Nanosphere Loaded With Curcumin Inhibits the Gastrointestinal Cell Death Signaling Pathway Induced by the Foodborne Pathogen Vibrio vulnificus

Abstract
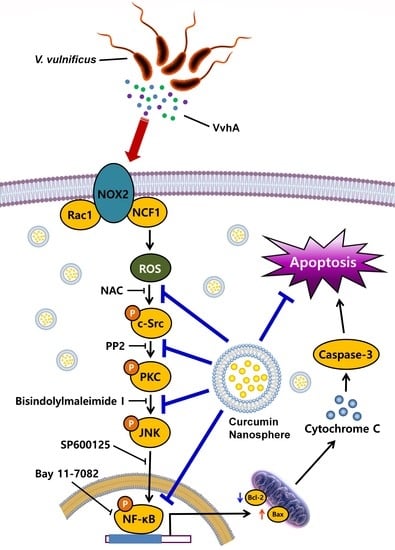
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cells
2.3. Purification of the Recombinant Protein (r)VvhA
2.4. Preparation of Curcumin Nanosphere (CN)
2.5. Ultraviolet-Visible Spectroscopy (UV-Vis) Analysis
2.6. Field Emission Scanning Electron Microscope (FE-SEM) Measurement
2.7. Fourier-Transform Infrared Spectroscopy (FT-IR) Measurement
2.8. Cell Viability Assay
2.9. Cell Number Count
2.10. Reactive Oxygen Species (ROS) Detection
2.11. Immunofluorescence Analysis
2.12. Western Blot Analysis
2.13. Ileal-Ligated Mouse Model
2.14. Apoptosis Detection
2.15. Flow Cytometry
2.16. Solubility Analysis
2.17. Statistical Analysis
3. Results
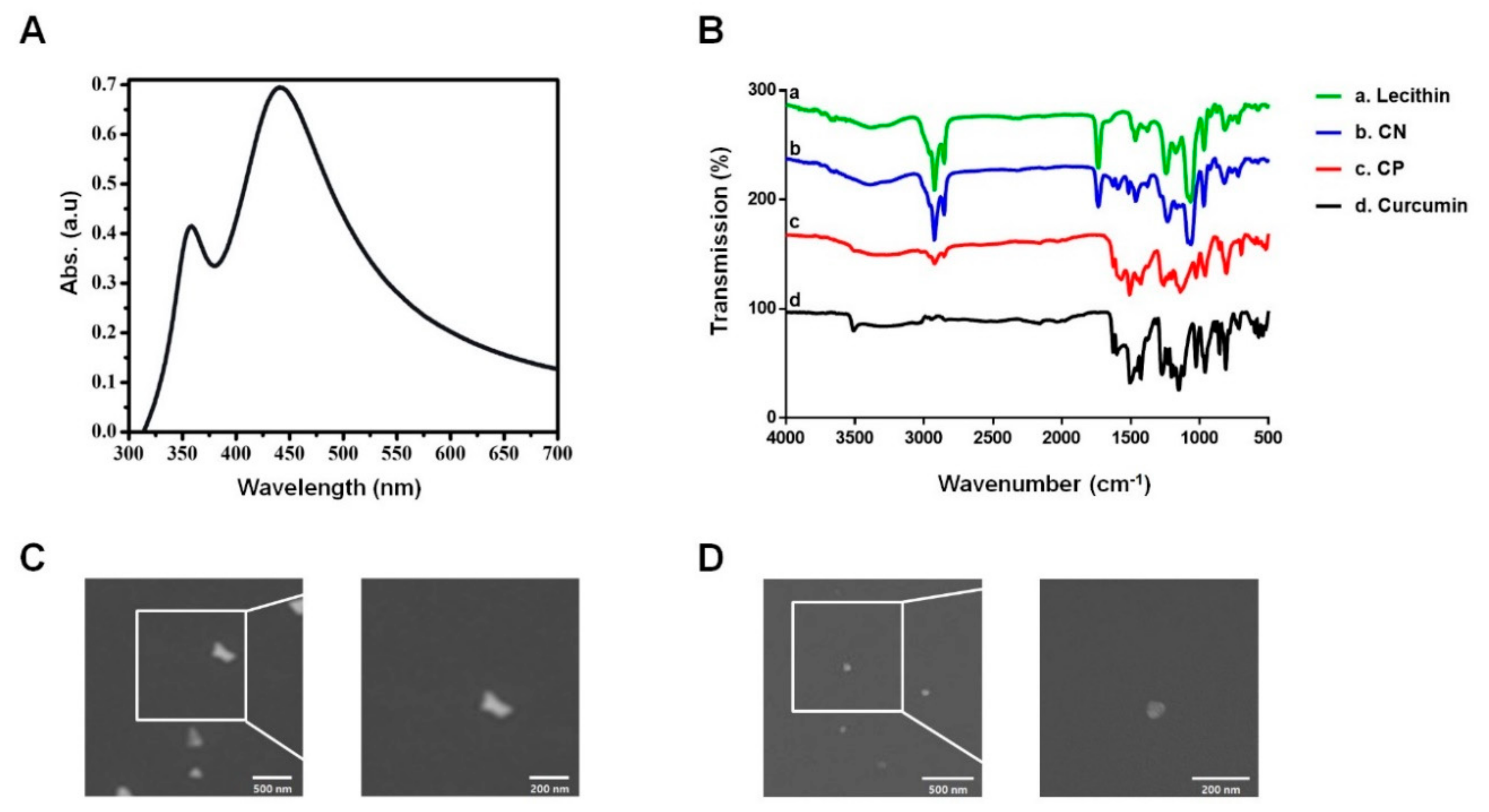
3.1. Characterization of the Curcumin Nanosphere (CN)
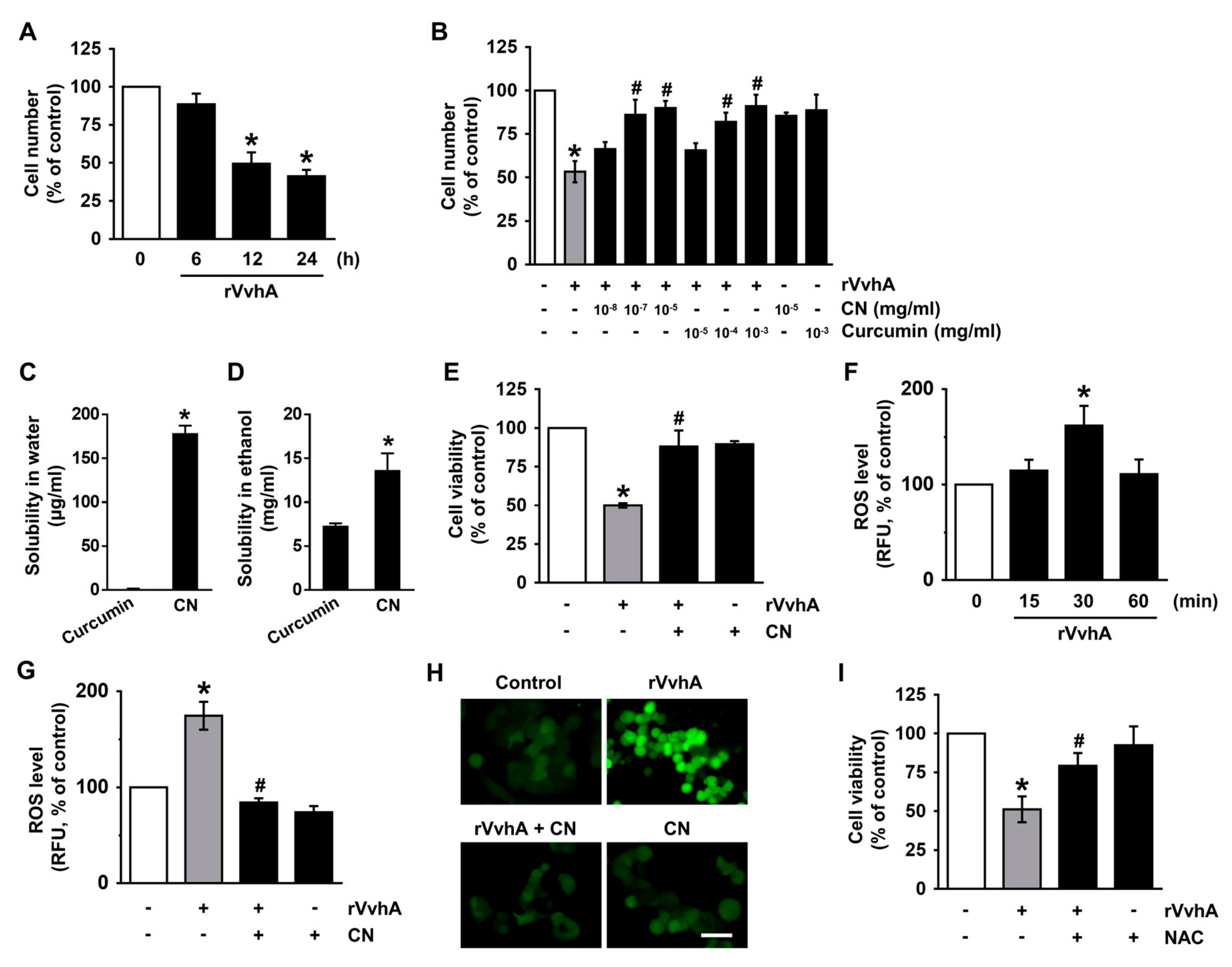
3.2. CN Has An Inhibitory Effect on the Production of ROS Responsible for Cytotoxicity Caused by V. Vulnificus, VvhA
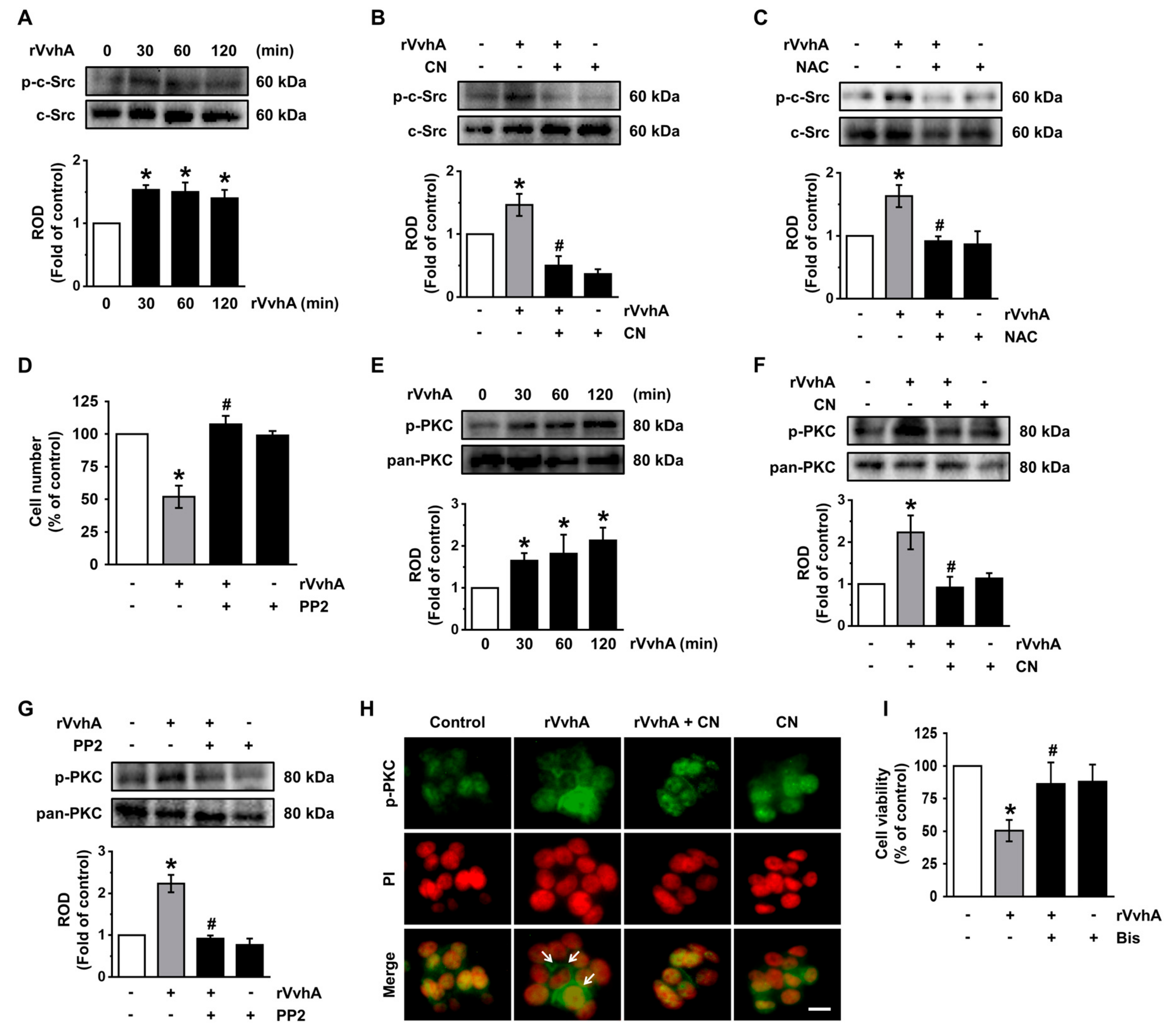
3.3. CN Regulates the Activation of c-Src and PKC Induced by rVvhA
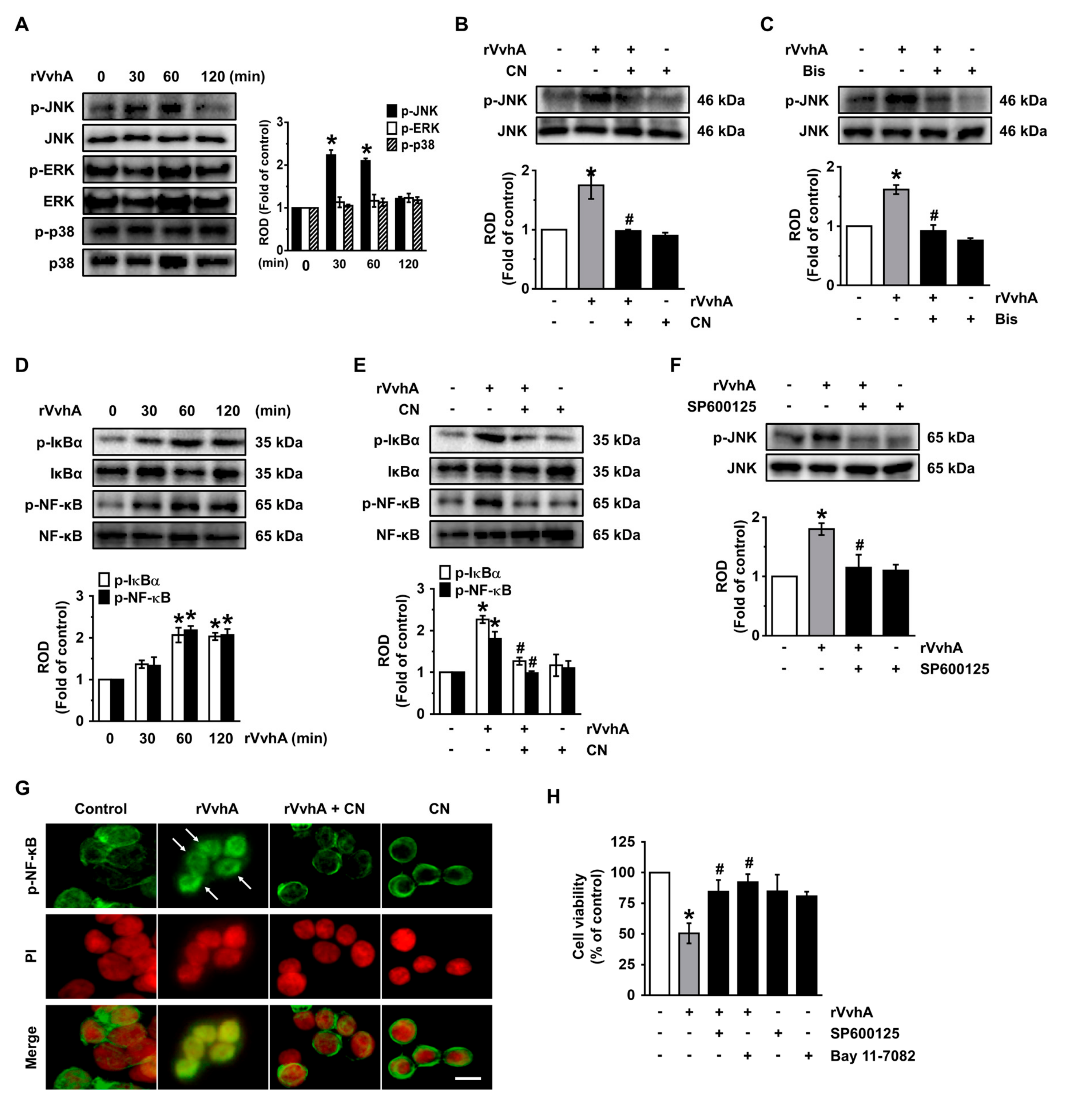
3.4. CN Uniquely Regulates the JNK/NF-κB Pathway Responsible for Cell Death Caused by rVvhA
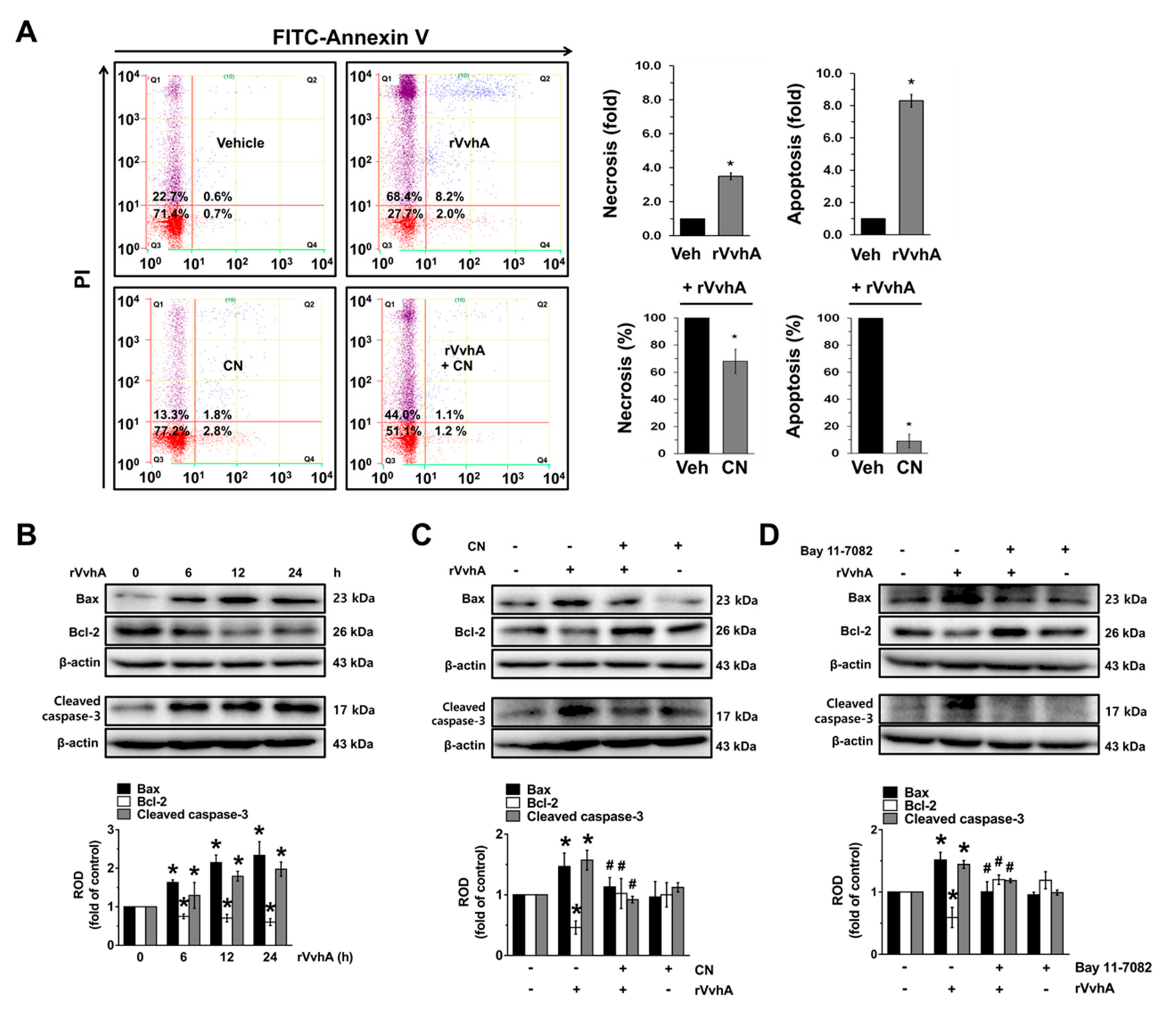
3.5. The Role of CN on Apoptotic Cell Death Induced by rVvhA
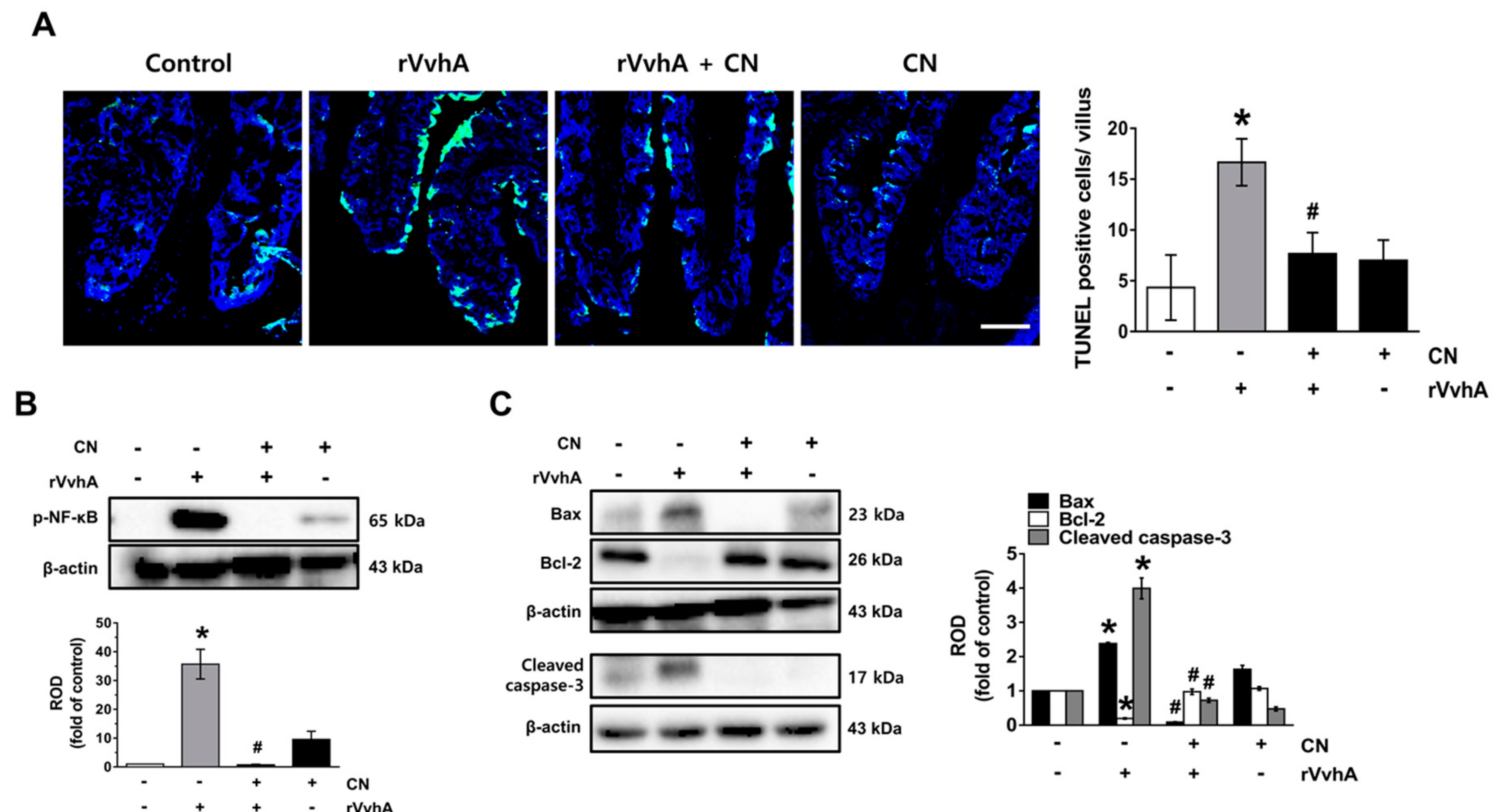
3.6. CN Functionally Blocks Apoptotic Responses Caused by rVvhA in Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations:
References
- Li, G.; Wang, M.Y. The role of Vibrio vulnificus virulence factors and regulators in its infection-induced sepsis. Folia Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Lee, S.J.; Lim, H.S.; Kim, J.S.; Jang, K.K.; Choi, S.H.; Han, H.J. Vibrio vulnificus VvhA induces autophagy-related cell death through the lipid raft-dependent c-Src/NOX signaling pathway. Sci. Rep. 2016, 6, 27080. [Google Scholar] [CrossRef]
- Jeong, H.G.; Satchell, K.J. Additive function of Vibrio vulnificus MARTXVv and VvhA cytolysins promotes rapid growth and epithelial tissue necrosis during intestinal infection. PLoS Pathog. 2012, 8, e1002581. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Park, J.P.; Lim, K.T.; Lee, S.J. Intestinal epithelial cell apoptosis due to a hemolytic toxin from Vibrio vulnificus and protection by a 36kDa glycoprotein from Rhus verniciflua Stokes. Food Chem. Toxicol. 2019, 125, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Jung, Y.H.; Song, E.J.; Jang, K.K.; Choi, S.H.; Han, H.J. Vibrio vulnificus VvpE stimulates IL-1β production by the hypomethylation of the IL-1β promoter and NF-κB activation via lipid raft-dependent ANXA2 recruitment and reactive oxygen species signaling in intestinal epithelial cells. J. Immunol. 2015, 195, 2282–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.M.; Park, J.P.; Jung, Y.H.; Lee, H.J.; Kim, J.S.; Choi, G.E.; Han, H.J.; Lee, S.J. Melatonin restores Muc2 depletion induced by V. vulnificus VvpM via melatonin receptor 2 coupling with Gαq. J. Biomed. Sci. 2020, 27, 21. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, H.J.; Jung, Y.H.; Kim, J.S.; Choi, S.H.; Han, H.J. Melatonin inhibits apoptotic cell death induced by Vibrio vulnificus VvhA via melatonin receptor 2 coupling with NCF-1. Cell Death Dis. 2018, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Meng, X.; Li, Y.; Zhao, C.N.; Tang, G.Y.; Li, H.B. Antibacterial and antifungal activities of spices. Int. J. Mol. Sci. 2017, 18, 1283. [Google Scholar] [CrossRef] [Green Version]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “curecumin”: From kitchen to clinic. Biochem. Pharmacol. 2008, 75, 787–809. [Google Scholar] [CrossRef] [Green Version]
- Dulbecco, P.; Savarino, V. Therapeutic potential of curcumin in digestive diseases. World J. Gastroenterol. 2013, 19, 9256–9270. [Google Scholar] [CrossRef]
- Gera, M.; Sharma, N.; Ghosh, M.; Huynh, D.L.; Lee, S.J.; Min, T.; Kwon, T.; Jeong, D.K. Nanoformulations of curcumin: An emerging paradigm for improved remedial application. Oncotarget 2017, 8, 66680–66698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.B.; Lee, Y.M.; Park, M.K.; Min, T.; Lee, S.J. Effects of anti-ecotoxicological curcumin nanospheres on feed efficiency and fecal odor in mice. J. Environ. Sci. Int. 2019, 28, 183–189. [Google Scholar] [CrossRef]
- Ohno, M.; Nishida, A.; Sugitani, Y.; Nishino, K.; Inatomi, O.; Sugimoto, M.; Kawahara, M.; Andoh, A. Nanoparticle curcumin ameliorates experimental colitis via modulation of gut microbiota and induction of regulatory T cells. PLoS ONE 2017, 12, e0185999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Jung, Y.H.; Oh, S.Y.; Jang, K.K.; Lee, H.S.; Choi, S.H.; Han, H.J. Vibrio vulnificus VvpE inhibits mucin 2 expression by hypermethylation via lipid raft-mediated ROS signaling in intestinal epithelial cells. Cell Death Dis. 2015, 6, e1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramera, E.I.; Konteles, S.J.; Karathanosa, V.T. Microencapsulation of curcumin in cells of Saccharomyces cerevisiae. Food Chem. 2011, 125, 892–902. [Google Scholar] [CrossRef]
- Schmitter, T.; Pils, S.; Weibel, S.; Agerer, F.; Peterson, L.; Buntru, A.; Kopp, K.; Hauck, C.R. Opa proteins of pathogenic neisseriae initiate Src kinase-dependent or lipid raft-mediated uptake via distinct human carcinoembryonic antigen-related cell adhesion molecule isoforms. Infect. Immun. 2007, 75, 4116–4126. [Google Scholar] [CrossRef] [Green Version]
- Brandt, D.; Gimona, M.; Hillmann, M.; Haller, H.; Mischak, H. Protein kinase C induces actin reorganization via a Src- and Rho-dependent pathway. J. Biol. Chem. 2002, 277, 20903–20910. [Google Scholar] [CrossRef] [Green Version]
- Yousuf, M.A.; Lee, J.S.; Zhou, X.; Ramke, M.; Lee, J.Y.; Chodosh, J.; Rajaiya, J. Protein kinase C signaling in adenoviral infection. Biochemistry 2016, 55, 5938–5946. [Google Scholar] [CrossRef]
- Jang, B.C.; Lim, K.J.; Paik, J.H.; Kwon, Y.K.; Shin, S.W.; Kim, S.C.; Jung, T.Y.; Kwon, T.K.; Cho, J.W.; Baek, W.K.; et al. Up-regulation of human β-defensin 2 by interleukin-1β in A549 cells: Involvement of PI3K, PKC, p38 MAPK, JNK, and NF-κB. Biochem. Biophys. Res. Commun. 2004, 320, 1026–1033. [Google Scholar] [CrossRef]
- Park, K.A.; Byun, H.S.; Won, M.; Yang, K.J.; Shin, S.; Piao, L.; Kim, J.M.; Yoon, W.H.; Junn, E.; Park, J.; et al. Sustained activation of protein kinase C downregulates nuclear factor-κB signaling by dissociation of IKK-γ and Hsp90 complex in human colonic epithelial cells. Carcinogenesis 2007, 28, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Baeuerle, P.A.; Baltimore, D. NF-κB: Ten years after. Cell 1996, 87, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.H.; Chang, F.Y. Development and characterization of eucalyptol microemulsions for topic delivery of curcumin. Chem. Pharm. Bull. 2011, 59, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zou, L.Q.; Niu, J.; Liu, W.; Peng, S.F.; Liu, C.M. The stability, sustained release and cellular antioxidant activity of curcumin nanoliposomes. Molecules 2015, 20, 14293–14311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Jung, Y.H.; Oh, S.Y.; Song, E.J.; Choi, S.H.; Han, H.J. Vibrio vulnificus VvhA induces NF-κB-dependent mitochondrial cell death via lipid raft-mediated ROS production in intestinal epithelial cells. Cell Death Dis. 2015, 6, 1655. [Google Scholar] [CrossRef] [Green Version]
- Kaul, S.; Blackford, J.A., Jr.; Cho, S.; Simons, S.S., Jr. Ubc9 is a novel modulator of the induction properties of glucocorticoid receptors. J. Biol. Chem. 2002, 277, 12541–12549. [Google Scholar] [CrossRef] [Green Version]
- Crane, J.K.; Vezina, C.M. Externalization of host cell protein kinase C during enteropathogenic Escherichia coli infection. Cell Death Differ. 2005, 12, 115–127. [Google Scholar] [CrossRef]
- Monturiol-Gross, L.; Flores-Diaz, M.; Pineda-Padilla, M.J.; Castro-Castro, A.C.; Alape-Giron, A. Clostridium perfringens phospholipase C induced ROS production and cytotoxicity require PKC, MEK1 and NF-κB activation. PLoS ONE 2014, 9, e86475. [Google Scholar] [CrossRef]
- Li, J.D.; Feng, W.; Gallup, M.; Kim, J.H.; Gum, J.; Kim, Y.; Basbaum, C. Activation of NF-κB via a Src-dependent Ras-MAPK-pp90rsk pathway is required for Pseudomonas aeruginosa-induced mucin overproduction in epithelial cells. Proc. Natl. Acad. Sci. USA 1998, 95, 5718–5723. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.C.; Wu, M.L.; Huang, K.C.; Lin, W.W. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc. Res. 2008, 80, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Ki, M.R.; Lee, H.R.; Goo, M.J.; Hong, I.H.; Do, S.H.; Jeong, D.H.; Yang, H.J.; Yuan, D.W.; Park, J.K.; Jeong, K.S. Differential regulation of ERK1/2 and p38 MAP kinases in VacA-induced apoptosis of gastric epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G635–G647. [Google Scholar] [CrossRef]
- Kohchi, C.; Inagawa, H.; Nishizawa, T.; Soma, G. ROS and innate immunity. Anticancer Res. 2009, 29, 817–821. [Google Scholar] [PubMed]
- Rahman, M.M.; McFadden, G. Modulation of NF-κB signalling by microbial pathogens. Nat. Rev. Microbiol. 2011, 9, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Olson, D.J.; Sharma, T.; Vadlamudi, R.K.; Kumar, R. Butyric acid induces apoptosis by up-regulating Bax expression via stimulation of the c-Jun n-terminal kinase/activation protein-1 pathway in human colon cancer cells. Gastroenterology 2001, 120, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, J.C.; Arnoult, D.; Youle, R.J. Control of mitochondrial permeability by Bcl-2 family members. Biochim. Biophys. Acta 2004, 1644, 107–113. [Google Scholar] [CrossRef]
- Hildebrand, A.; Pohl, M.; Bhakdi, S. Staphylococcus aureus α-toxin. Dual mechanism of binding to target cells. J. Biol. Chem. 1991, 266, 17195–17200. [Google Scholar]
- Nougayrede, J.P.; Donnenberg, M.S. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell. Microbiol. 2004, 6, 1097–1111. [Google Scholar] [CrossRef]
- Jeong, T.C.; Kim, H.J.; Park, J.I.; Ha, C.S.; Park, J.D.; Kim, S.I.; Roh, J.K. Protective effects of red ginseng saponins against carbon tetrachloride-induced hepatotoxicity in Sprague Dawley rats. Planta Med. 1997, 63, 136–140. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain or Plasmid | Relevant Haracteristics a | Reference or Source |
|---|---|---|
| Bacterial strains | ||
| V. vulnificus | ||
| M06-24/O | Clinical isolate; virulent; WT | Laboratory collection |
| CMM111 | M06-24/O vvhA:Pks1201; elastase deficient; vvhA mutant | [15] |
| E. coli | ||
| BL21 (DE3) | F− ompT hsdSB (rB−mB−) gal dcm (DE3) | Laboratory collection |
| Plasmids | ||
| pET29a (+) | His6 tag fusion expression vector; Kmr | Novagen |
| pKS1201 | pET29a (+) with VvhBA; Kmr | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-Y.; Lee, Y.-M.; Kim, D.-W.; Min, T.; Lee, S.-J. Nanosphere Loaded With Curcumin Inhibits the Gastrointestinal Cell Death Signaling Pathway Induced by the Foodborne Pathogen Vibrio vulnificus. Cells 2020, 9, 631. https://doi.org/10.3390/cells9030631
Kim J-Y, Lee Y-M, Kim D-W, Min T, Lee S-J. Nanosphere Loaded With Curcumin Inhibits the Gastrointestinal Cell Death Signaling Pathway Induced by the Foodborne Pathogen Vibrio vulnificus. Cells. 2020; 9(3):631. https://doi.org/10.3390/cells9030631
Chicago/Turabian StyleKim, Ji-Yun, Young-Min Lee, Do-Wan Kim, Taesun Min, and Sei-Jung Lee. 2020. "Nanosphere Loaded With Curcumin Inhibits the Gastrointestinal Cell Death Signaling Pathway Induced by the Foodborne Pathogen Vibrio vulnificus" Cells 9, no. 3: 631. https://doi.org/10.3390/cells9030631