Human P2X7 Receptor Causes Cycle Arrest in RPMI-8226 Myeloma Cells to Alter the Interaction with Osteoblasts and Osteoclasts

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
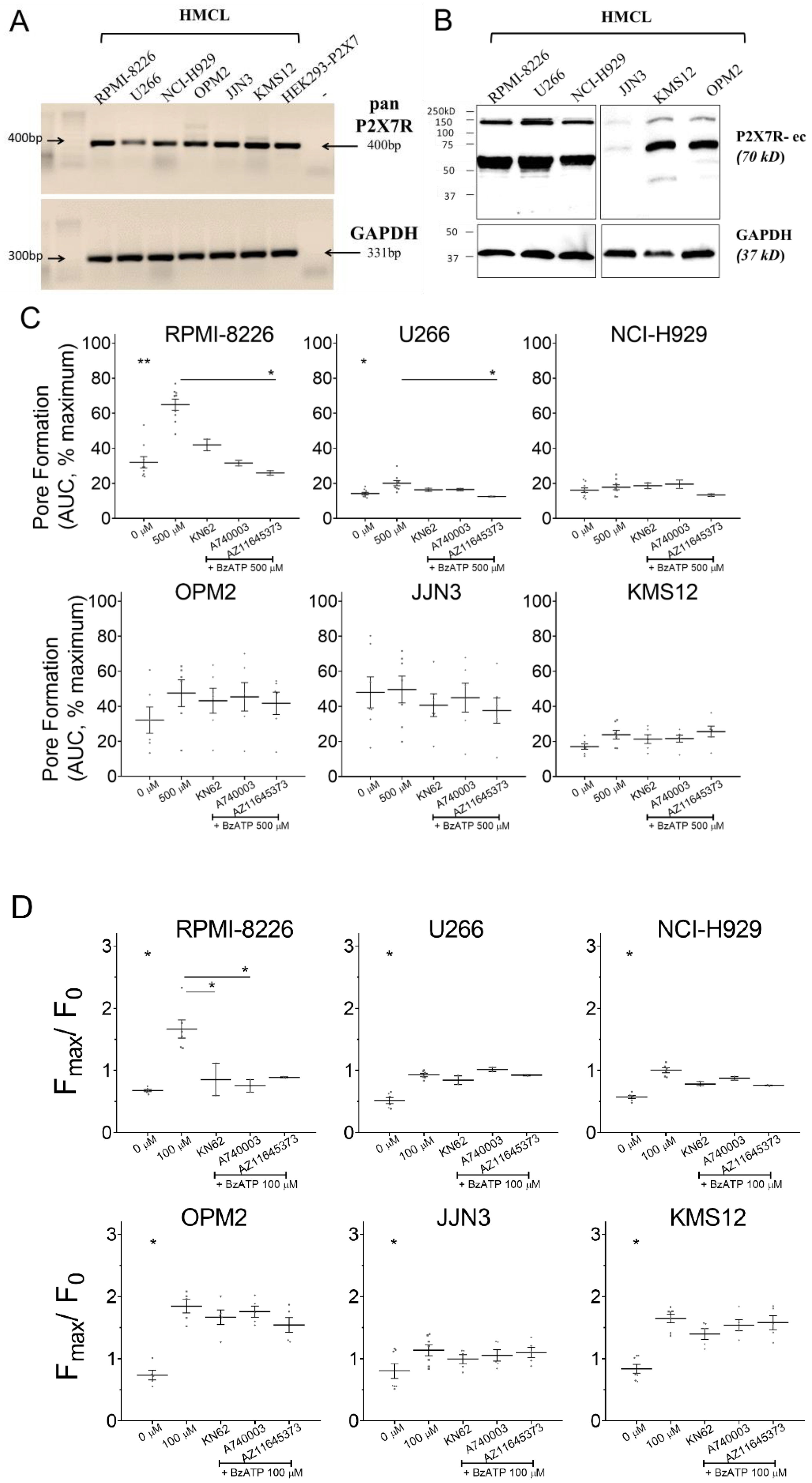
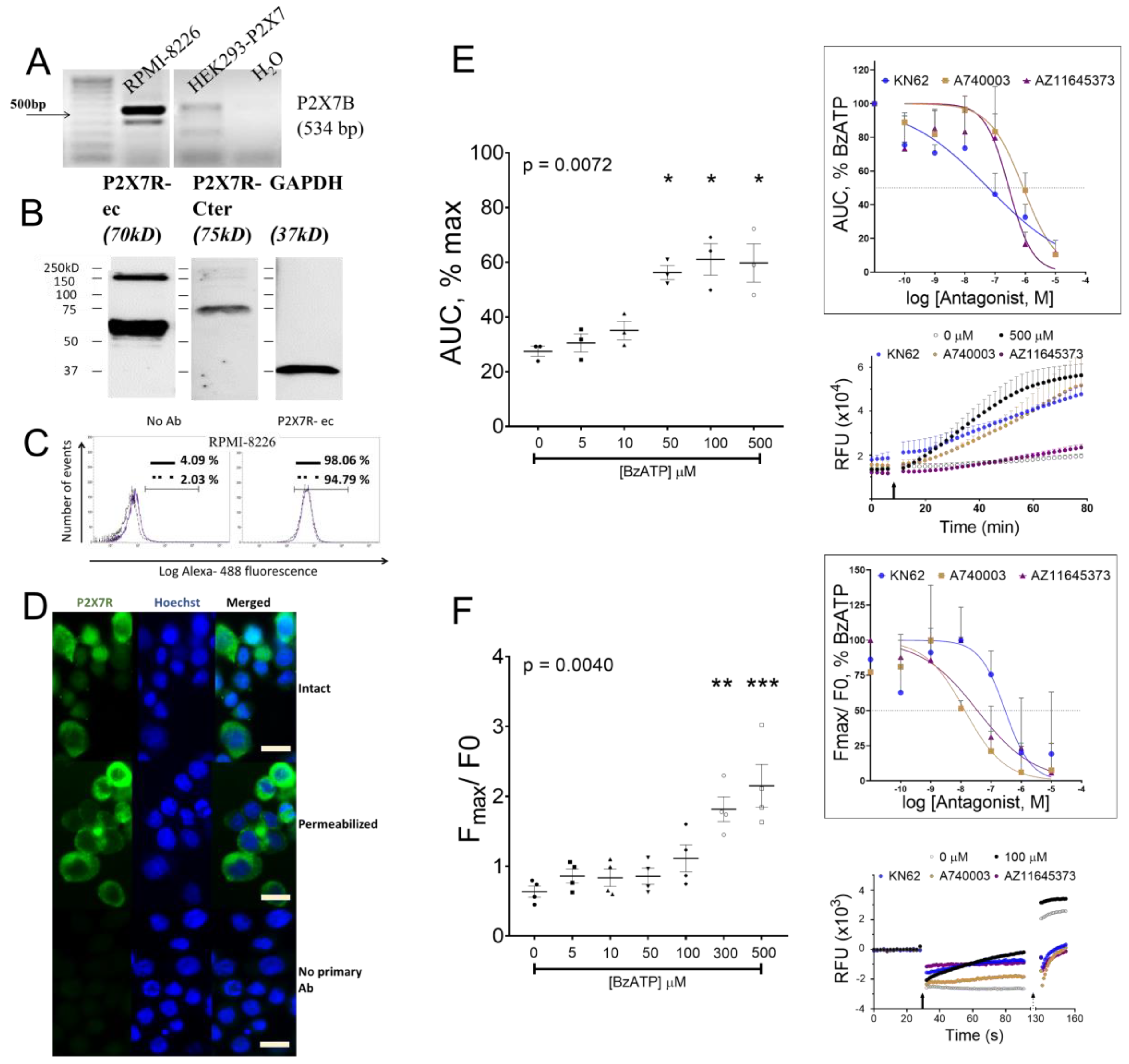
3.1. P2X7 Receptor Was Expressed and Induced Calcium Influx on HMCL
3.2. P2X7 Receptor Expression, Function, and Signaling on RPMI-8226
3.3. P2X7 Receptor Restricted Number of Viable RPMI-8226 by Cell Cycle Arrest and Not Apoptosis
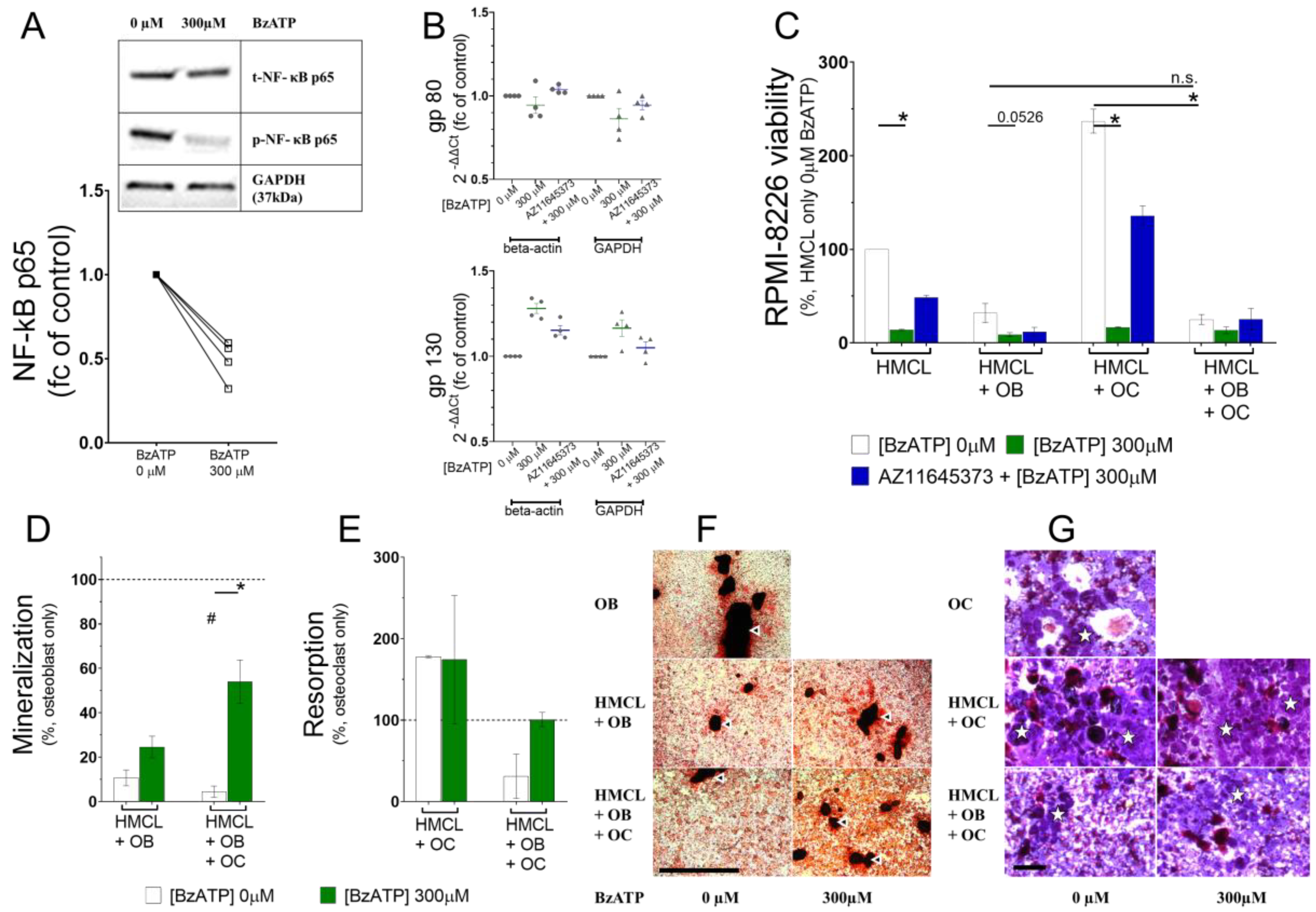
3.4. P2X7 Receptor Activation Deregulated Phosphorylation of NF-κB in RPMI-8226
3.5. P2X7 Receptor Restricted RPMI-8226 Growth even in the Presence of the Bone Cells and Altered RPMI-8226–Osteoblast and RPMI-8226–Osteoclast Interactions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marino, S.; Roodman, G.D. Multiple Myeloma and Bone: The Fatal Interaction. Cold Spring Harb. Perspect. Med. 2018, 8, a031286. [Google Scholar] [CrossRef] [PubMed]
- Silbermann, R.; Roodman, G.D. Current Controversies in the Management of Myeloma Bone Disease. J. Cell Physiol. 2016, 231, 2374–2379. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Gorgun, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci. Adv. 2018, 2, 2398212818817494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G.; Verkhratsky, A. Long-term (trophic) purinergic signalling: Purinoceptors control cell proliferation, differentiation and death. Cell Death Dis. 2010, 1, e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Andrade Mello, P.; Coutinho-Silva, R.; Savio, L.E.B. Multifaceted effects of extracellular adenosine triphosphate and adenosine in the Tumor-Host interaction and therapeutic perspectives. Front. Immunol. 2017, 8, 1526. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F. P2RX7: A receptor with a split personality in inflammation and cancer. Mol. Cell. Oncol. 2016, 3, e1010937. [Google Scholar] [CrossRef] [Green Version]
- Helenius, M.; Jalkanen, S.; Yegutkin, G. Enzyme-coupled assays for simultaneous detection of nanomolar ATP, ADP, AMP, adenosine, inosine and pyrophosphate concentrations in extracellular fluids. Biochim. Biophys. Acta 2012, 1823, 1967–1975. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.M.; Buchakjian, M.R.; Dubyak, G.R. Colocalization of ATP release sites and ecto-ATPase activity at the extracellular surface of human astrocytes. J. Biol. Chem. 2003, 278, 23331–23342. [Google Scholar] [CrossRef] [Green Version]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Pinton, P.; Falzoni, S. Assessing Extracellular ATP as Danger Signal In Vivo: The pmeLuc System. Methods Mol. Biol. 2016, 1417, 115–129. [Google Scholar] [CrossRef]
- Burnstock, G. Introduction to Purinergic Signaling. Methods Mol. Biol. 2020, 2041, 1–15. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2018, 14, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara, R.; Adinolfi, E.; Harwood, C.A.; Philpott, M.; Barden, J.A.; Di Virgilio, F.; McNulty, S. P2X7 in Cancer: From Molecular Mechanisms to Therapeutics. Front. Pharm. 2020, 11, 793. [Google Scholar] [CrossRef]
- Adinolfi, E.; Capece, M.; Franceschini, A.; Falzoni, S.; Giuliani, A.L.; Rotondo, A.; Sarti, A.C.; Bonora, M.; Syberg, S.; Corigliano, D.; et al. Accelerated tumor progression in mice lacking the ATP receptor P2X7. Cancer Res. 2015, 75, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Gorodeski, G.I. P2X7 -mediated chemoprevention of epithelial cancers. Expert Opin. Targets 2009, 13, 1313–1332. [Google Scholar] [CrossRef]
- Farrell, A.W.; Gadeock, S.; Pupovac, A.; Wang, B.; Jalilian, I.; Ranson, M.; Sluyter, R. P2X7 receptor activation induces cell death and CD23 shedding in human RPMI 8226 multiple myeloma cells. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 1173–1182. [Google Scholar] [CrossRef]
- Pupovac, A.; Foster, C.M.; Sluyter, R. Human P2X7 receptor activation induces the rapid shedding of CXCL16. Biochem. Biophys. Res. Commun. 2013, 432, 626–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupovac, A.; Geraghty, N.J.; Watson, D.; Sluyter, R. Activation of the P2X7 receptor induces the rapid shedding of CD23 from human and murine B cells. Immunol. Cell Biol. 2015, 93, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Falk, S.; Schwab, S.D.; Frosig-Jorgensen, M.; Clausen, R.P.; Dickenson, A.H.; Heegaard, A.M. P2X7 receptor-mediated analgesia in cancer-induced bone pain. Neuroscience 2015, 291, 93–105. [Google Scholar] [CrossRef]
- Falk, S.; Appel, C.K.; Bennedbaek, H.B.; Al-Dihaissy, T.; Unger, A.; Dinkel, K.; Heegaard, A.M. Chronic high dose P2X7 receptor inhibition exacerbates cancer-induced bone pain. Eur. J. Pharm. 2019, 845, 48–55. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Mechanisms and Pain. Adv. Pharm. 2016, 75, 91–137. [Google Scholar] [CrossRef]
- Agrawal, A.; Henriksen, Z.; Syberg, S.; Petersen, S.; Aslan, D.; Solgaard, M.; Nissen, N.; Larsen, T.K.; Schwarz, P.; Steinberg, T.H.; et al. P2X7Rs are involved in cell death, growth and cellular signaling in primary human osteoblasts. Bone 2017, 95, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Gallagher, J.A.; Gartland, A. Human osteoclast culture and phenotypic characterization. Methods Mol. Biol. 2012, 806, 357–375. [Google Scholar] [CrossRef]
- Giuliani, A.L.; Colognesi, D.; Ricco, T.; Roncato, C.; Capece, M.; Amoroso, F.; Wang, Q.G.; De Marchi, E.; Gartland, A.; Di Virgilio, F.; et al. Trophic activity of human P2X7 receptor isoforms A and B in osteosarcoma. PLoS ONE 2014, 9, e107224. [Google Scholar] [CrossRef]
- Buell, G.; Chessell, I.P.; Michel, A.D.; Collo, G.; Salazzo, M.; Herren, S.; Gretener, D.; Grahames, C.; Kaur, R.; Kosco-Vilbois, M.H.; et al. Blockade of human P2X7 receptor function with a monoclonal antibody. Blood 1998, 92, 3521–3528. [Google Scholar] [CrossRef]
- Adinolfi, E.; Cirillo, M.; Woltersdorf, R.; Falzoni, S.; Chiozzi, P.; Pellegatti, P.; Callegari, M.G.; Sandona, D.; Markwardt, F.; Schmalzing, G.; et al. Trophic activity of a naturally occurring truncated isoform of the P2X7 receptor. FASEB J. 2010, 24, 3393–3404. [Google Scholar] [CrossRef]
- Cheewatrakoolpong, B.; Gilchrest, H.; Anthes, J.C.; Greenfeder, S. Identification and characterization of splice variants of the human P2X7 ATP channel. Biochem. Biophys. Res. Commun. 2005, 332, 17–27. [Google Scholar] [CrossRef]
- Feng, Y.H.; Li, X.; Wang, L.; Zhou, L.; Gorodeski, G.I. A truncated P2X7 receptor variant (P2X7-j) endogenously expressed in cervical cancer cells antagonizes the full-length P2X7 receptor through hetero-oligomerization. J. Biol. Chem. 2006, 281, 17228–17237. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Samways, D.S.; Wolf, K.; Bowles, E.A.; Richards, J.P.; Bruno, J.; Dutertre, S.; DiPaolo, R.J.; Egan, T.M. Quantifying Ca2+ current and permeability in ATP-gated P2X7 receptors. J. Biol. Chem. 2015, 290, 7930–7942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, N.; Colla, S.; Morandi, F.; Rizzoli, V. The RANK/RANK ligand system is involved in interleukin-6 and interleukin-11 up-regulation by human myeloma cells in the bone marrow microenvironment. Haematologica 2004, 89, 1118–1123. [Google Scholar] [PubMed]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. (Lond) 2012, 122, 143–159. [Google Scholar] [CrossRef] [Green Version]
- Dechow, T.; Steidle, S.; Gotze, K.S.; Rudelius, M.; Behnke, K.; Pechloff, K.; Kratzat, S.; Bullinger, L.; Fend, F.; Soberon, V.; et al. GP130 activation induces myeloma and collaborates with MYC. J. Clin. Investig. 2014, 124, 5263–5274. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.; Gunther, A.; Klausz, K.; Staudinger, M.; Peipp, M.; Penas, E.M.; Rose-John, S.; Wijdenes, J.; Gramatzki, M. Due to interleukin-6 type cytokine redundancy only glycoprotein 130 receptor blockade efficiently inhibits myeloma growth. Haematologica 2017, 102, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Gougelet, A.; Mansuy, A.; Blay, J.Y.; Alberti, L.; Vermot-Desroches, C. Lymphoma and myeloma cell resistance to cytotoxic agents and ionizing radiations is not affected by exposure to anti-IL-6 antibody. PLoS ONE 2009, 4, e8026. [Google Scholar] [CrossRef]
- Schwabe, M.; Brini, A.T.; Bosco, M.C.; Rubboli, F.; Egawa, M.; Zhao, J.; Princler, G.L.; Kung, H.F. Disruption by interferon-alpha of an autocrine interleukin-6 growth loop in IL-6-dependent U266 myeloma cells by homologous and heterologous down-regulation of the IL-6 receptor alpha- and beta-chains. J. Clin. Investig. 1994, 94, 2317–2325. [Google Scholar] [CrossRef]
- Sluyter, R.; Stokes, L. Significance of P2X7 receptor variants to human health and disease. Recent Pat. Dna Gene Seq. 2011, 5, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paneesha, S.; Starczynski, J.; Pepper, C.; Delgado, J.; Hooper, L.; Fegan, C.; Pratt, G. The P2X7 receptor gene polymorphism 1513 A-->C has no effect on clinical prognostic markers and survival in multiple myeloma. Leuk. Lymphoma 2006, 47, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Vangsted, A.J.; Klausen, T.W.; Gimsing, P.; Abildgaard, N.; Andersen, N.F.; Gang, A.O.; Holmstrom, M.; Gregersen, H.; Vogel, U.; Schwarz, P.; et al. Genetic variants in the P2RX7 gene are associated with risk of multiple myeloma. Eur. J. Haematol. 2014, 93, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Callegari, M.G.; Ferrari, D.; Bolognesi, C.; Minelli, M.; Wieckowski, M.R.; Pinton, P.; Rizzuto, R.; Di Virgilio, F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol. Biol. Cell 2005, 16, 3260–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.M.; Oliphant, C.J.; Hassan, S.; Peille, A.L.; Bronsert, P.; Falzoni, S.; Di Virgilio, F.; McNulty, S.; Lara, R. ATP in the tumour microenvironment drives expression of nf P2X7, a key mediator of cancer cell survival. Oncogene 2019, 38, 194–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shupp, A.B.; Kolb, A.D.; Mukhopadhyay, D.; Bussard, K.M. Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers 2018, 10, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, R.E.; Lawson, M.A.; Buckle, C.H.; Snowden, J.A.; Chantry, A.D. Myeloma bone disease: Pathogenesis, current treatments and future targets. Br. Med. Bull. 2014, 111, 117–138. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Gartland, A. P2X7 receptors: Role in bone cell formation and function. J. Mol. Endocrinol. 2015, 54, R75–R88. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Agrawal, A.; Jorgensen, N.R.; Gartland, A. P2X7 receptor regulates osteoclast function and bone loss in a mouse model of osteoporosis. Sci. Rep. 2018, 8, 3507. [Google Scholar] [CrossRef]
- Gilbert, S.M.; Gidley Baird, A.; Glazer, S.; Barden, J.A.; Glazer, A.; Teh, L.C.; King, J. A phase I clinical trial demonstrates that nf P2X7 -targeted antibodies provide a novel, safe and tolerable topical therapy for basal cell carcinoma. Br. J. Derm. 2017, 177, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Salvestrini, V.; Orecchioni, S.; Talarico, G.; Reggiani, F.; Mazzetti, C.; Bertolini, F.; Orioli, E.; Adinolfi, E.; Di Virgilio, F.; Pezzi, A.; et al. Extracellular ATP induces apoptosis through P2X7R activation in acute myeloid leukemia cells but not in normal hematopoietic stem cells. Oncotarget 2017, 8, 5895–5908. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RPMI-8226 | U226 | NCI-H929 | OPM2 | JJN3 | KMS12 | |
|---|---|---|---|---|---|---|
| AUC of maximum, % mean ± SEM (n) | ||||||
| 0 µM | 32.03 ± 3.25 (9) | 14.21 ± 0.74 (9) | 16.10 ± 1.32 (9) | 33.21 ± 6.40 (7) | 47.96 ± 8.81 (7) | 17.06 ± 1.52 (7) |
| 500 µM | 64.92 ± 3.16 (9) | 20.11 ± 1.48 (9) | 17.82 ± 1.41 (9) | 44.95 ± 6.94 (7) | 49.59 ± 7.66 (7) | 23.82 ± 2.44 (7) |
| p-Value | 0.0067 | 0.0203 | 0.5092 | 0.1622 | 0.8143 | 0.1081 |
| 10 µM KN62 + 500 µM BzATP | 41.98 ± 3.28 (2) | 16.35 ± 0.89 (2) | 18.64 ± 1.58 (2) | 43.20 ± 7.16 (6) | 40.70 ± 6.47 (6) | 21.30 ± 2.48 (6) |
| p-Value | 0.3615 | 0.5025 | 0.7681 | 0.5836 | 0.5300 | 0.6135 |
| 10 µM A740003 + 500 µM BzATP | 31.58 ± 1.62 (2) | 16.50 ± 0.68 (2) | 19.57 ± 2.37 (2) | 45.42 ± 8.11 (6) | 44.95 ± 8.22 (6) | 21.60 ± 2.02 (6) |
| p-Value | 0.1688 | 0.5025 | 0.6214 | 0.6810 | 0.7351 | 0.6227 |
| 10 µM AZ11645373 + 500 µM BzATP | 25.94 ± 1.29 (2) | 12.51 ± 0.21 (2) | 13.35 ± 0.74 (2) | 41.73 ± 6.38 (6) | 37.61 ± 7.24 (6) | 25.65 ± 3.08 (6) |
| p-Value | 0.0353 | 0.0459 | 0.3020 | 0.5107 | 0.3190 | 0.6495 |
| Fmax/F0, mean ± SEM (n) | ||||||
| 0 µM BzATP | 0.678 ± 0.019 (6) | 0.516 ± 0.141 (7) | 0.598 ± 0.033 (7) | 0.737 ± 0.077 (5) | 0.803 ± 0.120 (7) | 0.840 ± 0.072 (7) |
| 100 µM BzATP | 1.668 ± 0.146 (6) | 0.916 ± 0.027 (7) | 1.056 ± 0.062 (7) | 1.845 ± 0.108 (5) | 1.136 ± 0.089 (7) | 1.647 ± 0.069 (7) |
| p-Value | 0.0008 | 0.0041 | 0.0001 | 0.0009 | 0.0413 | 0.0002 |
| 1 µM KN62 + 100 µM BzATP | 0.853 ± 0.258 (2) | 0.853 ± 0.041 (3) | 0.905 ± 0.124 (3) | 1.668 ± 0.117 (5) | 0.994 ± 0.073 (5) | 1.397 ± 0.087 (5) |
| p-Value | 0.0389 | 0.4791 | 0.1355 | 0.4143 | 0.2287 | 0.1818 |
| 1 µM A740003 + 100 µM BzATP | 0.753 ± 0.102 (2) | 0.988 ± 0.035 (3) | 1.017 ± 0.143 (3) | 1.758 ± 0.089 (5) | 1.054 ± 0.092 (5) | 1.539 ± 0.189 (5) |
| p-Value | 0.0389 | 0.3389 | 0.4908 | 0.7310 | 0.5473 | 0.6221 |
| 1 µM AZ11645373 + 100 µM BzATP | 0.890 ± 0.010 (2) | 0.940 ± 0.016 (3) | 0.926 ± 0.166 (3) | 1.544 ± 0.120 (5) | 1.103 ± 0.082 (5) | 1.580 ± 0.112 (5) |
| p-Value | 0.2513 | 0.9847 | 0.1079 | 0.1829 | 0.8098 | 0.8009 |
| Bold represents significant p-Value | ||||||
| HMCL | HMCL + OB | p-Value | HMCL + OC | p-Value | HMCL + OB + OC | p-Value | |
|---|---|---|---|---|---|---|---|
| Viability, % mean ± SEM (n = 3) | |||||||
| 0 µM | 100 ± 0.01 | 31.93 ± 10.08 | 0.1403 | 237.0 ± 12.75 | 0.3073 | 24.95 ± 5.31 | 0.1123 |
| 300 µM | 13.78 ± 0.79 | 8.81 ± 2.12 | 16.59 ± 0.74 | 13.33 ± 3.63 | |||
| p-value | 0.0070 | 0.0526 | 0.0073 | 0.1797 | |||
| 10 µM AZ11645373 + 300 µM BzATP | 48.47 ± 2.30 | 11.71 ± 4.83 | 135.8 ± 10.46 | 25.20 ± 11.54 | |||
| p-value | 0.1779 | 0.4561 | 0.1797 | 0.3711 | |||
| Mineralization, % of OB mean ± SEM (n = 4) | |||||||
| 0 µM | - | 10.65 ± 3.54 | 0.0579 | - | 4.44 ± 2.54 | 0.0038 | |
| 300 µM | 24.57 ± 4.91 | 53.94 ± 9.84 | |||||
| p-value | 0.1143 | 0.0286 | |||||
| Resorption, % of OC mean ± SEM (n = 2) | |||||||
| 0 µM | - | - | 177.9 ± 1.36 | 0.2781 | 31.09 ± 27.21 | 0.2781 | |
| 300 µM | 174.3 ± 78.82 | 100.8 ± 8.72 | |||||
| p-value | >0.999 | 0.333 | |||||
| Bold represents significant p-Value | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agrawal, A.; S. Kruse, L.; J. Vangsted, A.; Gartland, A.; R. Jørgensen, N. Human P2X7 Receptor Causes Cycle Arrest in RPMI-8226 Myeloma Cells to Alter the Interaction with Osteoblasts and Osteoclasts. Cells 2020, 9, 2341. https://doi.org/10.3390/cells9112341
Agrawal A, S. Kruse L, J. Vangsted A, Gartland A, R. Jørgensen N. Human P2X7 Receptor Causes Cycle Arrest in RPMI-8226 Myeloma Cells to Alter the Interaction with Osteoblasts and Osteoclasts. Cells. 2020; 9(11):2341. https://doi.org/10.3390/cells9112341
Chicago/Turabian StyleAgrawal, Ankita, Lars S. Kruse, Annette J. Vangsted, Alison Gartland, and Niklas R. Jørgensen. 2020. "Human P2X7 Receptor Causes Cycle Arrest in RPMI-8226 Myeloma Cells to Alter the Interaction with Osteoblasts and Osteoclasts" Cells 9, no. 11: 2341. https://doi.org/10.3390/cells9112341