Development of Artificial Plasma Membranes Derived Nanovesicles Suitable for Drugs Encapsulation


 , , and
, , and
Abstract
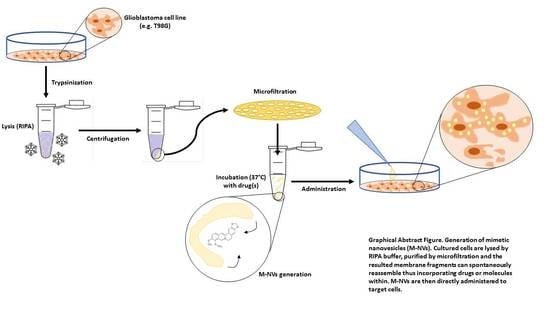
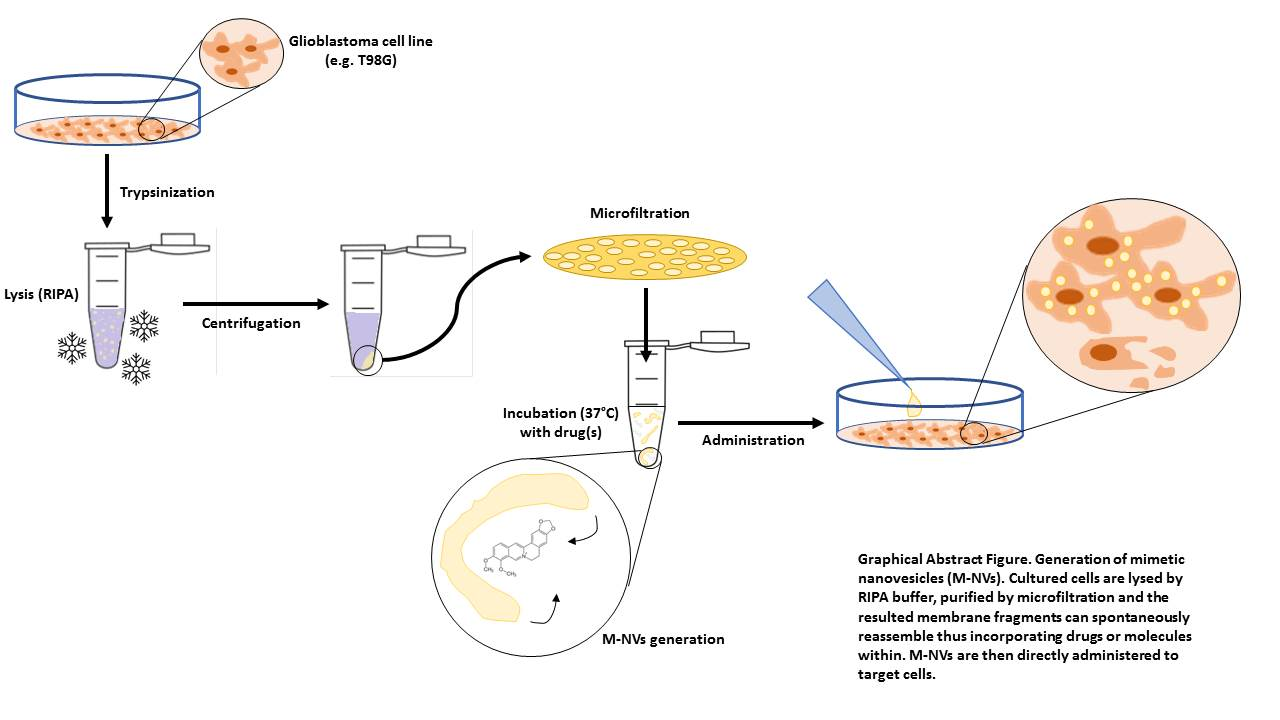
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
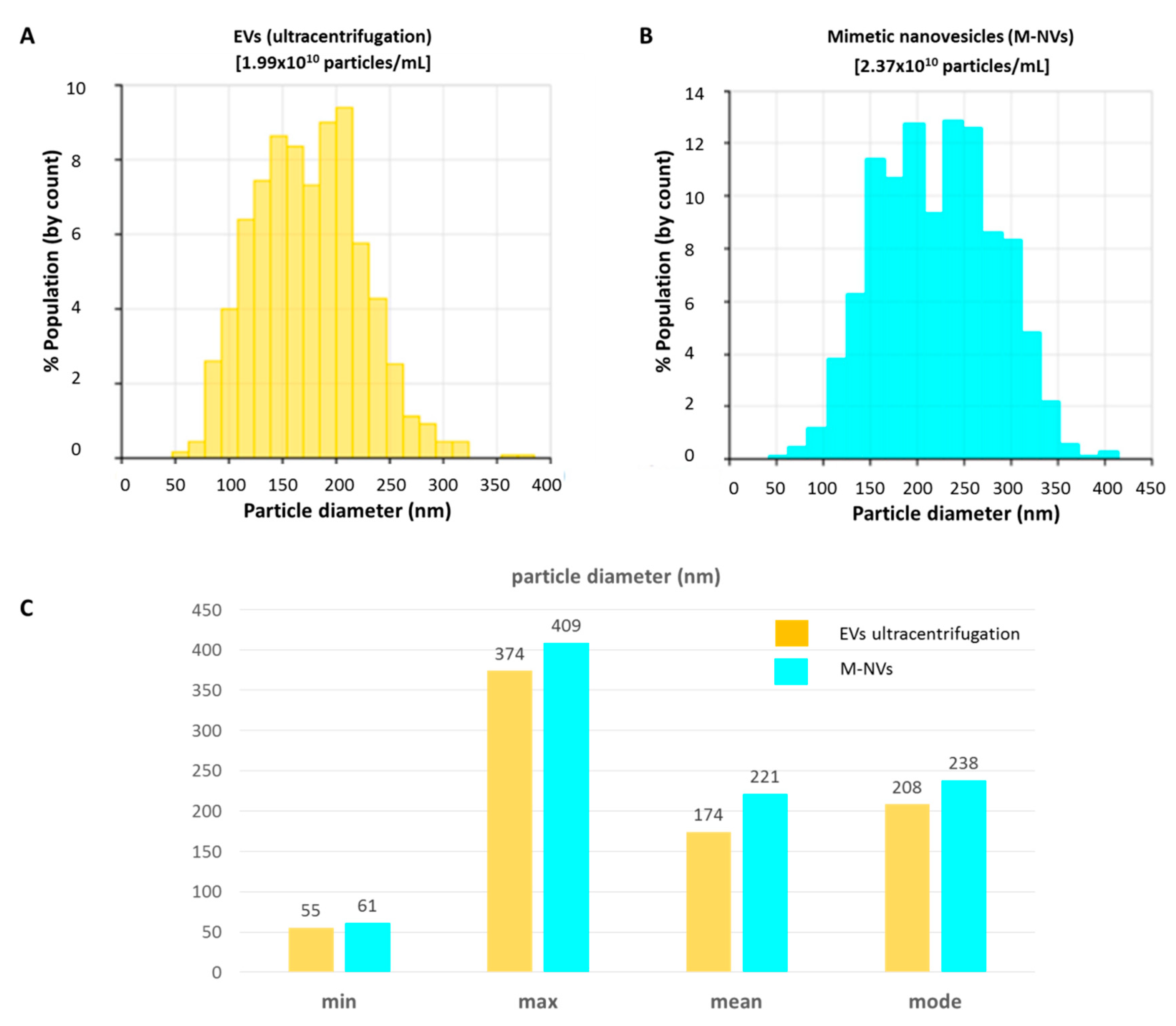
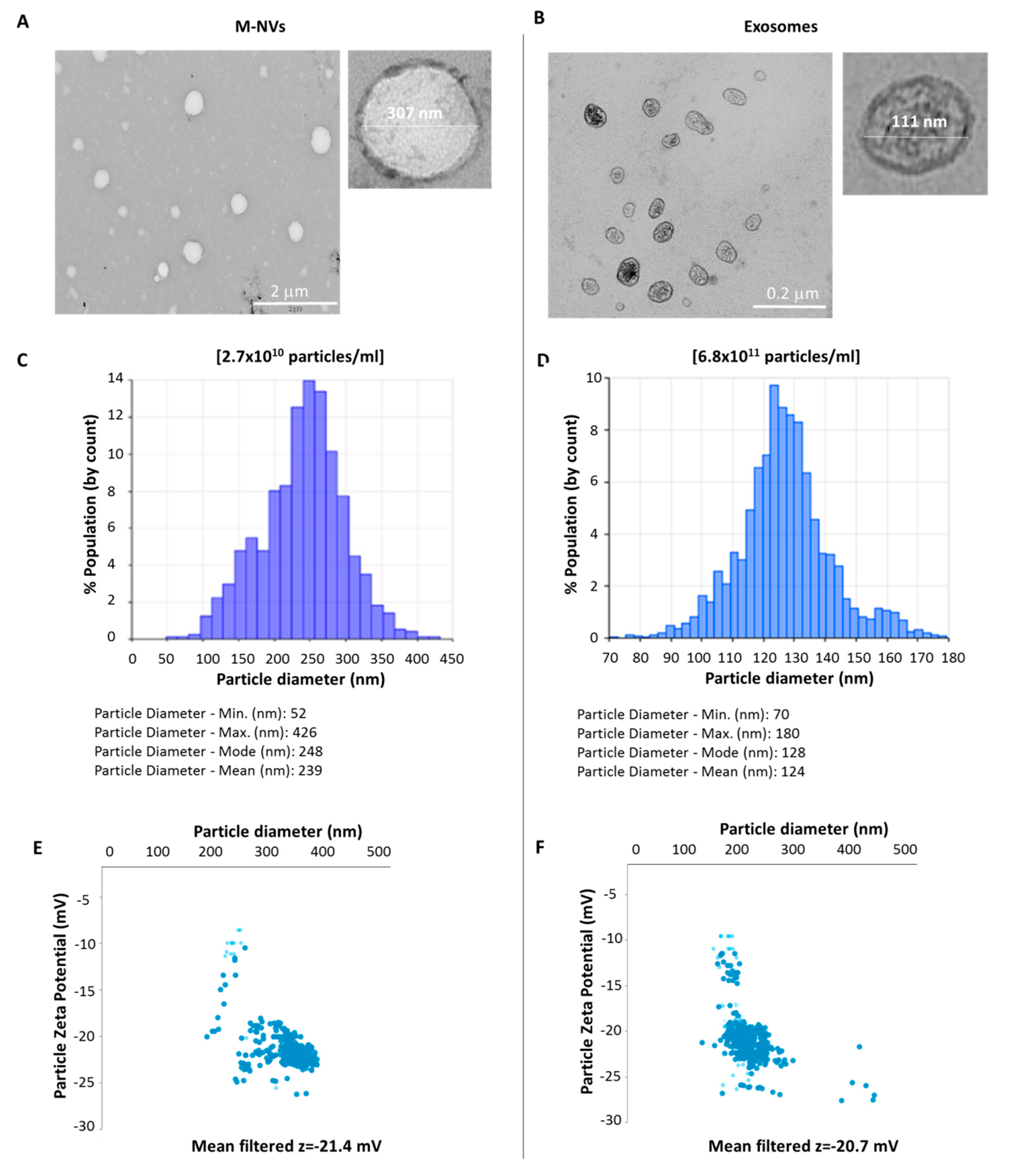
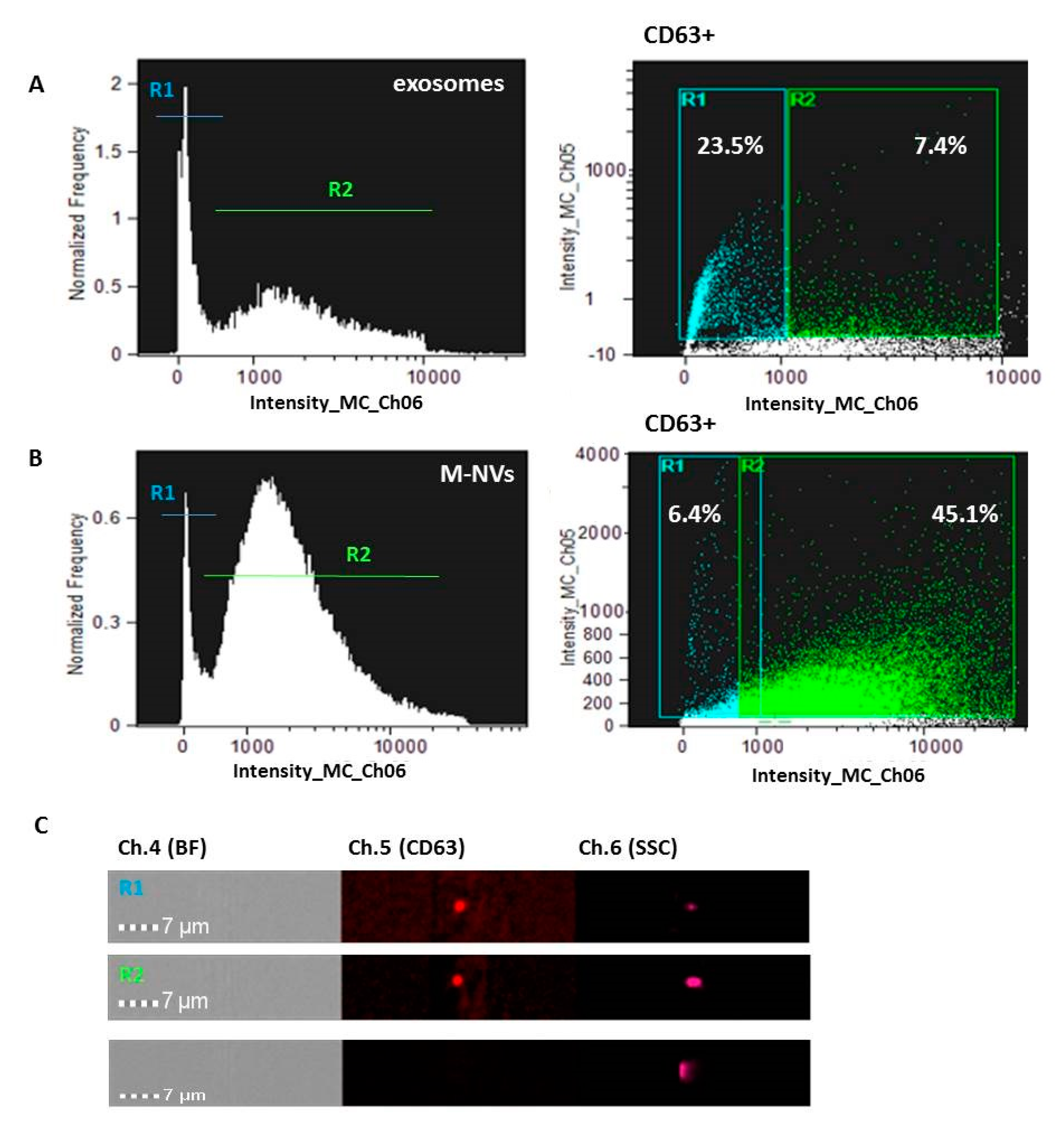
2.1. Isolation and Characterization of Extracellular Vesicles (EVs)
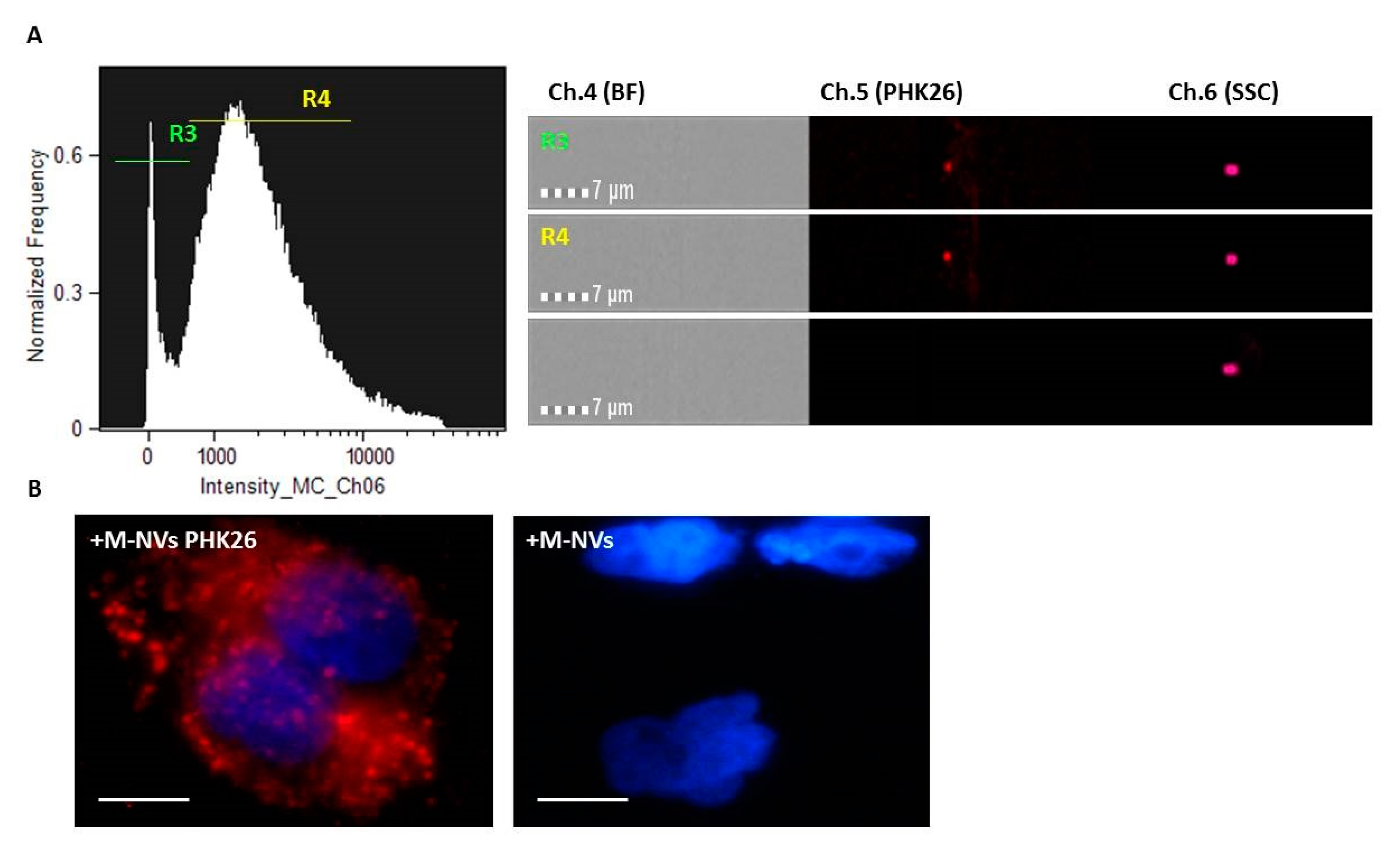
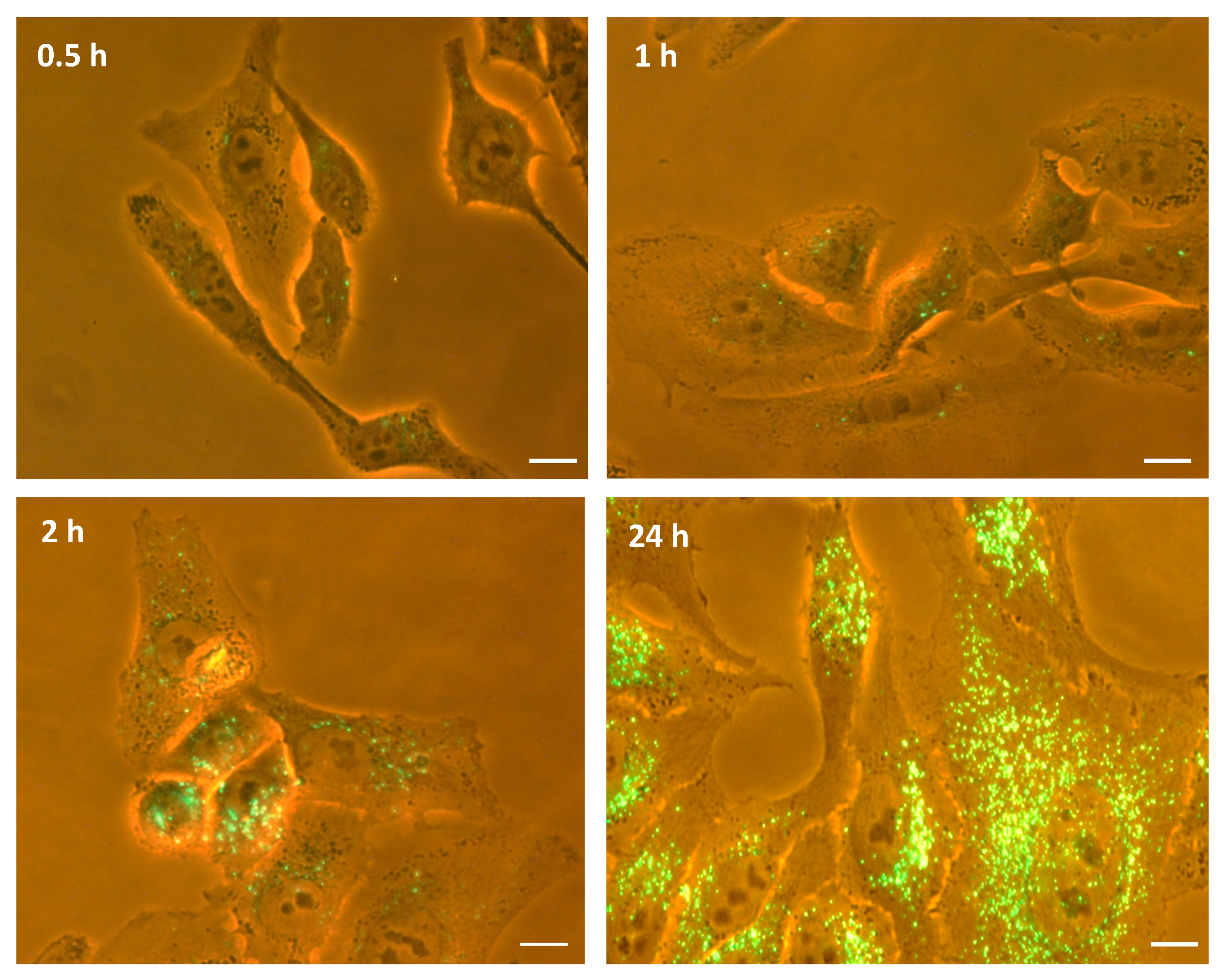
2.2. Generation of M-NVs
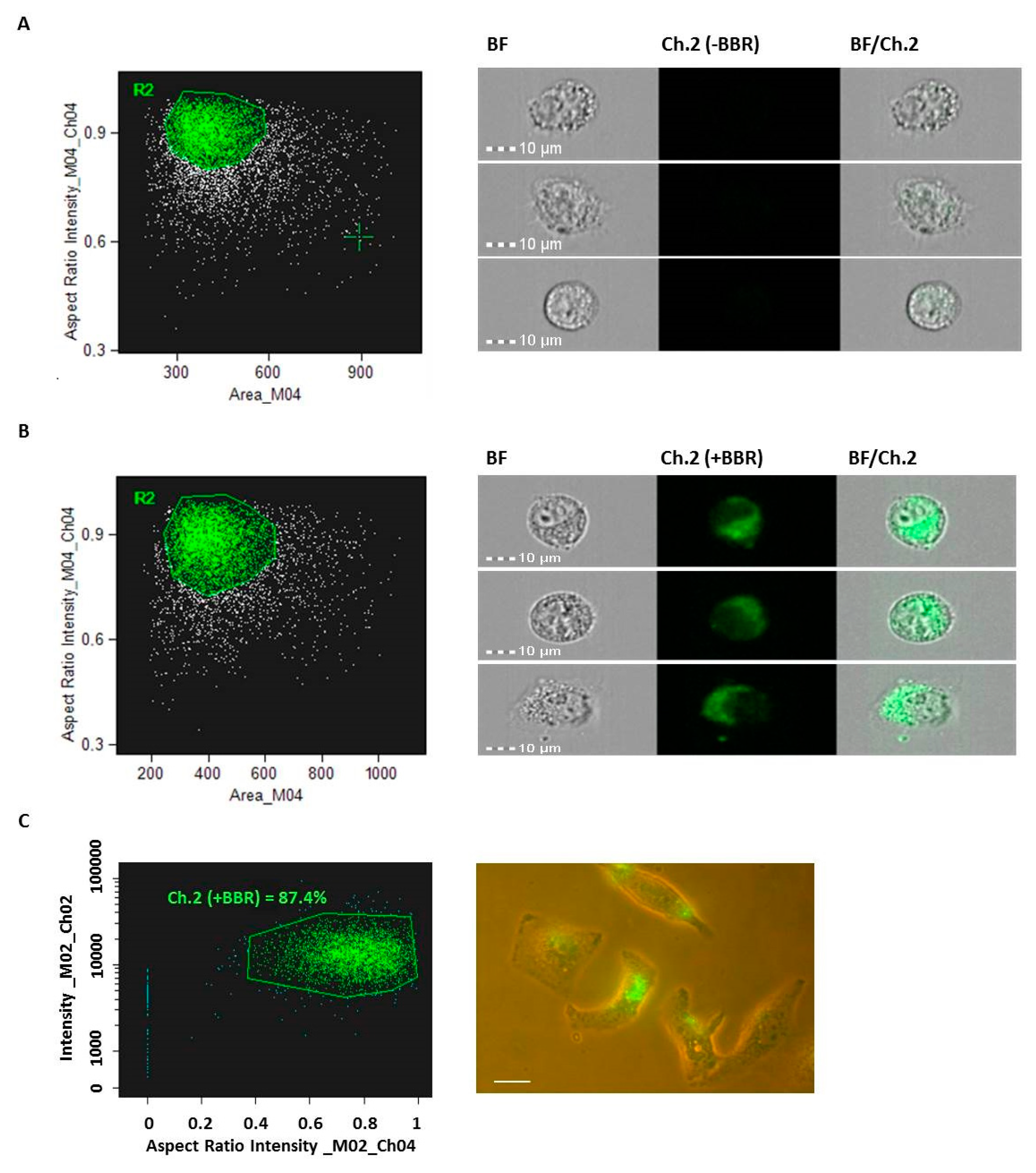
2.3. CD63 and PHK Membranes Staining of M-NVs or Exosomes
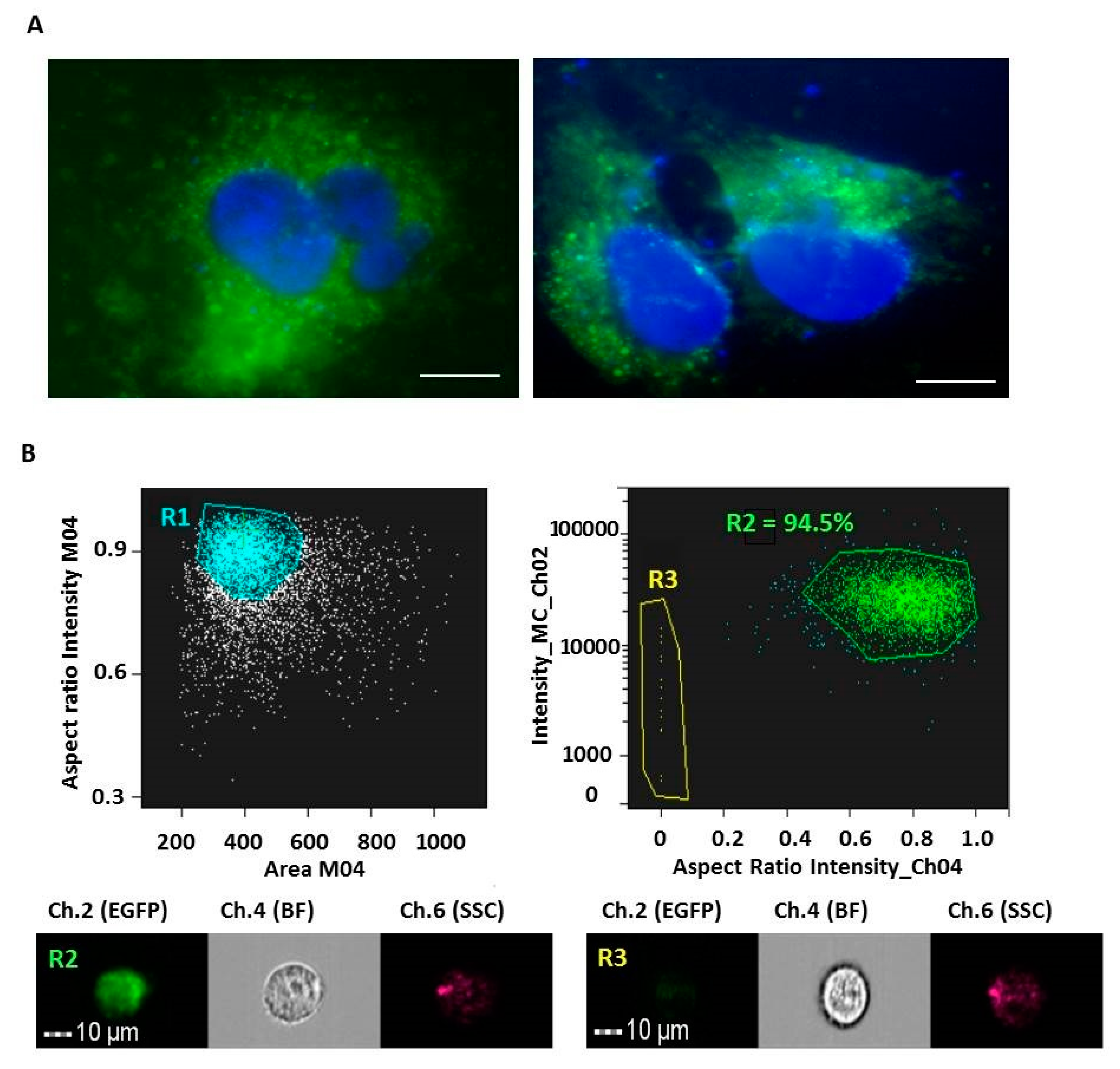
2.4. pEGFP Electroporation of M-NVs
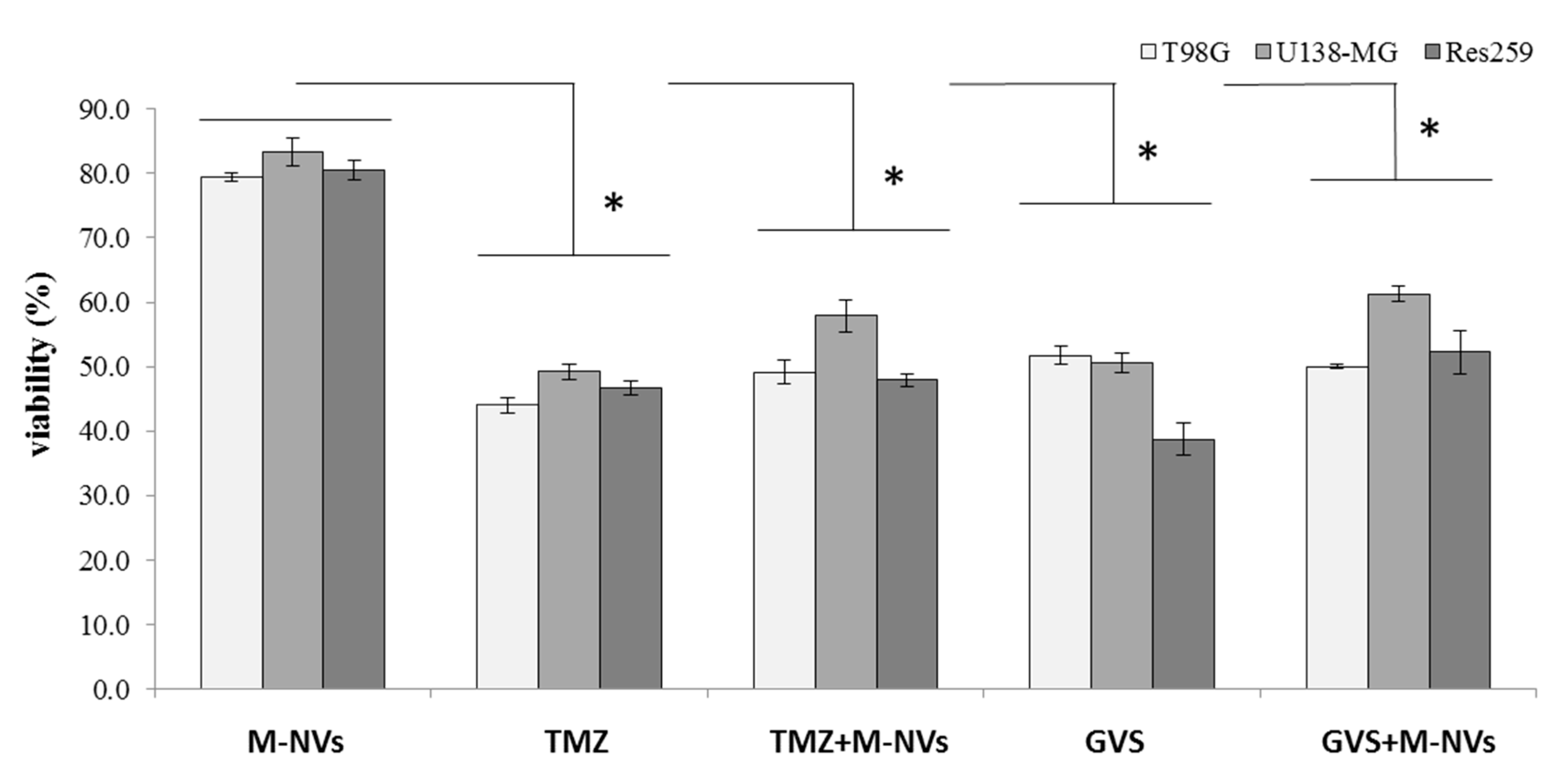
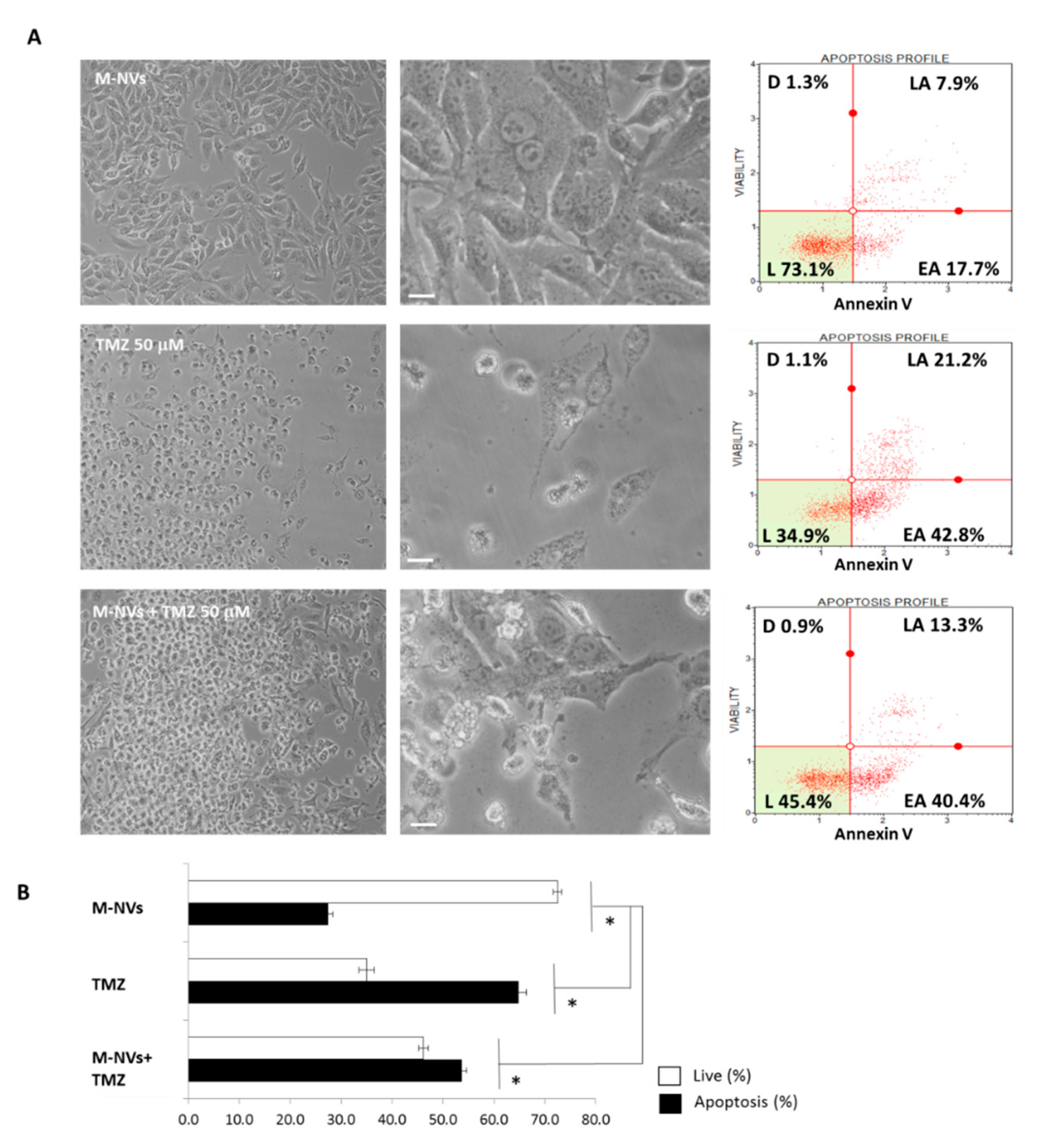
2.5. Berberine, Temozolomide or Givinostat In Vitro Treatments
2.6. Viability and Apoptosis Cytofluorimetric Analysis
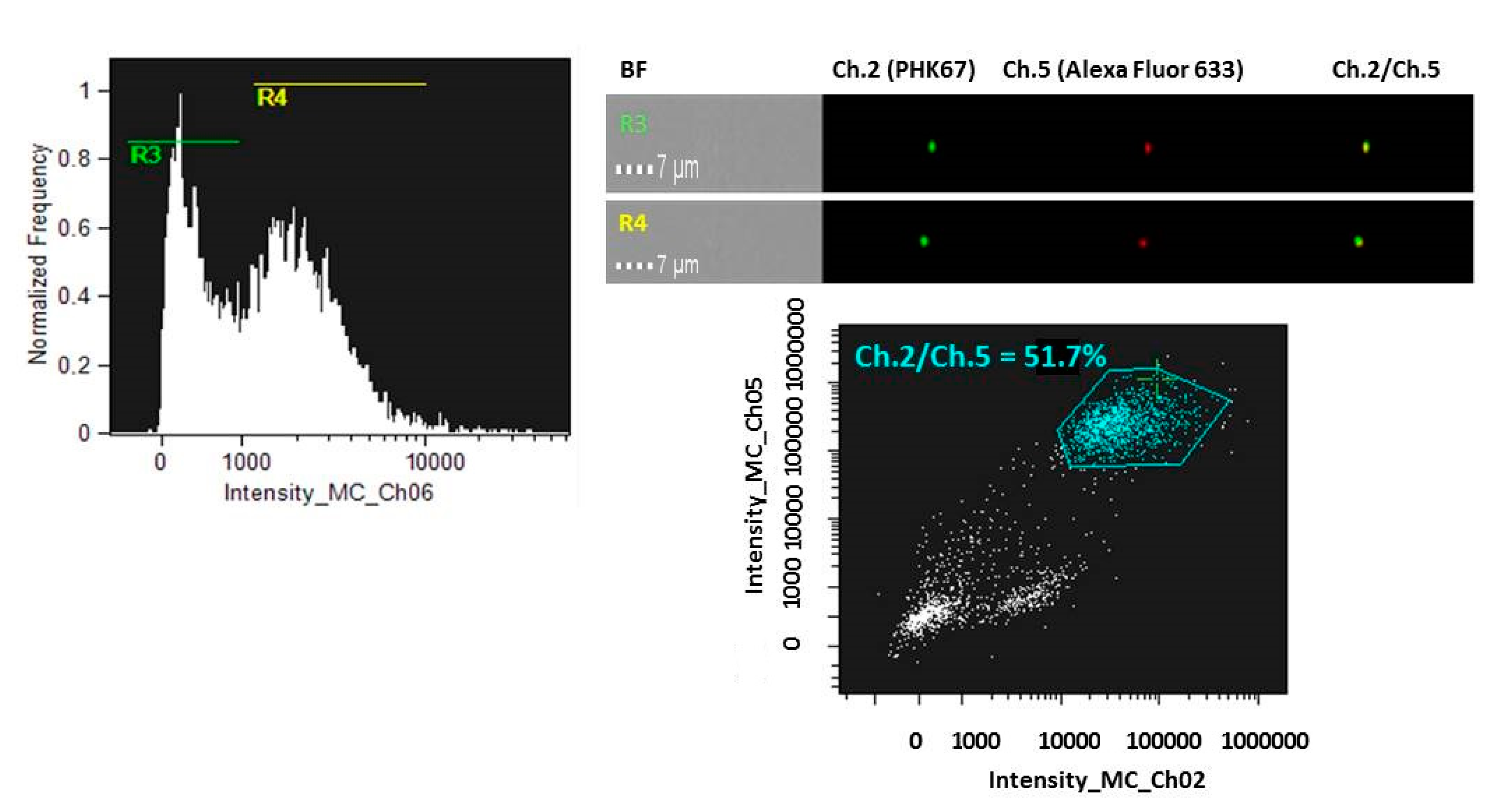
2.7. Imagestream Analysis
2.8. Statistical Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hugel, B.; Martínez, M.C.; Kunzelmann, C.; Freyssinet, J.M. Membrane microparticles: Two sides of the coin. Physiology (Bethesda) 2005, 20, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Soderland, C.; Horstman, L.L.; Ahn, Y.S. Endothelial cells release phenotypically and quantitatively distinct microparticles inactivation and apoptosis. Thromb. Res. 2003, 109, 175–180. [Google Scholar] [CrossRef]
- Mause, S.F.; Weber, C. Microparticles: Protagonists of a novel communication network for intercellular information exchange. Circ. Res. 2010, 107, 1047–1057. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Camacho, L.; Guerrero, P.; Marchetti, D. MicroRNA and protein profiling of brain metastasis competent cell-derived exosomes. PLoS ONE 2013, 8, e73790. [Google Scholar] [CrossRef] [Green Version]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Wolf, P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- Krause, M.; Samoylenko, A.; Vainio, S.J. Exosomes as renal inductive signals in health and disease, and their application as diagnostic markers and therapeutic agents. Front. Cell Dev. Biol. 2015, 3, 65. [Google Scholar] [CrossRef] [Green Version]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef]
- Holder, B.S.; Tower, C.L.; Jones, C.J.; Aplin, J.D.; Abrahams, V.M. Heightened pro-inflammatory effect of preeclamptic placental microvesicles on peripheral blood immune cells in humans. Biol. Reprod. 2012, 86, 103. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, N.; Tan, S.; Boudreau, L.H.; Cramb, C.; Subbaiah, R.; Lahey, L.; Albert, A.; Shnayder, R.; Gobezie, R.; Nigrovic, P.A.; et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Mol. Med. 2013, 5, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Prakash, P.S.; Caldwell, C.C.; Lentsch, A.B.; Pritts, T.A.; Robinson, B.R. Human microparticles generated during sepsis in patients with critical illness are neutrophil-derived and modulate the immune response. J. Trauma Acute Care Surg. 2012, 73, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Valenti, R.; Huber, V.; Iero, M.; Filipazzi, P.; Parmiani, G.; Rivoltini, L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res. 2007, 67, 2912–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, R.; Cabeza, L.; Perazzoli, G.; Jimenez-Lopez, J.; García-Pinel, B.; Melguizo, C.; Prados, J. Nanoformulations for glioblastoma multiforme: A new hope for treatment. Future Med. Chem. 2019, 11, 2459–2480. [Google Scholar] [CrossRef]
- Diaz, R.J.; Ali, S.; Qadir, M.G.; De La Fuente, M.I.; Ivan, M.E.; Komotar, R.J. The role of bevacizumab in the treatment of glioblastoma. J. Neurooncol. 2017, 133, 455–467. [Google Scholar] [CrossRef]
- Mujokoro, B.; Adabi, M.; Sadroddiny, E.; Adabi, M.; Khosravani, M. Nano-structures mediated co-delivery of therapeutic agents for glioblastoma treatment: A review. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 69, 1092–1102. [Google Scholar] [CrossRef]
- Šamec, N.; Zottel, A.; Videtič Paska, A.; Jovčevska, I. Nanomedicine and Immunotherapy: A Step Further towards Precision Medicine for Glioblastoma. Molecules 2020, 25, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zottel, A.; Videtič Paska, A.; Jovčevska, I. Nanotechnology Meets Oncology: Nanomaterials in Brain Cancer Research, Diagnosis and Therapy. Materials (Basel) 2019, 12, 1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; Relano-Gines, A.; Resina, S.; Brillaud, E.; Casanova, D.; Vincent, C.; Hamela, C.; Poupeau, S.; Laffont, M.; Gabelle, A.; et al. Systemic delivery of siRNA down regulates brain prion protein and ameliorates neuropathology in prion disorder. PLoS ONE 2014, 9, e88797. [Google Scholar] [CrossRef] [PubMed]
- Shigemoto-Kuroda, T.; Oh, J.Y.; Kim, D.K.; Jeong, H.J.; Park, S.Y.; Lee, H.J.; Park, J.W.; Kim, T.W.; An, S.Y.; Prockop, D.J.; et al. MSC-derived Extracellular Vesicles Attenuate Immune Responses in Two Autoimmune Murine Models: Type 1 Diabetes and Uveoretinitis. Stem Cell Rep. 2017, 8, 1214–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooijmans, S.A.A.; Schiffelers, R.M.; Zarovni, N.; Vago, R. Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharmacol. Res. 2016, 111, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Nasiri Kenari, A.; Cheng, L.; Hill, A.F. Methods for loading therapeutics into extracellular vesicles and generating extracellular vesicles mimetic-nanovesicles. Methods 2020, 177, 103–113. [Google Scholar] [CrossRef]
- Nasiri Kenari, A.; Kastaniegaard, K.; Greening, D.W.; Shambrook, M.; Stensballe, A.; Cheng, L.; Hill, A.F. Proteomic and Post-Translational Modification Profiling of Exosome-Mimetic Nanovesicles Compared to Exosomes. Proteomics 2019, 19, e1800161. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.C.; Rui, B.Y.; Wang, Q.Y.; Zhou, D.; Zhang, Y.; Guo, S.C. Extracellular vesicle-mimetic nanovesicles transport LncRNA-H19 as competing endogenous RNA for the treatment of diabetic wounds. Drug Deliv. 2018, 25, 241–255. [Google Scholar] [CrossRef] [Green Version]
- García-Manrique, P.; Matos, M.; Gutiérrez, G.; Pazos, C.; Blanco-López, M.C. Therapeutic biomaterials based on extracellular vesicles: Classification of bio-engineering and mimetic preparation routes. J. Extracell. Vesicles 2018, 7, 1422676. [Google Scholar] [CrossRef] [Green Version]
- Bobola, M.S.; Silber, J.R.; Ellenbogen, R.G.; Geyer, J.R.; Blank, A.; Goff, R.D. O6-methylguanine-DNA methyltransferase, O6-benzylguanine, and resistance to clinical alkylators in pediatric primary brain tumor cell lines. Clin. Cancer Res. 2005, 11, 2747–2755. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Xu, R.; Greening, D.W.; Zhu, H.J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Comincini, S.; Manai, F.; Meazza, C.; Pagani, S.; Martinelli, C.; Pasqua, N.; Pelizzo, G.; Biggiogera, M.; Bozzola, M. Identification of Autophagy-Related Genes and Their Regulatory miRNAs Associated with Celiac Disease in Children. Int. J. Mol. Sci. 2017, 18, 391. [Google Scholar] [CrossRef]
- Weatherall, E.; Willmott, G.R. Applications of tunable resistive pulse sensing. Analyst 2015, 140, 3318–3334. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, C.; Gabriele, F.; Manai, F.; Ciccone, R.; Novara, F.; Sauta, E.; Bellazzi, R.; Patane, M.; Moroni, I.; Paterra, R.; et al. The Search for Molecular Markers in a Gene-Orphan Case Study of a Pediatric Spinal Cord Pilocytic Astrocytoma. Cancer Genom. Proteom. 2020, 17, 117–130. [Google Scholar] [CrossRef]
- Palumbo, S.; Tini, P.; Toscano, M.; Allavena, G.; Angeletti, F.; Manai, F.; Miracco, C.; Comincini, S.; Pirtoli, L. Combined EGFR and autophagy modulation impairs cell migration and enhances radiosensitivity in human glioblastoma cells. J. Cell. Physiol. 2014, 229, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Sbalchiero, E.; Azzalin, A.; Palumbo, S.; Barbieri, G.; Arias, A.; Simonelli, L.; Ferretti, L.; Comincini, S. Altered cellular distribution and sub-cellular sorting of doppel (Dpl) protein in human astrocytoma cell lines. Cell. Oncol. 2008, 30, 337–347. [Google Scholar] [PubMed]
- Angeletti, F.; Fossati, G.; Pattarozzi, A.; Würth, R.; Solari, A.; Daga, A.; Masiello, I.; Barbieri, F.; Florio, T.; Comincini, S. Inhibition of the Autophagy Pathway Synergistically Potentiates the Cytotoxic Activity of Givinostat (ITF2357) on Human Glioblastoma Cancer Stem Cells. Front. Mol. Neurosci. 2016, 9, 107. [Google Scholar] [CrossRef] [Green Version]
- Manai, F.; Azzalin, A.; Morandi, M.; Riccardi, V.; Zanoletti, L.; Dei Giudici, M.; Gabriele, F.; Martinelli, C.; Bozzola, M.; Comincini, S. Trehalose Modulates Autophagy Process to Counteract Gliadin Cytotoxicity in an In Vitro Celiac Disease Model. Cells 2019, 8, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görgens, A.; Bremer, M.; Ferrer-Tur, R.; Murke, F.; Tertel, T.; Horn, P.A.; Thalmann, S.; Welsh, J.A.; Probst, C.; Guerin, C.; et al. Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence-tagged vesicles as biological reference material. J. Extracell. Vesicles 2019, 8, 1587567. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, C.; Di Vizio, D.; Sahoo, S.; Théry, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef]
- Shelke, G.V.; Lässer, C.; Gho, Y.S.; Lötvall, J. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Lee, Y.; Johansson, H.J.; Mäger, I.; Vader, P.; Nordin, J.Z.; Wiklander, O.P.; Lehtiö, J.; Wood, M.J.; Andaloussi, S.E. Serum-free culture alters the quantity and protein composition of neuroblastoma-derived extracellular vesicles. J. Extracell. Vesicles 2015, 4, 26883. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Subedi, P.; Schneider, M.; Philipp, J.; Azimzadeh, O.; Metzger, F.; Moertl, S.; Atkinson, M.J.; Tapio, S. Comparison of methods to isolate proteins from extracellular vesicles for mass spectrometry-based proteomic analyses. Anal. Biochem. 2019, 584, 113390. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.; Vader, P.; van Dommelen, S.M.; van Solinge, W.W.; Schiffelers, R.M. Exosome mimetics: A novel class of drug delivery systems. Int. J. Nanomed. 2012, 7, 1525–1541. [Google Scholar]
- Kan, A.; Mohamedali, A.; Tan, S.H.; Cheruku, H.R.; Slapetova, I.; Lee, L.Y.; Baker, M.S. An improved method for the detection and enrichment of low-abundant membrane and lipid raft-residing proteins. J. Proteom. 2013, 79, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, S.; Gangadaran, P.; Rajendran, R.L.; Zhu, L.; Oh, J.M.; Lee, H.W.; Gopal, A.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; et al. A New Approach for Loading Anticancer Drugs into Mesenchymal Stem Cell-Derived Exosome Mimetics for Cancer Therapy. Front. Pharmacol. 2018, 9, 1116. [Google Scholar] [CrossRef]
- Zarovni, N.; Corrado, A.; Guazzi, P.; Zocco, D.; Lari, E.; Radano, G.; Muhhina, J.; Fondelli, C.; Gavrilova, J.; Chiesi, A. Integrated isolation and quantitative analysis of exosome shuttled proteins and nucleic acids using immunocapture approaches. Methods 2015, 87, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.T.; Kim, J.; Jang, S.C.; Choi, E.J.; Gho, Y.S.; Park, J. Microfluidic filtration system to isolate extracellular vesicles from blood. Lab Chip 2012, 12, 5202–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragovic, R.A.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.J.; Hole, P.; Carr, B.; Redman, C.W.; Harris, A.L.; Dobson, P.J.; et al. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomedicine 2011, 7, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, L.M.; Lombardi, P.; Tillhon, M.; Scovassi, A.I. Berberine, an epiphany against cancer. Molecules 2014, 9, 12349–12357. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinelli, C.; Gabriele, F.; Dini, E.; Carriero, F.; Bresciani, G.; Slivinschi, B.; Dei Giudici, M.; Zanoletti, L.; Manai, F.; Paolillo, M.; et al. Development of Artificial Plasma Membranes Derived Nanovesicles Suitable for Drugs Encapsulation. Cells 2020, 9, 1626. https://doi.org/10.3390/cells9071626
Martinelli C, Gabriele F, Dini E, Carriero F, Bresciani G, Slivinschi B, Dei Giudici M, Zanoletti L, Manai F, Paolillo M, et al. Development of Artificial Plasma Membranes Derived Nanovesicles Suitable for Drugs Encapsulation. Cells. 2020; 9(7):1626. https://doi.org/10.3390/cells9071626
Chicago/Turabian StyleMartinelli, Carolina, Fabio Gabriele, Elena Dini, Francesca Carriero, Giorgia Bresciani, Bianca Slivinschi, Marco Dei Giudici, Lisa Zanoletti, Federico Manai, Mayra Paolillo, and et al. 2020. "Development of Artificial Plasma Membranes Derived Nanovesicles Suitable for Drugs Encapsulation" Cells 9, no. 7: 1626. https://doi.org/10.3390/cells9071626