High Content Screening Using New U2OS Reporter Cell Models Identifies Harmol Hydrochloride as a Selective and Competitive Antagonist of the Androgen Receptor

 ,
,  and
and 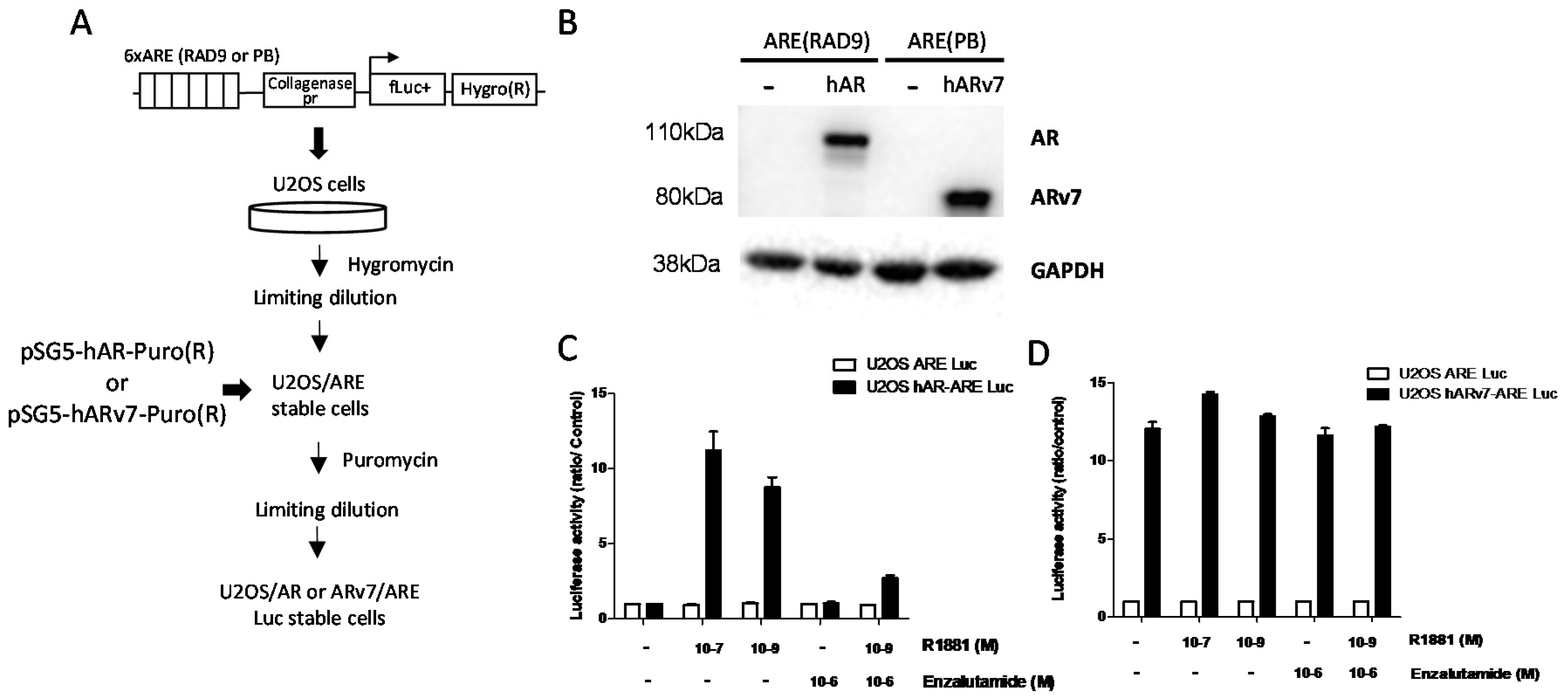{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Compound Library and Reagents
2.3. Stable Cell Lines
2.4. Transactivation Assays
2.5. Cytotoxicity Assays in Spheroid Cultures
2.6. RT-qPCR
2.7. Western Blotting
2.8. Whole-Cell AR Competitive Binding Assays
3. Results
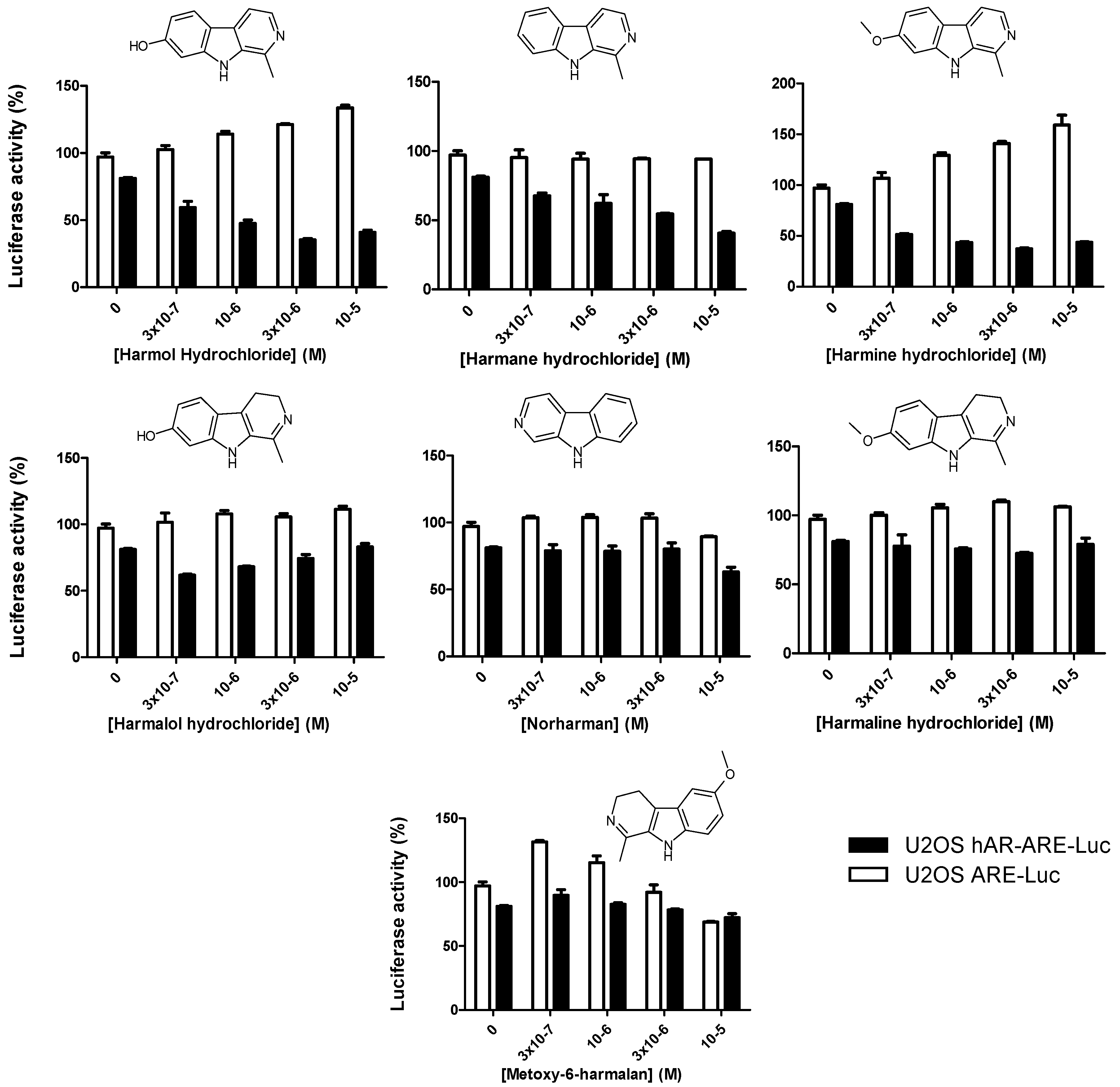
3.1. High-Throughput Screening of AR and ARv7 Inhibitors
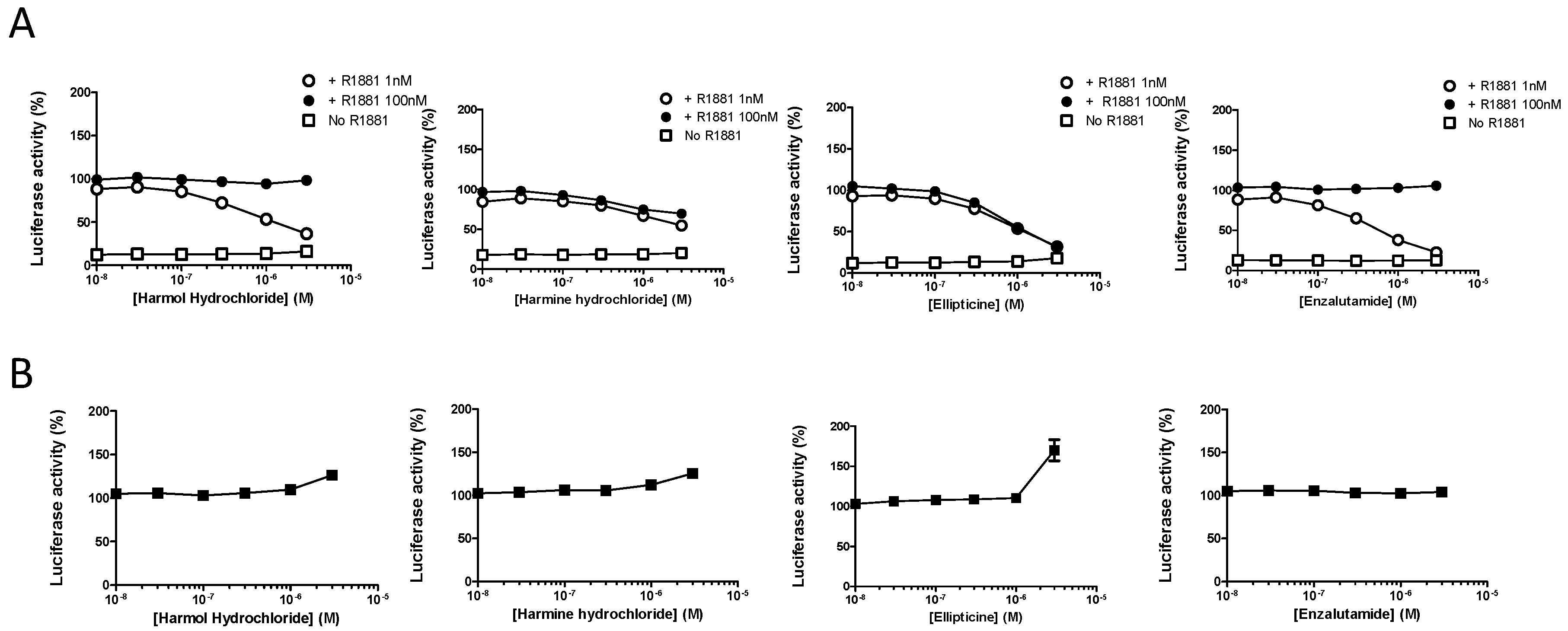
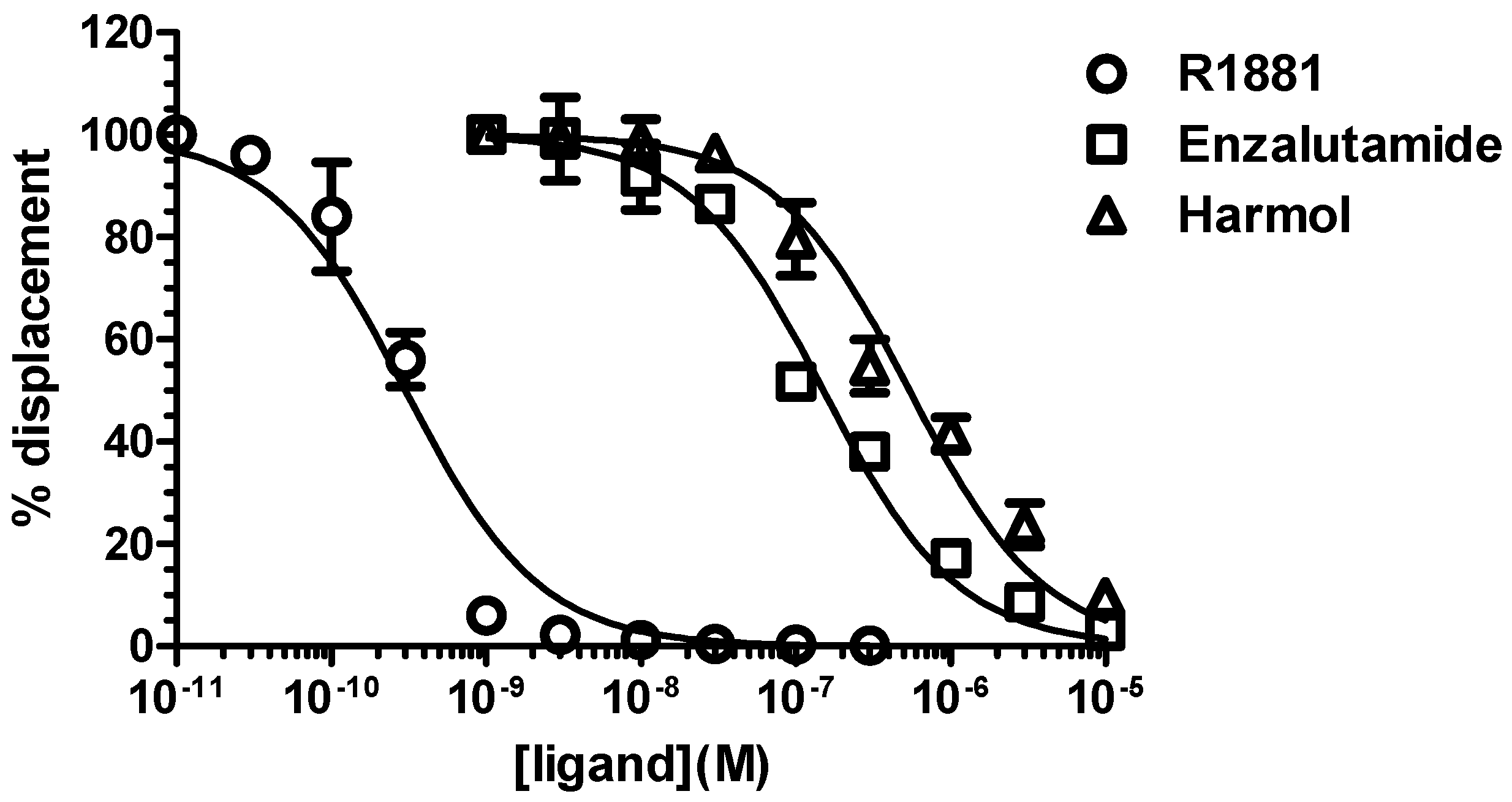
3.2. Harmol Hydrochloride is a Competitive Antagonist of Full-Length AR
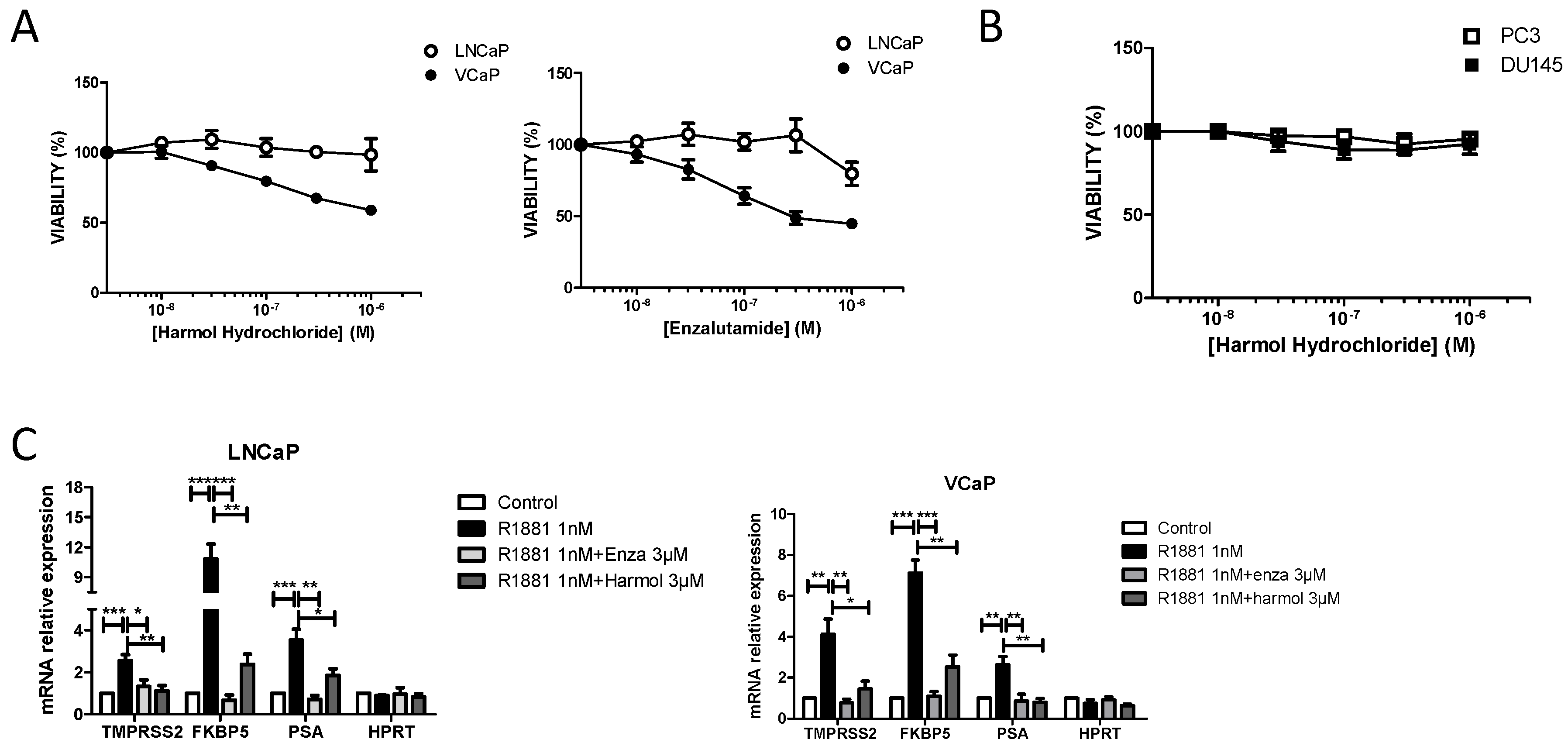
3.3. Harmol Hydrochloride Inhibits the Growth of AR Positive Prostate Cancer Cells
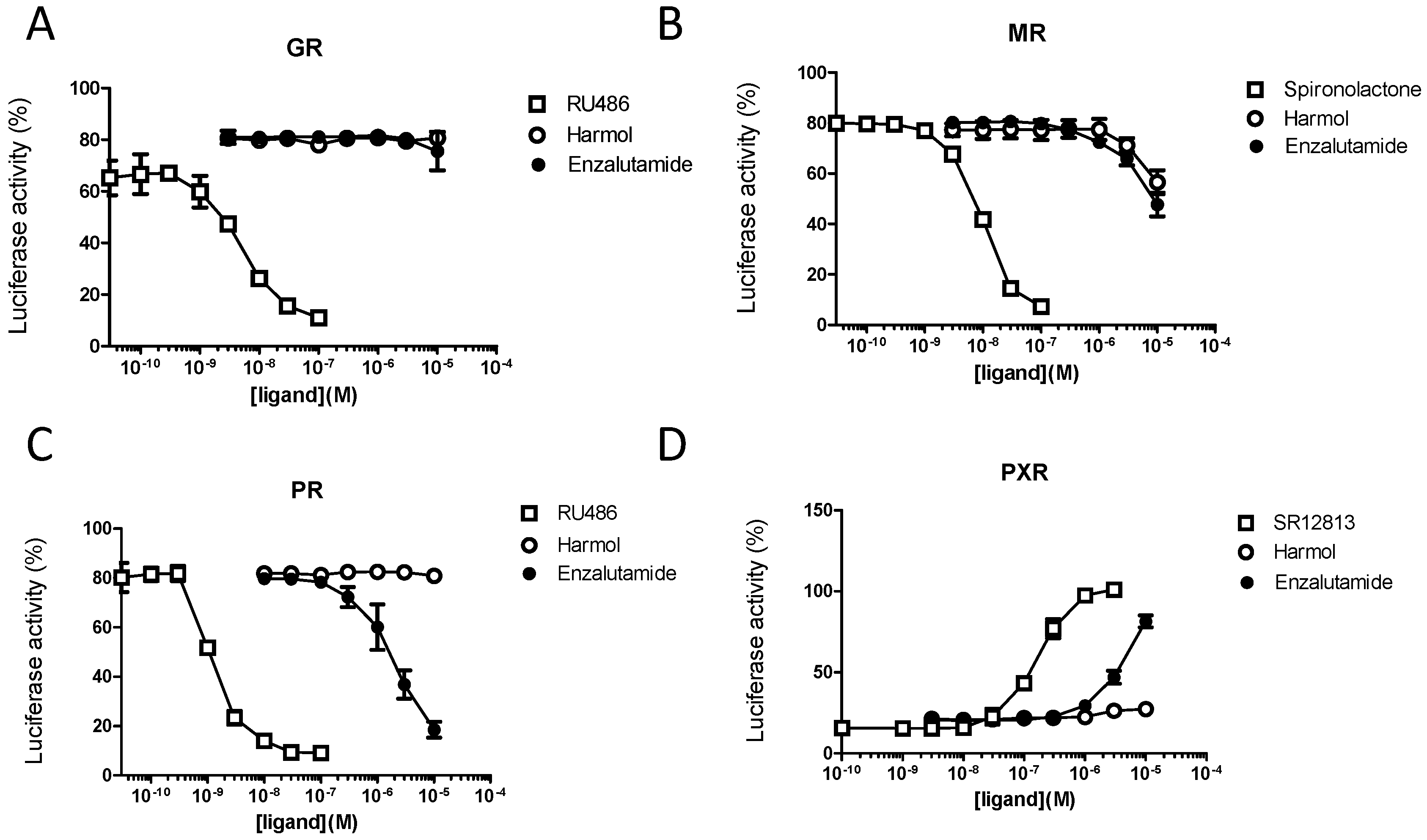
3.4. Harmol Hydrochloride Is a “Selective” Antagonist of the Androgen Receptor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Culig, Z. Androgen Receptor Coactivators in Regulation of Growth and Differentiation in Prostate Cancer. J. Cell. Physiol. 2016, 231, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Buttigliero, C.; Tucci, M.; Bertaglia, V.; Vignani, F.; Bironzo, P.; Di Maio, M.; Scagliotti, G.V. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat. Rev. 2015, 41, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Vander Ark, A.; Cao, J.; Li, X. Mechanisms and Approaches for Overcoming Enzalutamide Resistance in Prostate Cancer. Front. Oncol. 2018, 8, 180. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Goto, T.; Akamatsu, S.; Yamasaki, T.; Inoue, T.; Ogawa, O.; Kobayashi, T. Castration-Resistant Prostate Cancer Refractory to Second-Generation Androgen Receptor Axis-Targeted Agents: Opportunities and Challenges. Cancers 2018, 10, 345. [Google Scholar] [CrossRef] [Green Version]
- Culig, Z. Molecular Mechanisms of Enzalutamide Resistance in Prostate Cancer. Curr. Mol. Biol. Rep. 2017, 3, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Crona, D.J.; Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers 2017, 9, 67. [Google Scholar] [CrossRef]
- Attard, G.; Antonarakis, E.S. Prostate cancer: AR aberrations and resistance to abiraterone or enzalutamide. Nat. Rev. Urol. 2016, 13, 697–698. [Google Scholar] [CrossRef]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef]
- Hara, T.; Miyazaki, J.; Araki, H.; Yamaoka, M.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003, 63, 149–153. [Google Scholar]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steketee, K.; Timmerman, L.; Ziel-van der Made, A.C.J.; Doesburg, P.; Brinkmann, A.O.; Trapman, J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int. J. Cancer 2002, 100, 309–317. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- Cruz, E.C.S.; Carecho, A.R.; Saidel, M.E.; Montanari, C.A.; Leitão, A. In silico selection and cell-based characterization of selective and bioactive compounds for androgen-dependent prostate cancer cell. Bioorg. Med. Chem. Lett. 2017, 27, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Wahl, J.; Smieško, M. Endocrine Disruption at the Androgen Receptor: Employing Molecular Dynamics and Docking for Improved Virtual Screening and Toxicity Prediction. Int. J. Mol. Sci. 2018, 19, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.; Fu, W.; Zhang, M.; Wang, E.; Shan, L.; Chai, X.; Pang, J.; Wang, X.; Xu, X.; Xu, L.; et al. Novel androgen receptor antagonist identified by structure-based virtual screening, structural optimization, and biological evaluation. Eur. J. Med. Chem. 2020, 192, 112156. [Google Scholar] [CrossRef]
- Li, H.; Ren, X.; Leblanc, E.; Frewin, K.; Rennie, P.S.; Cherkasov, A. Identification of novel androgen receptor antagonists using structure- and ligand-based methods. J. Chem. Inf. Model. 2013, 53, 123–130. [Google Scholar] [CrossRef]
- Hao, G.-F.; Yang, G.-F.; Zhan, C.-G. Structure-based methods for predicting target mutation-induced drug resistance and rational drug design to overcome the problem. Drug Discov. Today 2012, 17, 1121–1126. [Google Scholar] [CrossRef] [Green Version]
- Divakar, S.; Saravanan, K.; Karthikeyan, P.; Elancheran, R.; Kabilan, S.; Balasubramanian, K.K.; Devi, R.; Kotoky, J.; Ramanathan, M. Iminoenamine based novel androgen receptor antagonist exhibited anti-prostate cancer activity in androgen independent prostate cancer cells through inhibition of AKT pathway. Chem. Biol. Interact. 2017, 275, 22–34. [Google Scholar] [CrossRef]
- Johnston, P.A.; Nguyen, M.M.; Dar, J.A.; Ai, J.; Wang, Y.; Masoodi, K.Z.; Shun, T.; Shinde, S.; Camarco, D.P.; Hua, Y.; et al. Development and Implementation of a High-Throughput High-Content Screening Assay to Identify Inhibitors of Androgen Receptor Nuclear Localization in Castration-Resistant Prostate Cancer Cells. Assay Drug Dev. Technol. 2016, 14, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.K.; Skoda, E.M.; Zhou, J.; Parrinello, E.; Wang, D.; O’Malley, K.; Eyer, B.R.; Kazancioglu, M.; Eisermann, K.; Johnston, P.A.; et al. Small Molecule Antagonists of the Nuclear Androgen Receptor for the Treatment of Castration-Resistant Prostate Cancer. ACS Med. Chem. Lett. 2016, 7, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Hieronymus, H.; Lamb, J.; Ross, K.N.; Peng, X.P.; Clement, C.; Rodina, A.; Nieto, M.; Du, J.; Stegmaier, K.; Raj, S.M.; et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 2006, 10, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.O.; Bolton, E.C.; Huang, Y.; Feau, C.; Guy, R.K.; Yamamoto, K.R.; Hann, B.; Diamond, M.I. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 7233–7238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.O.; Diamond, M.I. A cellular conformation-based screen for androgen receptor inhibitors. ACS Chem. Biol. 2008, 3, 412–418. [Google Scholar] [CrossRef]
- Sonneveld, E.; Jansen, H.J.; Riteco, J.A.C.; Brouwer, A.; van der Burg, B. Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol. Sci. Off. J. Soc. Toxicol. 2005, 83, 136–148. [Google Scholar] [CrossRef]
- Grimaldi, M.; Boulahtouf, A.; Toporova, L.; Balaguer, P. Functional profiling of bisphenols for nuclear receptors. Toxicology 2019, 420, 39–45. [Google Scholar] [CrossRef]
- Vinggaard, A.M.; Niemelä, J.; Wedebye, E.B.; Jensen, G.E. Screening of 397 chemicals and development of a quantitative structure--activity relationship model for androgen receptor antagonism. Chem. Res. Toxicol. 2008, 21, 813–823. [Google Scholar] [CrossRef]
- Ferroni, C.; Varchi, G. Non-Steroidal Androgen Receptor Antagonists and Prostate Cancer: A Survey on Chemical Structures Binding this Fast-Mutating Target. Curr. Med. Chem. 2019, 26, 6053–6073. [Google Scholar] [CrossRef]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. Natural Compounds in Prostate Cancer Prevention and Treatment: Mechanisms of Action and Molecular Targets. Cells 2020, 9, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Térouanne, B.; Tahiri, B.; Georget, V.; Belon, C.; Poujol, N.; Avances, C.; Orio, F.; Balaguer, P.; Sultan, C. A stable prostatic bioluminescent cell line to investigate androgen and antiandrogen effects. Mol. Cell. Endocrinol. 2000, 160, 39–49. [Google Scholar] [CrossRef]
- Delfosse, V.; Dendele, B.; Huet, T.; Grimaldi, M.; Boulahtouf, A.; Gerbal-Chaloin, S.; Beucher, B.; Roecklin, D.; Muller, C.; Rahmani, R.; et al. Synergistic activation of human pregnane X receptor by binary cocktails of pharmaceutical and environmental compounds. Nat. Commun. 2015, 6, 8089. [Google Scholar] [CrossRef] [PubMed]
- Delfosse, V.; Grimaldi, M.; Pons, J.-L.; Boulahtouf, A.; le Maire, A.; Cavailles, V.; Labesse, G.; Bourguet, W.; Balaguer, P. Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes. Proc. Natl. Acad. Sci. USA 2012, 109, 14930–14935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dart, D.A.; Ashelford, K.; Jiang, W.G. AR mRNA stability is increased with AR-antagonist resistance via 3’UTR variants. Endocr. Connect. 2020, 9, 9–19. [Google Scholar] [CrossRef]
- Kuruma, H.; Matsumoto, H.; Shiota, M.; Bishop, J.; Lamoureux, F.; Thomas, C.; Briere, D.; Los, G.; Gleave, M.; Fanjul, A.; et al. A novel antiandrogen, Compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, H.; Yamamoto, Y.; Shiota, M.; Kuruma, H.; Beraldi, E.; Matsuyama, H.; Zoubeidi, A.; Gleave, M. Cotargeting Androgen Receptor and Clusterin Delays Castrate-Resistant Prostate Cancer Progression by Inhibiting Adaptive Stress Response and AR Stability. Cancer Res. 2013, 73, 5206–5217. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Morales, C.; Cooke, L.S.; Johnson, B.; Somer, B.; Mahadevan, D. Reciprocal feedback inhibition of the androgen receptor and PI3K as a novel therapy for castrate-sensitive and -resistant prostate cancer. Oncotarget 2015, 6, 41976–41987. [Google Scholar] [CrossRef]
- Sun, W.; Li, L.; Du, Z.; Quan, Z.; Yuan, M.; Cheng, H.; Gao, Y.; Luo, C.; Wu, X. Combination of phospholipase Cε knockdown with GANT61 sensitizes castration-resistant prostate cancer cells to enzalutamide by suppressing the androgen receptor signaling pathway. Oncol. Rep. 2019, 41, 2689–2702. [Google Scholar] [CrossRef]
- Blankvoort, B.M.; de Groene, E.M.; van Meeteren-Kreikamp, A.P.; Witkamp, R.F.; Rodenburg, R.J.; Aarts, J.M. Development of an androgen reporter gene assay (AR-LUX) utilizing a human cell line with an endogenously regulated androgen receptor. Anal. Biochem. 2001, 298, 93–102. [Google Scholar] [CrossRef]
- de Gooyer, M.E.; Deckers, G.H.; Schoonen, W.G.E.J.; Verheul, H.A.M.; Kloosterboer, H.J. Receptor profiling and endocrine interactions of tibolone. Steroids 2003, 68, 21–30. [Google Scholar] [CrossRef]
- Paris, F.; Balaguer, P.; Térouanne, B.; Servant, N.; Lacoste, C.; Cravedi, J.-P.; Nicolas, J.-C.; Sultan, C. Phenylphenols, biphenols, bisphenol-A and 4-tert-octylphenol exhibit alpha and beta estrogen activities and antiandrogen activity in reporter cell lines. Mol. Cell. Endocrinol. 2002, 193, 43–49. [Google Scholar] [CrossRef]
- Wilson, V.S.; Bobseine, K.; Lambright, C.R.; Gray, L.E. A novel cell line, MDA-kb2, that stably expresses an androgen- and glucocorticoid-responsive reporter for the detection of hormone receptor agonists and antagonists. Toxicol. Sci. Off. J. Soc. Toxicol. 2002, 66, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015, 2, e17. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, U.; Bengtson, C.; Repenthin, G.; Schillinger, E. Stable transfection of androgen receptor and MMTV-CAT into mammalian cells: Inhibition of cat expression by anti-androgens. J. Steroid Biochem. Mol. Biol. 1992, 42, 787–793. [Google Scholar] [CrossRef]
- Kim, H.-J.; Park, Y.I.; Dong, M.-S. Comparison of prostate cancer cell lines for androgen receptor-mediated reporter gene assays. Toxicol. In Vitro 2006, 20, 1159–1167. [Google Scholar] [CrossRef]
- Hartig, P.C.; Bobseine, K.L.; Britt, B.H.; Cardon, M.C.; Lambright, C.R.; Wilson, V.S.; Gray, L.E. Development of two androgen receptor assays using adenoviral transduction of MMTV-luc reporter and/or hAR for endocrine screening. Toxicol. Sci. Off. J. Soc. Toxicol. 2002, 66, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Stiborová, M.; Frei, E. Ellipticines as DNA-targeted chemotherapeutics. Curr. Med. Chem. 2014, 21, 575–591. [Google Scholar] [CrossRef]
- Waki, H.; Park, K.W.; Mitro, N.; Pei, L.; Damoiseaux, R.; Wilpitz, D.C.; Reue, K.; Saez, E.; Tontonoz, P. The small molecule harmine is an antidiabetic cell-type-specific regulator of PPARgamma expression. Cell Metab. 2007, 5, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Braestrup, C.; Nielsen, M. Discovery of beta-carboline ligands for benzodiazepine receptors. Psychopharmacol. Ser. 1993, 11, 1–6. [Google Scholar]
- Hadjipavlou-Litina, D.; Garg, R.; Hansch, C. Comparative quantitative structure-activity relationship studies (QSAR) on non-benzodiazepine compounds binding to benzodiazepine receptor (BzR). Chem. Rev. 2004, 104, 3751–3794. [Google Scholar] [CrossRef] [PubMed]
- Bribes, E.; Carrière, D.; Goubet, C.; Galiègue, S.; Casellas, P.; Simony-Lafontaine, J. Immunohistochemical assessment of the peripheral benzodiazepine receptor in human tissues. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2004, 52, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecchin, E.; De Mattia, E.; Toffoli, G. Nuclear receptors and drug metabolism for the personalization of cancer therapy. Expert Opin. Drug Metab. Toxicol. 2016, 12, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.-E.; Efstathiou, E.; Rathkopf, D.; Shelkey, J.; Yu, E.Y.; Alumkal, J.; et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. Lancet Lond. Engl. 2010, 375, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, J.A.; Ouatas, T.; Krauwinkel, W.; Ohtsu, Y.; van der Walt, J.-S.; Beddo, V.; de Vries, M.; Mordenti, J. Clinical Pharmacokinetic Studies of Enzalutamide. Clin. Pharmacokinet. 2015, 54, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Benoist, G.E.; Hendriks, R.J.; Mulders, P.F.A.; Gerritsen, W.R.; Somford, D.M.; Schalken, J.A.; van Oort, I.M.; Burger, D.M.; van Erp, N.P. Pharmacokinetic Aspects of the Two Novel Oral Drugs Used for Metastatic Castration-Resistant Prostate Cancer: Abiraterone Acetate and Enzalutamide. Clin. Pharmacokinet. 2016, 55, 1369–1380. [Google Scholar] [CrossRef] [Green Version]
- Riba, J.; McIlhenny, E.H.; Valle, M.; Bouso, J.C.; Barker, S.A. Metabolism and disposition of N,N-dimethyltryptamine and harmala alkaloids after oral administration of ayahuasca. Drug Test. Anal. 2012, 4, 610–616. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Radek, J.T.; Meyers, M.J.; Nettles, K.W.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Agard, D.A.; Greene, G.L. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat. Struct. Biol. 2002, 9, 359–364. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dellal, H.; Boulahtouf, A.; Alaterre, E.; Cuenant, A.; Grimaldi, M.; Bourguet, W.; Gongora, C.; Balaguer, P.; Pourquier, P. High Content Screening Using New U2OS Reporter Cell Models Identifies Harmol Hydrochloride as a Selective and Competitive Antagonist of the Androgen Receptor. Cells 2020, 9, 1469. https://doi.org/10.3390/cells9061469
Dellal H, Boulahtouf A, Alaterre E, Cuenant A, Grimaldi M, Bourguet W, Gongora C, Balaguer P, Pourquier P. High Content Screening Using New U2OS Reporter Cell Models Identifies Harmol Hydrochloride as a Selective and Competitive Antagonist of the Androgen Receptor. Cells. 2020; 9(6):1469. https://doi.org/10.3390/cells9061469
Chicago/Turabian StyleDellal, Hadjer, Abdelhay Boulahtouf, Elina Alaterre, Alice Cuenant, Marina Grimaldi, William Bourguet, Céline Gongora, Patrick Balaguer, and Philippe Pourquier. 2020. "High Content Screening Using New U2OS Reporter Cell Models Identifies Harmol Hydrochloride as a Selective and Competitive Antagonist of the Androgen Receptor" Cells 9, no. 6: 1469. https://doi.org/10.3390/cells9061469