Knockout of the Mitochondrial Calcium Uniporter Strongly Suppresses Stimulus-Metabolism Coupling in Pancreatic Acinar Cells but Does Not Reduce Severity of Experimental Acute Pancreatitis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Cells
2.2. Materials
2.3. Ca2+ Measurements
2.4. NADH and FAD+ Fluorescence Measurements
2.5. Oxygen Consumption Measurements
2.6. Cell Death Assays
2.7. Experimental AP Models
2.8. Histopathology
2.9. Immunoblotting
2.10. Enzyme Activity and IL-6 Measurement
2.11. Statistical Analysis
3. Results
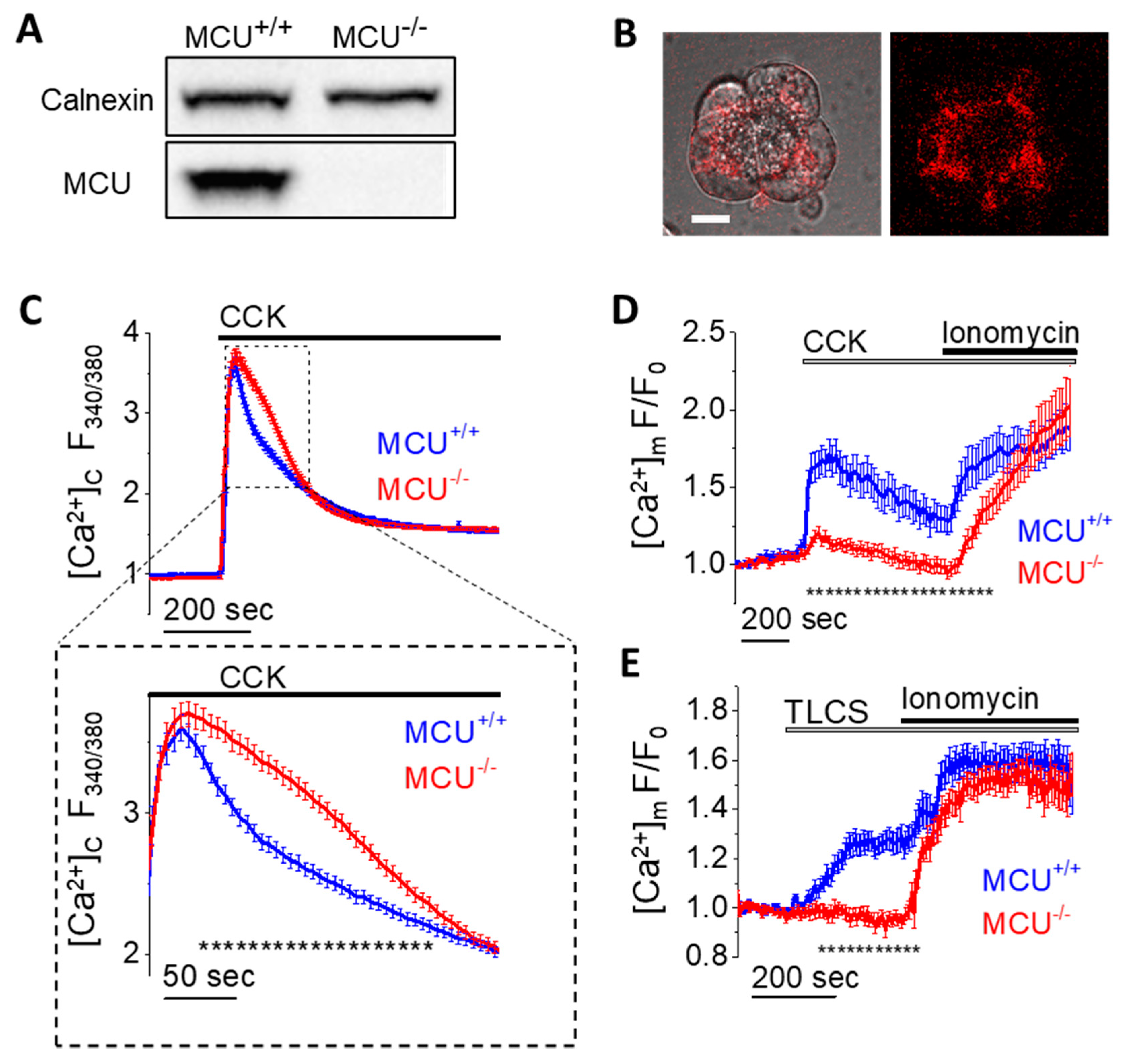
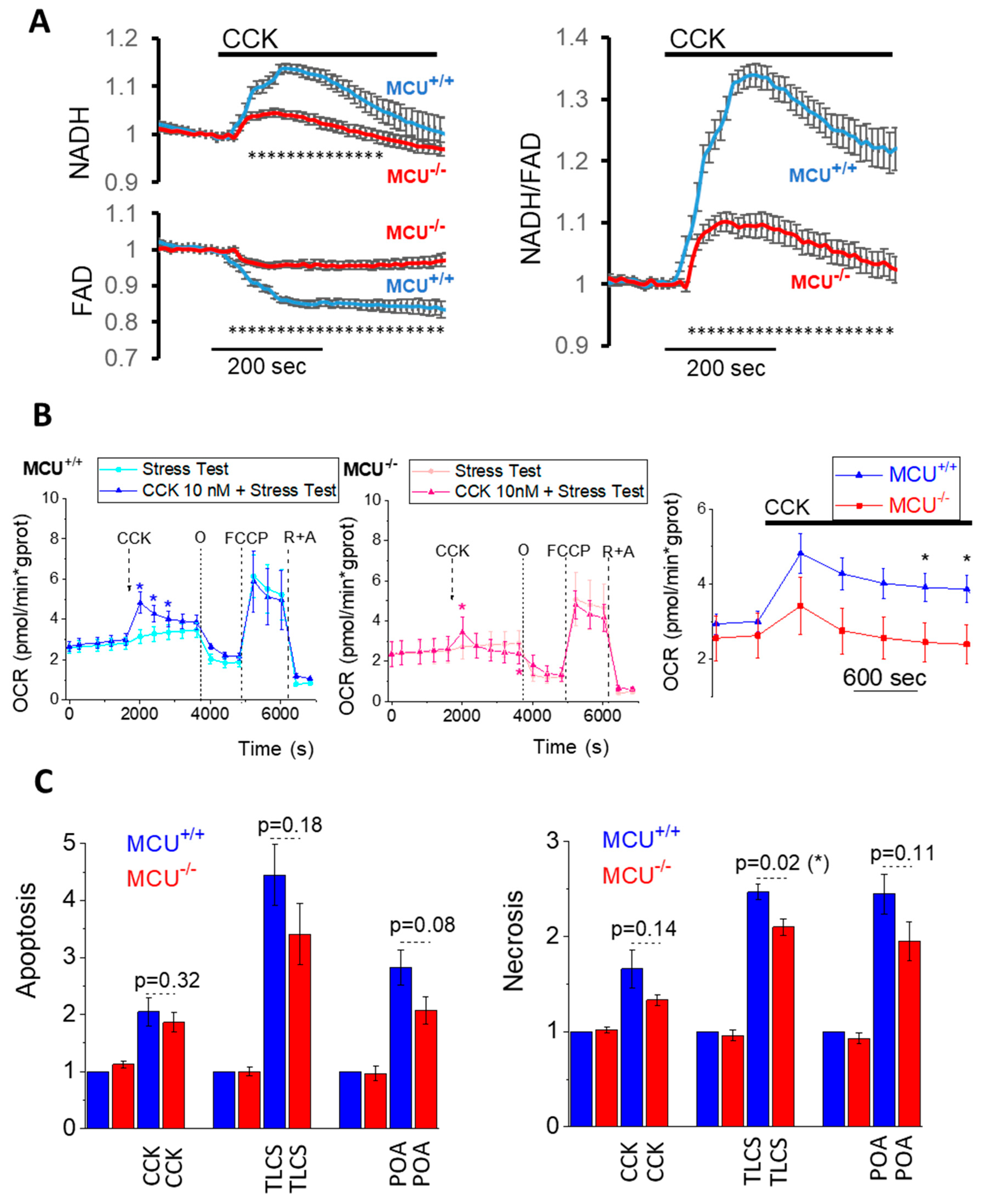
3.1. MCU Knockout Suppresses Mitochondrial Ca2+ Responses and Its Downstream Effects
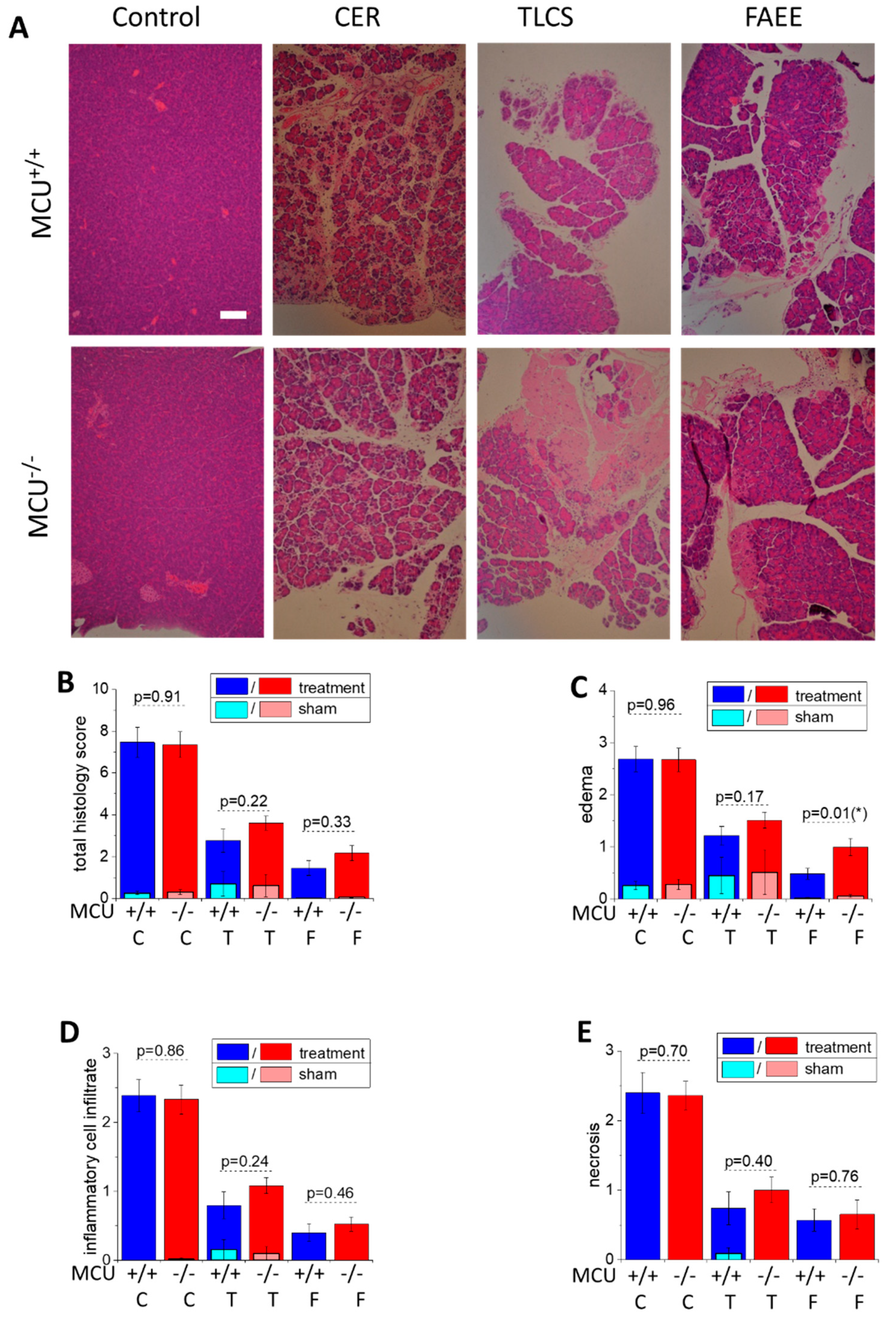
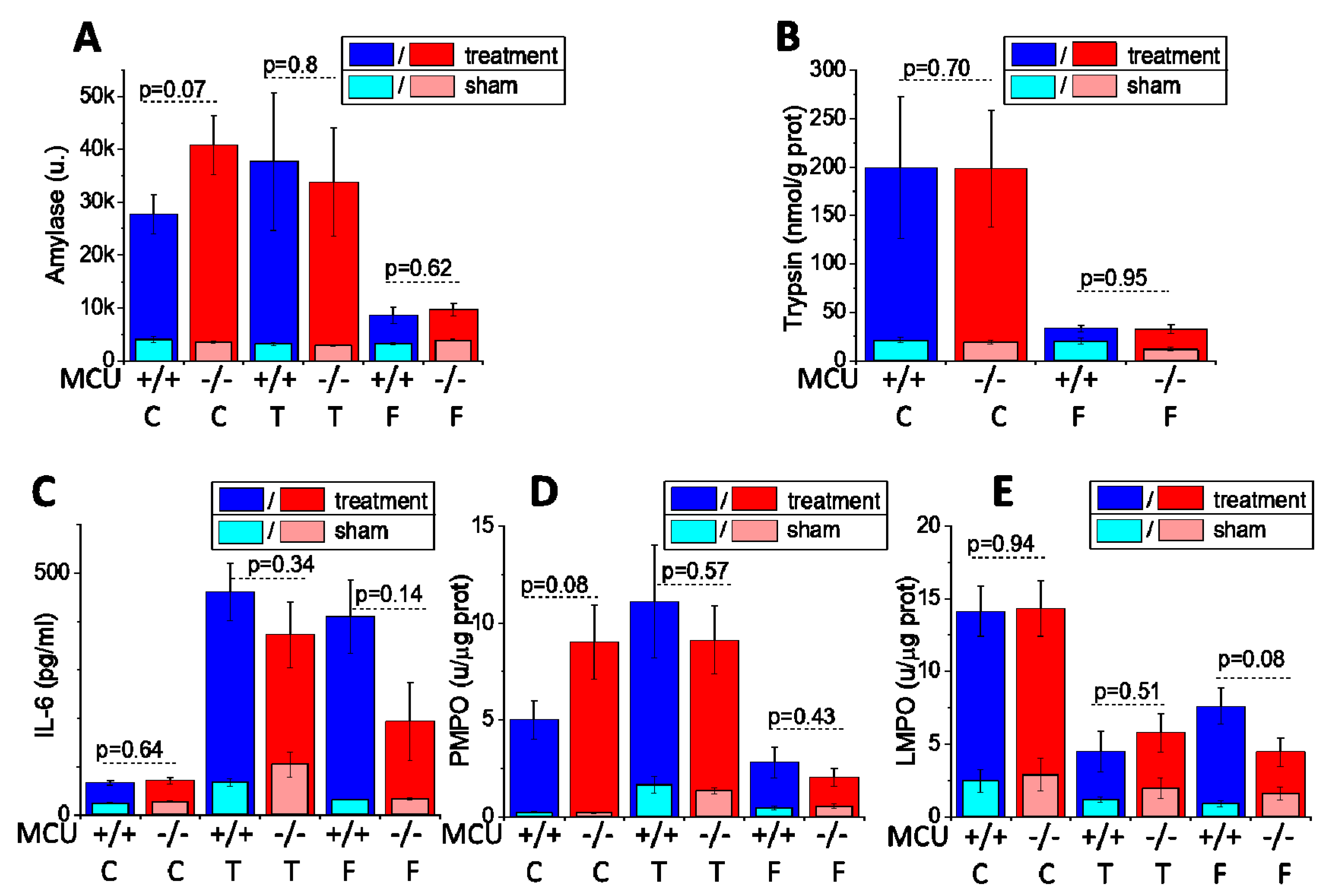
3.2. Knockout of the Mitochondrial Calcium Uniporter Does not Reduce Severity of Experimental Acute Pancreatitis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Petrov, M.S.; Yadav, D. Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 175–184. [Google Scholar] [CrossRef]
- Forsmark, C.E.; Vege, S.S.; Wilcox, C.M. Acute Pancreatitis. New Engl. J. Med. 2016, 375, 1972–1981. [Google Scholar] [CrossRef]
- Schild, L.; Stanarius, A.; Wolf, G.; Augustin, W.; Halangk, W. Induction of permeability transition in pancreatic mitochondria by cerulein in rats. Mol. Cell. Biochem. 1999, 195, 191–197. [Google Scholar] [CrossRef]
- Mukherjee, R.; Criddle, D.; Gukvoskaya, A.; Pandol, S.; Petersen, O.; Sutton, R.; Gukovskaya, A. Mitochondrial injury in pancreatitis. Cell Calcium 2008, 44, 14–23. [Google Scholar] [CrossRef]
- Shalbueva, N.; Mareninova, O.A.; Gerloff, A.; Yuan, J.; Waldron, R.T.; Pandol, S.J.; Gukovskaya, A. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 2012, 144, 437–446.e6. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.; Mareninova, O.A.; Odinokova, I.V.; Huang, W.; Murphy, J.; Chvanov, M.; Javed, M.A.; Wen, L.; Booth, D.M.; Cane, M.; et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: Inhibition prevents acute pancreatitis by protecting production of ATP. Gut 2015, 65, 1333–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, O.H.; Tepikin, A. Polarized Calcium Signaling in Exocrine Gland Cells. Annu. Rev. Physiol. 2008, 70, 273–299. [Google Scholar] [CrossRef]
- Yule, D.I. Ca2+ Signaling in Pancreatic Acinar Cells. In The Pancreapedia: Exocrine Pancreas Knowledge Base; University of Michigan Library: Ann Arbor, MI, USA, 2015. [Google Scholar] [CrossRef]
- Williams, J.A. Cholecystokinin (CCK) Regulation of Pancreatic Acinar Cells: Physiological Actions and Signal Transduction Mechanisms. Compr. Physiol. 2019, 9, 535–564. [Google Scholar] [CrossRef]
- Park, M.K.; Ashby, M.C.; Erdemli, G.; Petersen, O.H.; Tepikin, A. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001, 20, 1863–1874. [Google Scholar] [CrossRef] [Green Version]
- Voronina, S.; Sukhomlin, T.; Johnson, P.R.; Erdemli, G.; Petersen, O.H.; Tepikin, A. Correlation of NADH and Ca2+signals in mouse pancreatic acinar cells. J. Physiol. 2002, 539, 41–52. [Google Scholar] [CrossRef]
- Hajnoczky, G.; Robb-Gaspers, L.D.; Seitz, M.B.; Thomas, A.P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 1995, 82, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Voronina, S.G.; Barrow, S.L.; Simpson, A.W.; Gerasimenko, J.V.; Xavier, G.D.S.; Rutter, G.A.; Petersen, O.H.; Tepikin, A. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 2010, 138, 1976–1987. [Google Scholar] [CrossRef]
- Krüger, B.; Albrecht, E.; Lerch, M.M. The Role of Intracellular Calcium Signaling in Premature Protease Activation and the Onset of Pancreatitis. Am. J. Pathol. 2000, 157, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Laukkarinen, J.M.; Van Acker, G.J.D.; Weiss, E.R.; Steer, M.L.; Perides, G. A mouse model of acute biliary pancreatitis induced by retrograde pancreatic duct infusion of Na-taurocholate. Gut 2007, 56, 1590–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.; Sutton, R.; Jenkins, S.; Petersen, O. Progressive disruption of acinar cell calcium signaling is an early feature of cerulein-induced pancreatitis in mice. Gastroenterology 1996, 111, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Booth, D.M.; Cane, M.; Chvanov, M.; Javed, M.A.; Elliott, V.L.; Armstrong, J.A.; Dingsdale, H.; Cash, N.; Li, Y.; et al. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 2013, 63, 1313–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimenko, J.V.; Gerasimenko, J.V.; Petersen, O.H. The role of Ca2+ in the pathophysiology of pancreatitis. J. Physiol. 2013, 592, 269–280. [Google Scholar] [CrossRef]
- Maléth, J.; Hegyi, P. Ca2+ toxicity and mitochondrial damage in acute pancreatitis: Translational overview. Philos. Trans. R. Soc. B Boil. Sci. 2016, 371. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.J.; Papachristou, G.I. New insights into acute pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 479–496. [Google Scholar] [CrossRef]
- Weber, H.; Jonas, L.; Hühns, S.; Schuff-Werner, P. Dysregulation of the calpain-calpastatin system plays a role in the development of cerulein-induced acute pancreatitis in the rat. Am. J. Physiol. Liver Physiol. 2004, 286, G932–G941. [Google Scholar] [CrossRef]
- Watanabe, O.; Baccino, F.M.; Steer, M.L.; Meldolesi, J. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: Early morphological changes during development of experimental pancreatitis. Am. J. Physiol. Liver Physiol. 1984, 246, G457–G467. [Google Scholar] [CrossRef] [PubMed]
- Saluja, A.; Hashimoto, S.; Saluja, M.; Powers, R.E.; Meldolesi, J.; Steer, M.L. Subcellular redistribution of lysosomal enzymes during caerulein-induced pancreatitis. Am. J. Physiol. Liver Physiol. 1987, 253, G508–G516. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, M.; Prior, I.A.; Voronina, S.G.; Barrow, S.L.; Woodsmith, J.D.; Gerasimenko, J.V.; Petersen, O.H.; Tepikin, A. Activation of trypsinogen in large endocytic vacuoles of pancreatic acinar cells. Proc. Natl. Acad. Sci. USA 2007, 104, 5674–5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolai, S.; Liang, T.; Orabi, A.I.; Holmyard, D.; Xie, L.; Greitzer-Antes, D.; Kang, Y.; Xie, H.; Javed, T.A.; Lam, P.P.; et al. Pancreatitis-Induced Depletion of Syntaxin 2 Promotes Autophagy and Increases Basolateral Exocytosis. Gastroenterology 2018, 154, 1805–1821. [Google Scholar] [CrossRef] [PubMed]
- Muili, K.A.; Wang, N.; Orabi, A.I.; Sarwar, S.; Luo, Y.; Javed, T.A.; Eisses, J.F.; Mahmood, S.M.; Jin, S.; Singh, V.P.; et al. Bile Acids Induce Pancreatic Acinar Cell Injury and Pancreatitis by Activating Calcineurin*. J. Boil. Chem. 2012, 288, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Javed, T.A.; Yimlamai, D.; Mukherjee, A.; Xiao, X.; Husain, S.Z. Transient High Pressure in Pancreatic Ducts Promotes Inflammation and Alters Tight Junctions via Calcineurin Signaling in Mice. Gastroenterology 2018, 155, 1250–1263. [Google Scholar] [CrossRef]
- Otani, T.; Chepilko, S.M.; Grendell, J.H.; Gorelick, F.S. Codistribution of TAP and the granule membrane protein GRAMP-92 in rat caerulein-induced pancreatitis. Am. J. Physiol. Content 1998, 275, G999–G1009. [Google Scholar] [CrossRef]
- Halangk, W.; Lerch, M.M.; Brandt-Nedelev, B.; Roth, W.; Ruthenbuerger, M.; Reinheckel, T.; Domschke, W.; Lippert, H.; Peters, C.; Deussing, J.M. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Investig. 2000, 106, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Chvanov, M.; De Faveri, F.; Moore, D.; Sherwood, M.; Awais, M.; Voronina, S.; Sutton, R.; Criddle, D.N.; Haynes, L.; Tepikin, A. Intracellular rupture, exocytosis and actin interaction of endocytic vacuoles in pancreatic acinar cells: Initiating events in acute pancreatitis. J. Physiol. 2018, 596, 2547–2564. [Google Scholar] [CrossRef] [Green Version]
- Mareninova, O.A.; Hermann, K.; French, S.W.; O’Konski, M.S.; Pandol, S.J.; Webster, P.; Erickson, A.H.; Katunuma, N.; Gorelick, F.S.; Gukovsky, I.; et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J. Clin. Investig. 2009, 119, 3340–3355. [Google Scholar] [CrossRef] [Green Version]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2017, 154, 689–703. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. Biochemistry: A pore way to die. Nature 2005, 434, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, G.; Sharpe, J.A.; Sundier, S.Y.; Duchen, M.R. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann. New York Acad. Sci. 2015, 1350, 107–116. [Google Scholar] [CrossRef]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.M.; Gerasimenko, O.V. Calcium Elevation in Mitochondria Is the Main Ca2+Requirement for Mitochondrial Permeability Transition Pore (mPTP) Opening. J. Boil. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef] [Green Version]
- Odinokova, I.V.; Sung, K.-F.; Mareninova, O.A.; Hermann, K.; Evtodienko, Y.; Andreyev, A.Y.; Gukovsky, I.; Gukovskaya, A.S. Mechanisms regulating cytochrome c release in pancreatic mitochondria. Gut 2008, 58, 431–442. [Google Scholar] [CrossRef]
- Booth, D.M.; Murphy, J.A.; Mukherjee, R.; Awais, M.; Neoptolemos, J.P.; Gerasimenko, J.V.; Tepikin, A.; Petersen, O.H.; Sutton, R.; Criddle, D.N. Reactive Oxygen Species Induced by Bile Acid Induce Apoptosis and Protect Against Necrosis in Pancreatic Acinar Cells. Gastroenterology 2011, 140, 2116–2125. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Reane, D.V.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflügers Arch. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Tan, Y.-W.; Hagenston, A.M.; Martel, M.-A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.A.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soman, S.; Keatinge, M.; Moein, M.; Da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1 − / − zebrafish. Eur. J. Neurosci. 2016, 45, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.; Pavlov, E.V.; Robertson, G. Tamoxifen-induced knockdown of the mitochondrial calcium uniporter in Thy1-expressing neurons protects mice from hypoxic/ischemic brain injury. Cell Death Dis. 2018, 9, 606. [Google Scholar] [CrossRef] [Green Version]
- Soman, S.; Bazała, M.; Keatinge, M.; Bandmann, O.; Kuznicki, J. Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson’s disease. Boil. Open 2019, 8, bio044347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebag, S.C.; Koval, O.M.; Paschke, J.D.; Winters, C.J.; Comellas, A.P.; Grumbach, I.M. Inhibition of the mitochondrial calcium uniporter prevents IL-13 and allergen-mediated airway epithelial apoptosis and loss of barrier function. Exp. Cell Res. 2017, 362, 400–411. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Furuno, T.; Shinkai, N.; Inoh, Y.; Nakanishi, M. Impaired expression of the mitochondrial calcium uniporter suppresses mast cell degranulation. Mol. Cell. Biochem. 2015, 410, 215–221. [Google Scholar] [CrossRef]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef]
- Panahi, G.; Pasalar, P.; Zare, M.; Rizzuto, R.; Meshkani, R. MCU-knockdown attenuates high glucose-induced inflammation through regulating MAPKs/NF-κB pathways and ROS production in HepG2 cells. PLoS ONE 2018, 13, e0196580. [Google Scholar] [CrossRef] [Green Version]
- Tedesco, S.; Scattolini, V.; Albiero, M.; Bortolozzi, M.; Avogaro, A.; Cignarella, A.; Fadini, G.P. Mitochondrial Calcium Uptake Is Instrumental to Alternative Macrophage Polarization and Phagocytic Activity. Int. J. Mol. Sci. 2019, 20, 4966. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerboom, J.; Calderón, N.C.; Tian, L.; Wabnig, S.; Prigge, M.; Tolö, J.; Gordus, A.; Orger, M.B.; Severi, K.; Macklin, J.J.; et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 2013, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.A.; Cash, N.J.; Ouyang, Y.; Morton, J.C.; Chvanov, M.; Latawiec, D.; Awais, M.; Tepikin, A.; Sutton, R.; Criddle, D.N. Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin D, resulting in an apoptosis-to-necrosis shift. J. Boil. Chem. 2018, 293, 8032–8047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Cane, M.; Mukherjee, R.; Szatmary, P.; Zhang, X.; Elliott, V.; Ouyang, Y.; Chvanov, M.; Latawiec, D.; Wen, L.; et al. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+release. Gut 2015, 66, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F.A. Molecular Insights into the Mechanism of Necroptosis: The Necrosome As a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kroemer, G. Secondary Necrosis: Accidental No More. Trends Cancer 2017, 3, 1–2. [Google Scholar] [CrossRef]
- Mayerle, J.; Sendler, M.; Lerch, M.M. Secretagogue (Caerulein) Induced Pancreatitis in Rodents; University of Michigan Library: Ann Arbor, MI, USA, 2013. [Google Scholar]
- Ou, X.; Cheng, Z.; Liu, T.; Tang, Z.; Huang, W.; Szatmary, P.; Zheng, S.; Sutton, R.; Toh, C.H.; Zhang, N.; et al. Circulating Histone Levels Reflect Disease Severity in Animal Models of Acute Pancreatitis. Pancreas 2015, 44, 1089–1095. [Google Scholar] [CrossRef]
- Nathan, J.D.; Romac, J.; Peng, R.Y.; Peyton, M.; Macdonald, R.J.; Liddle, R.A. Transgenic expression of pancreatic secretory trypsin inhibitor-I ameliorates secretagogue-induced pancreatitis in mice. Gastroenterology 2005, 128, 717–727. [Google Scholar] [CrossRef]
- Dawra, R.; Ku, Y.S.; Sharif, R.; Dhaulakhandi, D.; Phillips, P.; Dudeja, V.; Saluja, A.K. An Improved Method for Extracting Myeloperoxidase and Determining Its Activity in the Pancreas and Lungs During Pancreatitis. Pancreas 2008, 37, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Boil. Chem. 1985, 260, 3440–3450. [Google Scholar]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Glancy, B.; Balaban, R.S. Role of Mitochondrial Ca2+in the Regulation of Cellular Energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [Green Version]
- Bondarenko, A.I.; Jean-Quartier, C.; Parichatikanond, W.; Alam, M.R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca(2+) uniporter (MCU)-dependent and MCU-independent Ca(2+) channels coexist in the inner mitochondrial membrane. Pflügers Arch. 2013, 466, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.; Brustovetsky, T.; Rysted, J.E.; Lin, Z.; Usachev, Y.M.; Brustovetsky, N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Boil. Chem. 2018, 293, 15652–15663. [Google Scholar] [CrossRef] [Green Version]
- Samanta, K.; Mirams, G.R.; Parekh, A.B. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat. Commun. 2018, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Menazza, S.; Holmström, K.; Parks, R.J.; Liu, J.C.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gukovskaya, A.S.; Gukovsky, I.; Jung, Y.; Mouria, M.; Pandol, S.J. Cholecystokinin Induces Caspase Activation and Mitochondrial Dysfunction in Pancreatic Acinar Cells. J. Boil. Chem. 2002, 277, 22595–22604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raraty, M.; Ward, J.; Erdemli, G.; Vaillant, C.; Neoptolemos, J.P.; Sutton, R.; Petersen, O.H. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc. Natl. Acad. Sci. USA 2000, 97, 13126–13131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria: I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria: III. Transitional Ca2+ release. Arch. Biochem. Biophys. 1979, 195, 468–477. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria: II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Parks, R.J.; Murphy, E.; Liu, J.C. Mitochondrial Permeability Transition Pore and Calcium Handling. Adv. Struct. Saf. Stud. 2018, 187–196. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Tóth, E.; Maléth, J.; Závogyán, N.; Fanczal, J.; Grassalkovich, A.; Erdős, R.; Pallagi, P.; Horváth, G.; Tretter, L.; Bálint, E.R.; et al. Novel mitochondrial transition pore inhibitor N -methyl-4-isoleucine cyclosporin is a new therapeutic option in acute pancreatitis. J. Physiol. 2019, 597, 5879–5898. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, T.P.; Wu, Y.; Mei-Ling, A.J.; Koval, O.M.; Wilson, N.R.; Luczak, E.D.; Wang, Q.; Chen, B.; Gao, Z.; Zhu, Z.; et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 2015, 112, 9129–9134. [Google Scholar] [CrossRef] [Green Version]
- Parks, R.J.; Menazza, S.; Holmström, K.; Amanakis, G.; Fergusson, M.; Ma, H.; Aponte, A.M.; Bernardi, P.; Finkel, T.; Murphy, E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc. Res. 2019, 115, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Malla, S.R.; Krueger, B.; Wartmann, T.; Sendler, M.; Mahajan, U.M.; Weiss, F.U.; Thiel, F.G.; De Boni, C.; Gorelick, F.S.; Halangk, W.; et al. Early trypsin activation develops independently of autophagy in caerulein-induced pancreatitis in mice. Cell. Mol. Life Sci. 2019, 77, 1811–1825. [Google Scholar] [CrossRef]
- De Faveri, F.; Chvanov, M.; Voronina, S.; Moore, D.; Pollock, L.; Haynes, L.; Awais, M.; Beckett, A.; Mayer, U.; Sutton, R.; et al. LAP-like non-canonical autophagy and evolution of endocytic vacuoles in pancreatic acinar cells. Autophagy 2019, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Grasso, D.; Ropolo, A.; Ré, A.L.; Boggio, V.; Molejón, M.I.; Iovanna, J.L.; Gonzalez, C.; Urrutia, R.; Vaccaro, M. Zymophagy, a Novel Selective Autophagy Pathway Mediated by VMP1-USP9x-p62, Prevents Pancreatic Cell Death. J. Biol. Chem. 2011, 286, 8308–8324. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chvanov, M.; Voronina, S.; Zhang, X.; Telnova, S.; Chard, R.; Ouyang, Y.; Armstrong, J.; Tanton, H.; Awais, M.; Latawiec, D.; et al. Knockout of the Mitochondrial Calcium Uniporter Strongly Suppresses Stimulus-Metabolism Coupling in Pancreatic Acinar Cells but Does Not Reduce Severity of Experimental Acute Pancreatitis. Cells 2020, 9, 1407. https://doi.org/10.3390/cells9061407
Chvanov M, Voronina S, Zhang X, Telnova S, Chard R, Ouyang Y, Armstrong J, Tanton H, Awais M, Latawiec D, et al. Knockout of the Mitochondrial Calcium Uniporter Strongly Suppresses Stimulus-Metabolism Coupling in Pancreatic Acinar Cells but Does Not Reduce Severity of Experimental Acute Pancreatitis. Cells. 2020; 9(6):1407. https://doi.org/10.3390/cells9061407
Chicago/Turabian StyleChvanov, Michael, Svetlana Voronina, Xiaoying Zhang, Svetlana Telnova, Robert Chard, Yulin Ouyang, Jane Armstrong, Helen Tanton, Muhammad Awais, Diane Latawiec, and et al. 2020. "Knockout of the Mitochondrial Calcium Uniporter Strongly Suppresses Stimulus-Metabolism Coupling in Pancreatic Acinar Cells but Does Not Reduce Severity of Experimental Acute Pancreatitis" Cells 9, no. 6: 1407. https://doi.org/10.3390/cells9061407