Transcriptional Regulator TonEBP Mediates Oxidative Damages in Ischemic Kidney Injury

 , and
, and
Abstract
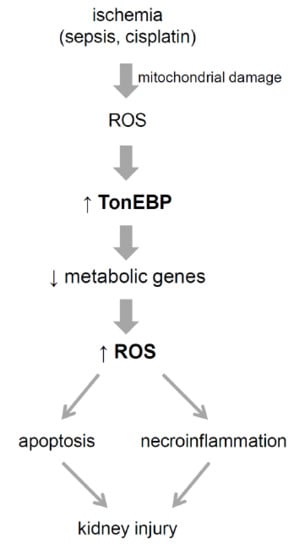
:
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Immunoblot Analysis
2.3. RNA Extraction and Quantitative Real-Time PCR
2.4. Histopathology Examination
2.5. Immunohistochemistry
2.6. In Situ TdT-Mediated dUTP-biotin Nick End Labeling Assay (TUNEL) Assay
2.7. Analyses of Blood and Urine
2.8. HK-2 Cells
2.9. Cell Viability Assay
2.10. Annexin-V/propidium Iodide (PI) Double Staining Assay
2.11. Microarray Analysis
2.12. Visualization of ROS
2.13. Statistical Analysis
3. Results
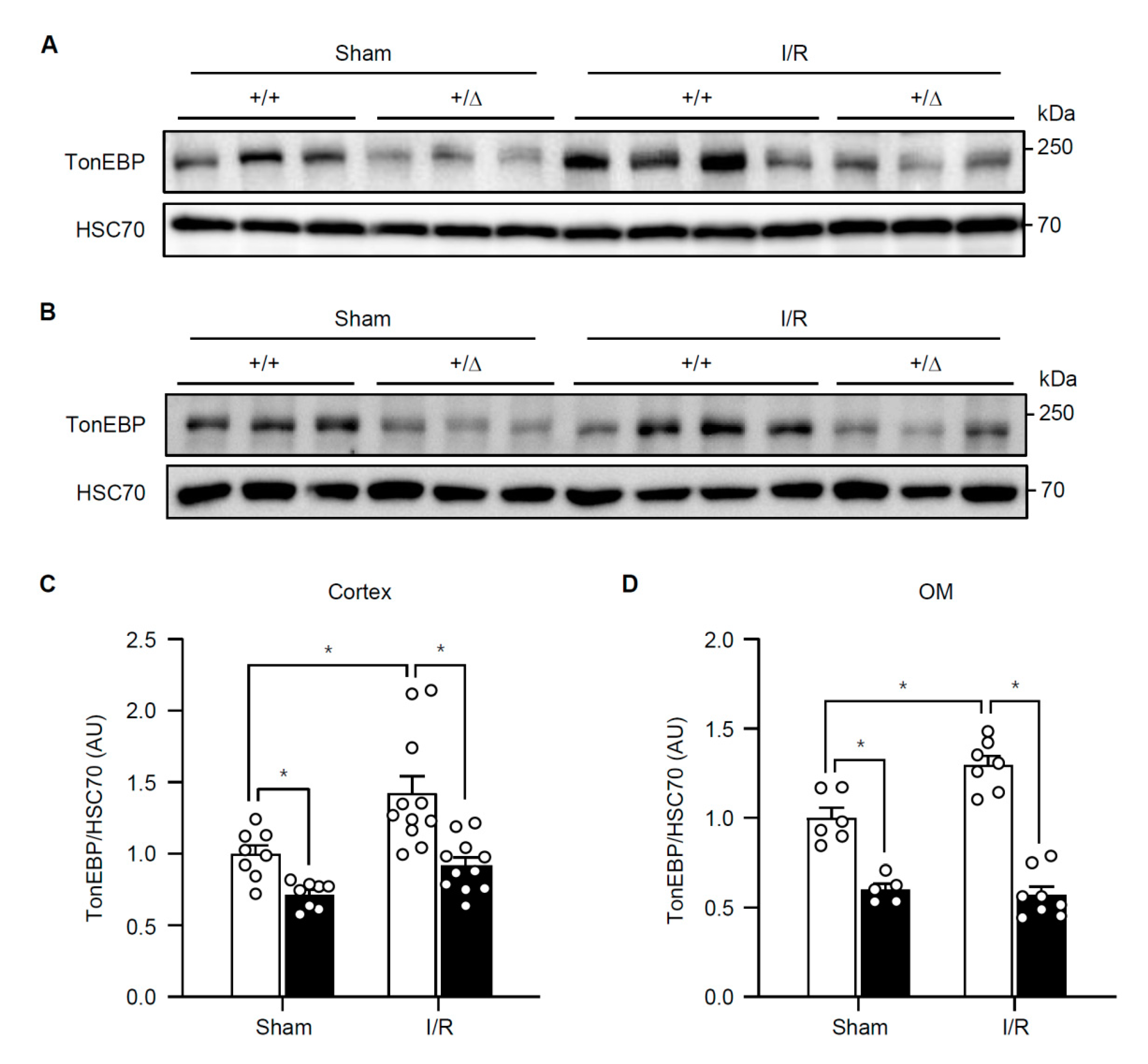
3.1. Renal TonEBP is Induced in Response to Ischemic Insult in Association with Inflammation and Tissue Injury
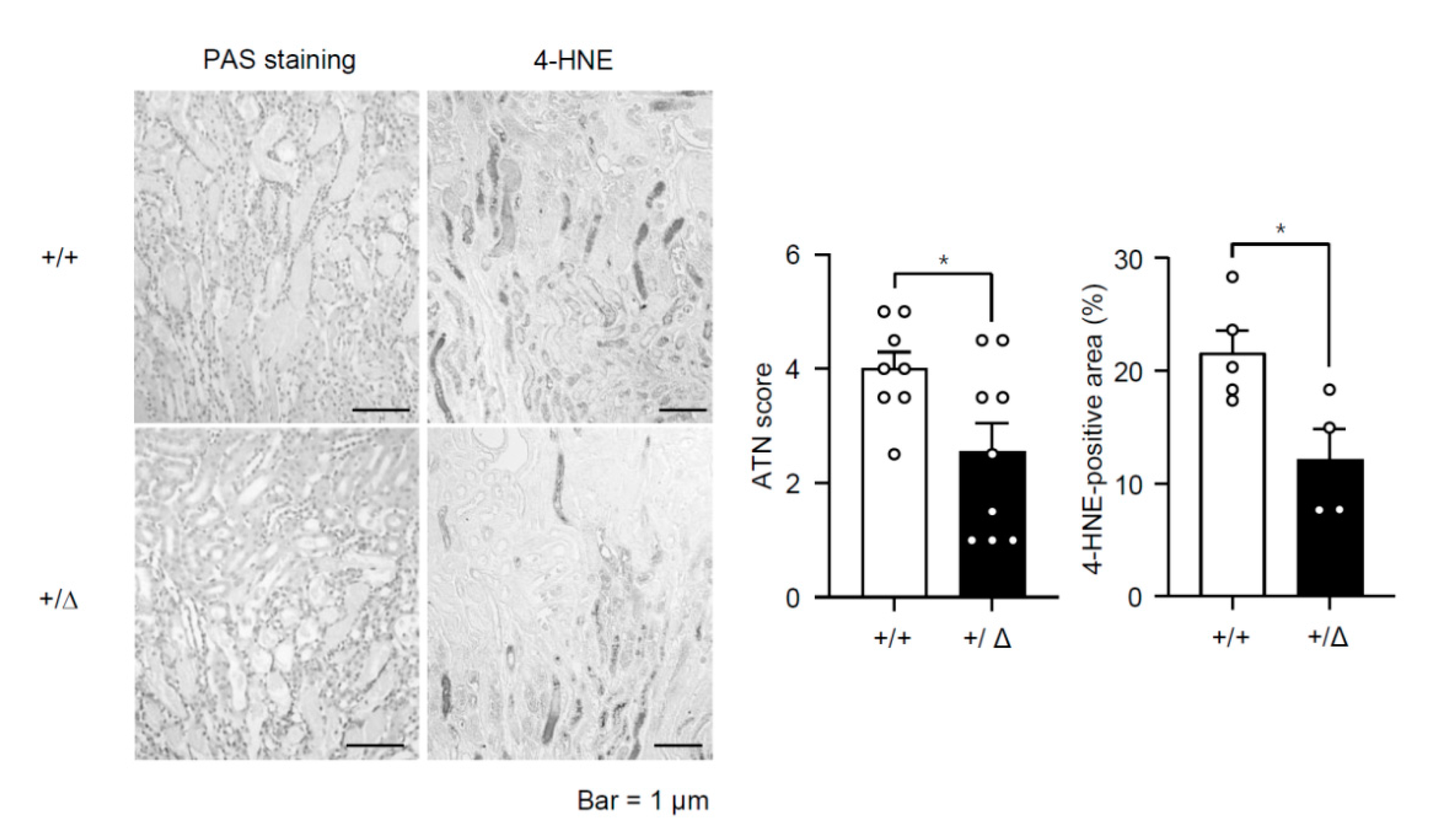
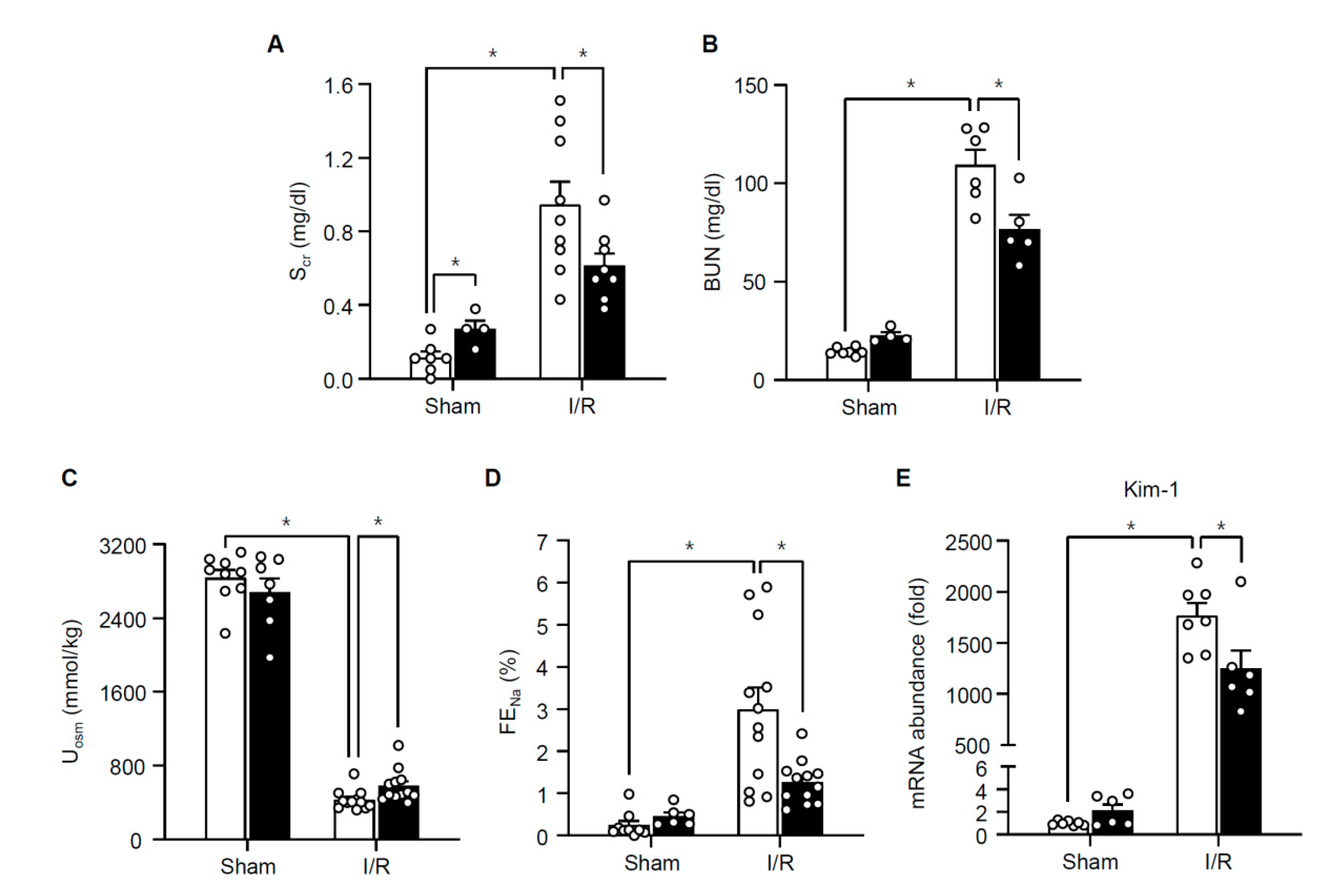
3.2. TonEBP Deficiency Protects Kidneys from Ischemia-Induced Inflammation and Tissue Injury
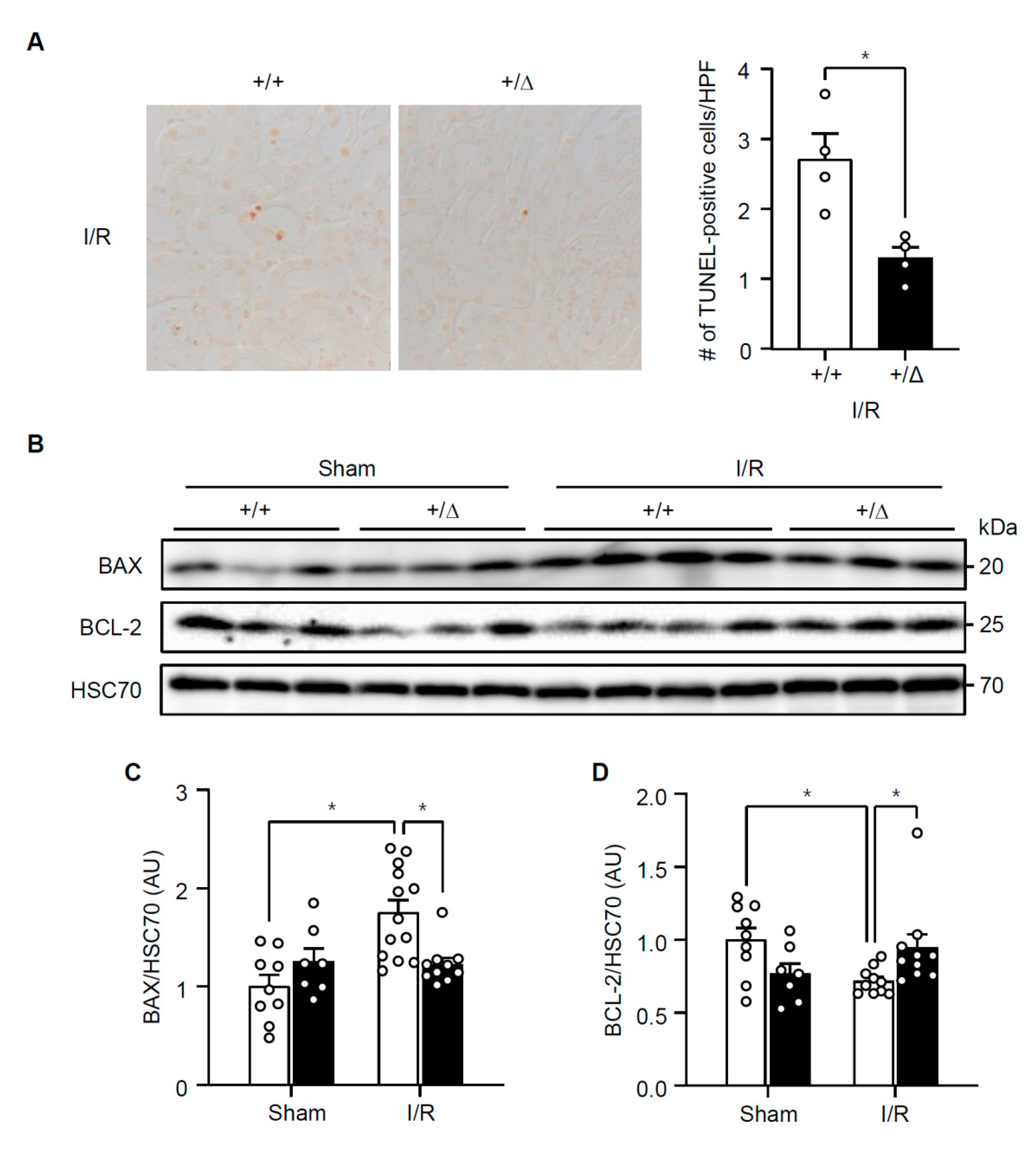
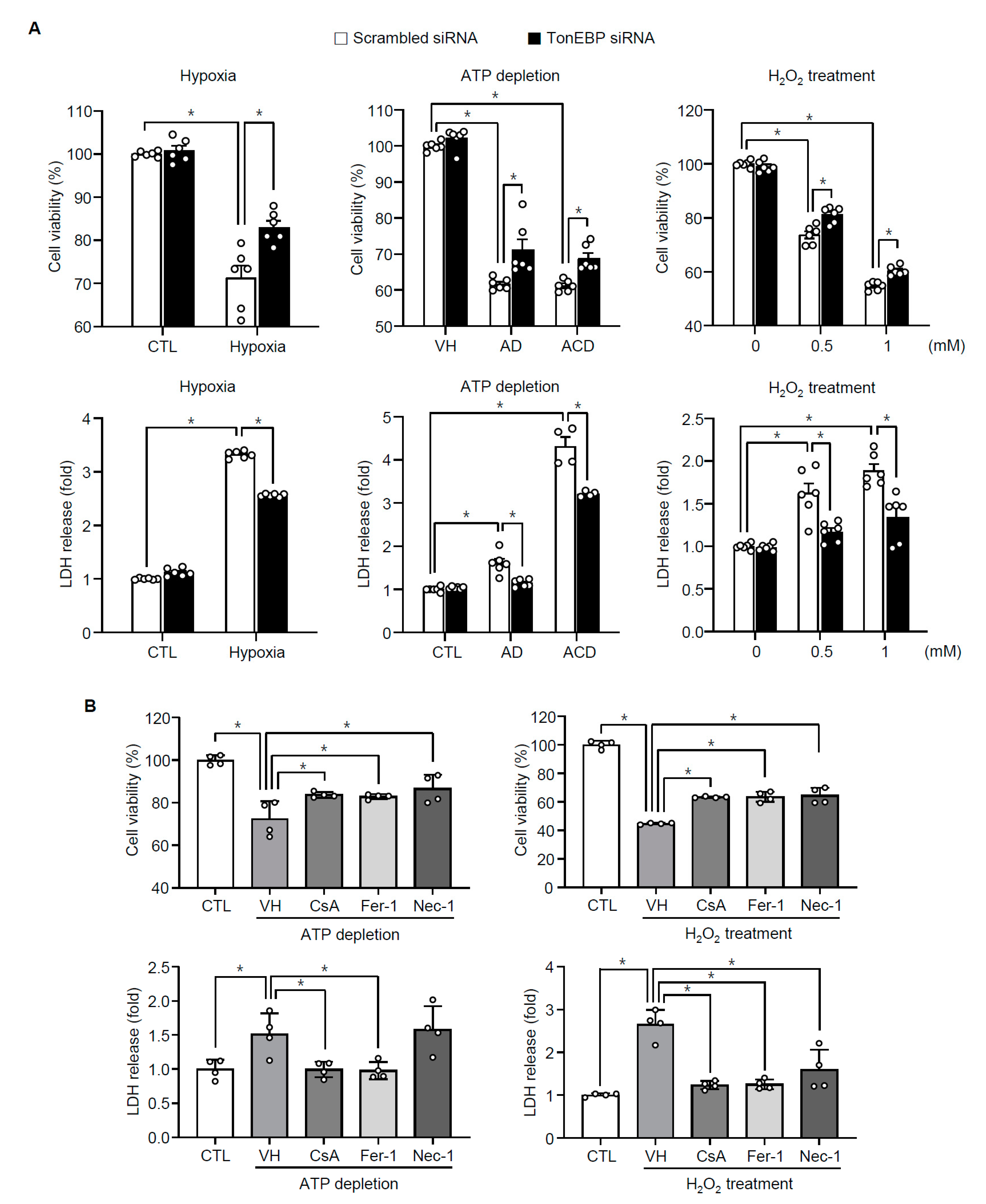
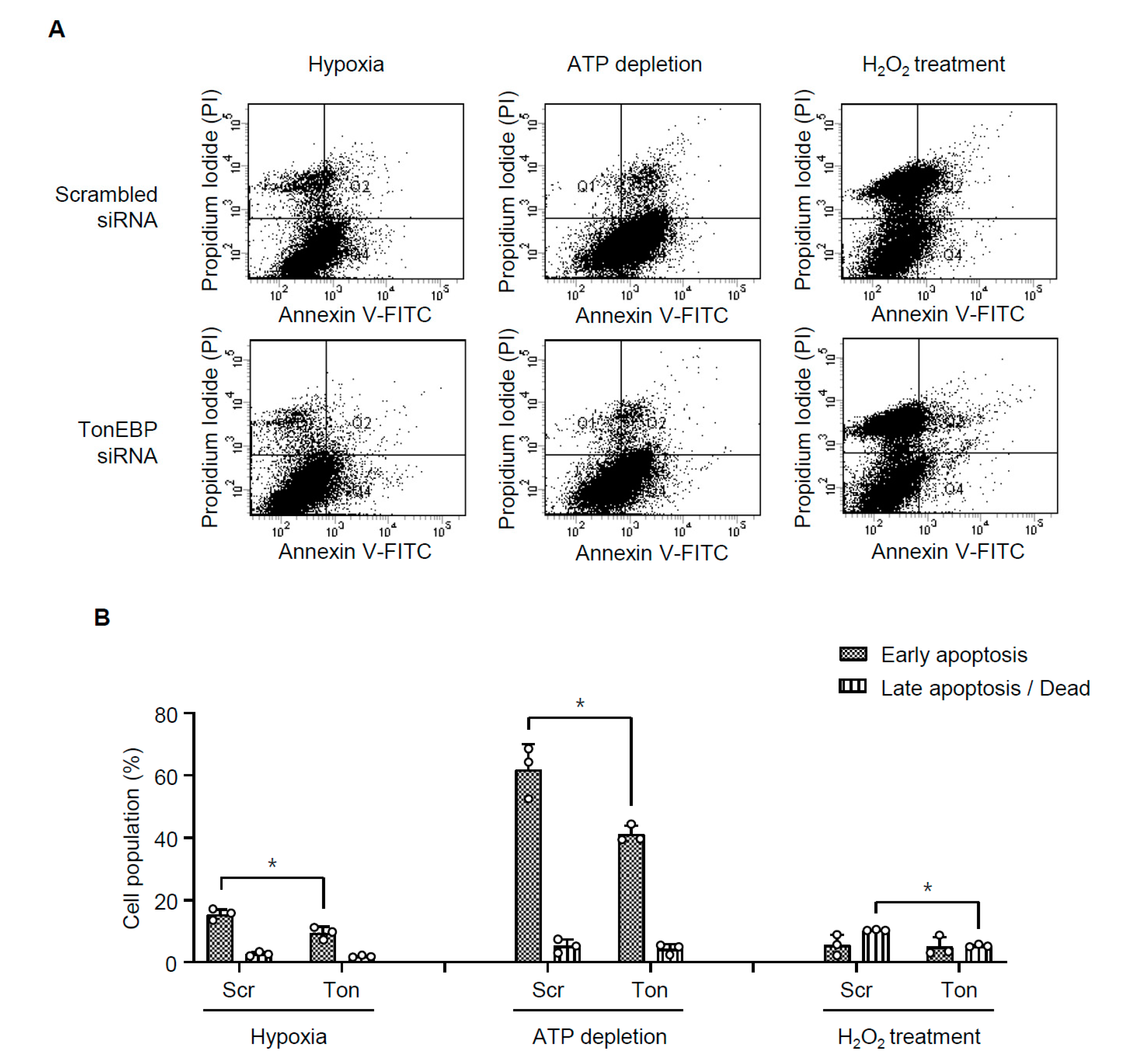
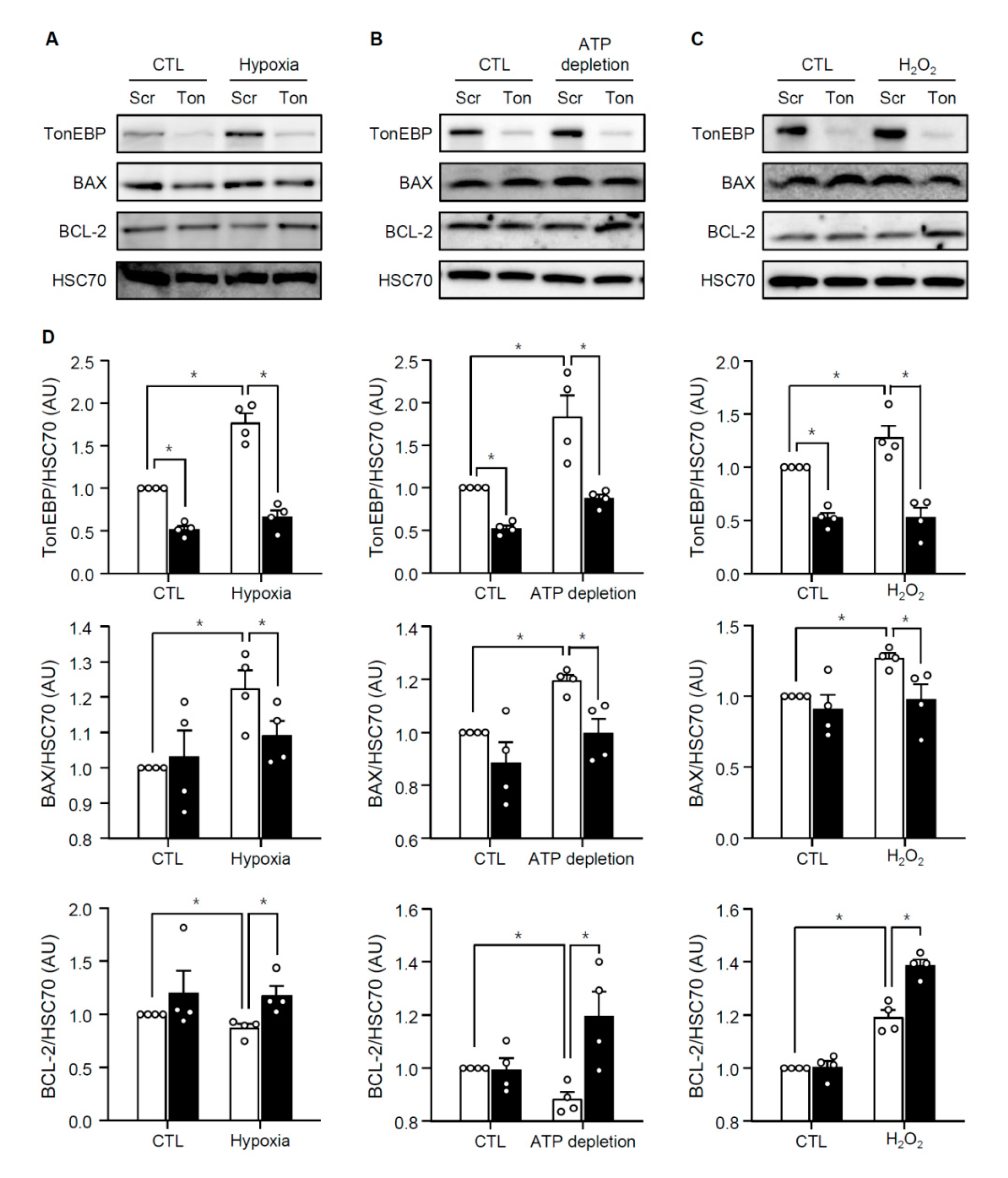
3.3. TonEBP Mediates Renal Tubular Cell Death in Response to Ischemic Insult
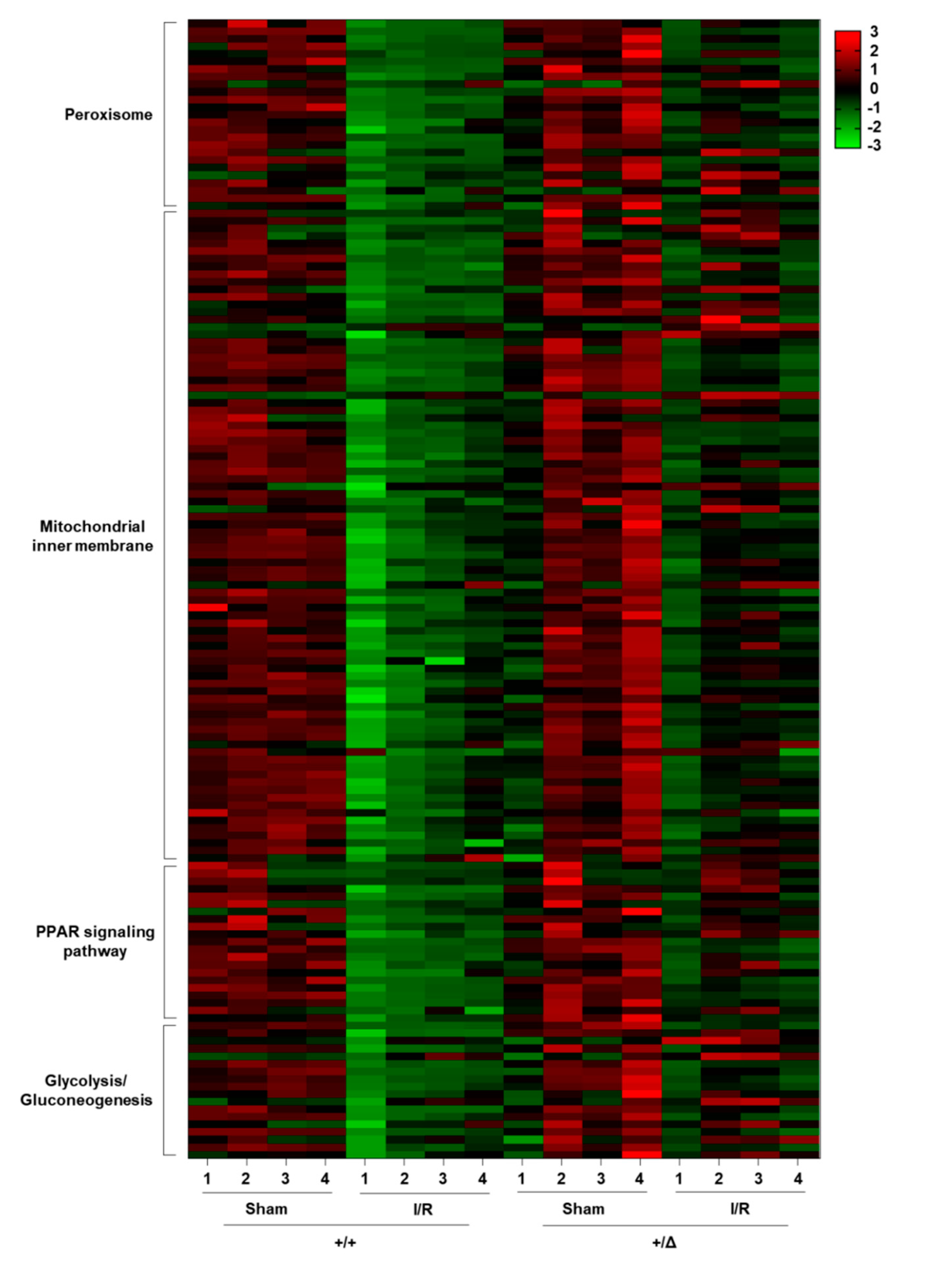
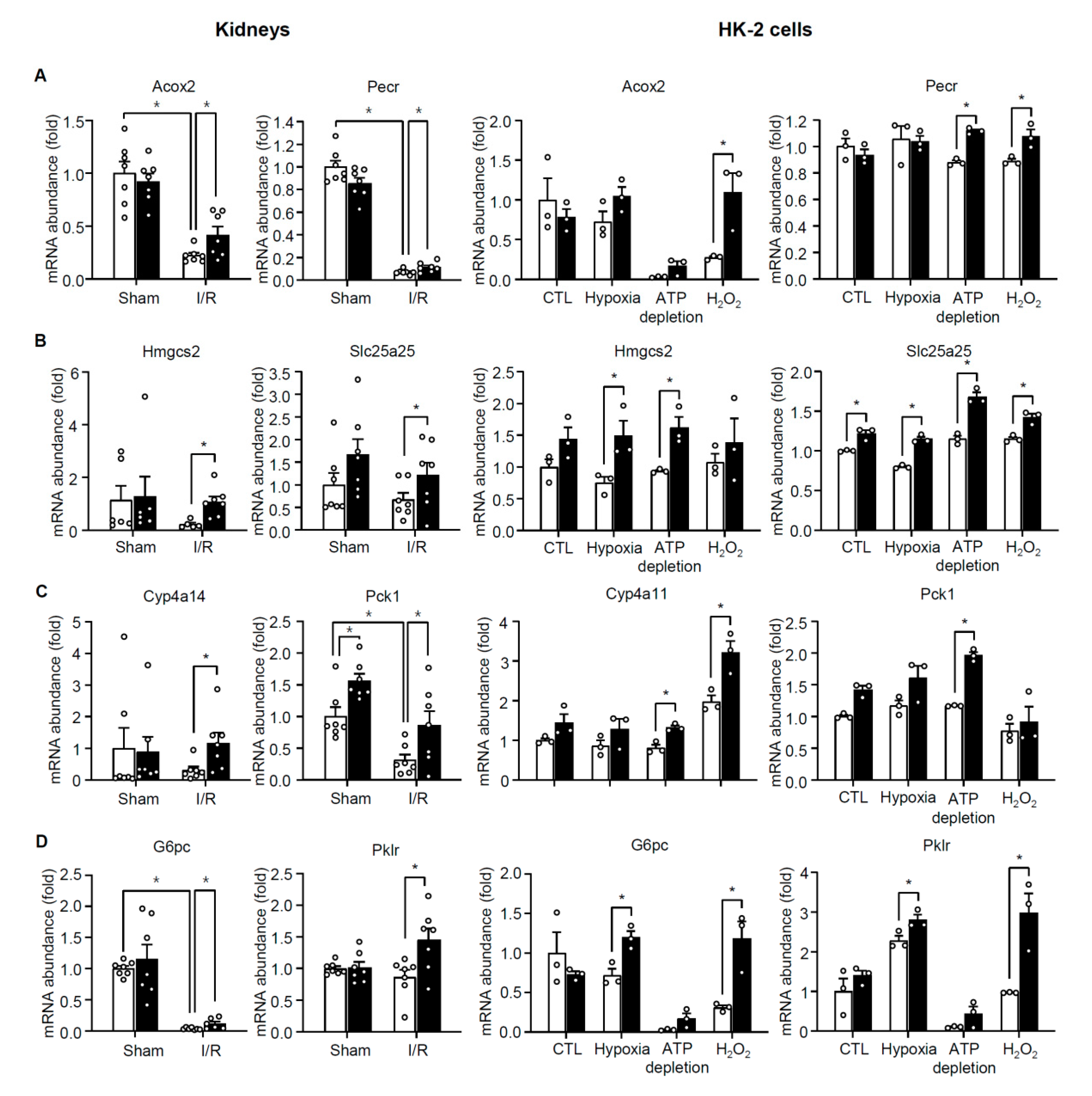
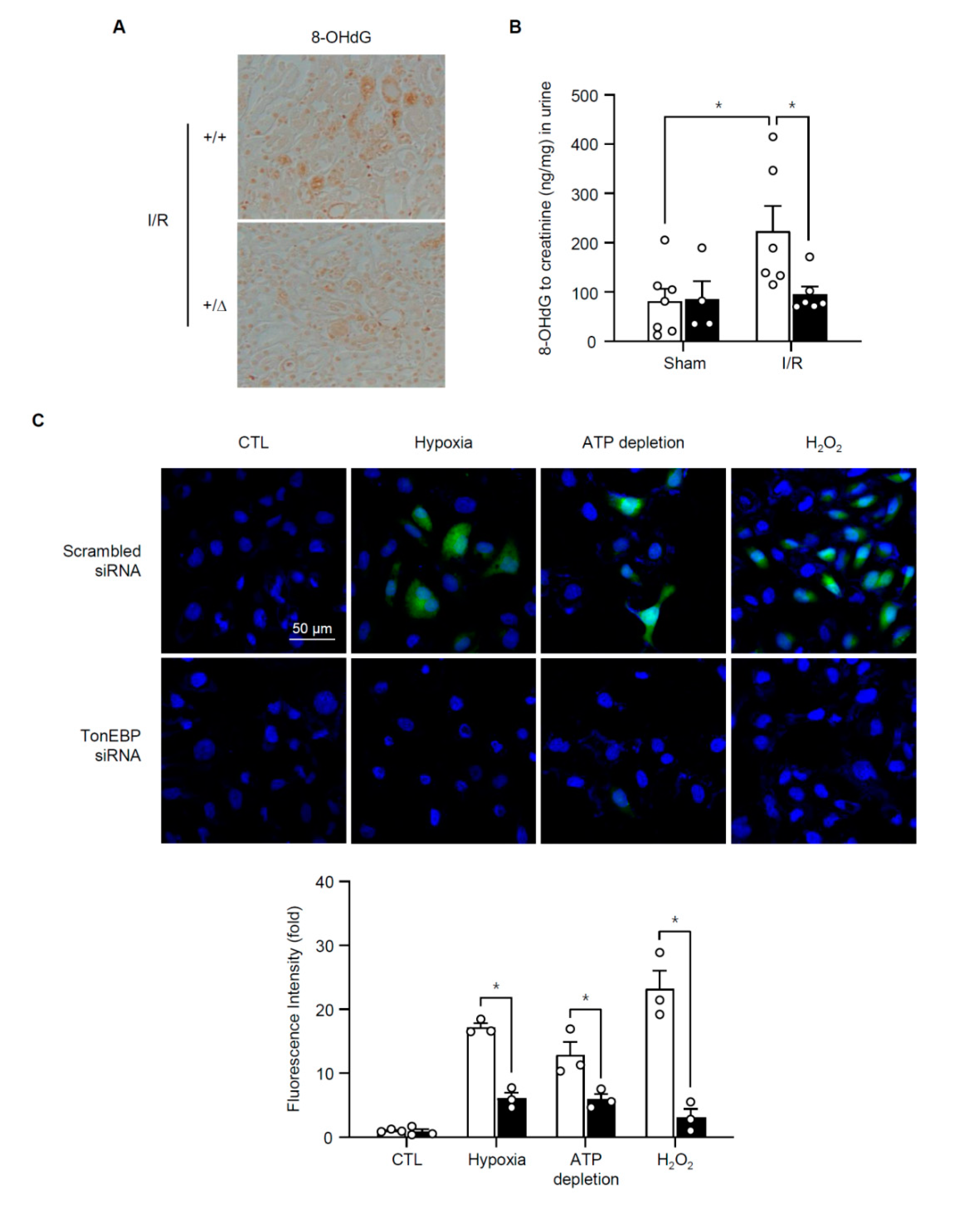
3.4. TonEBP Promotes Oxidative Stress via Suppression of Metabolic Genes
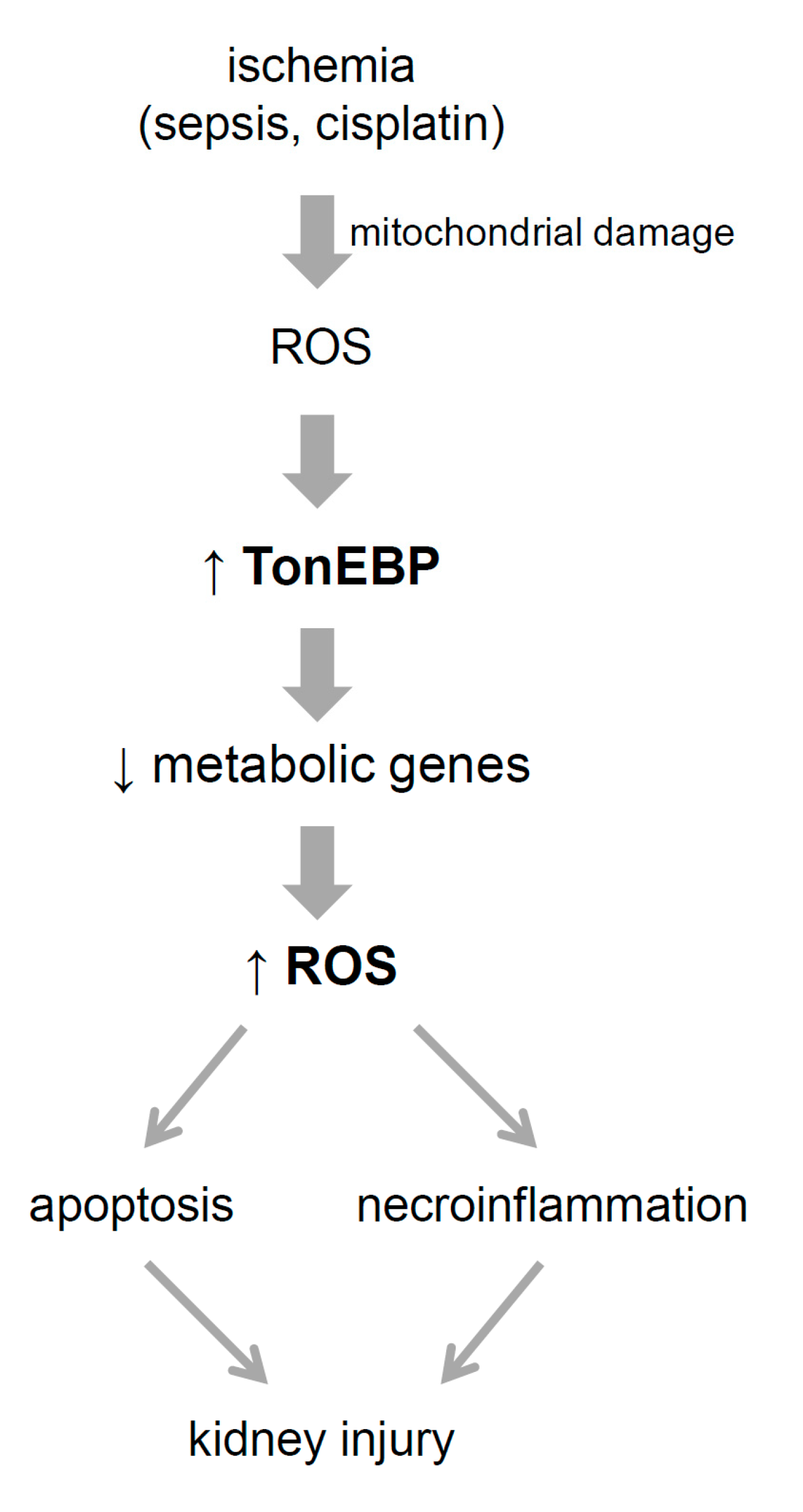
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Mehta, R.L.; Cerda, J.; Burdmann, E.A.; Tonelli, M.; Garcia-Garcia, G.; Jha, V.; Susantitaphong, P.; Rocco, M.; Vanholder, R.; Sever, M.S.; et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): A human rights case for nephrology. Lancet 2015, 385, 2616–2643. [Google Scholar] [CrossRef]
- Xue, J.L.; Daniels, F.; Star, R.A.; Kimmel, P.L.; Eggers, P.W.; Molitoris, B.A.; Himmelfarb, J.; Collins, A.J. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J. Am. Soc. Nephrol. 2006, 17, 1135–1142. [Google Scholar] [CrossRef]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulay, S.R.; Linkermann, A.; Anders, H.J. Necroinflammation in Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Brasen, J.H.; Himmerkus, N.; Liu, S.; Huber, T.B.; Kunzendorf, U.; Krautwald, S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012, 81, 751–761. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Devalaraja-Narashimha, K.; Diener, A.M.; Padanilam, B.J. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal Physiol. 2009, 297, F749–F759. [Google Scholar] [CrossRef] [Green Version]
- Miyakawa, H.; Woo, S.K.; Dahl, S.C.; Handler, J.S.; Kwon, H.M. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc. Natl. Acad. Sci. USA 1999, 96, 2538–2542. [Google Scholar] [CrossRef]
- Lee, H.H.; Sanada, S.; An, S.M.; Ye, B.J.; Lee, J.H.; Seo, Y.K.; Lee, C.; Lee-Kwon, W.; Kuper, C.; Neuhofer, W.; et al. LPS-induced NFkappaB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci. Rep. 2016, 6, 24921. [Google Scholar] [CrossRef]
- Lee, J.H.; Suh, J.H.; Choi, S.Y.; Kang, H.J.; Lee, H.H.; Ye, B.J.; Lee, G.R.; Jung, S.W.; Kim, C.J.; Lee-Kwon, W.; et al. Tonicity-responsive enhancer-binding protein promotes hepatocellular carcinogenesis, recurrence and metastasis. Gut 2019, 68, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, H.H.; Ye, B.J.; Lee-Kwon, W.; Choi, S.Y.; Kwon, H.M. TonEBP suppresses adipogenesis and insulin sensitivity by blocking epigenetic transition of PPARgamma2. Sci. Rep. 2015, 5, 10937. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Lee, H.H.; Lee, J.H.; Ye, B.J.; Yoo, E.J.; Kang, H.J.; Jung, G.W.; An, S.M.; Lee-Kwon, W.; Chiong, M.; et al. TonEBP suppresses IL-10-mediated immunomodulation. Sci. Rep. 2016, 6, 25726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, E.J.; Lee, H.H.; Ye, B.J.; Lee, J.H.; Lee, C.Y.; Kang, H.J.; Jeong, G.W.; Park, H.; Lim, S.W.; Lee-Kwon, W.; et al. TonEBP Suppresses the HO-1 Gene by Blocking Recruitment of Nrf2 to Its Promoter. Front. Immunol. 2019, 10, 850. [Google Scholar] [CrossRef]
- Lee, H.H.; An, S.M.; Ye, B.J.; Lee, J.H.; Yoo, E.J.; Jeong, G.W.; Kang, H.J.; Alfadda, A.A.; Lim, S.W.; Park, J.; et al. TonEBP/NFAT5 promotes obesity and insulin resistance by epigenetic suppression of white adipose tissue beiging. Nat. Commun. 2019, 10, 3536. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lim, S.W.; Salimi, S.; Yoo, E.J.; Lee-Kwon, W.; Lee, H.H.; Lee, J.H.; Mitchell, B.D.; Sanada, S.; Parsa, A.; et al. Tonicity-Responsive Enhancer-Binding Protein Mediates Hyperglycemia-Induced Inflammation and Vascular and Renal Injury. J. Am. Soc. Nephrol. 2018, 29, 492–504. [Google Scholar] [CrossRef]
- Yoon, H.J.; You, S.; Yoo, S.A.; Kim, N.H.; Kwon, H.M.; Yoon, C.H.; Cho, C.S.; Hwang, D.; Kim, W.U. NF-AT5 is a critical regulator of inflammatory arthritis. Arthritis Rheum 2011, 63, 1843–1852. [Google Scholar] [CrossRef]
- Buxade, M.; Lunazzi, G.; Minguillon, J.; Iborra, S.; Berga-Bolanos, R.; Del Val, M.; Aramburu, J.; Lopez-Rodriguez, C. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J. Exp. Med. 2012, 209, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, S.; Suazo, C.; Santapau, D.; Perez, F.; Quiroz, M.; Carreno, J.E.; Illanes, S.; Lavandero, S.; Michea, L.; Irarrazabal, C.E. NFAT5 is activated by hypoxia: Role in ischemia and reperfusion in the rat kidney. PLoS ONE 2012, 7, e39665. [Google Scholar] [CrossRef]
- Go, W.Y.; Liu, X.; Roti, M.A.; Liu, F.; Ho, S.N. NFAT5/TonEBP mutant mice define osmotic stress as a critical feature of the lymphoid microenvironment. Proc. Natl. Acad. Sci. USA 2004, 101, 10673–10678. [Google Scholar] [CrossRef] [Green Version]
- Lien, Y.H.; Yong, K.C.; Cho, C.; Igarashi, S.; Lai, L.W. S1P(1)-selective agonist, SEW2871, ameliorates ischemic acute renal failure. Kidney Int. 2006, 69, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Sheen, M.R.; Kim, J.A.; Lim, S.W.; Jung, J.Y.; Han, K.H.; Jeon, U.S.; Park, S.H.; Kim, J.; Kwon, H.M. Interstitial tonicity controls TonEBP expression in the renal medulla. Kidney Int. 2009, 75, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology, C. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bajwa, A.; Rosin, D.L.; Chroscicki, P.; Lee, S.; Dondeti, K.; Ye, H.; Kinsey, G.R.; Stevens, B.K.; Jobin, K.; Kenwood, B.M.; et al. Sphingosine 1-phosphate receptor-1 enhances mitochondrial function and reduces cisplatin-induced tubule injury. J. Am. Soc. Nephrol. 2015, 26, 908–925. [Google Scholar] [CrossRef]
- Oh, C.J.; Ha, C.M.; Choi, Y.K.; Park, S.; Choe, M.S.; Jeoung, N.H.; Huh, Y.H.; Kim, H.J.; Kweon, H.S.; Lee, J.M.; et al. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 2017, 91, 880–895. [Google Scholar] [CrossRef]
- Ni, D.; Jiang, D.; Kutyreff, C.J.; Lai, J.; Yan, Y.; Barnhart, T.E.; Yu, B.; Im, H.J.; Kang, L.; Cho, S.Y.; et al. Molybdenum-based nanoclusters act as antioxidants and ameliorate acute kidney injury in mice. Nat. Commun. 2018, 9, 5421. [Google Scholar] [CrossRef]
- Nezu, M.; Souma, T.; Yu, L.; Suzuki, T.; Saigusa, D.; Ito, S.; Suzuki, N.; Yamamoto, M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017, 91, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham | I/R | |||
|---|---|---|---|---|
| +/+ | +/Δ | +/+ | +/Δ | |
| Cytokines & Chemokines | ||||
| IL-6 | 1.0 ± 0.3 | 1.5 ± 0.5 | 29.1 ± 6.5 # | 9.3 ± 2.5 #,* |
| IL-1β | 1.0 ± 0.1 | 1.2 ± 0.2 | 12.5 ± 1.0 # | 9.7 ± 1.3 #,* |
| IL-18 | 1.0 ± 0.1 | 0.9 ± 0.1 | 1.4 ± 0.2 | 1.0 ± 0.1 * |
| TNF-α | 1.0 ± 0.1 | 0.8 ± 1.0 * | 5.6 ± 0.5 # | 4.6 ± 0.4 # |
| IFN-γ | 1.0 ± 0.2 | 0.7 ± 0.1 | 1.7 ± 0.2 # | 1.1 ± 0.1 #,* |
| IP-10 | 1.0 ± 0.1 | 1.1 ± 0.2 | 3.2 ± 0.3 # | 2.5 ± 0.1 #,* |
| RANTES | 1.0 ± 0.1 | 0.7 ± 0.1 * | 1.1 ± 0.1 | 1.2 ± 0.2# |
| IL-10 | 1.0 ± 0.2 | 5.3 ± 1.7 * | 17.2 ± 2.2 # | 9.8 ± 1.8 #,* |
| MCP-1 | 1.0 ± 0.1 | 2.4 ± 0.8 * | 40.9 ± 5.1 # | 21.9 ± 2.7 #,* |
| Adhesion Molecules | ||||
| ICAM | 1.0 ± 0.1 | 1.6 ± 0.2 * | 1.7 ± 0.1 # | 1.4 ± 0.1 * |
| E-selectin | 1.0 ± 0.3 | 0.9 ± 0.1 | 3.1± 0.4 # | 1.8 ± 0.2 #,* |
| VCAM-1 | 1.0 ± 0.1 | 2.0 ± 0.3 * | 3.3 ± 0.4 # | 2.8 ± 0.2 # |
| Pathway | Number of Genes | FDR q-Value | Selected Genes in Cluster |
|---|---|---|---|
| Peroxisome | 25/77 | <0.001 | Acox2, Pecr, Dao, Hacl1, Amacr, Baat, Hao1, Mpv17l, Decr2, Agxt, Hao2, Abcd4 |
| Mitochondrial Inner Membrane | 86/291 | <0.001 | Hmgc2, Prodh2, Maob, Slc25a25, Slc25a42, Hsd3b3, Gatm, Slc25a10 |
| PPAR Signaling Pathways | 21/79 | 0.0035 | Cyp4a14, Hmgcs2, Pck1, Fabp3, Cyp4a32, Scd1, Acox2, Cyp4a10, Apoc3, Gyk |
| Glycolysis/Gluconeogenesis | 18/56 | 0.0037 | G6pc, Pck1, Pklr, Acss2, Pfkm, Aldh3b1, Pgam2, Aldoc, Gatm, Acss1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, E.J.; Lim, S.W.; Kang, H.J.; Park, H.; Yoon, S.; Nam, D.; Sanada, S.; Kwon, M.J.; Lee-Kwon, W.; Choi, S.Y.; et al. Transcriptional Regulator TonEBP Mediates Oxidative Damages in Ischemic Kidney Injury. Cells 2019, 8, 1284. https://doi.org/10.3390/cells8101284
Yoo EJ, Lim SW, Kang HJ, Park H, Yoon S, Nam D, Sanada S, Kwon MJ, Lee-Kwon W, Choi SY, et al. Transcriptional Regulator TonEBP Mediates Oxidative Damages in Ischemic Kidney Injury. Cells. 2019; 8(10):1284. https://doi.org/10.3390/cells8101284
Chicago/Turabian StyleYoo, Eun Jin, Sun Woo Lim, Hyun Je Kang, Hyun Park, Sora Yoon, Dougu Nam, Satoru Sanada, Mi Jin Kwon, Whaseon Lee-Kwon, Soo Youn Choi, and et al. 2019. "Transcriptional Regulator TonEBP Mediates Oxidative Damages in Ischemic Kidney Injury" Cells 8, no. 10: 1284. https://doi.org/10.3390/cells8101284