The Intrinsically Disordered W Protein Is Multifunctional during Henipavirus Infection, Disrupting Host Signalling Pathways and Nuclear Import

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
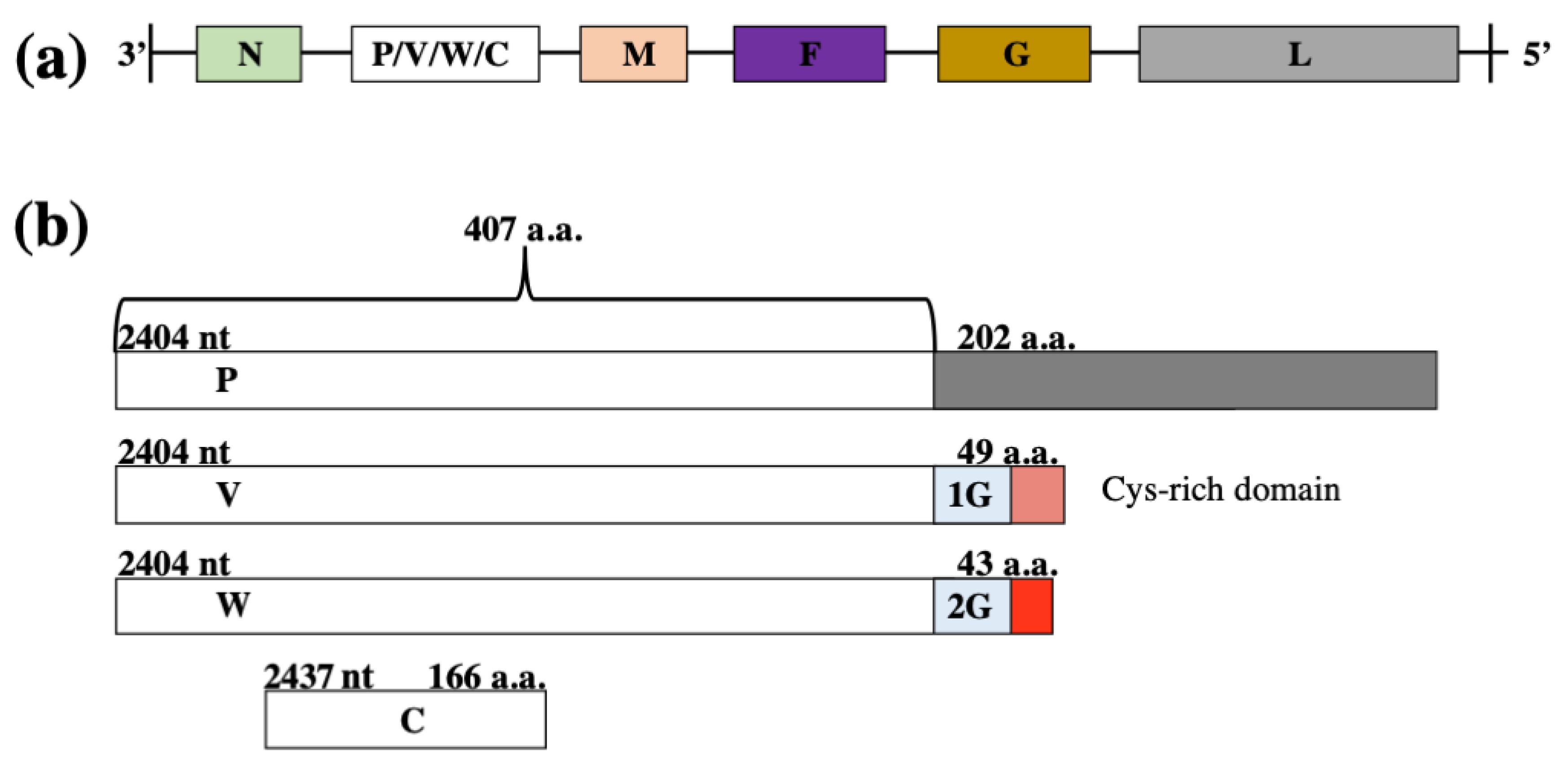
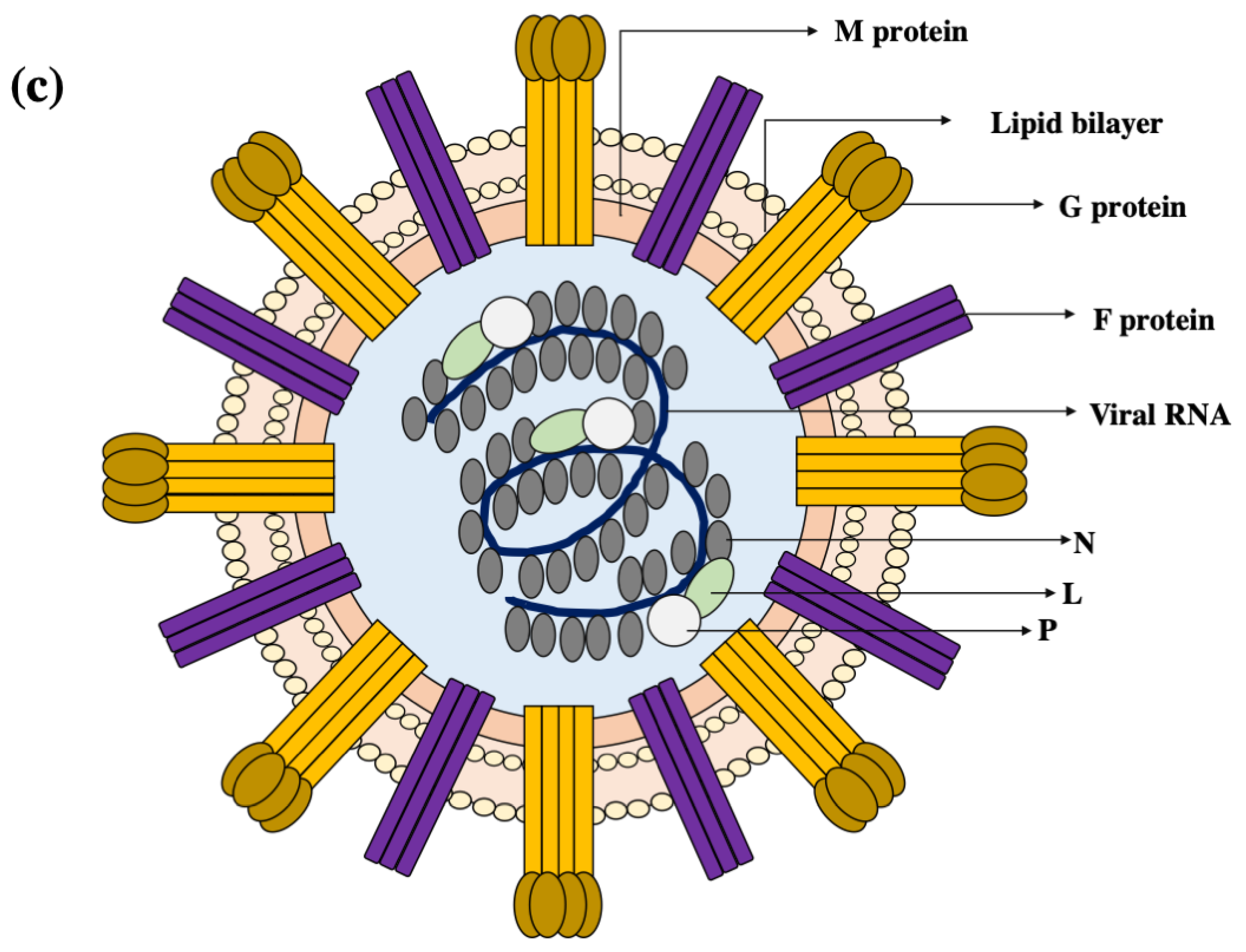
2. Virion Organisation
3. The Henipavirus Replication Cycle within the Host Cell
4. Innate Immune Antagonism and Virus Pathogenesis
5. W Protein Is Intrinsically Disordered and Binds Multiple Host Proteins Involved in the Suppression of Innate Immunity
5.1. W Binds Impα3 and Impα4 for Nuclear Import
5.2. W Protein Inhibits Type I IFN Response
5.3. W Protein Binds Host STAT to Create High Molecular Weight Complexes that Inhibit Type I IFN Signalling
5.4. W Binds Host 14-3-3 Proteins to Modulate Diverse Host Signalling Pathways
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, K.; Selleck, P.; Hooper, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Westbury, H.; Hiley, L.; Selvey, L.; Rodwell, B.; et al. A morbillivirus that caused fatal disease in horses and humans. Science 1995, 268, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Field, H.E. Hendra virus ecology and transmission. Curr. Opin. Virol. 2016, 16, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Ang, B.S.P.; Lim, T.C.C.; Wang, L. Nipah Virus Infection. J. Clin. Microbiol. 2018, 56, e01875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luby, S.P.; Hossain, M.J.; Gurley, E.S.; Ahmed, B.N.; Banu, S.; Khan, S.U.; Homaira, N.; Rota, P.A.; Rollin, P.E.; Comer, J.A.; et al. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg. Infect. Dis. 2009, 15, 1229–1235. [Google Scholar] [CrossRef]
- Goldsmith, C.S.; Whistler, T.; Rollin, P.E.; Ksiazek, T.G.; Rota, P.A.; Bellini, W.J.; Daszak, P.; Wong, K.T.; Shieh, W.J.; Zaki, S.R. Elucidation of Nipah virus morphogenesis and replication using ultrastructural and molecular approaches. Virus Res. 2003, 92, 89–98. [Google Scholar] [CrossRef]
- Patch, J.R.; Crameri, G.; Wang, L.-F.; Eaton, B.T.; Broder, C.C. Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol. J. 2007, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.F.; Yu, M.; Hansson, E.; Pritchard, L.I.; Shiell, B.; Michalski, W.P.; Eaton, B.T. The exceptionally large genome of Hendra virus: Support for creation of a new genus within the family Paramyxoviridae. J. Virol. 2000, 74, 9972–9979. [Google Scholar] [CrossRef] [Green Version]
- Enchéry, F.; Horvat, B. Understanding the Interaction between Henipaviruses and Their Natural Host, Fruit Bats: Paving the Way Toward Control of Highly Lethal Infection in Humans. Int. Rev. Immunol. 2017, 36, 1–14. [Google Scholar] [CrossRef]
- Hausmann, S.; Garcin, D.; Delenda, C.; Kolakofsky, D. The versatility of paramyxovirus RNA polymerase stuttering. J. Virol. 1999, 73, 5568–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.K.; Harcourt, B.H.; Mungall, B.A.; Tamin, A.; Peeples, M.E.; Bellini, W.J.; Rota, P.A. Determination of the henipavirus phosphoprotein gene mRNA editing frequencies and detection of the C, V and W proteins of Nipah virus in virus-infected cells. J. Gen. Virol. 2009, 90, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Battisti, A.J.; Meng, G.; Winkler, D.C.; McGinnes, L.W.; Plevka, P.; Steven, A.C.; Morrison, T.G.; Rossmann, M.G. Structure and assembly of a paramyxovirus matrix protein. Proc. Natl. Acad. Sci. USA 2012, 109, 13996–14000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Berger, B.; Kim, P.S. LearnCoil-VMF: Computational evidence for coiled-coil-like motifs in many viral membrane-fusion proteins. J. Mol. Biol. 1999, 290, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, J.P.; Daus, F.J.; van Oirschot, J.T. Sequence and structure alignment of Paramyxoviridae attachment proteins and discovery of enzymatic activity for a morbillivirus hemagglutinin. J. Virol. 1997, 71, 6155–6167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crennell, S.; Takimoto, T.; Portner, A.; Taylor, G. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol. 2000, 7, 1068–1074. [Google Scholar] [PubMed]
- Negrete, O.A.; Levroney, E.L.; Aguilar, H.C.; Bertolotti-Ciarlet, A.; Nazarian, R.; Tajyar, S.; Lee, B. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 2005, 436, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, H.C.; Iorio, R.M. Henipavirus membrane fusion and viral entry. Curr. Top. Microbiol. Immunol. 2012, 359, 79–94. [Google Scholar]
- Pernet, O.; Wang, Y.E.; Lee, B. Henipavirus receptor usage and tropism. Curr. Top. Microbiol. Immunol. 2012, 359, 59–78. [Google Scholar]
- Pentecost, M.; Vashisht, A.A.; Lester, T.; Voros, T.; Beaty, S.M.; Park, A.; Wang, Y.E.; Yun, T.E.; Freiberg, A.N.; Wohlschlegel, J.A.; et al. Evidence for Ubiquitin-Regulated Nuclear and Subnuclear Trafficking among Paramyxovirinae Matrix Proteins. PLoS Pathog. 2015, 11, e1004739. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.E.; Park, A.; Lake, M.; Pentecost, M.; Torres, B.; Yun, T.E.; Wolf, M.C.; Holbrook, M.R.; Freiberg, A.N.; Lee, B. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 2010, 6, e1001186. [Google Scholar] [CrossRef]
- Waning, D.L.; Schmitt, A.P.; Leser, G.P.; Lamb, R.A. Roles for the cytoplasmic tails of the fusion and hemagglutinin-neuraminidase proteins in budding of the paramyxovirus simian virus 5. J. Virol. 2002, 76, 9284–9297. [Google Scholar] [CrossRef] [Green Version]
- Pantua, H.D.; McGinnes, L.W.; Peeples, M.E.; Morrison, T.G. Requirements for the assembly and release of Newcastle disease virus-like particles. J. Virol. 2006, 80, 11062–11073. [Google Scholar] [CrossRef] [Green Version]
- Harrison, M.S.; Sakaguchi, T.; Schmitt, A.P. Paramyxovirus assembly and budding: Building particles that transmit infections. Int. J. Biochem. Cell Biol. 2010, 42, 1416–1429. [Google Scholar] [CrossRef] [Green Version]
- Ghildyal, R.; Mills, J.; Murray, M.; Vardaxis, N.; Meanger, J. Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J. Gen. Virol. 2002, 83, 753–757. [Google Scholar] [CrossRef]
- Goh, K.J.; Tan, C.T.; Chew, N.K.; Tan, P.S.; Kamarulzaman, A.; Sarji, S.A.; Wong, K.T.; Abdullah, B.J.; Chua, K.B.; Lam, S.K. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N. Engl. J. Med. 2000, 342, 1229–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.T.; Shieh, W.J.; Kumar, S.; Norain, K.; Abdullah, W.; Guarner, J.; Goldsmith, C.S.; Chua, K.B.; Lam, S.K.; Tan, C.T.; et al. Nipah virus infection: Pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am. J. Pathol. 2002, 161, 2153–2167. [Google Scholar] [CrossRef]
- Satterfield, B.A.; Cross, R.W.; Fenton, K.A.; Agans, K.N.; Basler, C.F.; Geisbert, T.W.; Mire, C.E. The immunomodulating V and W proteins of Nipah virus determine disease course. Nat. Commun. 2015, 6, 7483. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.L.; Cardenas, W.B.; Zamarin, D.; Palese, P.; Basler, C.F. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J. Virol. 2005, 79, 6078–6088. [Google Scholar] [CrossRef] [Green Version]
- Satterfield, B.A.; Cross, R.W.; Fenton, K.A.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Graber, J.; Basler, C.F.; Geisbert, T.W.; Mire, C.E. Nipah Virus C and W Proteins Contribute to Respiratory Disease in Ferrets. J. Virol. 2016, 90, 6326–6343. [Google Scholar] [CrossRef] [Green Version]
- Kolakofsky, D.; Roux, L.; Garcin, D.; Ruigrok, R.W.H. Paramyxovirus mRNA editing, the “rule of six” and error catastrophe: A hypothesis. J. Gen. Virol. 2005, 86, 1869–1877. [Google Scholar] [CrossRef]
- Curran, J.; Kolakofsky, D. Sendai virus P gene produces multiple proteins from overlapping open reading frames. Enzyme 1990, 44, 244–249. [Google Scholar] [CrossRef]
- Karlin, D.; Longhi, S.; Receveur, V.; Canard, B. The N-terminal domain of the phosphoprotein of Morbilliviruses belongs to the natively unfolded class of proteins. Virology 2002, 296, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Curran, J.; Boeck, R.; Lin-Marq, N.; Lupas, A.; Kolakofsky, D. Paramyxovirus phosphoproteins form homotrimers as determined by an epitope dilution assay, via predicted coiled coils. Virology 1995, 214, 139–149. [Google Scholar] [CrossRef]
- Jensen, M.R.; Yabukarski, F.; Communie, G.; Condamine, E.; Mas, C.; Volchkova, V.; Tarbouriech, N.; Bourhis, J.M.; Volchkov, V.; Blackledge, M.; et al. Structural Description of the Nipah Virus Phosphoprotein and Its Interaction with STAT1. Biophys. J. 2020. [Google Scholar] [CrossRef]
- Karlin, D.; Belshaw, R. Detecting remote sequence homology in disordered proteins: Discovery of conserved motifs in the N-termini of Mononegavirales phosphoproteins. PLoS ONE 2012, 7, e31719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.R.; Hoad, M.; Tsimbalyuk, S.; Menicucci, A.R.; Messaoudi, I.; Forwood, J.K.; Basler, C.F. Henipavirus W proteins interact with 14-3-3 to modulate host gene expression. J. Virol. 2020. [Google Scholar] [CrossRef]
- Parisien, J.P.; Lau, J.F.; Rodriguez, J.J.; Ulane, C.M.; Horvath, C.M. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J. Virol. 2002, 76, 4190–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.L.; García-Sastre, A.; Palese, P.; Basler, C.F. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J. Virol. 2004, 78, 5633–5641. [Google Scholar] [CrossRef] [Green Version]
- Erdős, G.; Dosztányi, Z. Analyzing Protein Disorder with IUPred2A. Curr. Protoc. Bioinform. 2020, 70, e99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.K.; Miller, D.; Aljofan, M.; Mungall, B.A.; Rollin, P.E.; Bellini, W.J.; Rota, P.A. Characterization of the antiviral and inflammatory responses against Nipah virus in endothelial cells and neurons. Virology 2010, 404, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Gil, L.; Vera-Velasco, N.M.; Mingarro, I. Exploring the Human-Nipah Virus Protein-Protein Interactome. J. Virol. 2017, 91, e01461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 2007, 8, 195–208. [Google Scholar] [CrossRef]
- Goldfarb, D.S.; Corbett, A.H.; Mason, D.A.; Harreman, M.T.; Adam, S.A. Importin alpha: A multipurpose nuclear-transport receptor. Trends Cell Biol. 2004, 14, 505–514. [Google Scholar] [CrossRef]
- Garcia-Sastre, A. Mechanisms of inhibition of the host interferon alpha/beta-mediated antiviral responses by viruses. Microbes Infect. 2002, 4, 647–655. [Google Scholar] [CrossRef]
- Smith, K.M.; Tsimbalyuk, S.; Edwards, M.R.; Cross, E.M.; Batra, J.; Soares da Costa, T.P.; Aragao, D.; Basler, C.F.; Forwood, J.K. Structural basis for importin alpha 3 specificity of W proteins in Hendra and Nipah viruses. Nat. Commun. 2018, 9, 3703. [Google Scholar] [CrossRef]
- Kumar, K.P.; McBride, K.M.; Weaver, B.K.; Dingwall, C.; Reich, N.C. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell Biol. 2000, 20, 4159–4168. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Fang, T.; Li, S.; Meng, K.; Guo, D. Bipartite Nuclear Localization Signal Controls Nuclear Import and DNA-Binding Activity of IFN Regulatory Factor 3. J. Immunol. 2015, 195, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braciale, T.J.; Hahn, Y.S. Immunity to viruses. Immunol. Rev. 2013, 255, 5–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.S.; Liu, H.M. The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses. Front. Immunol. 2018, 9, 3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Goraya, M.U.; Yuan, X.; Zhang, B.; Chiu, S.H.; Chen, J.L. Functional Involvement of Interferon-Inducible Transmembrane Proteins in Antiviral Immunity. Front. Microbiol. 2019, 10, 1097. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Garcia-Sastre, A. Viruses and the type I interferon antiviral system: Induction and evasion. Int. Rev. Immunol. 2002, 21, 305–337. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Mikulasova, A.; Martinez-Sobrido, L.; Paragas, J.; Mühlberger, E.; Bray, M.; Klenk, H.D.; Palese, P.; García-Sastre, A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003, 77, 7945–7956. [Google Scholar] [CrossRef] [Green Version]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Genin, P.; Mamane, Y.; Sgarbanti, M.; Battistini, A.; Harrington, W.J., Jr.; Barber, G.N.; Hiscott, J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 2001, 20, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Talon, J.; Horvath, C.M.; Polley, R.; Basler, C.F.; Muster, T.; Palese, P.; García-Sastre, A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 2000, 74, 7989–7996. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Condit, R.C.; Vijaysri, S.; Jacobs, B.; Williams, B.R.; Silverman, R.H. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002, 76, 5251–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, B.L.; Langland, J.O. When two strands are better than one: The mediators and modulators of the cellular responses to double-stranded RNA. Virology 1996, 219, 339–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.A. The immune response of Drosophila. Nature 2003, 426, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Grandvaux, N.; Servant, M.J.; tenOever, B.; Sen, G.C.; Balachandran, S.; Barber, G.N.; Lin, R.; Hiscott, J. Transcriptional profiling of interferon regulatory factor 3 target genes: Direct involvement in the regulation of interferon-stimulated genes. J. Virol. 2002, 76, 5532–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, K.L.; Smith, H.L.; Stark, G.R.; Sen, G.C. IRF-3-dependent, NFkappa B- and JNK-independent activation of the 561 and IFN-beta genes in response to double-stranded RNA. Proc. Natl. Acad. Sci. USA 2002, 99, 6322–6327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Iampietro, M.; Aurine, N.; Dhondt, K.P.; Dumont, C.; Pelissier, R.; Spanier, J.; Vallve, A.; Raoul, H.; Kalinke, U.; Horvat, B. Control of Nipah Virus Infection in Mice by the Host Adaptors Mitochondrial Antiviral Signaling Protein (MAVS) and Myeloid Differentiation Primary Response 88 (MyD88). J. Infect. Dis. 2020, 221, S401–S406. [Google Scholar] [CrossRef]
- He, B.; Paterson, R.G.; Stock, N.; Durbin, J.E.; Durbin, R.K.; Goodbourn, S.; Randall, R.E.; Lamb, R.A. Recovery of paramyxovirus simian virus 5 with a V protein lacking the conserved cysteine-rich domain: The multifunctional V protein blocks both interferon-beta induction and interferon signaling. Virology 2002, 303, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.; He, B.; Lamb, R.A.; Randall, R.E.; Goodbourn, S. The V proteins of simian virus 5 and other paramyxoviruses inhibit induction of interferon-beta. Virology 2002, 303, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, T.; Takeuchi, K.; Yokoo, J.; Gotoh, B. C and V proteins of Sendai virus target signaling pathways leading to IRF-3 activation for the negative regulation of interferon-beta production. Virology 2004, 325, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Kumthip, K.; Chusri, P.; Jilg, N.; Zhao, L.; Fusco, D.N.; Zhao, H.; Goto, K.; Cheng, D.; Schaefer, E.A.; Zhang, L.; et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J. Virol. 2012, 86, 8581–8591. [Google Scholar] [CrossRef] [Green Version]
- Sen, A.; Rott, L.; Phan, N.; Mukherjee, G.; Greenberg, H.B. Rotavirus NSP1 Protein Inhibits Interferon-Mediated STAT1 Activation. J. Virol. 2014, 88, 41. [Google Scholar] [CrossRef] [Green Version]
- Valmas, C.; Grosch, M.N.; Schümann, M.; Olejnik, J.; Martinez, O.; Best, S.M.; Krähling, V.; Basler, C.F.; Mühlberger, E. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. 2010, 6, e1000721. [Google Scholar] [CrossRef]
- Didcock, L.; Young, D.F.; Goodbourn, S.; Randall, R.E. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 1999, 73, 9928–9933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisien, J.-P.; Lau, J.F.; Rodriguez, J.J.; Sullivan, B.M.; Moscona, A.; Parks, G.D.; Lamb, R.A.; Horvath, C.M. The V Protein of Human Parainfluenza Virus 2 Antagonizes Type I Interferon Responses by Destabilizing Signal Transducer and Activator of Transcription 2. Virology 2001, 283, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, T.; Yokosawa, N.; Yokota, S.-I.; Fujii, N. C Terminal CYS-RICH Region of Mumps Virus Structural V Protein Correlates with Block of Interferon α and γ Signal Transduction Pathway through Decrease of STAT 1-α. Biochem. Biophys. Res. Commun. 2001, 283, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.J.; Parisien, J.-P.; Horvath, C.M. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 2002, 76, 11476–11483. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.J.; Cruz, C.D.; Horvath, C.M. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J. Virol. 2004, 78, 5358–5367. [Google Scholar] [CrossRef] [Green Version]
- Park, M.-S.; Shaw, M.L.; Muñoz-Jordan, J.; Cros, J.F.; Nakaya, T.; Bouvier, N.; Palese, P.; García-Sastre, A.; Basler, C.F. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol. 2003, 77, 1501–1511. [Google Scholar] [CrossRef] [Green Version]
- Ciancanelli, M.J.; Volchkova, V.A.; Shaw, M.L.; Volchkov, V.E.; Basler, C.F. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J. Virol. 2009, 83, 7828–7841. [Google Scholar] [CrossRef] [Green Version]
- Hagmaier, K.; Stock, N.; Goodbourn, S.; Wang, L.-F.; Randall, R. A single amino acid substitution in the V protein of Nipah virus alters its ability to block interferon signalling in cells from different species. J. Gen. Virol. 2006, 87, 3649–3653. [Google Scholar] [CrossRef]
- Satterfield, B.A.; Borisevich, V.; Foster, S.L.; Rodriguez, S.E.; Cross, R.W.; Fenton, K.A.; Agans, K.N.; Basler, C.F.; Geisbert, T.W.; Mire, C.E. Antagonism of STAT1 by Nipah virus P gene products modulates disease course but not lethal outcome in the ferret model. Sci. Rep. 2019, 9, 16710. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.A.; Goncalves, A.; et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 2012, 487, 486–490. [Google Scholar] [CrossRef]
- Aghazadeh, Y.; Papadopoulos, V. The role of the 14-3-3 protein family in health, disease, and drug development. Drug Discov. Today 2016, 21, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Subramanian, R.R.; Masters, S.C. 14-3-3 proteins: Structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 617–647. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Shen, Y.H.; Zhu, J. 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene 2001, 20, 6331–6338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, M.K.; Morrison, D.K. Unlocking the code of 14-3-3. J. Cell Sci. 2004, 117, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yang, H.; Liu, Y.C.; Jelinek, T.; Zhang, L.; Ruoslahti, E.; Fu, H. Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry 1999, 38, 12499–12504. [Google Scholar] [CrossRef]
- Lin, J.P.; Fan, Y.K.; Liu, H.M. The 14-3-3η chaperone protein promotes antiviral innate immunity via facilitating MDA5 oligomerization and intracellular redistribution. PLoS Pathog. 2019, 15, e1007582. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.M.; Loo, Y.M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3ε regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Riedl, W.; Acharya, D.; Lee, J.H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 2019, 26, 493–503. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsimbalyuk, S.; Cross, E.M.; Hoad, M.; Donnelly, C.M.; Roby, J.A.; Forwood, J.K. The Intrinsically Disordered W Protein Is Multifunctional during Henipavirus Infection, Disrupting Host Signalling Pathways and Nuclear Import. Cells 2020, 9, 1913. https://doi.org/10.3390/cells9081913
Tsimbalyuk S, Cross EM, Hoad M, Donnelly CM, Roby JA, Forwood JK. The Intrinsically Disordered W Protein Is Multifunctional during Henipavirus Infection, Disrupting Host Signalling Pathways and Nuclear Import. Cells. 2020; 9(8):1913. https://doi.org/10.3390/cells9081913
Chicago/Turabian StyleTsimbalyuk, Sofiya, Emily M. Cross, Mikayla Hoad, Camilla M. Donnelly, Justin A. Roby, and Jade K. Forwood. 2020. "The Intrinsically Disordered W Protein Is Multifunctional during Henipavirus Infection, Disrupting Host Signalling Pathways and Nuclear Import" Cells 9, no. 8: 1913. https://doi.org/10.3390/cells9081913