MIP-1α Expression Induced by Co-Stimulation of Human Monocytic Cells with Palmitate and TNF-α Involves the TLR4-IRF3 Pathway and Is Amplified by Oxidative Stress

 , , , ,
, , , ,  and
and 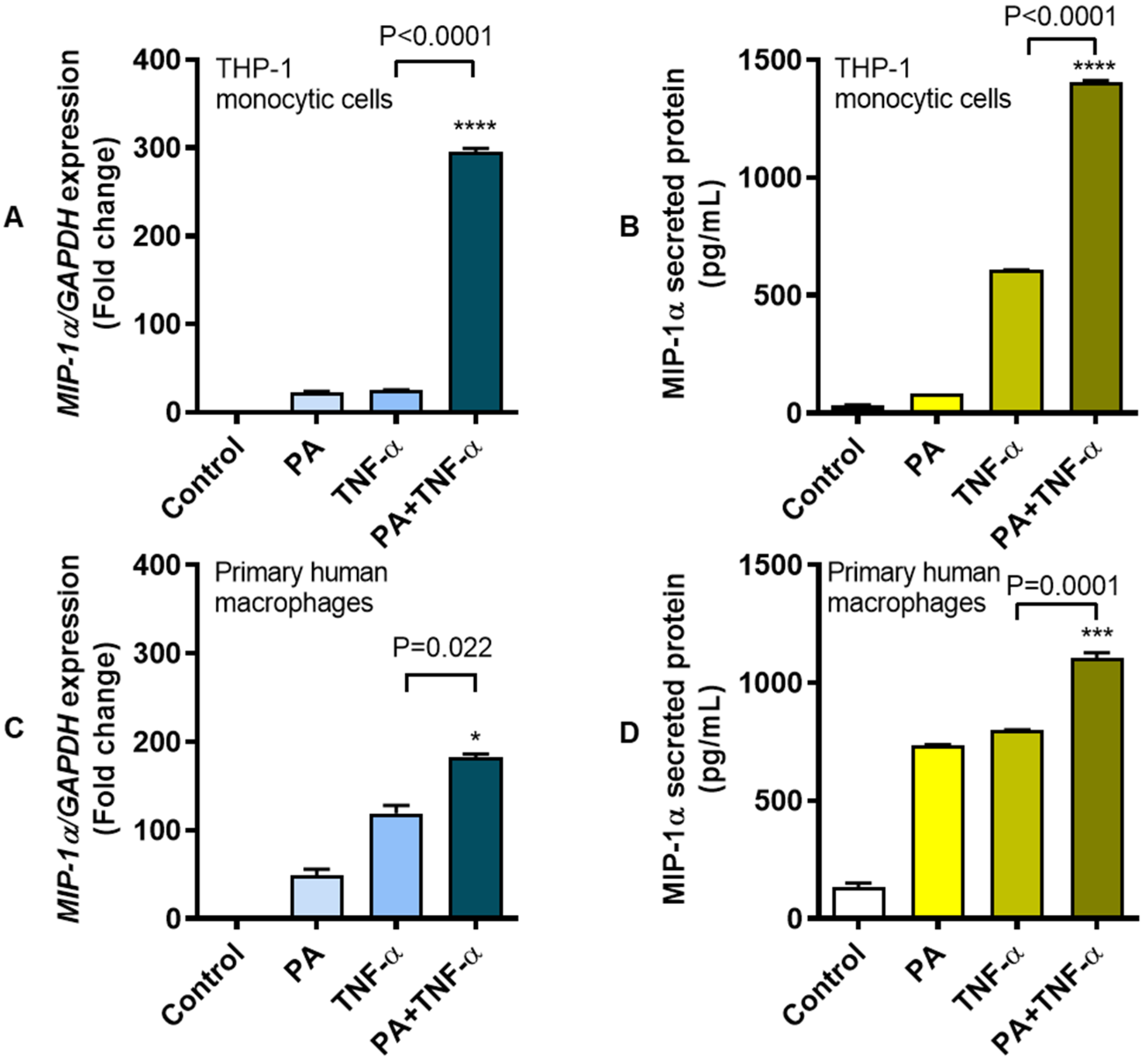{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Stimulations
2.2. Isolation of Human Peripheral Blood Mononuclear Cells (PBMC), Differentiation, and Stimulation of Primary Macrophages
2.3. siRNA-Mediated Genetic Suppression of TLR4 and IRF3
2.4. TLR4 Neutralization
2.5. Chemical Inhibition of TLR4-Mediated Signaling
2.6. Trafficking Inhibition
2.7. Induction of Oxidative Stress, ROS Measurement, and Cell Treatments with Anti-Oxidants/ROS Scavengers
2.8. Real-Time qRT-PCR
2.9. ELISA
2.10. Flow Cytometry
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
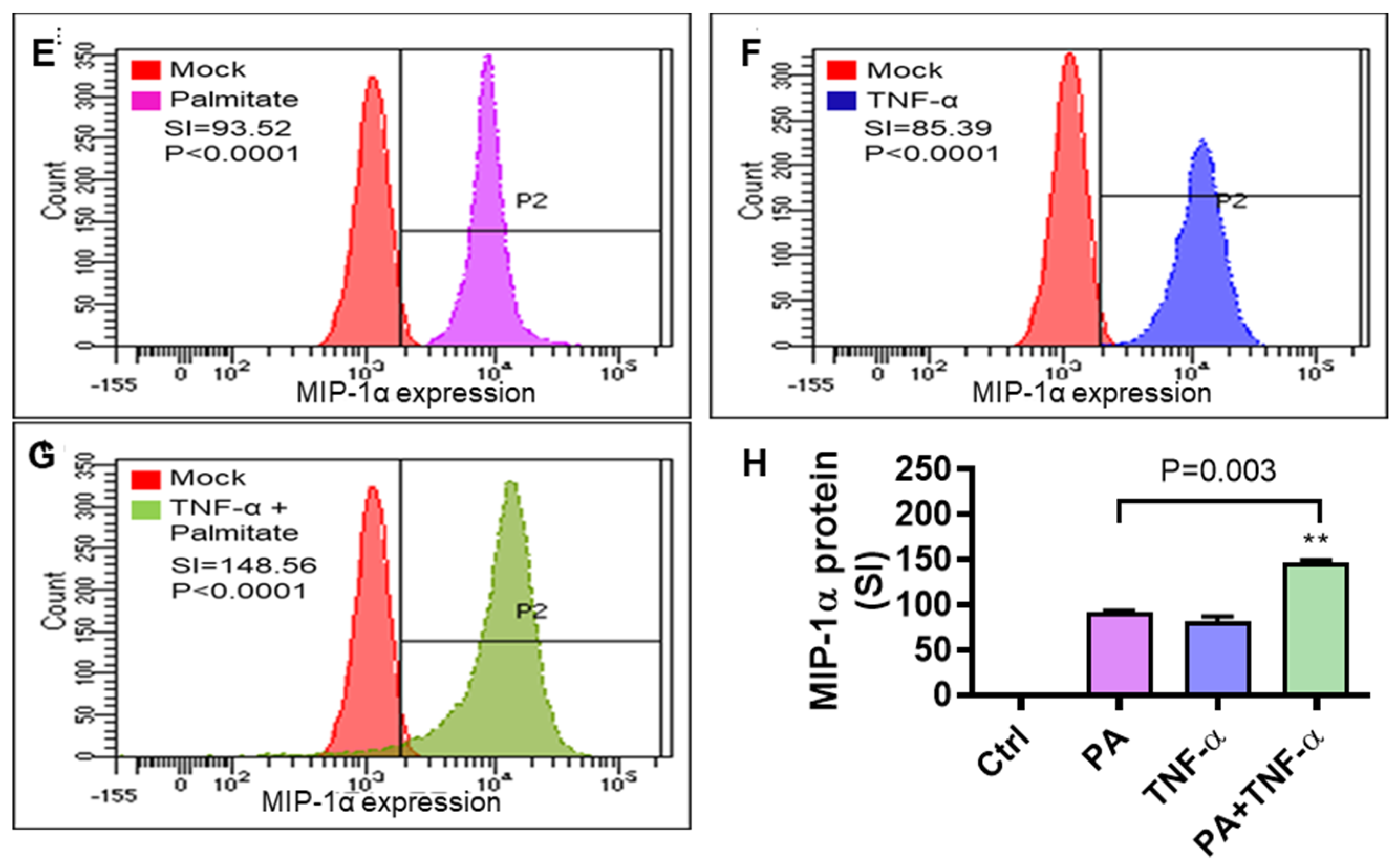
3.1. Increased MIP-1α Expression in THP-1 Cells and Primary Human Macrophages Co-Stimulated with Palmitate and TNF-α
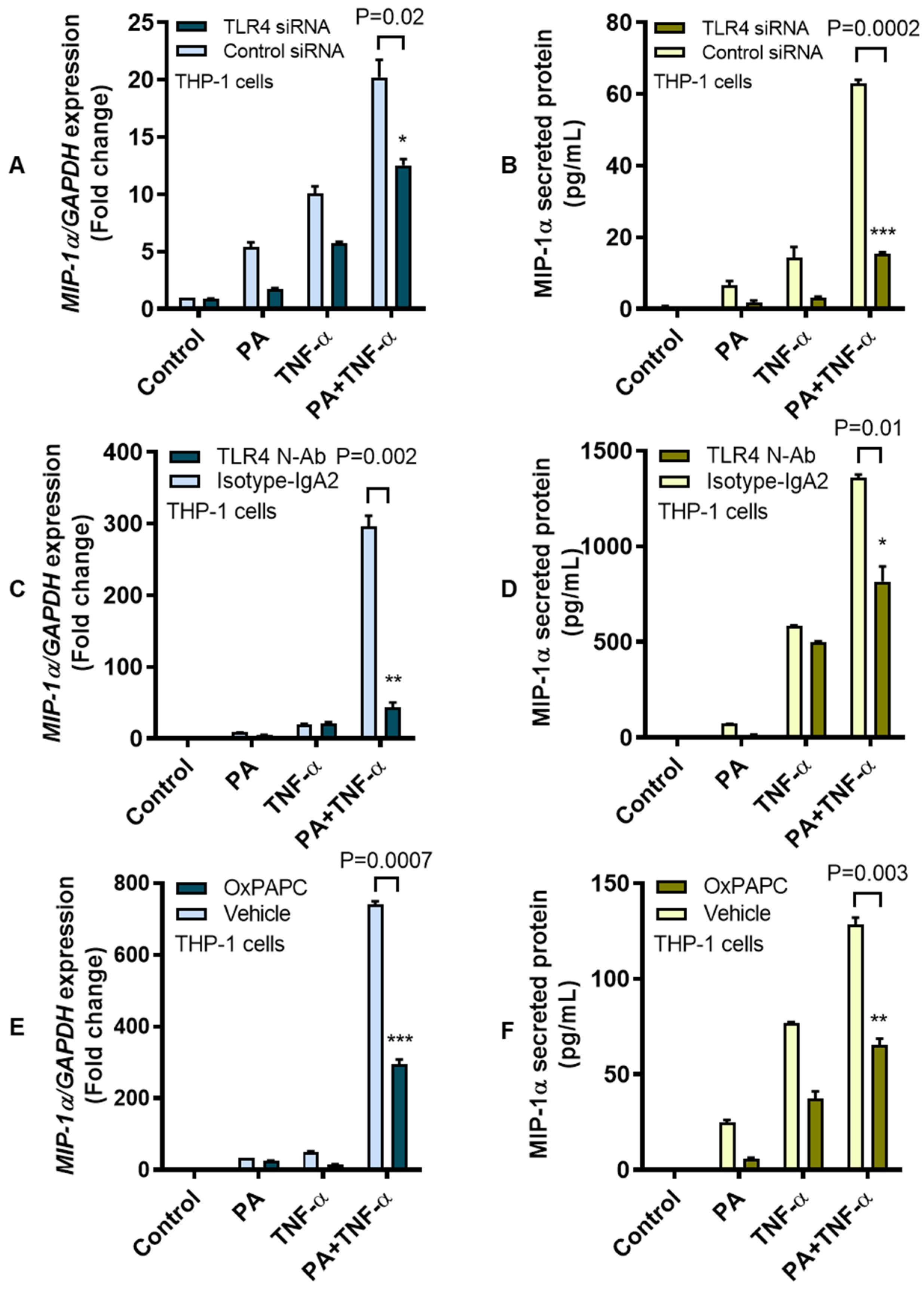
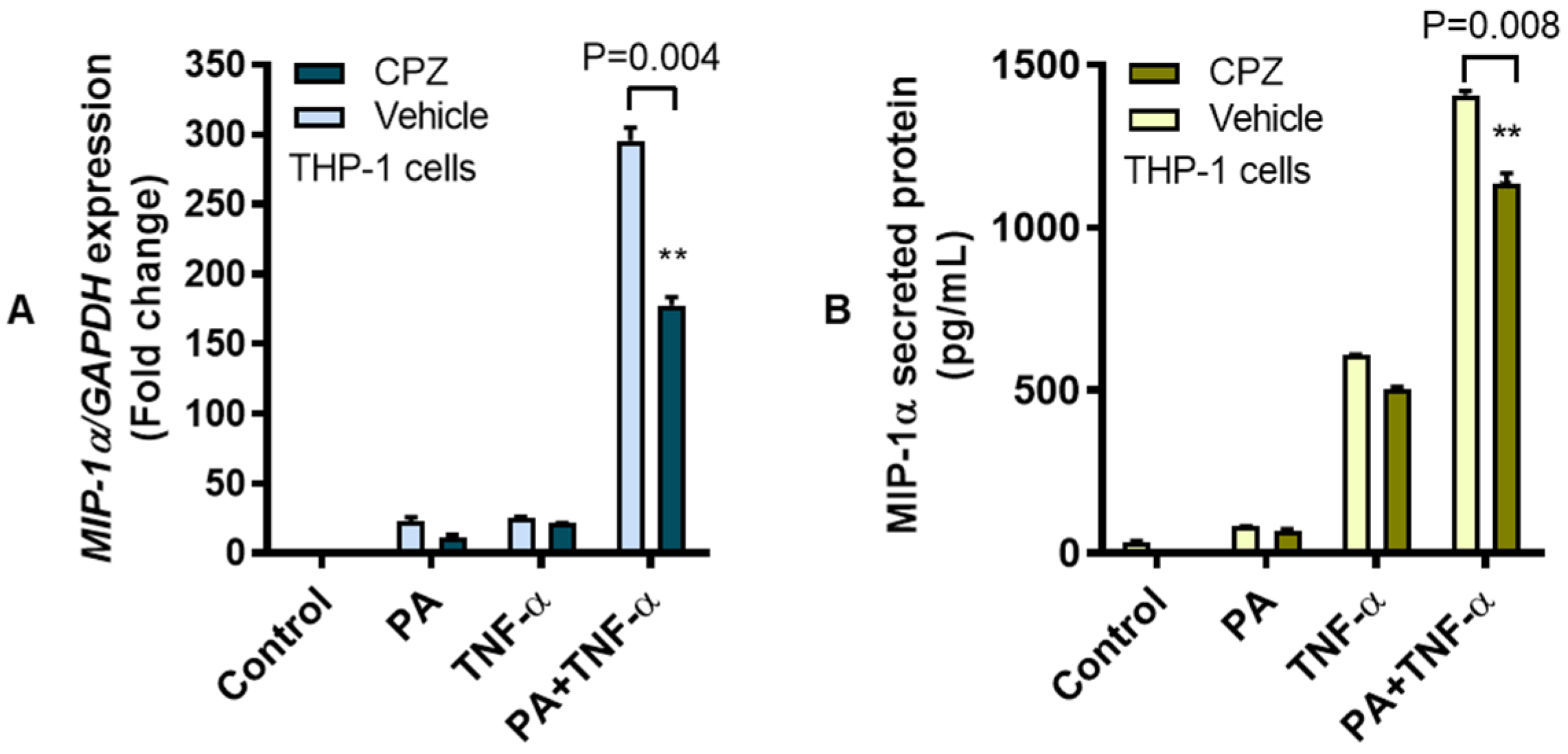
3.2. MIP-1α Co-Induction by Palmitate and TNF-α Involves the TLR4-Mediated Signaling and Clathrin-Mediated Endocytosis (CME)
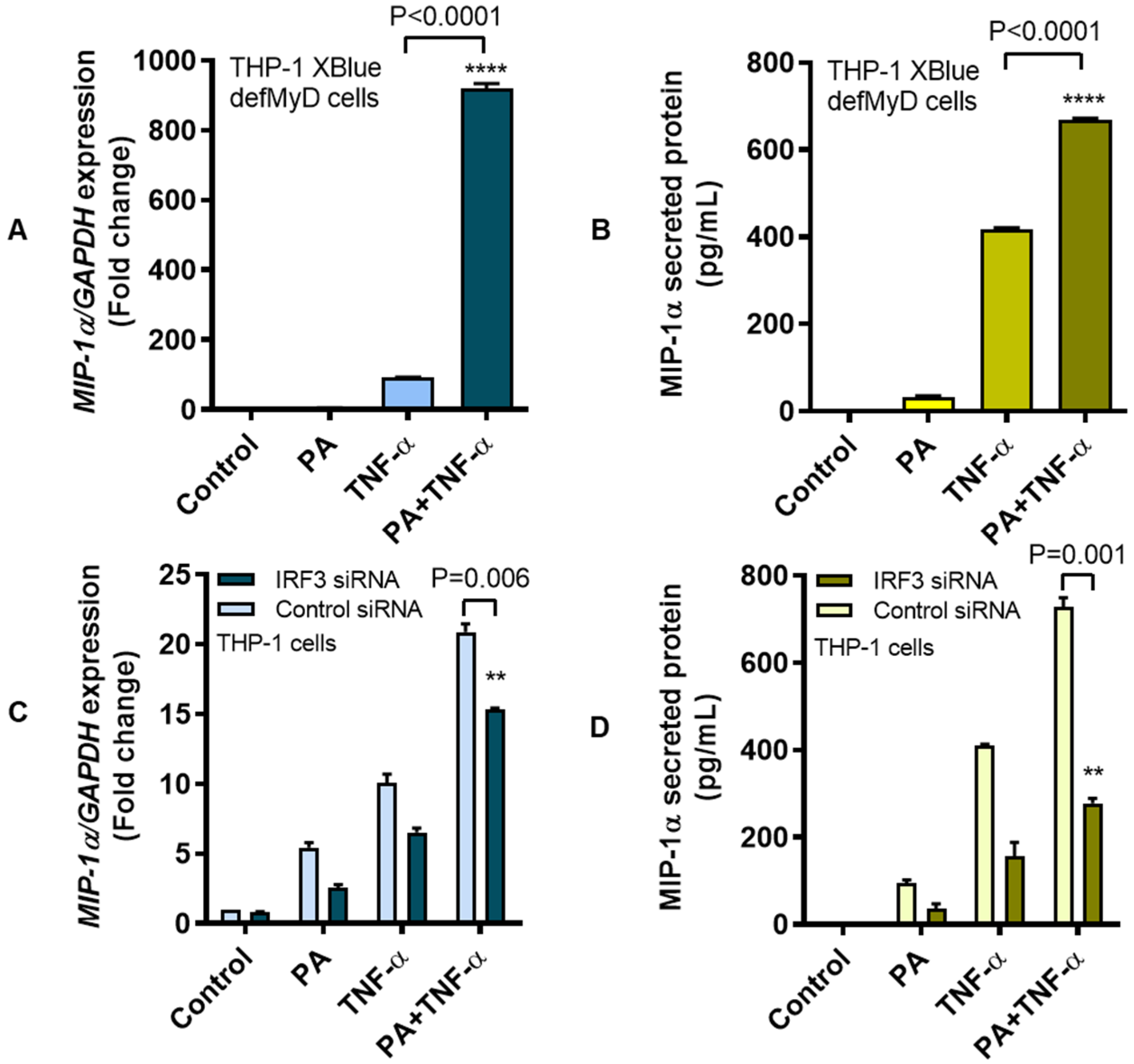
3.3. MIP-1α Co-Induced by Palmitate and TNF-α Involves the IRF3 Pathway
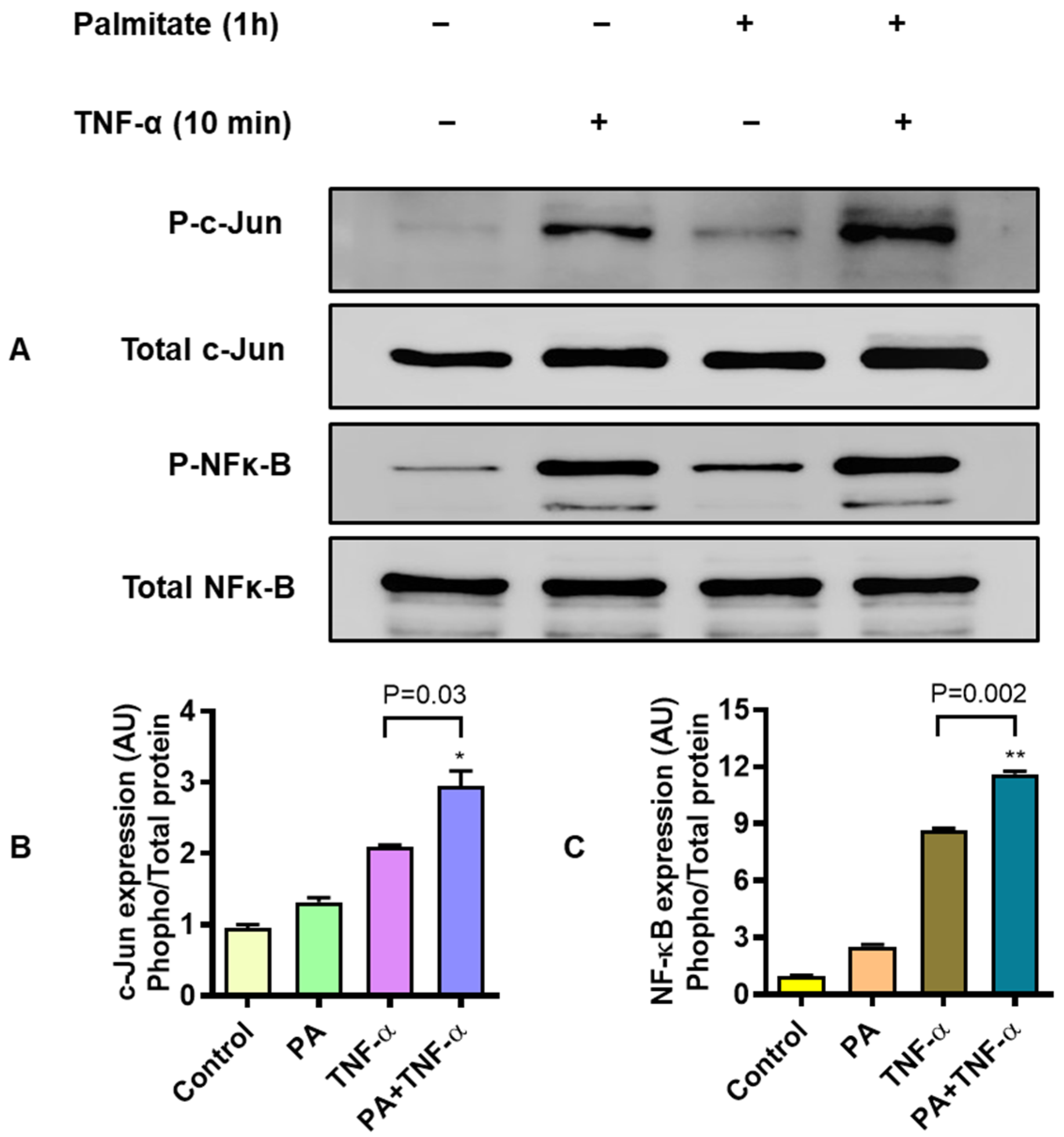
3.4. MIP-1α Co-Induced by Palmitate and TNF-α Involves Signaling Via c-Jun and NF-κB
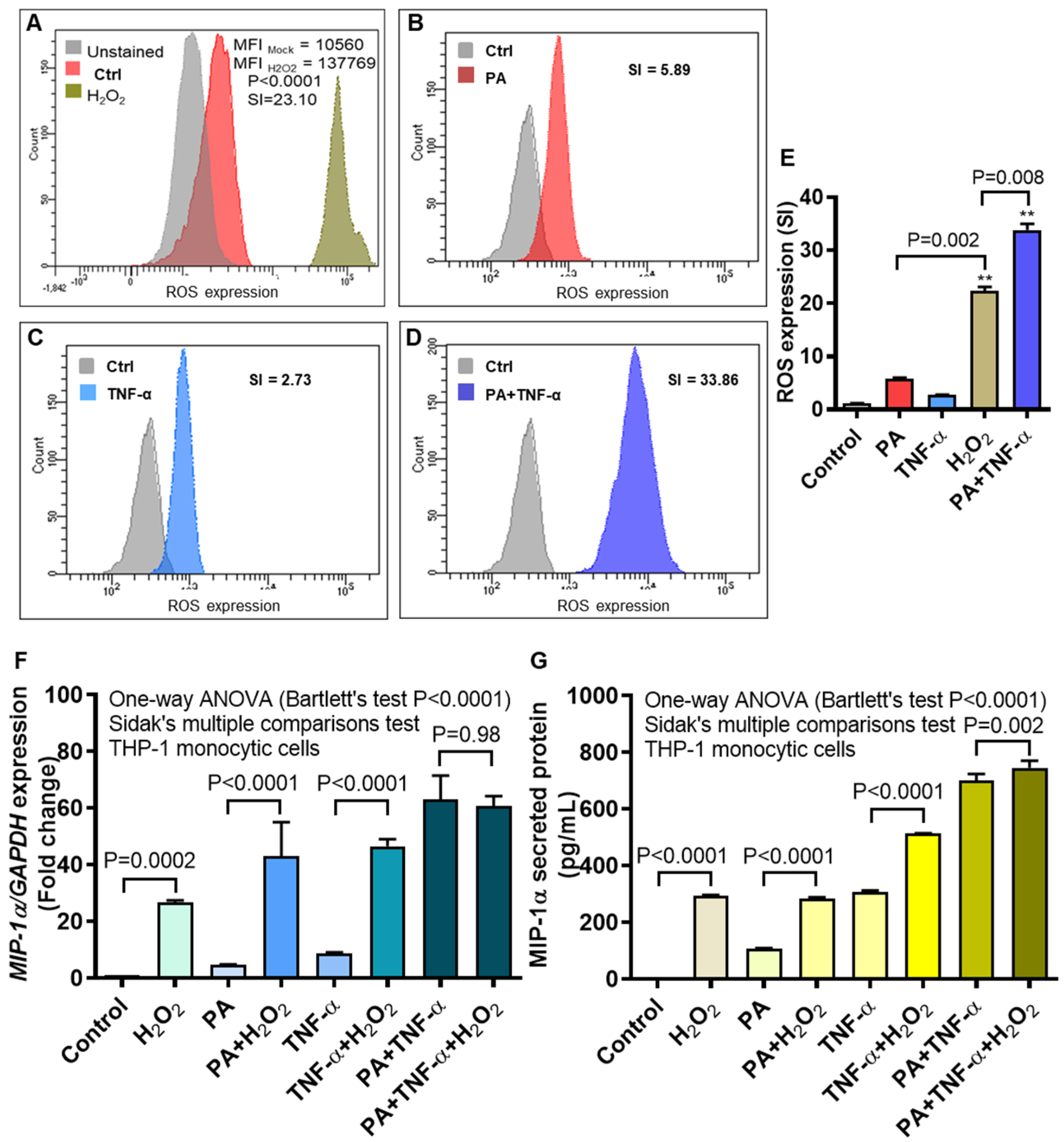
3.5. MIP-1α Expression Induced by Palmitate and/or TNF-α, in Presence or Absence of Oxidative Stress
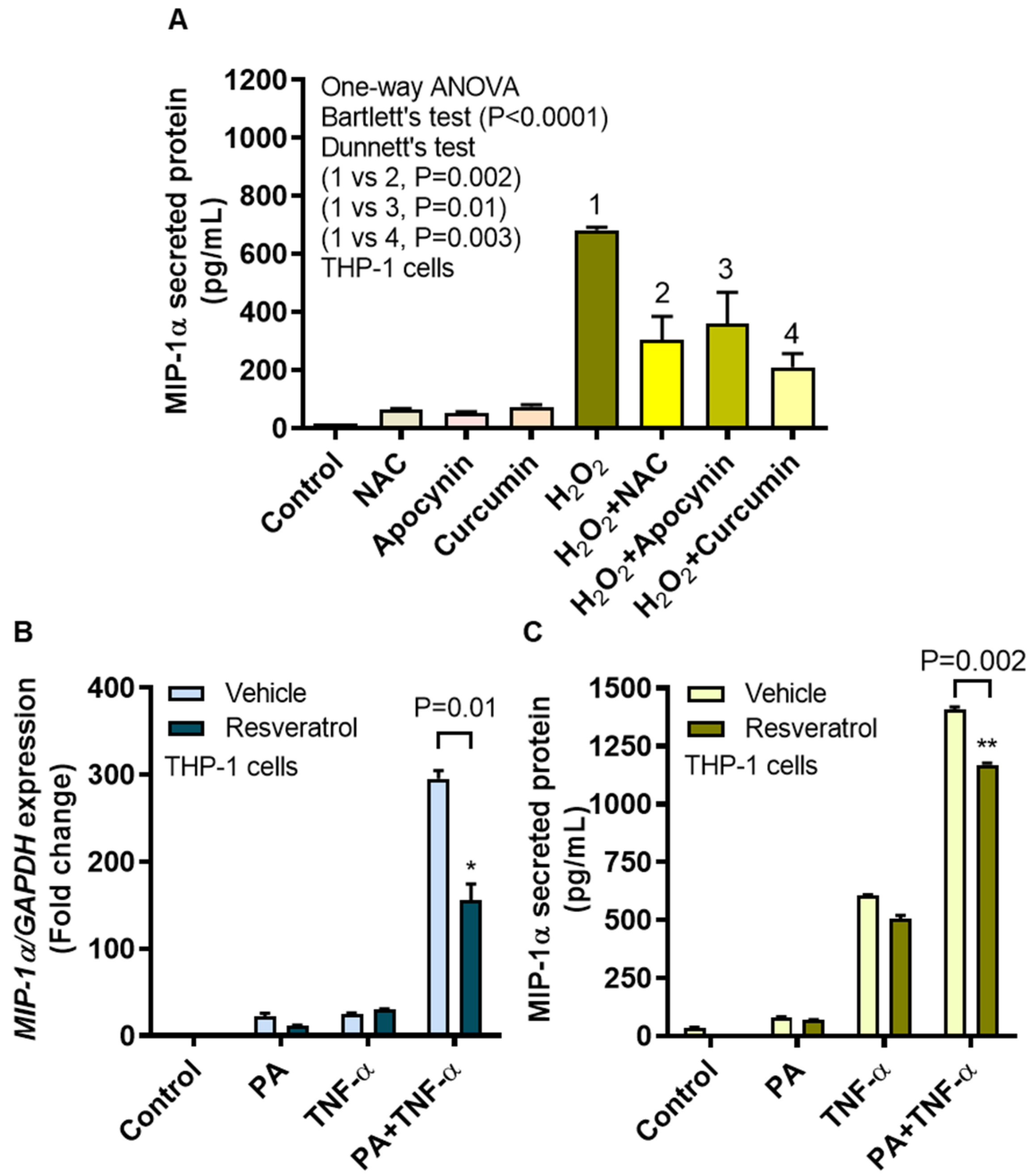
3.6. MIP-1α Induction by Oxidative Stress Is Counteracted by ROS/NF-κB Inhibitors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATCC | American Type Culture Collection |
| CME | Clathrin-mediated endocytosis |
| CPZ | Chlorpromazine |
| DCFH-DA | Dichloro-dihydro-fluorescein diacetate |
| ELISA | Enzyme-linked immunosorbent assay |
| FFAs | Free fatty acids |
| GSH | Reduced glutathione |
| H2O2 | Hydrogen peroxide |
| IRF3 | Interferon regulatory factor-3 |
| LPS | Lipopolysaccharide |
| MFI | Mean fluorescence intensity |
| MIP-1α | Macrophage inflammatory protein-1 |
| MIPs | Macrophage inflammatory proteins |
| MyD88 | Myeloid differentiation-88 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NIK | NF-κB-inducing kinase |
| NOX | NADPH oxidase |
| O2–• | Superoxide anion |
| OD | Optical density |
| OxPAPC | Oxidized phospholipids such as 1-palmitoyl-2-arachidonyl-snglycero-3-phosphorylcholine |
| PBMC | Peripheral blood mononuclear cells |
| qRT-PCR | Quantitative reverse transcription, polymerase chain reaction |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SI | Staining index |
| TLR4 | Toll-like receptor |
| TLRs | Toll-like receptors |
| TNFR | TNF receptor |
| TNF-α | Tumor necrosis factor-α |
| TRAM | TRIF related adaptor molecule |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
References
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci 2014, 16, 378–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca-Alaniz, M.H.; Takada, J.; Alonso-Vale, M.I.; Lima, F.B. Adipose tissue as an endocrine organ: From theory to practice. J. Pediatr. (Rio J.) 2007, 83, S192–S203. [Google Scholar] [CrossRef]
- Lavrovsky, Y.; Chatterjee, B.; Clark, R.A.; Roy, A.K. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp. Gerontol. 2000, 35, 521–532. [Google Scholar] [CrossRef]
- Ren, M.; Guo, Q.; Guo, L.; Lenz, M.; Qian, F.; Koenen, R.R.; Xu, H.; Schilling, A.B.; Weber, C.; Ye, R.D.; et al. Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J. 2010, 29, 3952–3966. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.A.; Dao, T.L.; Guignet, M.A.; Geddes, C.E.; Koemeter-Cox, A.I.; Kan, R.K. Increased expression of the chemokines CXCL1 and MIP-1alpha by resident brain cells precedes neutrophil infiltration in the brain following prolonged soman-induced status epilepticus in rats. J. Neuroinflamm. 2011, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Honey, K. CCL3 and CCL4 actively recruit CD8+ T cells. Nat. Rev. Immunol. 2006, 6, 427. [Google Scholar] [CrossRef]
- White, G.E.; Iqbal, A.J.; Greaves, D.R. CC chemokine receptors and chronic inflammation--therapeutic opportunities and pharmacological challenges. Pharmacol. Rev. 2013, 65, 47–89. [Google Scholar] [CrossRef] [Green Version]
- Jordan, L.A.; Fenner, B.F.; Davies, R.; Harvey, A.K.; Choy, E.H.; Errington, R.; Bokarewa, M.; Williams, A.S. 02.31 Targeted inhibition of macrophage inflammatory protein 1-alpha (ccl3) prevents pit formation by human osteoclasts and potently attenuates the erosion of bone in collagen-induced arthritis. Ann. Rheumat. Dis. 2017, 76, A21. [Google Scholar] [CrossRef]
- Amft, N.; Bowman, S.J. Chemokines and cell trafficking in Sjogren’s syndrome. Scand. J. Immunol. 2001, 54, 62–69. [Google Scholar] [CrossRef]
- Ping, D.; Jones, P.L.; Boss, J.M. TNF regulates the in vivo occupancy of both distal and proximal regulatory regions of the MCP-1/JE gene. Immunity 1996, 4, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Maeshima, N.; Fernandez, R.C. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell Infect. Microbiol. 2013, 3, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, R.; Al-Roub, A.; Kochumon, S.; Akther, N.; Thomas, R.; Kumari, M.; Koshy, M.S.; Tiss, A.; Hannun, Y.A.; Tuomilehto, J.; et al. The Synergy between Palmitate and TNF-alpha for CCL2 Production Is Dependent on the TRIF/IRF3 Pathway: Implications for Metabolic Inflammation. J. Immunol. 2018, 200, 3599–3611. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- O’Dea, E.; Hoffmann, A. The regulatory logic of the NF-kappaB signaling system. Cold Spring Harb. Perspect. Biol. 2010, 2, a000216. [Google Scholar] [CrossRef]
- Brasier, A.R. The NF-κB regulatory network. Cardiovasc. Toxicol. 2006, 6, 111–130. [Google Scholar] [CrossRef]
- Zandi, E.; Chen, Y.; Karin, M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: Discrimination between free and NF-kappaB-bound substrate. Science 1998, 281, 1360–1363. [Google Scholar] [CrossRef]
- Lyons, C.L.; Kennedy, E.B.; Roche, H.M. Metabolic Inflammation-Differential Modulation by Dietary Constituents. Nutrients 2016, 8, 247. [Google Scholar] [CrossRef]
- Ahmad, R.; Akhter, N.; Al-Roub, A.; Kochumon, S.; Wilson, A.; Thomas, R.; Ali, S.; Tuomilehto, J.; Sindhu, S. MIP-1alpha Induction by Palmitate in the Human Monocytic Cells Implicates TLR4 Signaling Mechanism. Cell Physiol. Biochem. 2019, 52, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Vazquez, I.; Fernandez-Veledo, S.; Kramer, D.K.; Vila-Bedmar, R.; Garcia-Guerra, L.; Lorenzo, M. Insulin resistance associated to obesity: The link TNF-alpha. Arch. Physiol. Biochem. 2008, 114, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.; Kiefer, F.W.; Zeyda, M.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; Zlabinger, G.J.; Stulnig, T.M. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J. Clin. Endocrinol. Metab. 2008, 93, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindhu, S.T.; Ahmad, R.; Morisset, R.; Ahmad, A.; Menezes, J. Peripheral blood cytotoxic gammadelta T lymphocytes from patients with human immunodeficiency virus type 1 infection and AIDS lyse uninfected CD4+ T cells, and their cytocidal potential correlates with viral load. J. Virol. 2003, 77, 1848–1855. [Google Scholar] [CrossRef] [Green Version]
- Erridge, C.; Kennedy, S.; Spickett, C.M.; Webb, D.J. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: Roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J. Biol. Chem. 2008, 283, 24748–24759. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [Green Version]
- Sindhu, S.; Akhter, N.; Kochumon, S.; Thomas, R.; Wilson, A.; Shenouda, S.; Tuomilehto, J.; Ahmad, R. Increased Expression of the Innate Immune Receptor TLR10 in Obesity and Type-2 Diabetes: Association with ROS-Mediated Oxidative Stress. Cell. Physiol. Biochem. 2018, 45, 572–590. [Google Scholar] [CrossRef]
- Britton, R.G.; Kovoor, C.; Brown, K. Direct molecular targets of resveratrol: Identifying key interactions to unlock complex mechanisms. Ann. N. Y. Acad. Sci. 2015, 1348, 124–133. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Deng, X.; Tamai, R. Mouse macrophages primed with alendronate down-regulate monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1alpha (MIP-1alpha) production in response to Toll-like receptor (TLR) 2 and TLR4 agonist via Smad3 activation. Int. Immunopharmacol. 2009, 9, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Wang, X.; Lantier, L.; Lyubetskaya, A.; Eguchi, J.; Kang, S.; Tenen, D.; Roh, H.C.; Kong, X.; Kazak, L.; et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Investig. 2016, 126, 2839–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haridas, V.; Shrivastava, A.; Su, J.; Yu, G.L.; Ni, J.; Liu, D.; Chen, S.F.; Ni, Y.; Ruben, S.M.; Gentz, R.; et al. VEGI, a new member of the TNF family activates nuclear factor-kappa B and c-Jun N-terminal kinase and modulates cell growth. Oncogene 1999, 18, 6496–6504. [Google Scholar] [CrossRef] [Green Version]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, L.; Cui, J.; Huoc, Z.; Xue, J.; Cui, H.; Mao, Q.; Yang, R. Resveratrol inhibits NF-kB signaling through suppression of p65 and IkappaB kinase activities. Pharmazie 2013, 68, 689–694. [Google Scholar]
- Pillon, N.J.; Azizi, P.M.; Li, Y.E.; Liu, J.; Wang, C.; Chan, K.L.; Hopperton, K.E.; Bazinet, R.P.; Heit, B.; Bilan, P.J.; et al. Palmitate-induced inflammatory pathways in human adipose microvascular endothelial cells promote monocyte adhesion and impair insulin transcytosis. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E35–E44. [Google Scholar] [CrossRef] [Green Version]
- Tzanavari, T.; Giannogonas, P.; Karalis, K.P. TNF-alpha and obesity. Curr. Dir. Autoimmun. 2010, 11, 145–156. [Google Scholar] [CrossRef]
- Chow, J.C.; Young, D.W.; Golenbock, D.T.; Christ, W.J.; Gusovsky, F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol.Chem. 1999, 274, 10689–10692. [Google Scholar] [CrossRef] [Green Version]
- Sindhu, S.; Kochumon, S.; Shenouda, S.; Wilson, A.; Al-Mulla, F.; Ahmad, R. The Cooperative Induction of CCL4 in Human Monocytic Cells by TNF-alpha and Palmitate Requires MyD88 and Involves MAPK/NF-kappaB Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 4658. [Google Scholar] [CrossRef] [Green Version]
- Takei, K.; Haucke, V. Clathrin-mediated endocytosis: Membrane factors pull the trigger. Trends Cell Biol. 2001, 11, 385–391. [Google Scholar] [CrossRef]
- Tatematsu, M.; Yoshida, R.; Morioka, Y.; Ishii, N.; Funami, K.; Watanabe, A.; Saeki, K.; Seya, T.; Matsumoto, M. Raftlin Controls Lipopolysaccharide-Induced TLR4 Internalization and TICAM-1 Signaling in a Cell Type-Specific Manner. J. Immunol. 2016, 196, 3865–3876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husebye, H.; Halaas, O.; Stenmark, H.; Tunheim, G.; Sandanger, O.; Bogen, B.; Brech, A.; Latz, E.; Espevik, T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006, 25, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuper, C.; Beck, F.X.; Neuhofer, W. Toll-like receptor 4 activates NF-kappaB and MAP kinase pathways to regulate expression of proinflammatory COX-2 in renal medullary collecting duct cells. Am. J. Physiol. Renal Physiol. 2012, 302, F38–F46. [Google Scholar] [CrossRef] [Green Version]
- Akhter, N.; Hasan, A.; Shenouda, S.; Wilson, A.; Kochumon, S.; Ali, S.; Tuomilehto, J.; Sindhu, S.; Ahmad, R. TLR4/MyD88 -mediated CCL2 production by lipopolysaccharide (endotoxin): Implications for metabolic inflammation. J. Diabetes Metab. Disord. 2018, 17, 77–84. [Google Scholar] [CrossRef]
- Medeiros, M.C.; Frasnelli, S.C.; Bastos Ade, S.; Orrico, S.R.; Rossa, C., Jr. Modulation of cell proliferation, survival and gene expression by RAGE and TLR signaling in cells of the innate and adaptive immune response: Role of p38 MAPK and NF-KB. J. Appl. Oral Sci. 2014, 22, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Abate, C.; Patel, L.; Rauscher, F.J., 3rd; Curran, T. Redox regulation of fos and jun DNA-binding activity in vitro. Science 1990, 249, 1157–1161. [Google Scholar] [CrossRef]
- Jaramillo, M.; Olivier, M. Hydrogen peroxide induces murine macrophage chemokine gene transcription via extracellular signal-regulated kinase- and cyclic adenosine 5’-monophosphate (cAMP)-dependent pathways: Involvement of NF-kappa B, activator protein 1, and cAMP response element binding protein. J. Immunol. 2002, 169, 7026–7038. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, J.; Delgado-Ramos, L.; Ayala, G.; Pelechano, V.; Medina, D.A.; Carrasco, F.; Gonzalez, R.; Andres-Leon, E.; Steinmetz, L.; Warringer, J.; et al. The cellular growth rate controls overall mRNA turnover, and modulates either transcription or degradation rates of particular gene regulons. Nucleic Acids Res. 2016, 44, 3643–3658. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, K.E.; Simpson, L.; Carter, J.; Hassenbein, D.; Leikauf, G.D. Ozone inhalation stimulates expression of a neutrophil chemotactic protein, macrophage inflammatory protein 2. Toxicol. Appl. Pharmacol. 1993, 119, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, L.H.; Parlakpinar, H.; Ozhan, O.; Ermis, N.; Polat, A.; Vardi, N.; Tanbek, K.; Yildiz, A.; Acet, A. Inhibition of NADPH oxidase by apocynin promotes myocardial antioxidant response and prevents isoproterenol-induced myocardial oxidative stress in rats. Free Radic. Res. 2017, 51, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Lestari, M.L.; Indrayanto, G. Curcumin. Profiles Drug Subst. Excip. Relat. Methodol. 2014, 39, 113–204. [Google Scholar] [CrossRef] [PubMed]
- Odewumi, C.O.; Latinwo, L.M.; Ruden, M.L.; Badisa, V.L.; Fils-Aime, S.; Badisa, R.B. Modulation of cytokines and chemokines expression by NAC in cadmium chloride treated human lung cells. Environ. Toxicol. 2016, 31, 1612–1619. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Vanaudenaerde, B.M.; Dupont, L.J.; Demedts, M.G.; Verleden, G.M. N-acetylcysteine reduces chemokine release via inhibition of p38 MAPK in human airway smooth muscle cells. Eur. Respir. J. 2003, 22, 43–49. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sindhu, S.; Akhter, N.; Wilson, A.; Thomas, R.; Arefanian, H.; Al Madhoun, A.; Al-Mulla, F.; Ahmad, R. MIP-1α Expression Induced by Co-Stimulation of Human Monocytic Cells with Palmitate and TNF-α Involves the TLR4-IRF3 Pathway and Is Amplified by Oxidative Stress. Cells 2020, 9, 1799. https://doi.org/10.3390/cells9081799
Sindhu S, Akhter N, Wilson A, Thomas R, Arefanian H, Al Madhoun A, Al-Mulla F, Ahmad R. MIP-1α Expression Induced by Co-Stimulation of Human Monocytic Cells with Palmitate and TNF-α Involves the TLR4-IRF3 Pathway and Is Amplified by Oxidative Stress. Cells. 2020; 9(8):1799. https://doi.org/10.3390/cells9081799
Chicago/Turabian StyleSindhu, Sardar, Nadeem Akhter, Ajit Wilson, Reeby Thomas, Hossein Arefanian, Ashraf Al Madhoun, Fahd Al-Mulla, and Rasheed Ahmad. 2020. "MIP-1α Expression Induced by Co-Stimulation of Human Monocytic Cells with Palmitate and TNF-α Involves the TLR4-IRF3 Pathway and Is Amplified by Oxidative Stress" Cells 9, no. 8: 1799. https://doi.org/10.3390/cells9081799