Activation of Astroglial Connexin Is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Agents
2.2. Preparation of Primary Astrocyte Culture
2.3. Study Designs
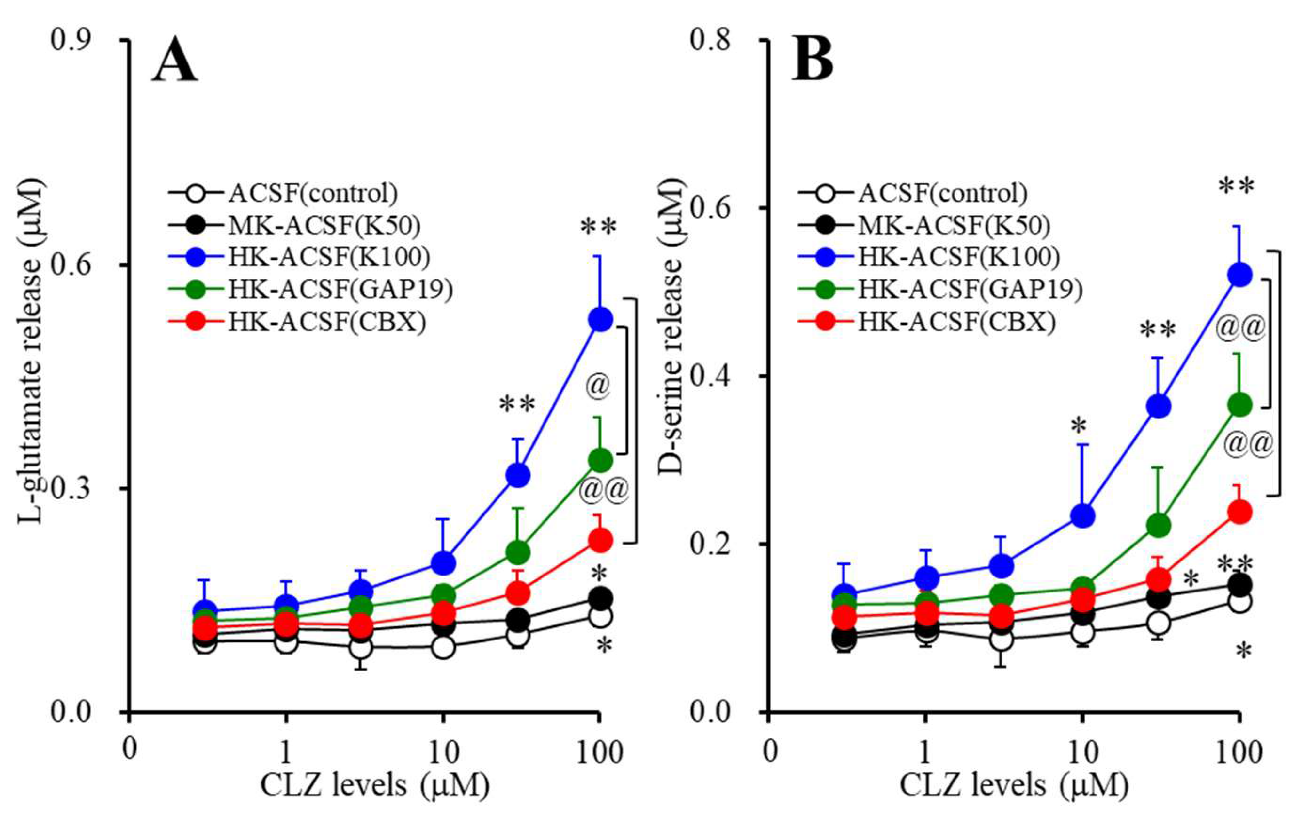
2.3.1. Concentration-Dependent Effects of Acute Administration of CLZ on Astroglial Releases of l-Glutamate and D-serine (Study_1)
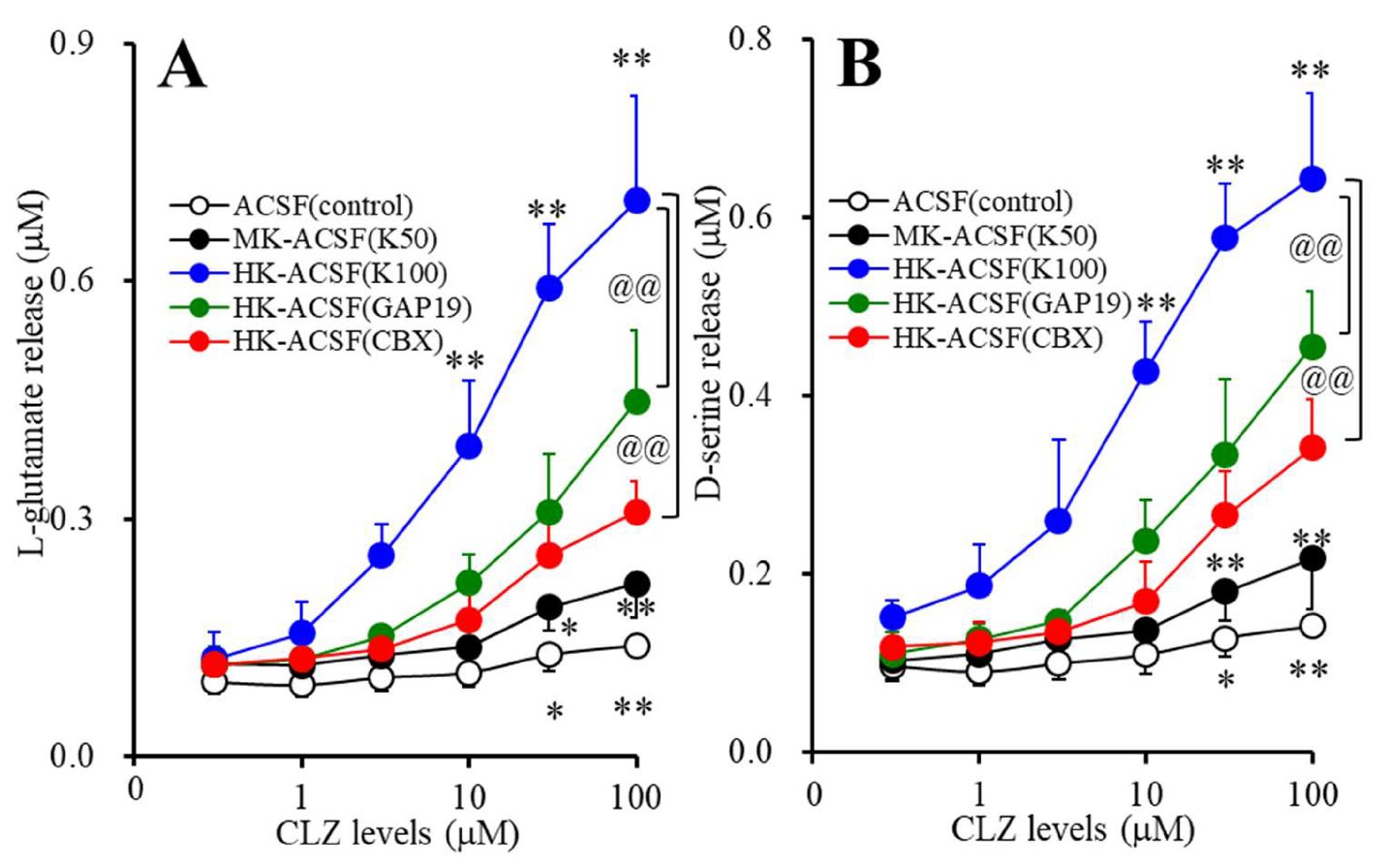
2.3.2. Concentration-Dependent Effects of Subchronic Administration of CLZ on Astroglial Releases of l-glutamate and d-serine (Study_2)
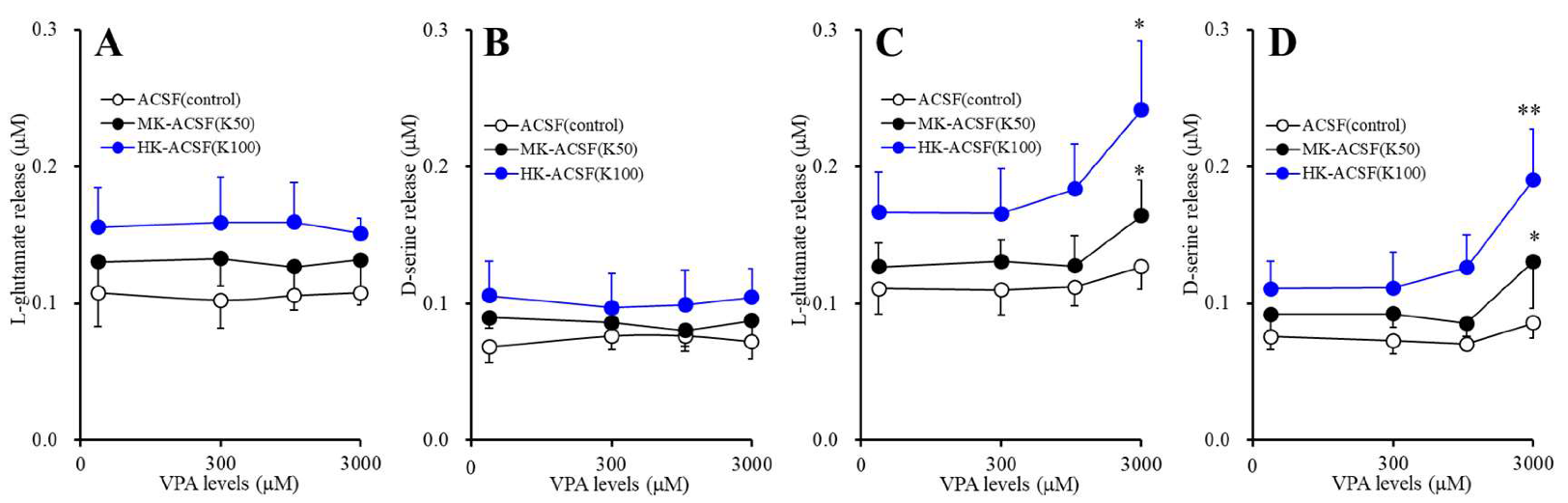
2.3.3. Concentration-Dependent Effects of Acute and Subchronic Administration of VPA on Astroglial Releases of l-glutamate and d-serine (Study_3)
2.3.4. Acute Effects of Therapeutic-Relevant Concentration of VPA on Astroglial Releases of l-glutamate and d-serine from Astrocytes Subchronically Administrated with CLZ (Study_4)
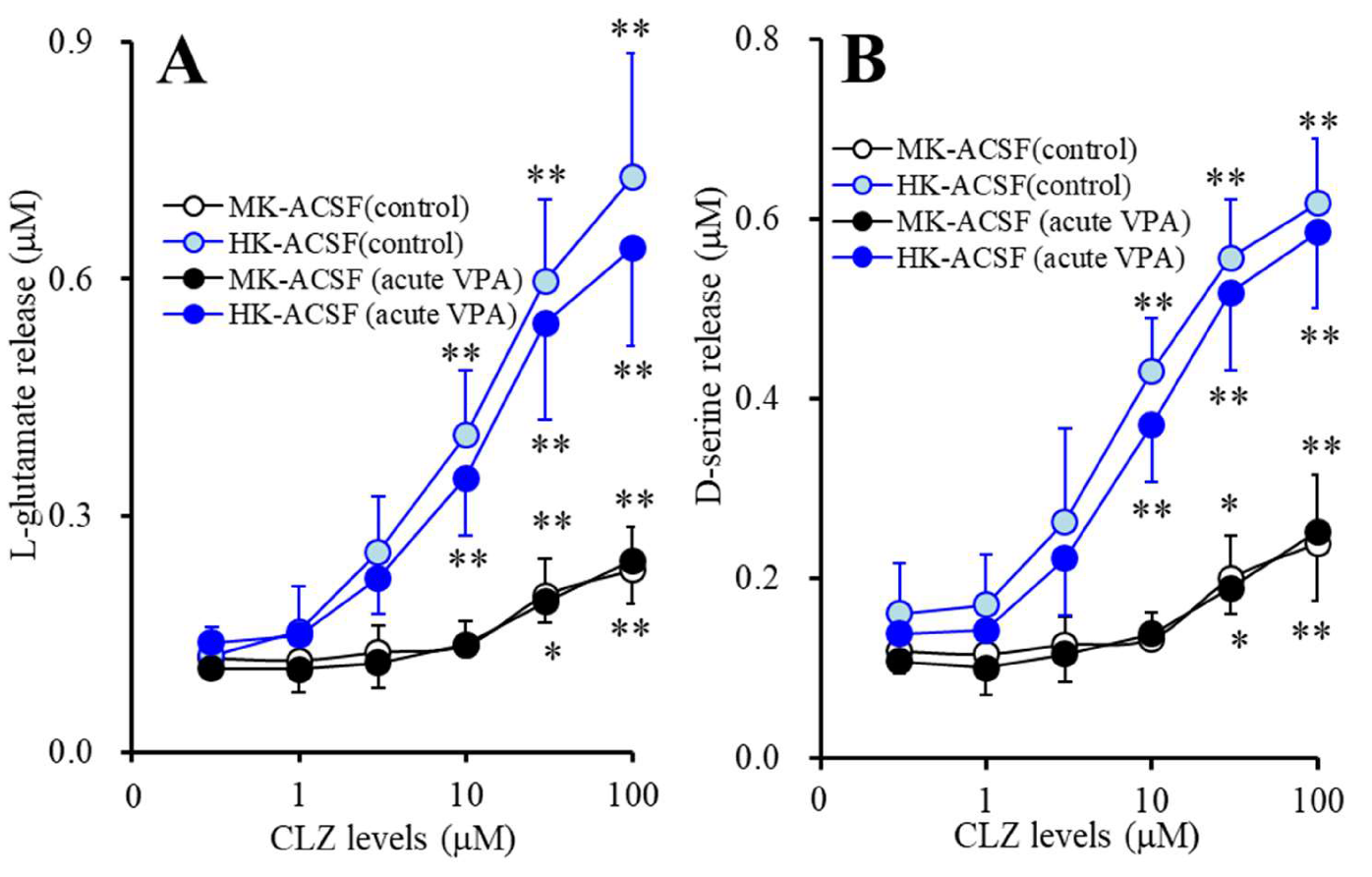
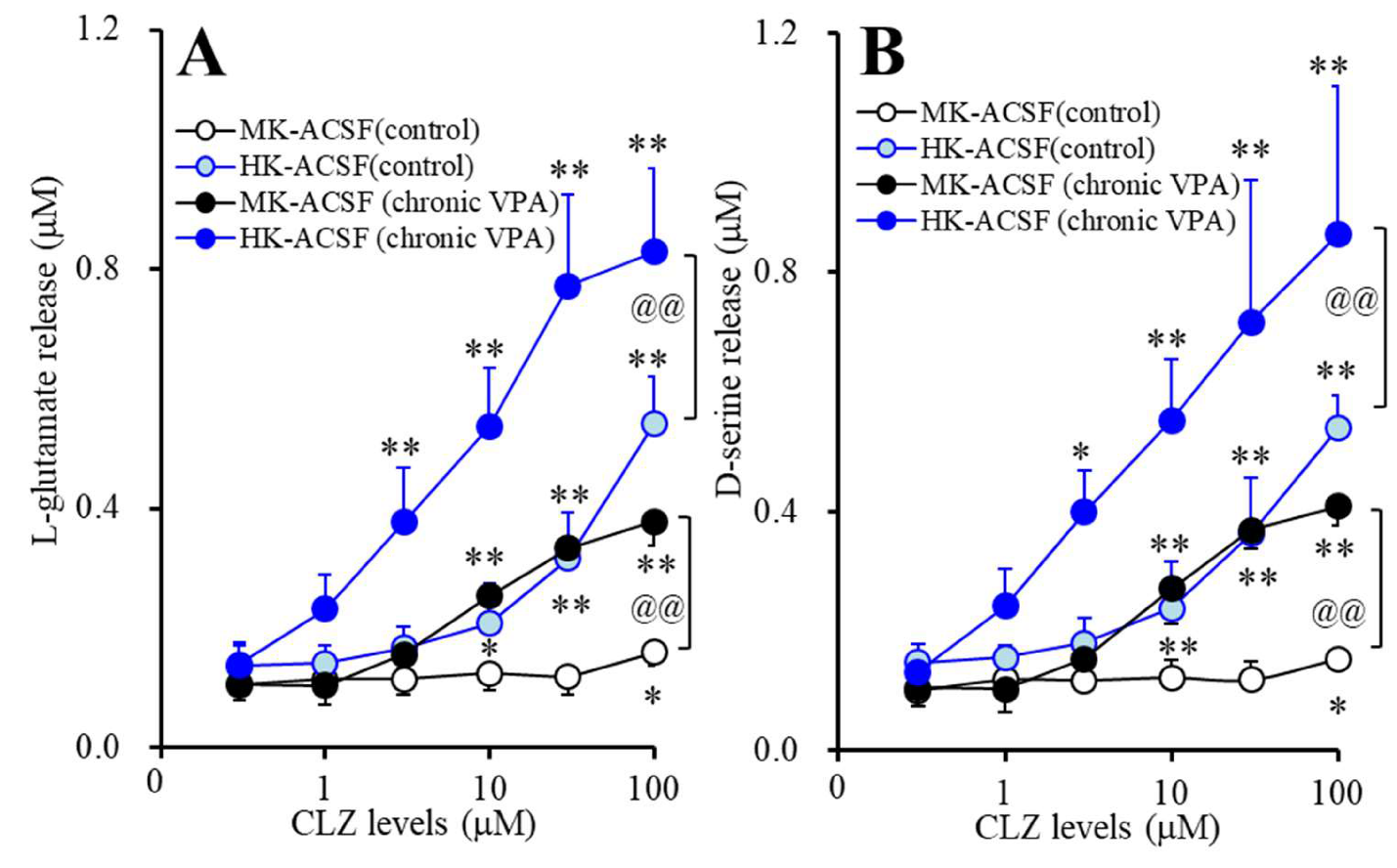
2.3.5. Acute Effects of CLZ on Astroglial Releases of l-glutamate and d-serine from Astrocytes Subchronically Administrated with VPA (Study_5)
2.4. Determination of Levels of l-glutamate and GABA
2.5. Simple Western Analysis
2.6. Statistical Analysis
3. Results
3.1. Concentration-Dependent Effects of Acute Administration of CLZ on Astroglial Releases of L-glutamate and D-serine (Study_1)
3.2. Concentration-Dependent Effects of Subchronic Administration of CLZ on Astroglial Releases of L-glutamate and D-serine (Study_2)
3.3. Concentration-Dependent Effects of Acute and Subchronic Administrations of VPA on Astroglial Releases of L-glutamate and D-serine (Study_3)
3.4. Acute Effects of Therapeutic-Relevant Concentration of VPA on Astroglial Releases of l-glutamate and d-serine from Astrocytes Subchronically Administrated with CLZ (Study_4)
3.5. Acute Effects of CLZ on Astroglial Releases of L-glutamate and D-serine from Astrocytes Subchronically Administrated with Therapeutic-Relevant Concentration of VPA (Study_5)
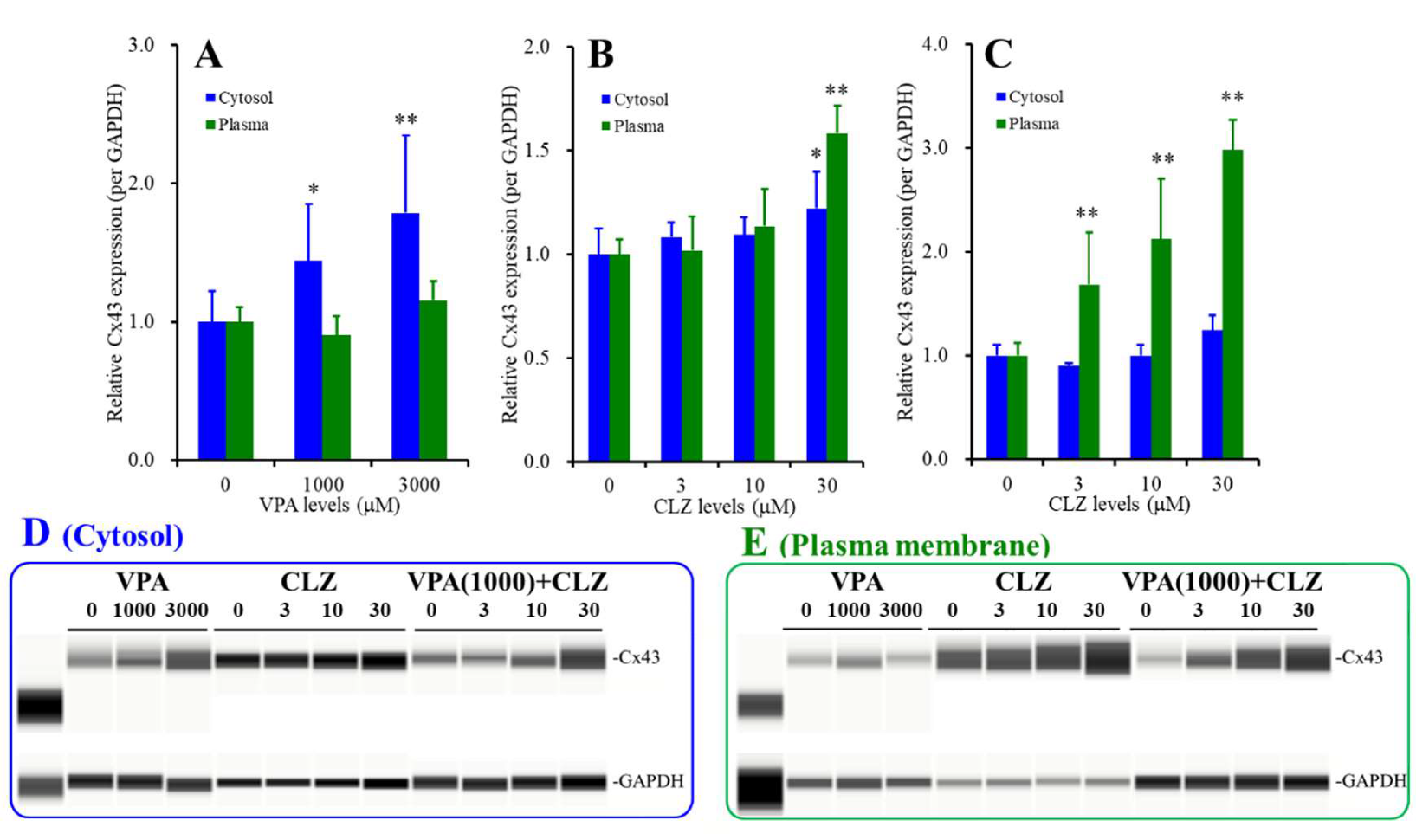
3.6. Interaction between VPA and CLZ on Cx43 Expression in Astrocytes (Studies_2,3,5)
4. Discussion
4.1. Effects of VPA and CLZ on Astroglial Transmission Associated with Cxs
4.2. Interaction between VPA and CLZ
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lieberman, J.A.; Bymaster, F.P.; Meltzer, H.Y.; Deutch, A.Y.; Duncan, G.E.; Marx, C.E.; Aprille, J.R.; Dwyer, D.S.; Li, X.M.; Mahadik, S.P.; et al. Antipsychotic drugs: Comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol. Rev. 2008, 60, 358–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meltzer, H.Y.; Huang, M. In vivo actions of atypical antipsychotic drug on serotonergic and dopaminergic systems. Prog. Brain Res. 2008, 172, 177–197. [Google Scholar] [PubMed]
- Okada, M.; Fukuyama, K.; Ueda, Y. Lurasidone inhibits NMDA receptor antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone Sub-Chronically Activates Serotonergic Transmission via Desensitization of 5-HT1A and 5-HT7 Receptors in Dorsal Raphe Nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc. Pharmacol. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [Green Version]
- Mitterauer, B. Loss of function of glial gap junctions may cause severe cognitive impairments in schizophrenia. Med. Hypotheses 2009, 73, 393–397. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Nakano, T.; Ueda, Y. Pharmacological Discrimination of Effects of MK801 on Thalamocortical, Mesothalamic, and Mesocortical Transmissions. Biomolecules 2019, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Javitt, D.C. Glutamate and schizophrenia: Phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar]
- Labrie, V.; Roder, J.C. The involvement of the NMDA receptor D-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci. Biobehav. Rev. 2010, 34, 351–372. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br. J. Pharmacol. 2018, 175, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, A.K.; Pinals, D.A.; Weingartner, H.; Sirocco, K.; Missar, C.D.; Pickar, D.; Breier, A. NMDA receptor function and human cognition: The effects of ketamine in healthy volunteers. Neuropsychopharmacology 1996, 14, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Pinals, D.A.; Adler, C.M.; Elman, I.; Clifton, A.; Pickar, D.; Breier, A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 1997, 17, 141–150. [Google Scholar] [CrossRef]
- Krystal, J.H.; D’Souza, D.C.; Mathalon, D.; Perry, E.; Belger, A.; Hoffman, R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: Toward a paradigm shift in medication development. Psychopharmacology 2003, 169, 215–233. [Google Scholar] [CrossRef]
- Farooq, S.; Choudry, A.; Cohen, D.; Naeem, F.; Ayub, M. Barriers to using clozapine in treatment-resistant schizophrenia: Systematic review. BJPsych Bull. 2019, 43, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.; Hazari, N.; Chakrabarti, S.; Avasthi, A. Association of Clozapine with Seizures: A Brief Report Involving 222 Patients Prescribed Clozapine. East Asian Arch. Psychiatry 2015, 25, 73–78. [Google Scholar]
- Farooq, S.; Taylor, M. Clozapine: Dangerous orphan or neglected friend? Br. J. Psychiatry 2011, 198, 247–249. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Xiang, Y.T.; Yang, X.H.; Xiang, Y.Q.; de Leon, J. Clozapine Augmentation With Antiepileptic Drugs for Treatment-Resistant Schizophrenia: A Meta-Analysis of Randomized Controlled Trials. J. Clin. Psychiatry 2017, 78, e498–e505. [Google Scholar] [CrossRef]
- David, M.; Taylor, T.R.E.B.; Young, A.H. The Maudsley Prescribing Guidelines in Psychiatry, 13th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2018. [Google Scholar]
- Ronaldson, K.J.; Fitzgerald, P.B.; Taylor, A.J.; Topliss, D.J.; Wolfe, R.; McNeil, J.J. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: A case-control study. Schizophrenia Res. 2012, 141, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Siskind, D.; McCartney, L.; Goldschlager, R.; Kisely, S. Clozapine v. first- and second-generation antipsychotics in treatment-refractory schizophrenia: Systematic review and meta-analysis. Br. J. Psychiatry 2016, 209, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, A.K.; Adler, C.M.; Kennison, S.D.; Elman, I.; Pickar, D.; Breier, A. Clozapine blunts N-methyl-D-aspartate antagonist-induced psychosis: A study with ketamine. Biol. Psychiatry 1997, 42, 664–668. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Lapato, A.S.; Tiwari-Woodruff, S.K. Connexins and pannexins: At the junction of neuro-glial homeostasis & disease. J. Neurosci. Res. 2018, 96, 31–44. [Google Scholar]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Liu, Z.Q.; Zhou, H.H.; Mao, X.Y. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed. Pharmacother. 2019, 109, 57–65. [Google Scholar] [CrossRef]
- Medina-Ceja, L.; Salazar-Sanchez, J.C.; Ortega-Ibarra, J.; Morales-Villagran, A. Connexins-Based Hemichannels/Channels and Their Relationship with Inflammation, Seizures and Epilepsy. Int. J. Mol. Sci. 2019, 20, 5976. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girao, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [Green Version]
- Dallerac, G.; Rouach, N. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef]
- Zhong, C.; Chang, H.; Wu, Y.; Zhou, L.; Wang, Y.; Wang, M.; Wu, P.; Qi, Z.; Zou, J. Up-regulated Cx43 phosphorylation at Ser368 prolongs QRS duration in myocarditis. J. Cell. Mol. Med. 2018, 22, 3537–3547. [Google Scholar] [CrossRef]
- Mylvaganam, S.; Ramani, M.; Krawczyk, M.; Carlen, P.L. Roles of gap junctions, connexins, and pannexins in epilepsy. Front. Physiol. 2014, 5, 172. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.M.; Wang, G.L.; Miao, J.; Feng, J.C. Effect of connexin 36 blockers on the neuronal cytoskeleton and synaptic plasticity in kainic acid-kindled rats. Transl. Neurosci. 2015, 6, 252–258. [Google Scholar] [CrossRef]
- Jin, M.; Dai, Y.; Xu, C.; Wang, Y.; Wang, S.; Chen, Z. Effects of meclofenamic acid on limbic epileptogenesis in mice kindling models. Neurosci. Lett. 2013, 543, 110–114. [Google Scholar] [CrossRef]
- Xiang, Y.Q.; Zhang, Z.J.; Weng, Y.Z.; Zhai, Y.M.; Li, W.B.; Cai, Z.J.; Tan, Q.R.; Wang, C.Y. Serum concentrations of clozapine and norclozapine in the prediction of relapse of patients with schizophrenia. Schizophrenia Res. 2006, 83, 201–210. [Google Scholar] [CrossRef]
- Varma, S.; Bishara, D.; Besag, F.M.; Taylor, D. Clozapine-related EEG changes and seizures: Dose and plasma-level relationships. Ther. Adv. Psychopharmacol. 2011, 1, 47–66. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Bultynck, G.; Leybaert, L. Connexin targeting peptides as inhibitors of voltage- and intracellular Ca2+-triggered Cx43 hemichannel opening. Neuropharmacology 2013, 75, 506–516. [Google Scholar] [CrossRef]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor. Int. J. Mol. Sci. 2019, 20, 3727. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine Combines Astroglial System Xc(-) Activation with Glutamate/NMDA Receptor Inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Tanahashi, S.; Hoshikawa, M.; Shinagawa, R.; Okada, M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology 2014, 76, 137–145. [Google Scholar] [CrossRef]
- Yamamura, S.; Hoshikawa, M.; Kato, D.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. ONO-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharmacol. 2013, 168, 1088–1100. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Dopamine D2 and serotonin 5-HT1A receptors mediate the actions of aripiprazole in mesocortical and mesoaccumbens transmission. Neuropharmacology 2012, 62, 765–774. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Nakagawa, M.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Tanii, H.; Shiroyama, T.; Okada, M. Effects of zotepine on extracellular levels of monoamine, GABA and glutamate in rat prefrontal cortex. Br. J. Pharmacol. 2009, 157, 656–665. [Google Scholar] [CrossRef]
- Yoshida, S.; Okada, M.; Zhu, G.; Kaneko, S. Carbamazepine prevents breakdown of neurotransmitter release induced by hyperactivation of ryanodine receptor. Neuropharmacology 2007, 52, 1538–1546. [Google Scholar] [CrossRef]
- Okada, M.; Yoshida, S.; Zhu, G.; Hirose, S.; Kaneko, S. Biphasic actions of topiramate on monoamine exocytosis associated with both soluble N-ethylmaleimide-sensitive factor attachment protein receptors and Ca(2+)-induced Ca(2+)-releasing systems. Neuroscience 2005, 134, 233–246. [Google Scholar] [CrossRef]
- Yoshida, S.; Yamamura, S.; Ohoyama, K.; Nakagawa, M.; Motomura, E.; Kaneko, S.; Okada, M. Effects of valproate on neurotransmission associated with ryanodine receptors. Neurosci. Res. 2010, 68, 322–328. [Google Scholar] [CrossRef]
- Zancan, M.; Malysz, T.; Moura, D.J.; Moras, A.M.; Steffens, L.; Rasia-Filho, A.A. Gap junctions and expression of Cx36, Cx43 and Cx45 in the posterodorsal medial amygdala of adult rats. Histol. Histopathol. 2019. [Google Scholar] [CrossRef]
- Flores, C.E.; Nannapaneni, S.; Davidson, K.G.; Yasumura, T.; Bennett, M.V.; Rash, J.E.; Pereda, A.E. Trafficking of gap junction channels at a vertebrate electrical synapse in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E573–E582. [Google Scholar] [CrossRef] [Green Version]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 2013, 1828, 118–133. [Google Scholar] [CrossRef] [Green Version]
- Fessler, E.B.; Chibane, F.L.; Wang, Z.; Chuang, D.M. Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery. Curr. Pharm. Des. 2013, 19, 5105–5120. [Google Scholar] [CrossRef]
- Hernandez, M.; Shao, Q.; Yang, X.J.; Luh, S.P.; Kandouz, M.; Batist, G.; Laird, D.W.; Alaoui-Jamali, M.A. A histone deacetylation-dependent mechanism for transcriptional repression of the gap junction gene cx43 in prostate cancer cells. Prostate 2006, 66, 1151–1161. [Google Scholar] [CrossRef]
- Ogawa, T.; Hayashi, T.; Tokunou, M.; Nakachi, K.; Trosko, J.E.; Chang, C.C.; Yorioka, N. Suberoylanilide hydroxamic acid enhances gap junctional intercellular communication via acetylation of histone containing connexin 43 gene locus. Cancer Res. 2005, 65, 9771–9778. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.; Akhtar, M.; Asklund, T.; Juliusson, B.; Almqvist, P.M.; Ekstrom, T.J. HDAC inhibition amplifies gap junction communication in neural progenitors: Potential for cell-mediated enzyme prodrug therapy. Exp. Cell Res. 2007, 313, 2958–2967. [Google Scholar] [CrossRef]
- Dambach, H.; Hinkerohe, D.; Prochnow, N.; Stienen, M.N.; Moinfar, Z.; Haase, C.G.; Hufnagel, A.; Faustmann, P.M. Glia and epilepsy: Experimental investigation of antiepileptic drugs in an astroglia/microglia co-culture model of inflammation. Epilepsia 2014, 55, 184–192. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Pandian, T.; Braun, N.N.; Haug, K. Chronic psychotropic drug treatment causes differential expression of connexin 43 and GFAP in frontal cortex of rats. Schizophrenia Res. 2008, 104, 127–134. [Google Scholar] [CrossRef]
- de la Fuente Revenga, M.; Ibi, D.; Cuddy, T.; Toneatti, R.; Kurita, M.; Ijaz, M.K.; Miles, M.F.; Wolstenholme, J.T.; Gonzalez-Maeso, J. Chronic clozapine treatment restrains via HDAC2 the performance of mGlu2 receptor agonism in a rodent model of antipsychotic activity. Neuropsychopharmacology 2019, 44, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Sutton, L.P.; Honardoust, D.; Mouyal, J.; Rajakumar, N.; Rushlow, W.J. Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J. Neurochem. 2007, 102, 153–169. [Google Scholar] [CrossRef]
- Pinacho, R.; Valdizan, E.M.; Pilar-Cuellar, F.; Prades, R.; Tarrago, T.; Haro, J.M.; Ferrer, I.; Ramos, B. Increased SP4 and SP1 transcription factor expression in the postmortem hippocampus of chronic schizophrenia. J. Psychiatr. Res. 2014, 58, 189–196. [Google Scholar] [CrossRef]
- Kontkanen, O.; Lakso, M.; Wong, G.; Castren, E. Chronic antipsychotic drug treatment induces long-lasting expression of fos and jun family genes and activator protein 1 complex in the rat prefrontal cortex. Neuropsychopharmacology 2002, 27, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Girao, H.; Catarino, S.; Pereira, P. Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp. Cell Res. 2009, 315, 3587–3597. [Google Scholar] [CrossRef]
- Chever, O.; Lee, C.Y.; Rouach, N. Astroglial connexin43 hemichannels tune basal excitatory synaptic transmission. J. Neurosci. 2014, 34, 11228–11232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontes, M.S.; van Veen, T.A.; de Bakker, J.M.; van Rijen, H.V. Functional consequences of abnormal Cx43 expression in the heart. Biochim. Biophys. Acta 2012, 1818, 2020–2029. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of Astroglial Connexin Is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine. Cells 2020, 9, 414. https://doi.org/10.3390/cells9020414
Fukuyama K, Okubo R, Murata M, Shiroyama T, Okada M. Activation of Astroglial Connexin Is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine. Cells. 2020; 9(2):414. https://doi.org/10.3390/cells9020414
Chicago/Turabian StyleFukuyama, Kouji, Ruri Okubo, Masahiko Murata, Takashi Shiroyama, and Motohiro Okada. 2020. "Activation of Astroglial Connexin Is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine" Cells 9, no. 2: 414. https://doi.org/10.3390/cells9020414