.; San, A.; Li, N.; Chen, S.-Q.; et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain 2011, 134, 3493–3501. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, A.R.; Jaffer, F.; Dale, R.C.; Labrum, R.; Erro, R.; Meyer, E.; Xiromerisiou, G.; Stamelou, M.; Walker, M.; Kullmann, D.; et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain 2015, 138, 3567–3580. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Grinton, B.E.; Kivity, S.; Afawi, Z.; Zuberi, S.M.; Hughes, J.N.; Pridmore, C.; Hodgson, B.L.; Iona, X.; Sadleir, L.G.; et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am. J. Hum. Genet. 2012, 90, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, S.; Yoshiura, K.; Kinoshita, A.; Kikuchi, T.; Nakane, Y.; Kato, N.; Sadamatsu, M.; Konishi, T.; Nagamitsu, S.; Matsuura, M.; et al. Mutations in PRRT2 responsible for paroxysmal kinesigenic dyskinesias also cause benign familial infantile convulsions. J. Hum. Genet. 2012, 57, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Valente, P.; Castroflorio, E.; Rossi, P.; Fadda, M.; Sterlini, B.; Cervigni, R.I.; Prestigio, C.; Giovedi, S.; Onofri, F.; Mura, E.; et al. PRRT2 Is a Key Component of the Ca(2+)-Dependent Neurotransmitter Release Machinery. Cell Rep. 2016, 15, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Fruscione, F.; Valente, P.; Sterlini, B.; Romei, A.; Baldassari, S.; Fadda, M.; Prestigio, C.; Giansante, G.; Sartorelli, J.; Rossi, P.; et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain 2018, 141, 1000–1016. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Tang, H.D.; Huang, X.J.; Zheng, L.; Liu, X.L.; Wang, T.; Wang, J.Y.; Cao, L.; Chen, S.D. PRRT2 truncated mutations lead to nonsense-mediated mRNA decay in Paroxysmal Kinesigenic Dyskinesia. Parkinsonism Relat. Disord. 2014, 20, 1399–1404. [Google Scholar] [CrossRef]
- Lee, H.Y.; Xu, Y.; Huang, Y.; Ahn, A.H.; Auburger, G.W.; Pandolfo, M.; Kwiecinski, H.; Grimes, D.A.; Lang, A.E.; Nielsen, J.E.; et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum. Mol. Genet. 2004, 13, 3161–3170. [Google Scholar] [CrossRef] [Green Version]
- Rainier, S.; Thomas, D.; Tokarz, D.; Ming, L.; Bui, M.; Plein, E.; Zhao, X.; Lemons, R.; Albin, R.; Delaney, C.; et al. Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch. Neurol. 2004, 61, 1025–1029. [Google Scholar] [CrossRef]
- Weller, C.M.; Leen, W.G.; Neville, B.G.; Duncan, J.S.; de Vries, B.; Geilenkirchen, M.A.; Haan, J.; Kamsteeg, E.J.; Ferrari, M.D.; van den Maagdenberg, A.M.; et al. A novel SLC2A1 mutation linking hemiplegic migraine with alternating hemiplegia of childhood. Cephalalgia 2015, 35, 10–15. [Google Scholar] [CrossRef]
- Seidner, G.; Alvarez, M.G.; Yeh, J.I.; O’Driscoll, K.R.; Klepper, J.; Stump, T.S.; Wang, D.; Spinner, N.B.; Birnbaum, M.J.; De Vivo, D.C. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat. Genet. 1998, 18, 188–191. [Google Scholar] [CrossRef]
- Kovermann, P.; Hessel, M.; Kortzak, D.; Jen, J.C.; Koch, J.; Fahlke, C.; Freilinger, T. Impaired K(+) binding to glial glutamate transporter EAAT1 in migraine. Sci. Rep. 2017, 7, 13913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jen, J.C.; Wan, J.; Palos, T.P.; Howard, B.D.; Baloh, R.W. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 2005, 65, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Van Paesschen, W.; Stalmans, I.; Horita, S.; Yamada, H.; Bergmans, B.A.; Legius, E.; Riant, F.; De Jonghe, P.; Li, Y.; et al. Defective membrane expression of the Na(+)-HCO(3)(-) cotransporter NBCe1 is associated with familial migraine. Proc. Natl. Acad. Sci. USA 2010, 107, 15963–15968. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, T.; Inatomi, J.; Sekine, T.; Cha, S.H.; Kanai, Y.; Kunimi, M.; Tsukamoto, K.; Satoh, H.; Shimadzu, M.; Tozawa, F.; et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat. Genet. 1999, 23, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, P.; Palumbo, O.; Operto, F.F.; Castellana, S.; Di Muro, E.; Leopne, M.P.; Biagini, T.; Pastorino, G.; Mazza, T.; Carella, M.; et al. Familial Hemiplegic Migraine: A new gene in an Italian family. Arch. Clin. Med. Case Rep. 2019, 3, 534–543. [Google Scholar] [CrossRef] [Green Version]
- Heinzen, E.L.; Swoboda, K.J.; Hitomi, Y.; Gurrieri, F.; Nicole, S.; de Vries, B.; Tiziano, F.D.; Fontaine, B.; Walley, N.M.; Heavin, S.; et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat. Genet. 2012, 44, 1030–1034. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.Y.; Markus, H.S. CADASIL: Migraine, Encephalopathy, Stroke and Their Inter-Relationships. PLoS ONE 2016, 11, e0157613. [Google Scholar] [CrossRef]
- Rossi, G.; Shambhu, S. Hemiplegic Migraine as the Initial Presentation of Biopsy Positive Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy. Cureus 2018, 10, e2631. [Google Scholar] [CrossRef] [Green Version]
- Maksemous, N.; Roy, B.; Smith, R.A.; Griffiths, L.R. Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol. Genet. Genom. Med. 2016, 4, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Maksemous, N.; Smith, R.A.; Sutherland, H.G.; Sampaio, H.; Griffiths, L.R. Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity. Int. J. Mol. Sci. 2018, 19, 3113. [Google Scholar] [CrossRef] [Green Version]
- Lafreniere, R.G.; Cader, M.Z.; Poulin, J.F.; Andres-Enguix, I.; Simoneau, M.; Gupta, N.; Boisvert, K.; Lafreniere, F.; McLaughlan, S.; Dube, M.P.; et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 2010, 16, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pascual, J.M.; Iserovich, P.; Yang, H.; Ma, L.; Kuang, K.; Zuniga, F.A.; Sun, R.P.; Swaroop, K.M.; Fischbarg, J.; et al. Functional studies of threonine 310 mutations in Glut1: T310I is pathogenic, causing Glut1 deficiency. J. Biol. Chem 2003, 278, 49015–49021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelzer, N.; Haan, J.; Stam, A.H.; Vijfhuizen, L.S.; Koelewijn, S.C.; Smagge, A.; de Vries, B.; Ferrari, M.D.; van den Maagdenberg, A.; Terwindt, G.M. Clinical spectrum of hemiplegic migraine and chances of finding a pathogenic mutation. Neurology 2018, 90, e575–e582. [Google Scholar] [CrossRef]
- Pelzer, N.; de Vries, B.; Kamphorst, J.T.; Vijfhuizen, L.S.; Ferrari, M.D.; Haan, J.; van den Maagdenberg, A.M.; Terwindt, G.M. PRRT2 and hemiplegic migraine: A complex association. Neurology 2014, 83, 288–290. [Google Scholar] [CrossRef]
- Jing, X.Y.; Li, X.H.; Yuan, P.; Deng, J.; Hu, B.; Wang, Y. A novel mutation and functional implications of 5 variants in the PRRT2 gene in 20 paroxysmal kinesigenic dyskinesia pedigrees. Parkinsonism Relat. Disord. 2013, 19, 639–642. [Google Scholar] [CrossRef]
- Adamczyk, A.; Gause, C.D.; Sattler, R.; Vidensky, S.; Rothstein, J.D.; Singer, H.; Wang, T. Genetic and functional studies of a missense variant in a glutamate transporter, SLC1A3, in Tourette syndrome. Psychiatr. Genet. 2011, 21, 90–97. [Google Scholar] [CrossRef]
- De Giorgis, V.; Veggiotti, P. GLUT1 deficiency syndrome 2013: Current state of the art. Seizure 2013, 22, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef]
- Mohammad, S.S.; Coman, D.; Calvert, S. Glucose transporter 1 deficiency syndrome and hemiplegic migraines as a dominant presenting clinical feature. J. Paediatr. Child. Health 2014, 50, 1025–1026. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, G.; Liu, R.; Wei, J.; Zhang-Negrerie, D.; Jian, X.; Gao, Q. Mechanistic Study of Human Glucose Transport Mediated by GLUT1. J. Chem. Inf. Model. 2016, 56, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Abuladze, N.; Azimov, R.; Newman, D.; Sassani, P.; Liu, W.; Tatishchev, S.; Pushkin, A.; Kurtz, I. Critical amino acid residues involved in the electrogenic sodium-bicarbonate cotransporter kNBC1-mediated transport. J. Physiol. 2005, 565, 717–730. [Google Scholar] [CrossRef]
- Sinning, A.; Hubner, C.A. Minireview: pH and synaptic transmission. FEBS Lett. 2013, 587, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Kors, E.E.; Terwindt, G.M.; Vermeulen, F.L.; Fitzsimons, R.B.; Jardine, P.E.; Heywood, P.; Love, S.; van den Maagdenberg, A.M.; Haan, J.; Frants, R.R.; et al. Delayed cerebral edema and fatal coma after minor head trauma: Role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 2001, 49, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Bassi, M.T.; Bresolin, N.; Tonelli, A.; Nazos, K.; Crippa, F.; Baschirotto, C.; Zucca, C.; Bersano, A.; Dolcetta, D.; Boneschi, F.M.; et al. A novel mutation in the ATP1A2 gene causes alternating hemiplegia of childhood. J. Med. Genet. 2004, 41, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Panagiotakaki, E.; De Grandis, E.; Stagnaro, M.; Heinzen, E.L.; Fons, C.; Sisodiya, S.; de Vries, B.; Goubau, C.; Weckhuysen, S.; Kemlink, D.; et al. Clinical profile of patients with ATP1A3 mutations in Alternating Hemiplegia of Childhood-a study of 155 patients. Orphanet. J. Rare Dis. 2015, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Keryanov, S.; Gardner, K.L. Physical mapping and characterization of the human Na,K-ATPase isoform, ATP1A4. Gene 2002, 292, 151–166. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, A.H.; Olofsson, I.; Chalmer, M.A.; Olesen, J.; Hansen, T.F. Higher burden of rare frameshift indels in genes related to synaptic transmission separate familial hemiplegic migraine from common types of migraine. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lin, Z.J.; Liu, L.; Xu, H.Q.; Shi, Y.W.; Yi, Y.H.; He, N.; Liao, W.P. Epilepsy-associated genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Dale, R.C.; Gardiner, A.; Branson, J.A.; Houlden, H. Benefit of carbamazepine in a patient with hemiplegic migraine associated with PRRT2 mutation. Dev. Med. Child. Neurol. 2014, 56, 910. [Google Scholar] [CrossRef] [Green Version]




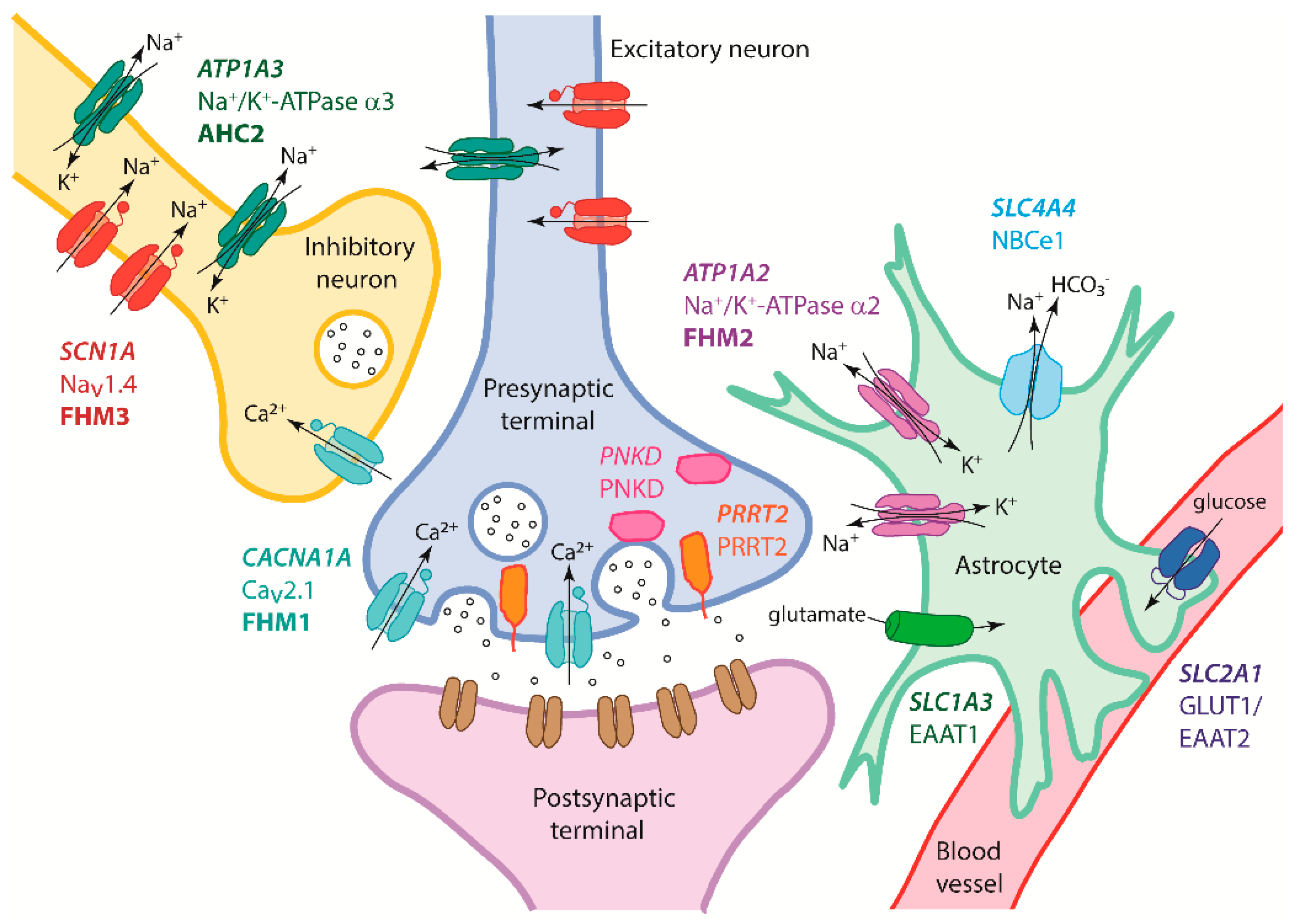{kind=link}
{kind=link}
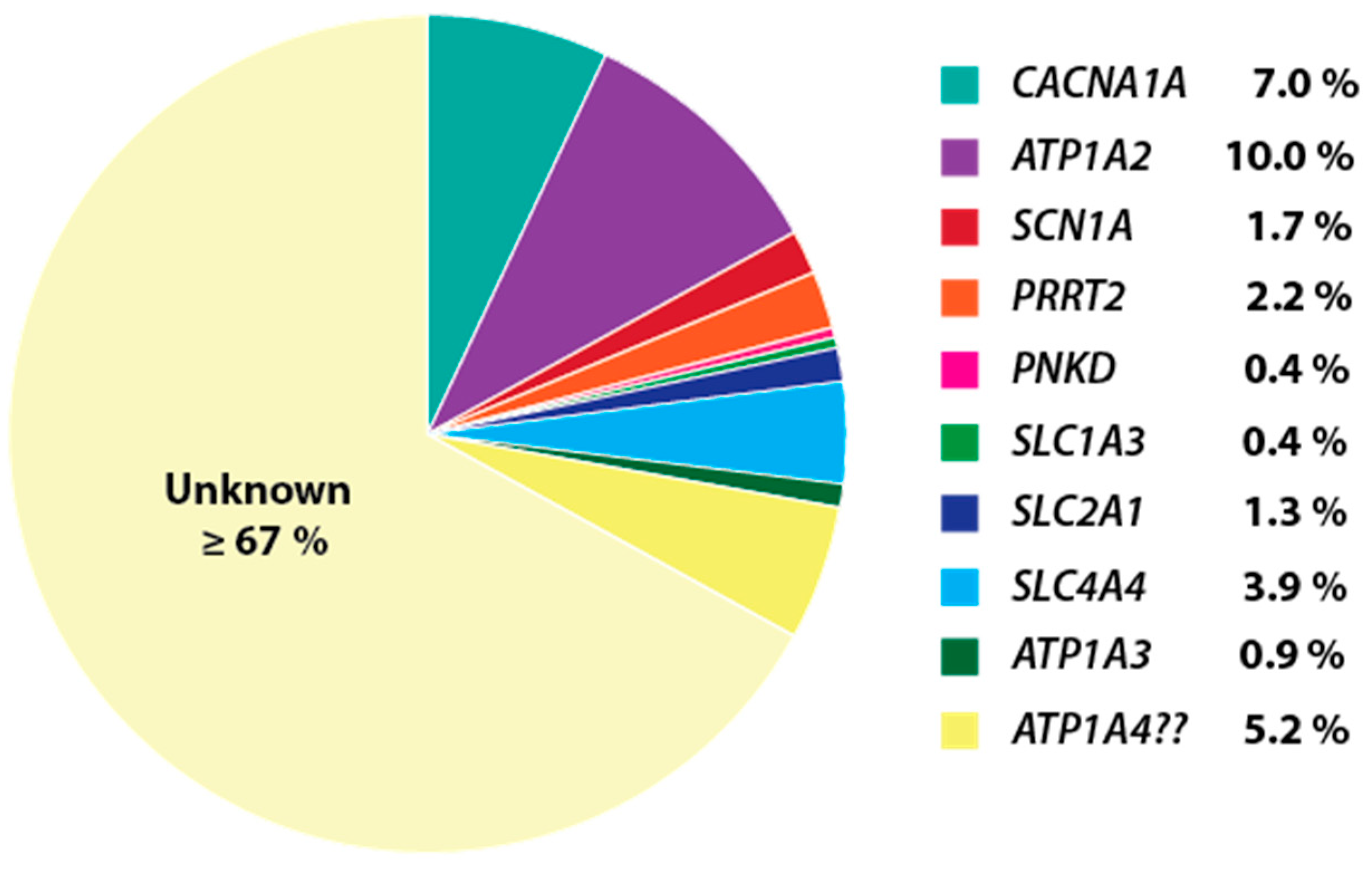{kind=link}
| Gene | Patient ID# | Locus | Transcript | Coding | Protein Change | dbSNP | MAF gnomAD (No. Alleles) | MAF gnomAD Eur NF (No. Alleles) | ClinVar (or LOVD) Annotation | SIFT a | Polyphen2 b | Mutation Taster c | Conservation d PhylopP/ PhastCons |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRRT2 | 224 225 | chr16:29824442 | NM_145239.3 | c.67G>A | p.Glu23Lys | rs140383655 | 0.0011 (306) | 0.0017 (219) | LB, VUS | T | B | P | 0.01/0.68 |
| 185 201 211 227 | chr16:29825022 | NM_145239.3 | c.647C>T | p.Pro216Leu | rs76335820 | 0.007 (1737) | 0.010 (1183) | B, LB | D | D | D | 3.75/0.99 | |
| 433 | chr16:29825025 | NM_145239.3 | c.650G>A | p.Arg217Gln | rs75497546 | 0.000008 (2) | 0.0000092 (1) | D | D | P | 1.94/0.75 | ||
| 137 | chr16:29825762 | NM_145239.3 | c.988G>A | p.Ala330Thr | rs757132796 | 0.0000042 (1) | 0.0000092 (1) | D | PD | D | 2.77/1 | ||
| 261 | chr16:29825888 | NM_001256442 | c.1114C>T | p.Leu372Phe | rs565298585 | 0.000046 (13) | 0.0001 (13) | D | B | P | 0.07/0.99 | ||
| PNKD | 32 | chr2:219209684 | NM_015488.5 | c.1140_1145delTATGCA | p.Met381_His382del | rs576363906 | 0.00061 (170) | 0.0010 (135) | LB | N/A | N/A | P | N/A |
| SLC1A3 | 135 155 159 196 | chr5:36677083 | NM_004172.5 | c.657G>C | p.Glu219Asp | rs2032892 | 0.023 (6697) | 0.0069 (887) | B, LB | T | B | P | |
| 107 | chr5:36680556 | NM_004172.5 | c.1154G>A | p.Arg385His | rs115702388 | 0.00026 (74) | 0.000061 (8) | B, LB | D | D | D | 6.24/0.998 | |
| SLC2A1 | 109 | chr1:43396789 | NM_006516.2 | c.203C>T | p.Ser68Leu | 0 | 0 | VUS | D | B | D | 3.79/0.997 | |
| 189 | chr1:43394924 | NM_006516.2 | c.929C>T | p.Thr310Ile | rs80359824 | 0 | 0 | D | D | D | 3.47/1 | ||
| 179 | chr1:43394873 | NM_006516.2 | c.972+7del (intronic) | p.Leu232PhefsTer? | rs531385270 | 0.00018 (52) | 0.00038 (50) | B, LB, VUS | N/A | N/A | D | 1.22/0.62 | |
| SLC4A4 | 136 | chr4:72205146 | NM_001098484.3 | c.313C>G | p.Pro105Ala | rs768913941 | 0.000056 (16) | 0.000092 (12) | D | B/PD | D | 6.09/1 | |
| 142 | chr4:72306423 | NM_001098484.3 | c.898A>G | p.Ile300Val | rs747159754 | 0.000053 (15) | 0.00010 (11) | T | B | D | 1.72/1 | ||
| 237 | chr4:72316925 | NM_001098484.3 | c.1229G>A | p.Gly410Glu | 0 | 0 | T | B | D | 5.76/1 | |||
| 120 179 | chr4:72338589 | NM_001098484.3 | c.1805A>G | p.Lys602Arg | rs72650362 | 0.0022 (609) | 0.0035 (455) | D | D | D | 5.00/1 | ||
| 202 232 | chr4:72363294 | NM_001098484.3 | c.2051A>T | p.Asn684Ile | rs35891845 | 0.0016 (443) | 0.0029 (368) | T | B | D | 2.58/1 | ||
| 128 | chr4:72399974 | NM_001098484.3 | c.2311C>T | p.Pro771Ser | rs140882617 | 0.0014 (414) | 0.0017 (231) | VUS | T | B | D | 2.55/1 | |
| 150 | chr4:72412109 | NM_001098484.3 | c.2485C>A | p.Leu829Ile | rs201643562 | 0.00036 (103) | 0.00056 (72) | VUS | D | D | D | 4.27/1 | |
| ATP1A3 | 165 | chr19:42474557 | NM_152296.5 | c.2401G>A | p.Asp801Asn | rs80356537 | 0 | 0 | Pathogenic for AHC2 | D | D | D | 4.83/1 |
| 87 | chr19:42474436 | NM_152296.5 | c.2443G>A | p.Glu815Lys | rs387907281 | 0 | 0 | Pathogenic for AHC2 | D | D | D | 4.77/1 | |
| ATP1A4 | 228 | chr1:160123000 | NM_144699.4 | c.193G>A | p.Val65Met | rs7549352 | 0.00087 (246) | 0.0015 (194) | D | PD | D | 2.14/0.95 | |
| 150 | chr1:160125005 | NM_144699.4 | c.378G>T | p.Gln126His | rs370755520 | 0.0013 (364) | 0 | D | B | D | 2.46/0.93 | ||
| 161 186 | chr1:160125859 | NM_144699.4 | c.436G>A | p.Val146Ile | rs41288133 | 0.0022 (623) | 0.0041 (539) | T | B | P | 0.322/0 | ||
| 186 | chr1:160129260 | NM_144699.4 | c.722A>G | p.His241Arg | rs151137285 | 0.0005 (140) | 0.0010 (129) | D | B | D | 4.453/1 | ||
| 189 | chr1:160133954 | NM_144699.4 | c.787C>T | p.Arg263Trp | rs146761116 | 0.00037 (105) | 0.00063 (81) | D | D | P | 1.41/0.95 | ||
| 133 144 204 | chr1:160133955 | NM_144699.4 | c.788G>A | p.Arg263Gln | rs76528638 | 0.014 (3969) | 0.0012 (159) | T | B | P | 0.83/0.92 | ||
| 141 188 | chr1:160134012 | NM_144699.4 | c.845C>T | p.Thr282Met | rs144463520 | 0.001 (307) | 0.0020 (268) | D | D | D | 5.19/1 | ||
| 92 | chr1:160136403 | NM_144699.4 | c.1133C>T | p.Thr378Met | rs150693480 | 0.000045 (13) | 0.000061 (8) | D | D | D | 5.09/1 | ||
| 90 | chr1:160136459 | NM_144699.4 | c.1189G>A | p.Ala397Thr | rs147875149 | 0.000050 (14) | 0.0000077 (1) | D | D | D | 1.84/1 | ||
| 144 | chr1:160141171 | NM_144699.4 | c.1622T>G | p.Met541Arg | rs16831482 | 0.000004 (1) | 0.0000087 (1) | D | PD | D | 1.63/0.87 | ||
| 144 | chr1:160141525 | NM_144699.4 | c.1832A>G | p.Lys611Arg | rs79938119 | 0.0041 (1180) | 0.000061 (4) | B, VUS (LOVD) | D | D | D | 2.64/1 | |
| 96 | chr1:160143962 | NM_144699.4 | c.2053G>C | p.Asp685His | rs144428770 | 0.00081 (228) | 0.0016 (207) | D | D | D | 3.49/0.96 | ||
| 124 | chr1:160146341 | NM_144699.4 | c.2539A>T | p.Thr847Ser | rs145873902 | 0.00016 (45) | 0.00026 (34) | T | B | P | 1.38/0.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutherland, H.G.; Maksemous, N.; Albury, C.L.; Ibrahim, O.; Smith, R.A.; Lea, R.A.; Haupt, L.M.; Jenkins, B.; Tsang, B.; Griffiths, L.R. Comprehensive Exonic Sequencing of Hemiplegic Migraine-Related Genes in a Cohort of Suspected Probands Identifies Known and Potential Pathogenic Variants. Cells 2020, 9, 2368. https://doi.org/10.3390/cells9112368
Sutherland HG, Maksemous N, Albury CL, Ibrahim O, Smith RA, Lea RA, Haupt LM, Jenkins B, Tsang B, Griffiths LR. Comprehensive Exonic Sequencing of Hemiplegic Migraine-Related Genes in a Cohort of Suspected Probands Identifies Known and Potential Pathogenic Variants. Cells. 2020; 9(11):2368. https://doi.org/10.3390/cells9112368
Chicago/Turabian StyleSutherland, Heidi G., Neven Maksemous, Cassie L. Albury, Omar Ibrahim, Robert A. Smith, Rod A. Lea, Larisa M. Haupt, Bronwyn Jenkins, Benjamin Tsang, and Lyn R. Griffiths. 2020. "Comprehensive Exonic Sequencing of Hemiplegic Migraine-Related Genes in a Cohort of Suspected Probands Identifies Known and Potential Pathogenic Variants" Cells 9, no. 11: 2368. https://doi.org/10.3390/cells9112368