Intrinsic Abnormalities of Cystic Fibrosis Airway Connective Tissue Revealed by an In Vitro 3D Stromal Model

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Source
2.2. Evaluation of Endogenous Collagen Production
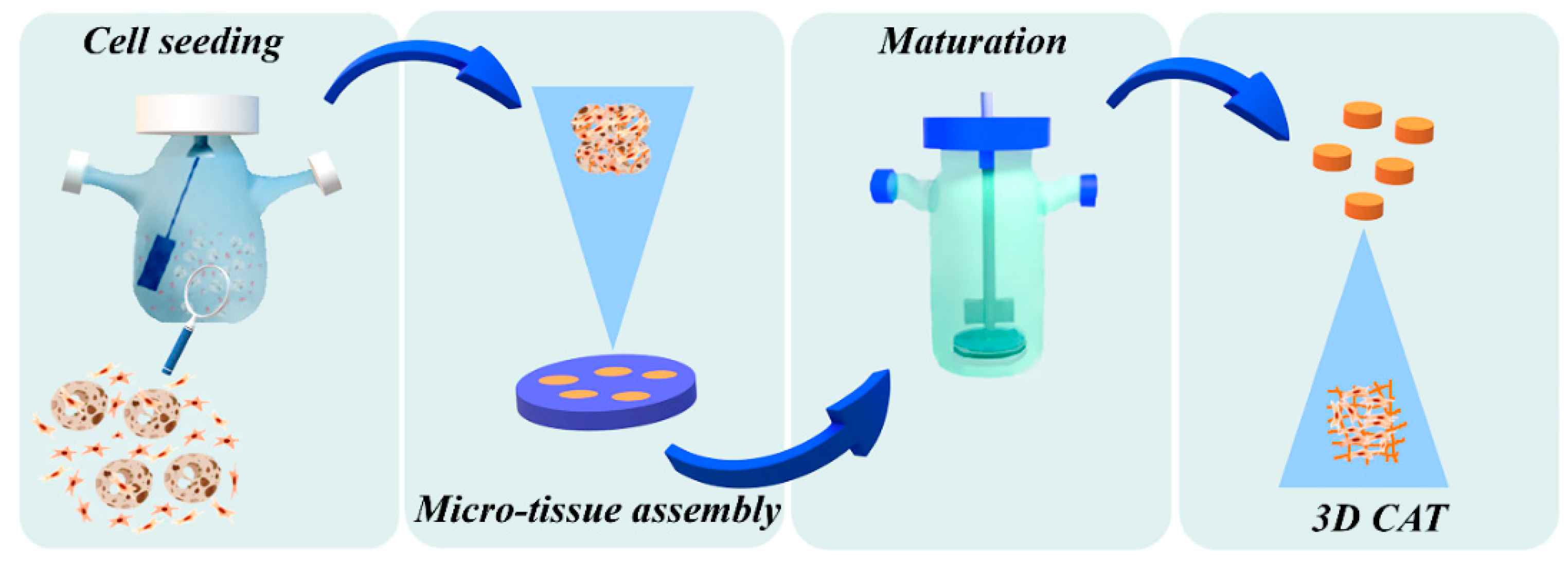
2.3. Micro-Scaffold Production
2.4. Micro-Tissues (µTPs) Production
2.5. Connective Airway Tissue (CAT) Production
2.6. Fluorescent Tissue Staining
2.7. Multiphoton Analysis
2.8. Collagen Fraction Quantification
2.9. Gray-Level Co-Occurrence Matrix Analysis: Correlation
2.10. Immunofluorescence on Tissue Sections
2.11. Protein Quantification
2.12. Mechanical Properties
2.13. RNA Extraction
2.14. QuantSeq 3’ mRNA Sequencing Library Preparation
2.15. QuantSeq 3’ mRNA Sequencing Data Processing and Analysis
2.16. Accession Codes
2.17. The Accession for the Reviewer
2.18. Statistical Analysis
3. Results
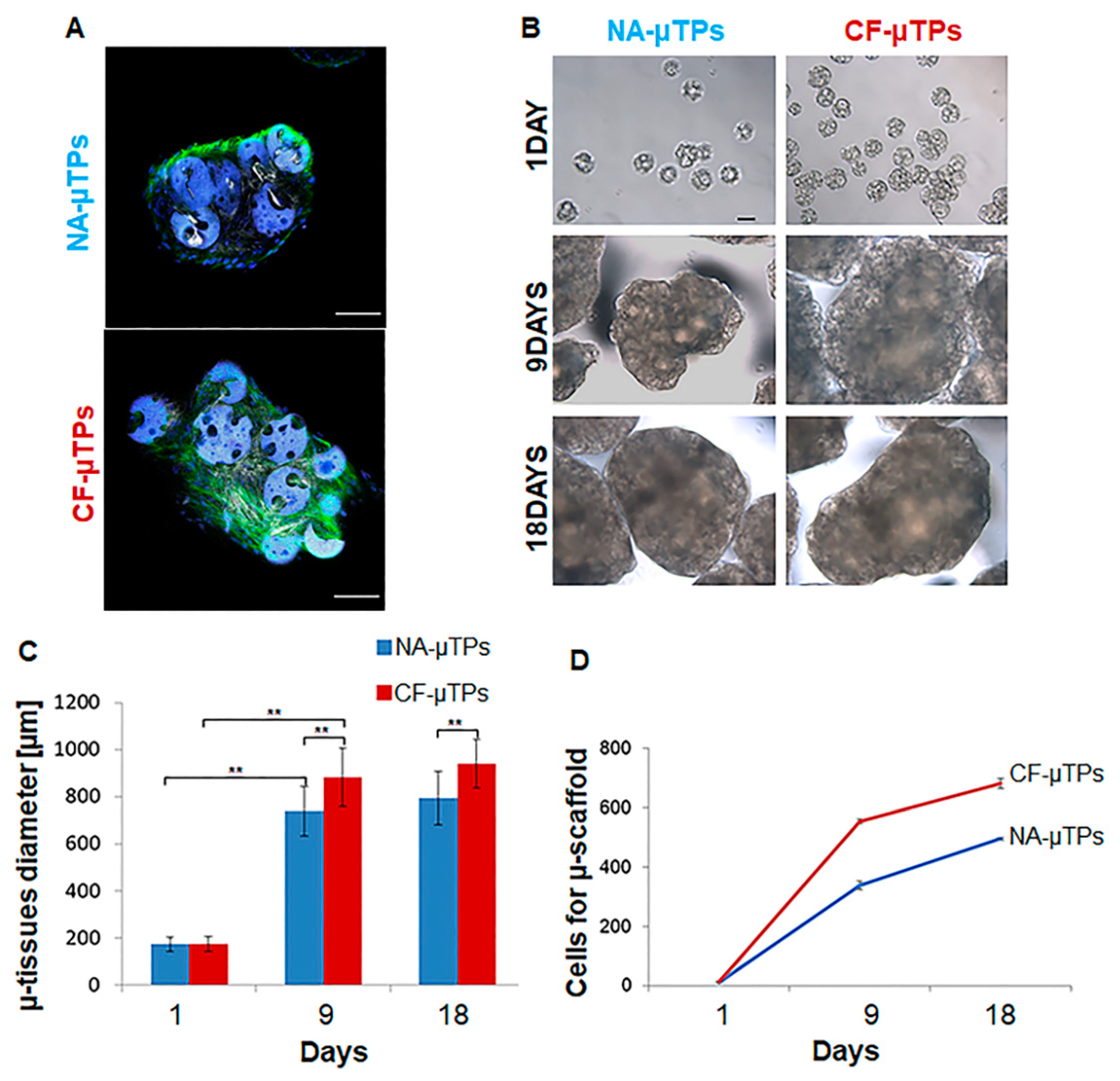
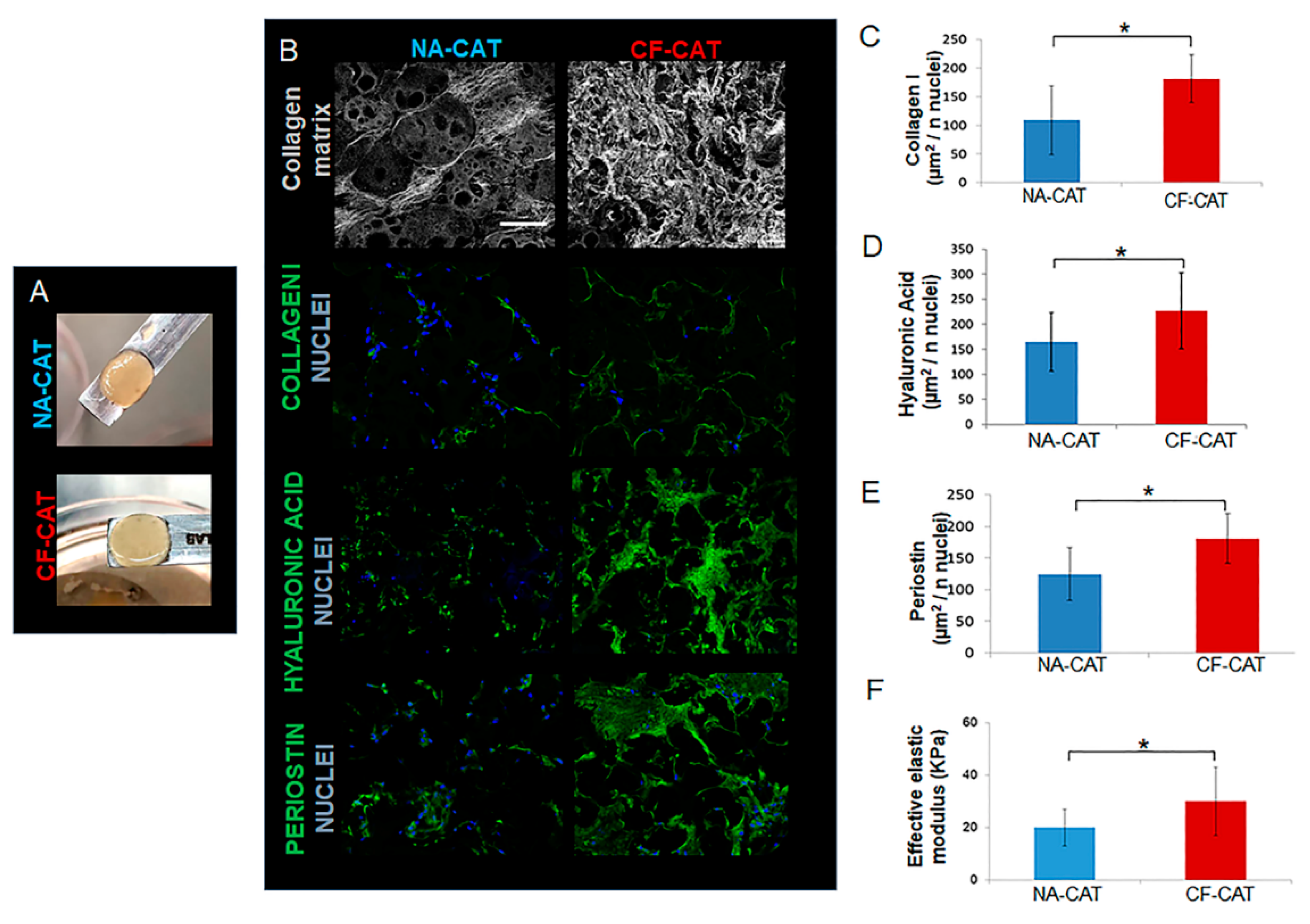
3.1. Bioengineering Bottom-Up Approach Leads to Connective Airway Tissue Equivalent Production
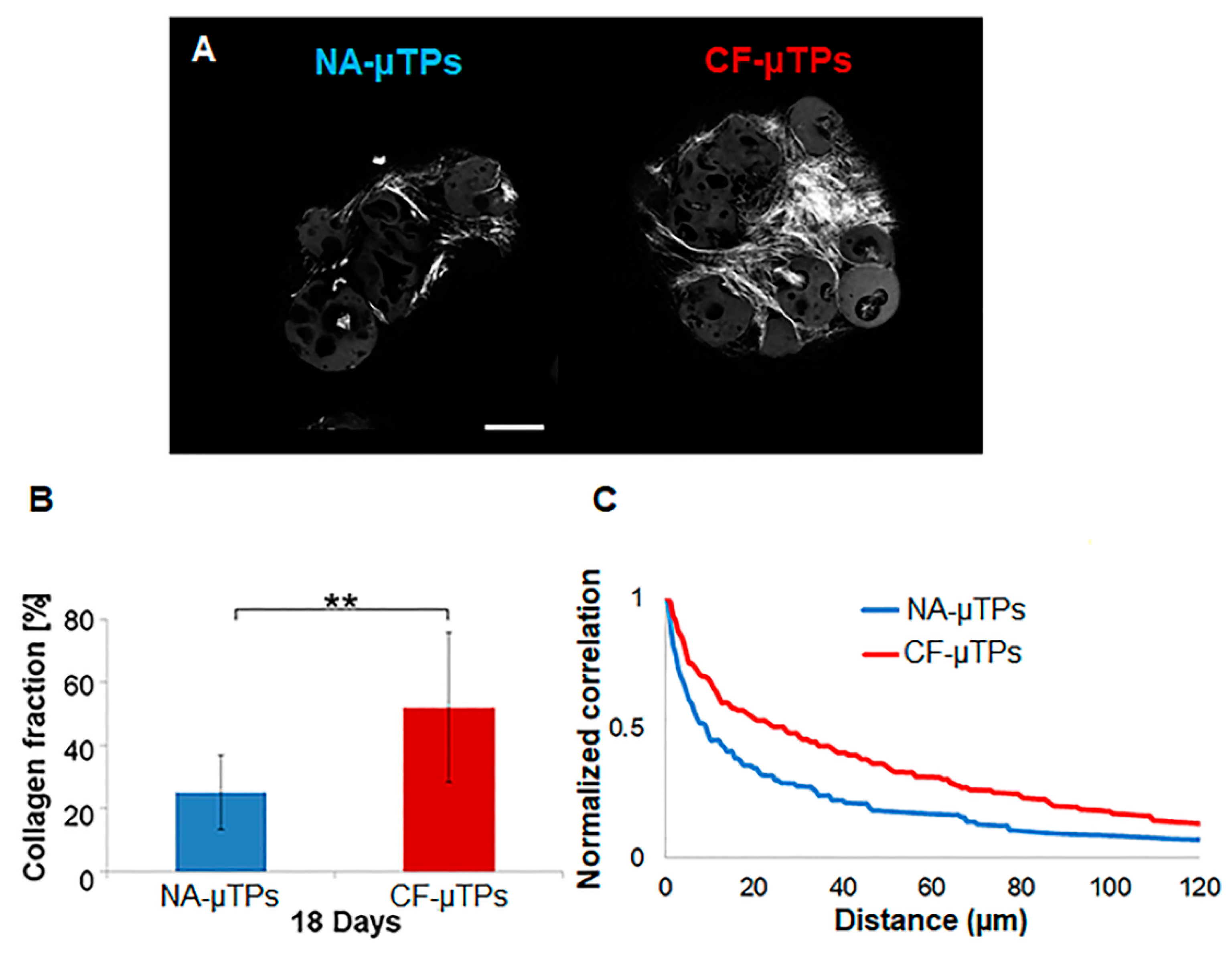
3.2. Cystic Fibrosis Connective Airway Tissues Overexpress Extracellular Matrix (ECM) Macromolecules Synthesis and Assembling
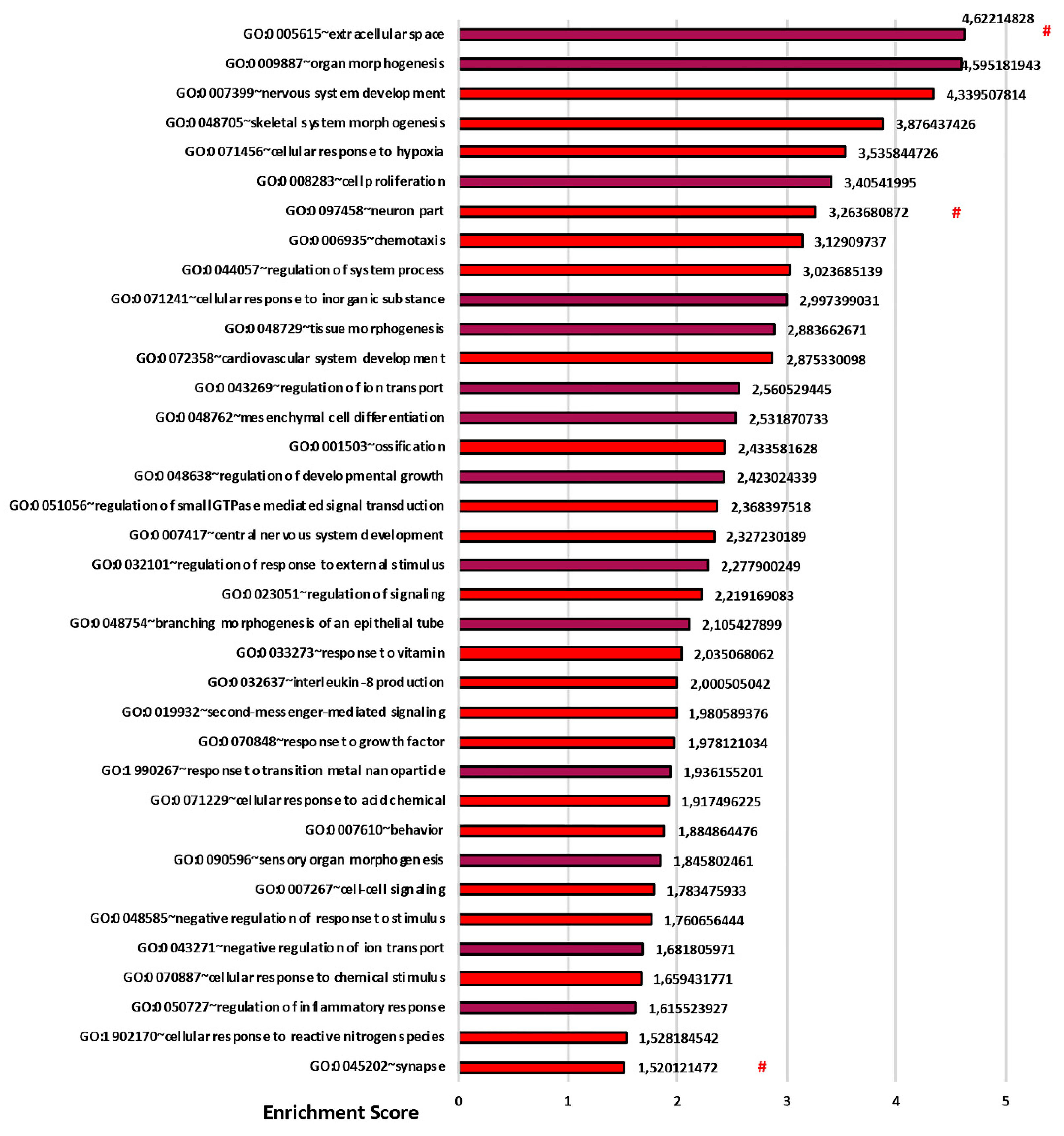
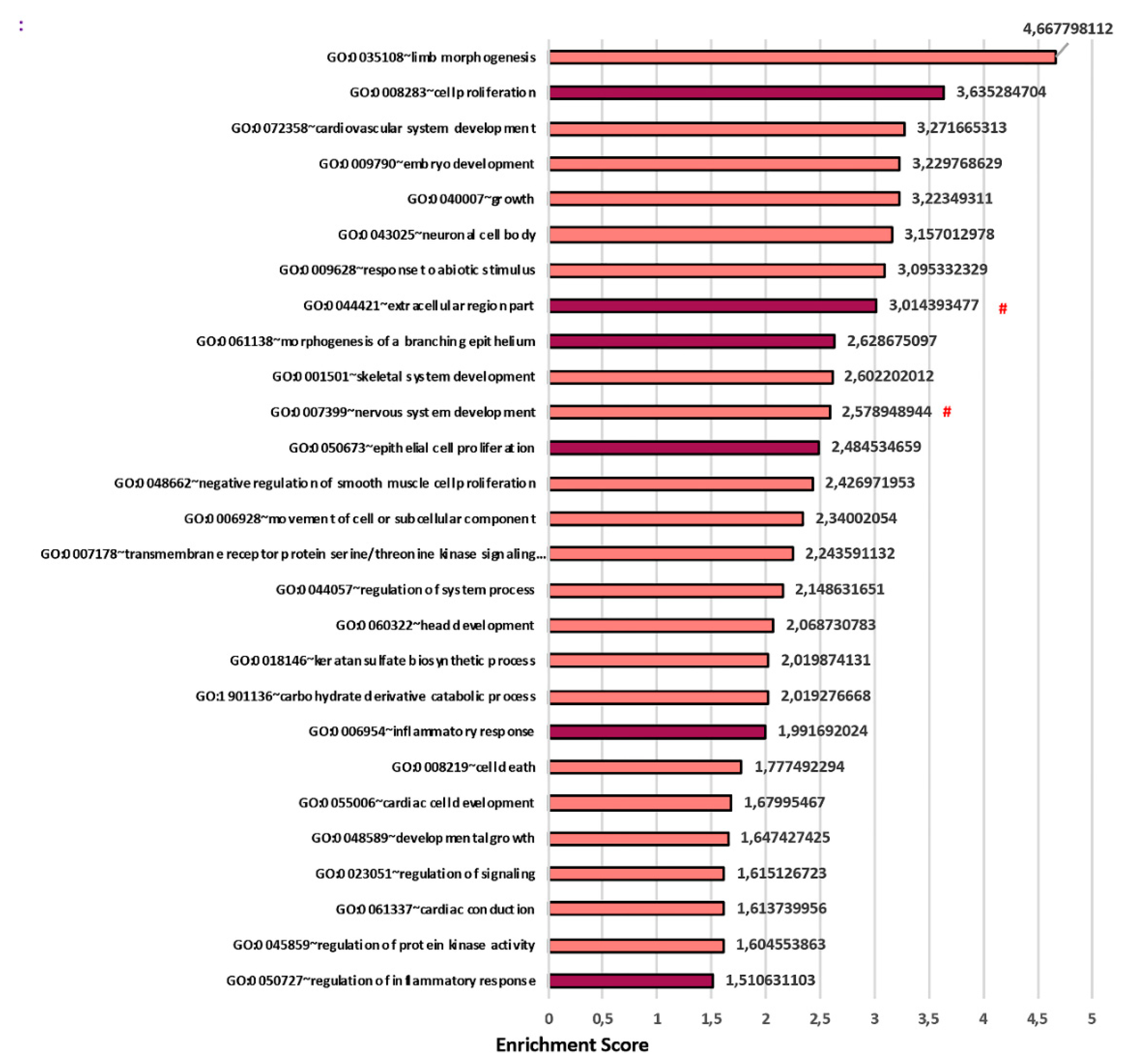
3.3. Transcriptomics Analysis Reveals a Fundamental Role of the 3D Stromal Environment in Up-Regulating Genes Involved in the Epithelial Morphogenesis and Inflammatory Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chunying, L.; Anjaparavanda, P.N. CFTR Chloride Channel in the Apical Compartments: Spatiotemporal Coupling to its Interacting Partners. Integr. Biol. 2010, 2, 161–177. [Google Scholar]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Prim. 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Rout-pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19, 136. [Google Scholar] [CrossRef]
- Awatade, N.T.; Wong, S.L.; Hewson, C.K.; Fawcett, L.K.; Kicic, A.; Jaffe, A.; Waters, S. Human Primary Epithelial Cell Models: Promising Tools in the Era of Cystic Fibrosis Personalized Medicine. Front. Pharmacol. 2018, 9, 1429. [Google Scholar] [CrossRef] [Green Version]
- Ikpa, P.T.; Bijvelds, M.J.C.; De Jonge, H.R. Cystic fibrosis: Toward personalized therapies. Int. J. Biochem. Cell Biol. 2014, 52, 192–200. [Google Scholar] [CrossRef]
- Pageau, S.C.; Sazonova, O.V.; Wong, J.Y.; Soto, A.M.; Sonnenschein, C. The effect of stromal components on the modulation of the phenotype of human bronchial epithelial cells in 3D culture. Biomaterials 2011, 32, 7169–7180. [Google Scholar] [CrossRef]
- Casale, C.; Imparato, G.; Urciuolo, F.; Netti, P.A. Endogenous human skin equivalent promotes in vitro morphogenesis of follicle-like structures. Biomaterials 2016, 101, 86–95. [Google Scholar] [CrossRef]
- Huaux, F.; Noel, S.; Dhooghe, B.; Panin, N.; Lo Re, S.; Lison, D.; Wallemacq, P.; Marbaix, E.; Scholte, B.J.; Lebecque, P.; et al. Dysregulated Proinflammatory and Fibrogenic Phenotype of Fibroblasts in Cystic Fibrosis. PLoS ONE 2013, 8, e64341. [Google Scholar] [CrossRef] [Green Version]
- Lavelle, G.M.; White, M.M.; Browne, N.; Mcelvaney, N.G.; Reeves, E.P. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. Biomed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggar, A.; Hector, A.; Bratcher, P.E.; Mall, M.A.; Griese, M.; Hartl, D. The role of matrix metalloproteases in cystic fibrosis lung disease. Eur. Respir. J. 2011, 38, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Regamey, N.; Jeffery, P.K.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 2011, 66, 624–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urciuolo, F.; Imparato, G.; Totaro, A.; Netti, P.A. Building a tissue in vitro from the bottom up: Implications in regenerative medicine. Methodist Debakey Cardiovasc. J. 2013, 9, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, K.; Buzsaky, F.; Mosbach, K. Growth of anchorage-dependent cells on macroporous microcarriers. Bio/technology 1986, 4, 989–990. [Google Scholar] [CrossRef]
- Imparato, G.; Urciuolo, F.; Casale, C.; Netti, P.A. The role of microscaffold properties in controlling the collagen assembly in 3D dermis equivalent using modular tissue engineering. Biomaterials 2013, 34, 7851–7861. [Google Scholar] [CrossRef]
- Mazio, C.; Casale, C.; Imparato, G.; Urciuolo, F.; Attanasio, C.; De Gregorio, M.; Rescigno, F.; Netti, P.A. Pre-vascularized dermis model for fast and functional anastomosis with host vasculature. Biomaterials 2019, 192, 159–170. [Google Scholar] [CrossRef]
- Urciuolo, F.; Imparato, G.; Palmiero, C.; Trilli, A.; Netti, P.A. Effect of process conditions on the growth of three-dimensional dermal-equivalent tissue obtained by microtissue precursor assembly. Tissue Eng. Part C. Methods 2011, 17, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Mazio, C.; Casale, C.; Imparato, G.; Urciuolo, F.; Netti, P.A. Recapitulating spatiotemporal tumor heterogeneity in vitro through engineered breast cancer microtissues. Acta Biomater. 2018, 73, 236–249. [Google Scholar] [CrossRef]
- Mattei, G.; Gruca, G.; Rijnveld, N.; Ahluwalia, A. The nano-epsilon dot method for strain rate viscoelastic characterisation of soft biomaterials by spherical nano-indentation. J. Mech. Behav. Biomed. Mater. 2015, 50, 150–159. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Qi, M.; Wang, R.; Jing, B.; Jian, F.; Ning, C.; Zhang, L. Prevalence and multilocus genotyping of Cryptosporidium andersoni in dairy cattle and He cattle in Xinjiang, China. Infect. Genet. Evol. 2016, 44, 313–317. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, M.; Kanehisa, M. Using the KEGG database resource. Curr. Protoc. Bioinform. 2012, 11, 1–12. [Google Scholar] [CrossRef]
- Wu, S.; Peng, Y.; Hu, L.; Zhang, X.; Li, H. Classification and recognition of texture collagen obtaining by multiphoton microscope with neural network analysis. J. Phys. Conf. Ser. 2016, 680, 012014. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Nadiarynkh, O.; Plotnikov, S.; Campagnola, P.J. Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat. Protoc. 2012, 7, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Imparato, C.; Casale, S.; Scamardella, F.; Urciuolo, M.; Bimonte, F.; Apone, G.; Colucci, P.A.; Netti, G. A novel engineered dermis for in vitro photodamage research. J. Tissue Eng. Regen. Med. 2016, 11, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Hristu, R.; Eftimie, L.G.; Stanciu, S.G.; Tranca, D.E.; Paun, B.; Sajin, M.; Stanciu, G.A. Quantitative second harmonic generation microscopy for the structural characterization of capsular collagen in thyroid neoplasms. Biomed. Opt. Express 2018, 9, 3923. [Google Scholar] [CrossRef] [PubMed]
- Cicchi, R.; Kapsokalyvas, D.; De Giorgi, V.; Maio, V.; Van Wiechen, A.; Massi, D.; Lotti, T.; Pavone, F.S. Scoring of collagen organization in healthy and diseased human dermis by multiphoton microscopy. J. Biophotonics 2010, 3, 34–43. [Google Scholar] [CrossRef]
- Panwar, P.; Lamour, G.; Mackenzie, N.C.W.; Yang, H.; Ko, F.; Li, H.; Brömme, D. Changes in structural-mechanical properties and degradability of collagen during aging-associated modifications. J. Biol. Chem. 2015, 290, 23291–23306. [Google Scholar] [CrossRef] [Green Version]
- Lauer, M.E.; Dweik, R.A.; Garantziotis, S.; Aronica, M.A. The Rise and Fall of Hyaluronan in Respiratory Diseases. Int. J. Cell Biol. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, D.N.; Moore, B.B. The role of periostin in lung fibrosis and airway remodeling. Cell. Mol. Life Sci. 2017, 74, 4305–4314. [Google Scholar] [CrossRef]
- Norris, R.A.; Damon, B.; Mironov, V.; Kasyanov, V.; Moreno-rodriguez, R.; Trusk, T.; Potts, J.D.; Richard, L.; Davis, J.; Hoffman, S.; et al. Periostin Regulates Collagen Fibrillogenesis. J. Cell. Biochem. 2007, 101, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Wells, R.G. Biochimica et Biophysica Acta Tissue mechanics and fibrosis. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 884–890. [Google Scholar] [CrossRef] [Green Version]
- Oshima, A.; Tanabe, H.; Yan, T.; Lowe, G.N.; Glackin, C.A.; Kudo, A. A Novel Mechanism for the Regulation of Osteoblast Differentiation: Transcription of Periostin, a Member of the Fasciclin I Family, is Regulated by the bHLH Transcription Factor, Twist. J. Cell. Biochem. 2002, 86, 792–804. [Google Scholar] [CrossRef]
- Qin, X.; Yan, M.; Zhang, J.; Wang, X.; Shen, Z.; Lv, Z. TGF β 3-mediated induction of Periostin facilitates head and neck cancer growth and is associated with metastasis. Sci. Rep. 2016, 6, 20587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Königshoff, M.; Balsara, N.; Pfaff, E.M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munguía-Reyes, A.; Balderas-Martínez, Y.I.; Becerril, C.; Checa, M.; Ramírez, R.; Ortiz, B.; Meléndez-Zajgla, J.; Pardo, A.; Selman, M. R-spondin-2 is upregulated in idiopathic pulmonary fibrosis and affects fibroblast behavior. Am. J. Respir. Cell Mol. Biol. 2018, 59, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Liang, J.; Liu, N.; Huan, C.; Zhang, Y.; Liu, W.; Kumar, M.; Xiao, R.; D’Armiento, J.; Metzger, D.; et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J. Clin. Investig. 2016, 126, 3063–3079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.F.; Wilson, C.L.; Chow, Y.H.; Schnapp, L.M. Role of integrin alpha8 in murine model of lung fibrosis. PLoS ONE 2018, 13, e0197937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Plaat, D.A.; de Jong, K.; Lahousse, L.; Faiz, A.; Vonk, J.M.; van Diemen, C.C.; Nedeljkovic, I.; Amin, N.; Brusselle, G.G.; Hofman, A.; et al. Genome-wide association study on the FEV 1 /FVC ratio in never-smokers identifies HHIP and FAM13A. J. Allergy Clin. Immunol. 2017, 139, 533–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hector, A.; Kormann, M.S.D.; Mack, I.; Latzin, P.; Casaulta, C.; Kieninger, E.; Zhou, Z.; Yildirim, A.O.; Bohla, A.; Rieber, N.; et al. The chitinase-like protein YKL-40 modulates cystic fibrosis lung disease. PLoS ONE 2011, 6, e24399. [Google Scholar] [CrossRef] [Green Version]
- Haase, H.; Hebel, S.; Engelhardt, G.; Rink, L. The biochemical effects of extracellular Zn2+ and other metal ions are severely affected by their speciation in cell culture media. Metallomics 2015, 7, 102–111. [Google Scholar] [CrossRef]
- Noble, P.W. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123–129. [Google Scholar] [CrossRef]
- Parrinello, S.; Coppe, J.; Krtolica, A.; Campisi, J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Shtilbans, V. Role of Stromal-Epithelial Interaction in the Formation and Development of Cancer Cells. Cancer Microenviron. 2013, 6, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brancato, V.; Comunanza, V.; Imparato, G.; Corà, D.; Urciuolo, F.; Noghero, A.; Bussolino, F.; Netti, P.A. Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer. Acta Biomater. 2017, 49, 152–166. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, V.; Corrado, B.; Sbrescia, S.; Sibilio, S.; Urciuolo, F.; Netti, P.A.; Imparato, G. Intestine-on-chip device increases ECM remodeling inducing faster epithelial cell differentiation. Biotechnol. Bioeng. 2020, 117, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.P.; Chmiel, J.F. Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 2015, 50, S39–S56. [Google Scholar] [CrossRef]
- Su, X.; Looney, M.R.; Su, H.; Lee, J.W.; Song, Y.; Matthay, M.A. Role of CFTR expressed by neutrophils in modulating acute lung inflammation and injury in mice. Inflamm. Res. 2011, 60, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A matricellular protein with multiple functions in cancer development and progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Kudo, A.; Kii, I. Periostin function in communication with extracellular matrices. J. Cell Commun. Signal. 2018, 12, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Ghatak, S.; Misra, S.; Norris, R.A.; Moreno-rodriguez, R.A.; Hoffman, S.; Levine, R.A.; Hascall, V.C.; Markwald, R.R. Periostin Induces Intracellular Cross-talk between Kinases and Hyaluronan in Atrioventricular Valvulogenesis. J. Biol. Chem. 2014, 289, 8545–8561. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, N.A.; Tao, R.; Chenoweth, J.G.; Brandtjen, A.; Mighdoll, M.I.; Genova, J.D.; McKay, R.D.; Jia, Y.; Weinberger, D.R.; Kleinman, J.E.; et al. Strong Components of Epigenetic Memory in Cultured Human Fibroblasts Related to Site of Origin and Donor Age. PLoS Genet. 2016, 12, e1005819. [Google Scholar] [CrossRef] [PubMed]
- Ozes, A.R.; Nephew, K.P. 3D culture adds an extra dimension to targeted epigenetic therapies Comment. Cell Cycle 2013, 12, 2173–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatangelo, M.D.; Garipov, A.; Li, H.; Conejo-Garcia, J.R.; Speicher, D.W.; Zhang, R. Three-dimensional culture sensitizes epithelial ovarian cancer cells to EZH2 methyltransferase inhibition. Cell Cycle 2013, 12, 2113–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.; De Sario, A. DNA methylation changes in cystic fibrosis: Cause or consequence? Clin. Genet. 2020, 1–7. [Google Scholar] [CrossRef]
- Magalhães, M.; Rivals, I.; Claustres, M.; Varilh, J.; Thomasset, M.; Bergougnoux, A.; Mely, L.; Leroy, S.; Corvol, H.; Guillot, L.; et al. DNA methylation at modifier genes of lung disease severity is altered in cystic fibrosis. Clin. Epigenetics 2017, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Helling, B.A.; Yang, I.V. Epigenetics in lung fibrosis: From pathobiology to treatment perspective. Physiol. Behav. 2015, 21, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.; Pardo, A. Stochastic age-related epigenetic drift in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1328–1330. [Google Scholar] [CrossRef]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial—Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Santoiemma, M. Epithelial-mesenchymal interactions: A fundamental Developmental Biology mechanism. Int. J. Dev. Biol. 2014, 58, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.J.; Lynch, T.J.; Engelhardt, J.F. Multipotent myoepithelial progenitor cells are born early during airway submucosal gland development. Am. J. Respir. Cell Mol. Biol. 2017, 56, 716–726. [Google Scholar] [CrossRef]
- Salinas, D.; Haggie, P.M.; Thiagarajah, J.R.; Song, Y.; Rosbe, K.; Finkbeiner, W.E.; Nielson, D.W.; Verkman, A.S. Submucosal gland dysfunction as a primary defect in cystic fibrosis. FASEB J. 2005, 19, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Hays, S.R.; Fahy, J.V. Characterizing mucous cell remodeling in cystic fibrosis: Relationship to neutrophils. Am. J. Respir. Crit. Care Med. 2006, 174, 1018–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inglis, S.K.; Wilson, S.M. Cystic fibrosis and airway submucosal glands. Pediatr. Pulmonol. 2005, 40, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J. Leukoc. Biol. 2006, 80, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebihara, N.; Matsuda, A.; Nakamura, S.; Matsuda, H.; Murakami, A. Role of the IL-6 classic-and trans-signaling pathways in corneal sterile inflammation and wound healing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8549–8557. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.Y.; Mun, S.K.; Rhee, S.; Kim, B.J. Epidermal growth factor improves the migration and contractility of aged fibroblasts cultured on 3D collagen matrices. Int. J. Mol. Med. 2015, 35, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Cole, M.A.; Quan, T.; Voorhees, J.J.; Fisher, G.J. Extracellular matrix regulation of fibroblast function: Redefining our perspective on skin aging. J. Cell Commun. Signal. 2018, 12, 35–43. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Code | Working Dilution | Unmasking |
|---|---|---|---|
| CollI (Rabbit) | ab34710 | 1:500 | Heat mediated (citrate buffer) |
| Hyaluronic Acid (Sheep) | ab53842 | 1:50 | Enzymatic (Trypsin) |
| Periostin (Rabbit) | ab14041 | 1:100 | Heat mediated (citrate buffer |
| Secondary Antibody | Code | Working Dilution |
|---|---|---|
| 488 Goat Anti-Rabbit | A11008 | 1:500 |
| 546 Donkey Anti-Sheep | A21098 | 1:500 |
| Sample | Correlation Length (λ) |
|---|---|
| NA-µTPs | 62.6254 ± 16.23 |
| CF-µTPs | 135.88 ± 43.31 |
| Gene Symbol | logFC_CF-HLF vs. N-HLF |
|---|---|
| TGFβ3 | 1.755 |
| HAS-2 | 2.062 |
| TWIST1 | 2.425 |
| WNT2 | 3.052 |
| SFRP2 | 3.695 |
| RSPO2 | 4.331 |
| TBX4 | 4.593 |
| ITGA8 | 4.874 |
| CHI3L1 | 5.027 |
| HHIP | 5.09 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazio, C.; Scognamiglio, L.S.; De Cegli, R.; Galietta, L.J.V.; Di Bernardo, D.; Casale, C.; Urciuolo, F.; Imparato, G.; Netti, P.A. Intrinsic Abnormalities of Cystic Fibrosis Airway Connective Tissue Revealed by an In Vitro 3D Stromal Model. Cells 2020, 9, 1371. https://doi.org/10.3390/cells9061371
Mazio C, Scognamiglio LS, De Cegli R, Galietta LJV, Di Bernardo D, Casale C, Urciuolo F, Imparato G, Netti PA. Intrinsic Abnormalities of Cystic Fibrosis Airway Connective Tissue Revealed by an In Vitro 3D Stromal Model. Cells. 2020; 9(6):1371. https://doi.org/10.3390/cells9061371
Chicago/Turabian StyleMazio, Claudia, Laura S. Scognamiglio, Rossella De Cegli, Luis J. V. Galietta, Diego Di Bernardo, Costantino Casale, Francesco Urciuolo, Giorgia Imparato, and Paolo A. Netti. 2020. "Intrinsic Abnormalities of Cystic Fibrosis Airway Connective Tissue Revealed by an In Vitro 3D Stromal Model" Cells 9, no. 6: 1371. https://doi.org/10.3390/cells9061371