Tubulin-Dependent Transport of Connexin-36 Potentiates the Size and Strength of Electrical Synapses

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Constructs
2.2. Cell Line, Cell Culture, and Transient Transfection
2.3. Protein Isolation and Immunoblot Analysis
2.4. In Vivo Affinity Capture of Biotinylated Tubulin Using BioID
2.5. Pharmacology
2.6. Electrophysiology
2.7. Fluorescence Microscopy and Image Processing
2.7.1. Fluorescent Imaging and Gap-Junction Plaque Measurements
2.7.2. Fluorescence Recovery After Photobleaching (FRAP)
2.7.3. Total Internal Reflection Fluorescence (TIRF).
2.8. Molecular Modeling
2.9. Statistics
3. Results
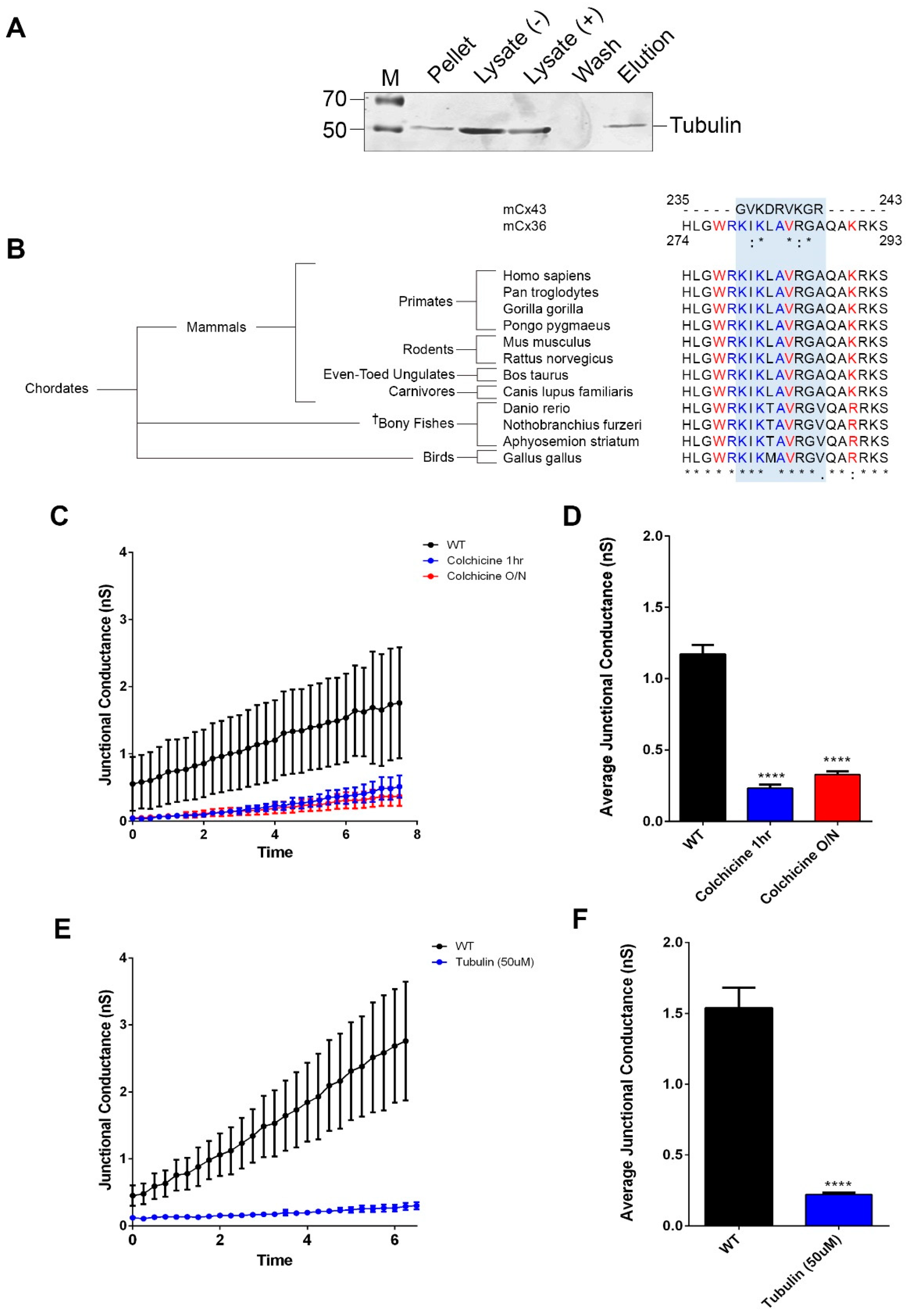
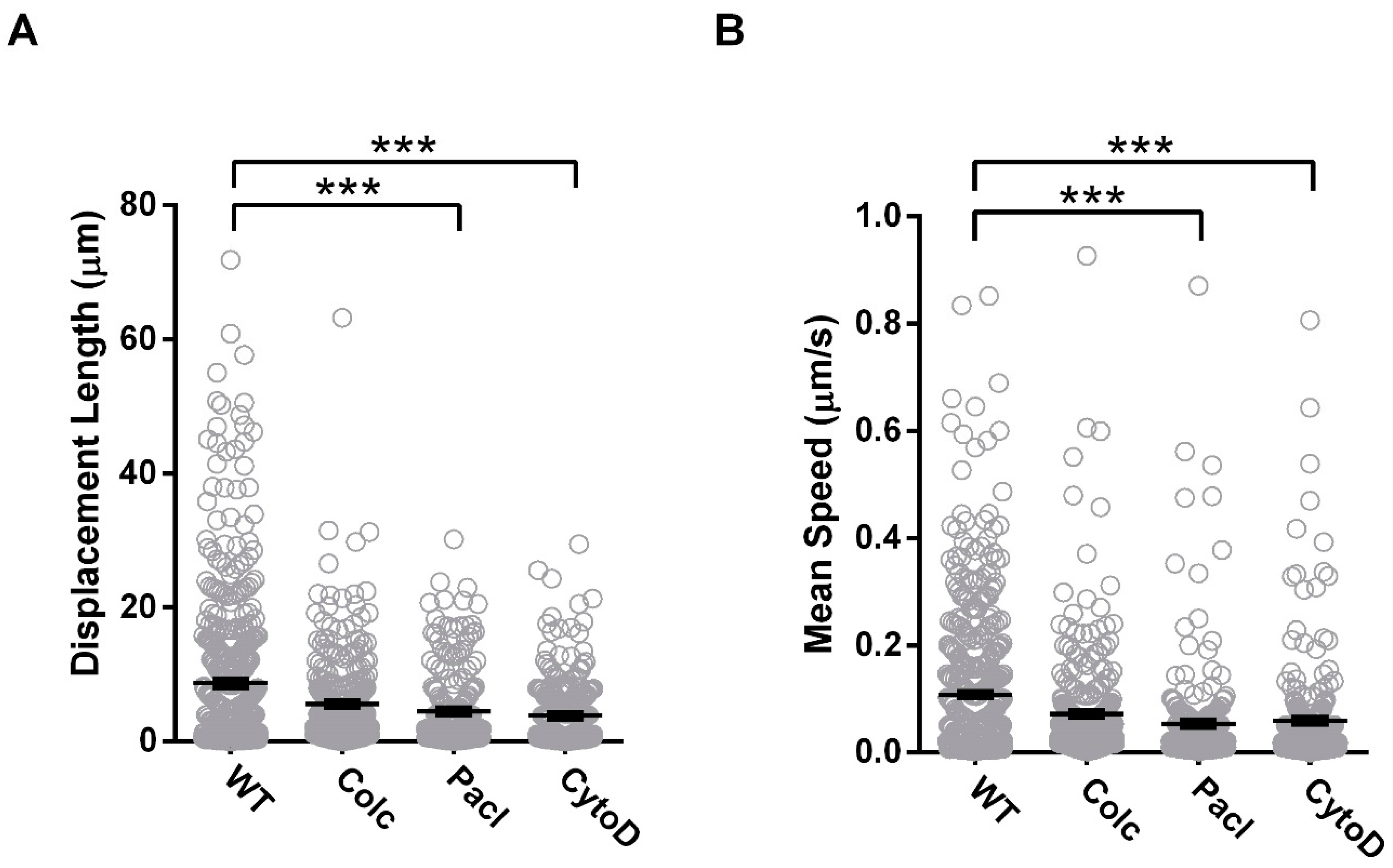
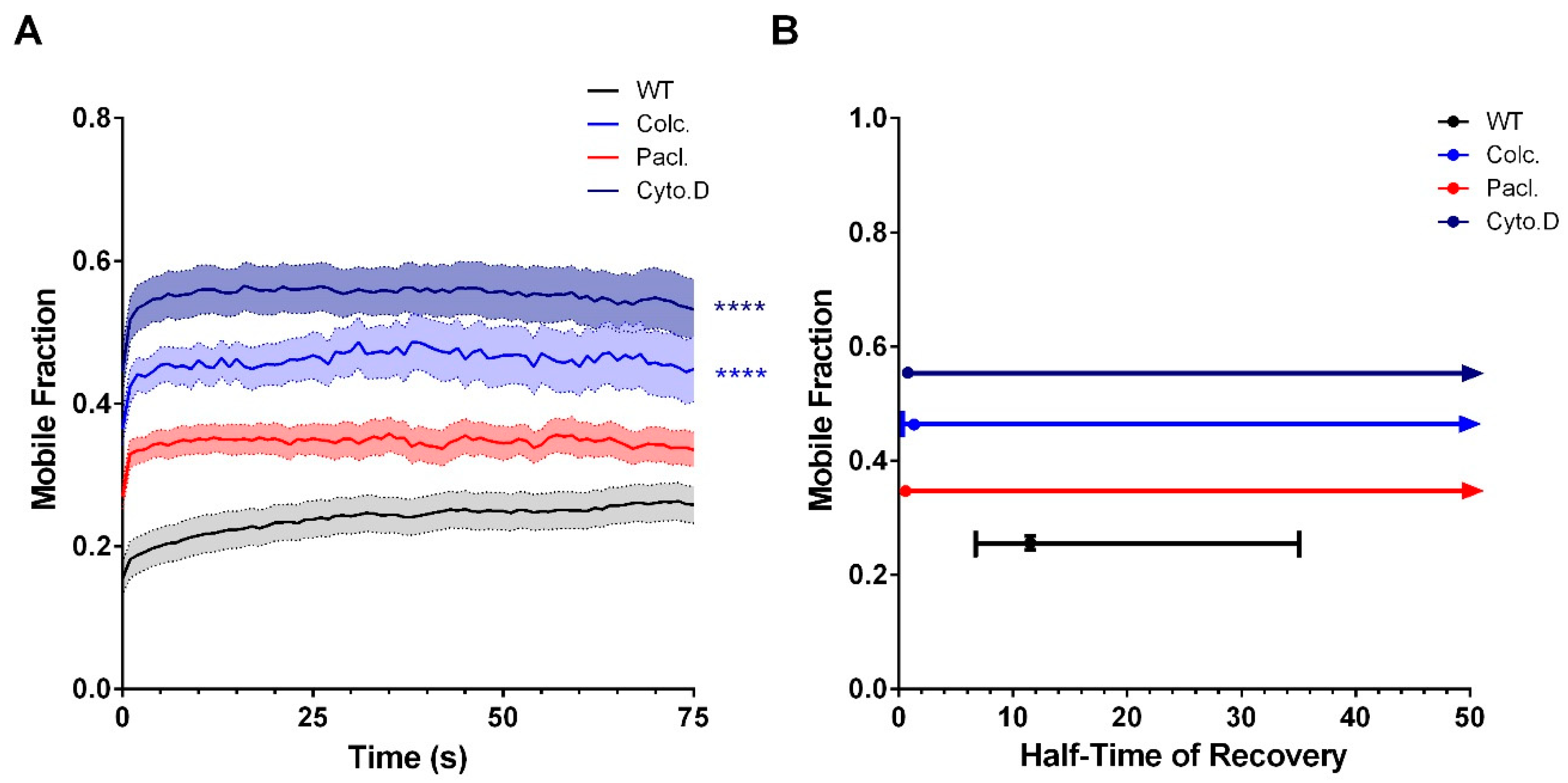
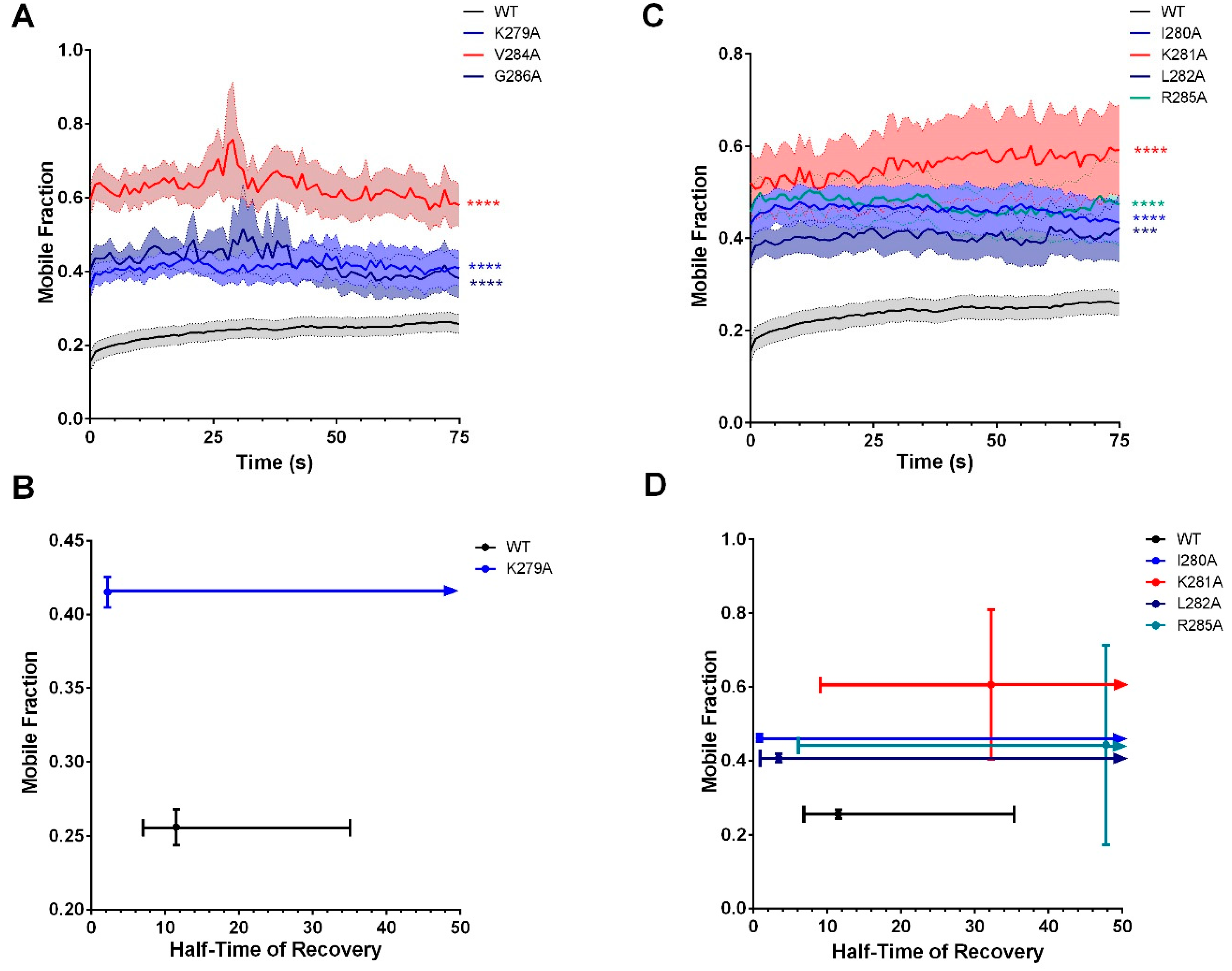
3.1. Cx36 Interaction with the Tubulin Cytoskeleton is Required for Electrical Plasticity
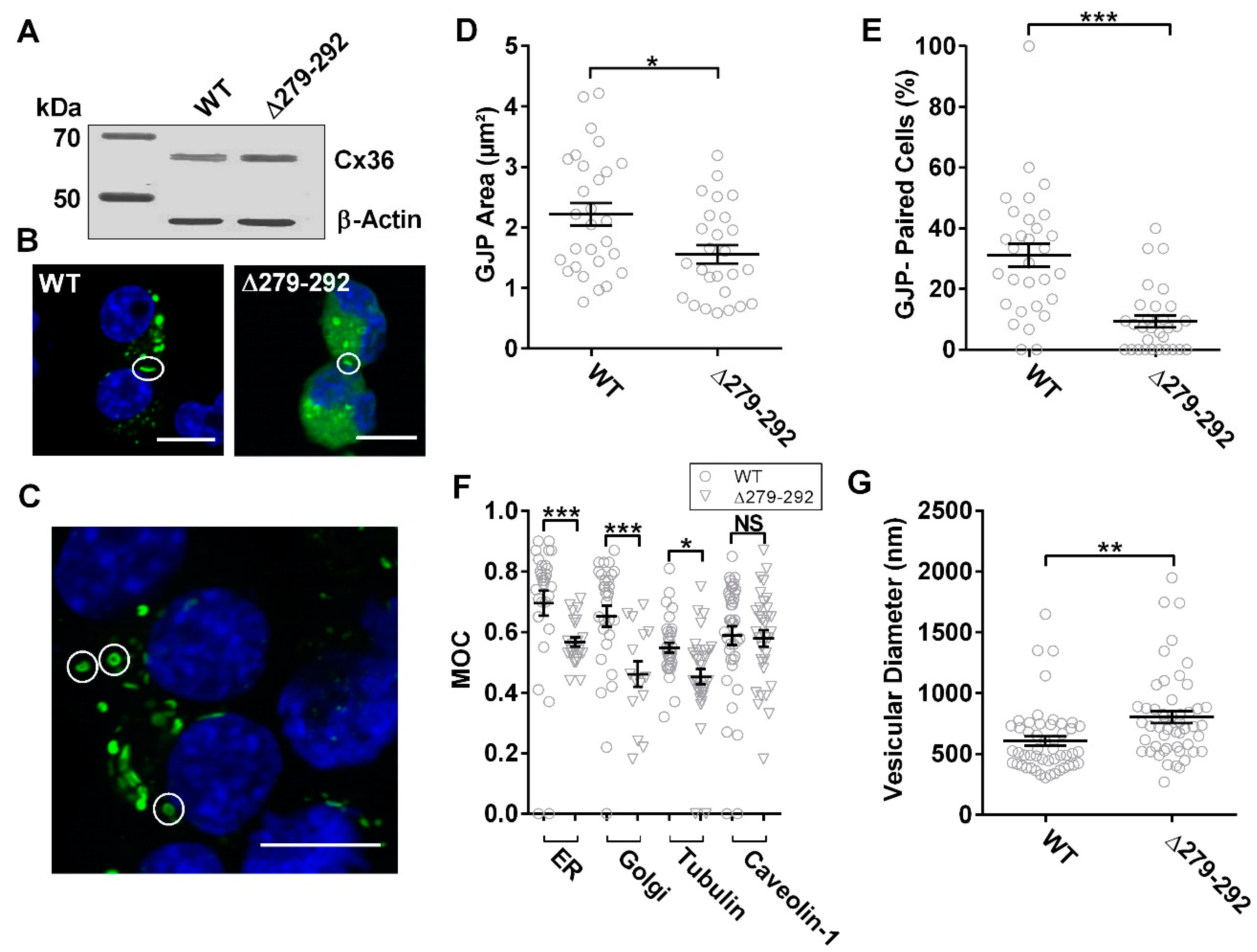
3.2. Inhibition of the Cx36-Tubulin Interaction Promotes Intracellular Localization
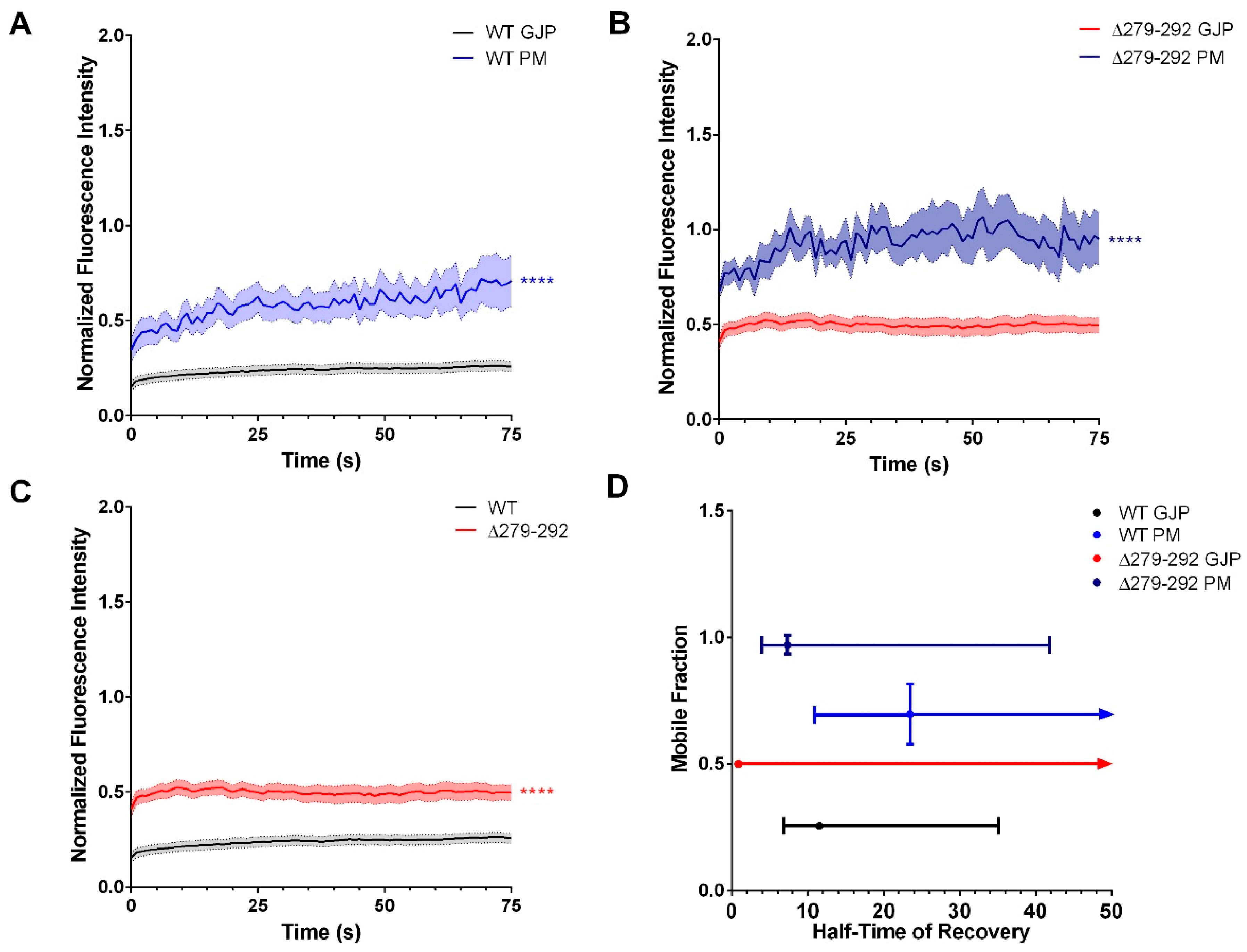
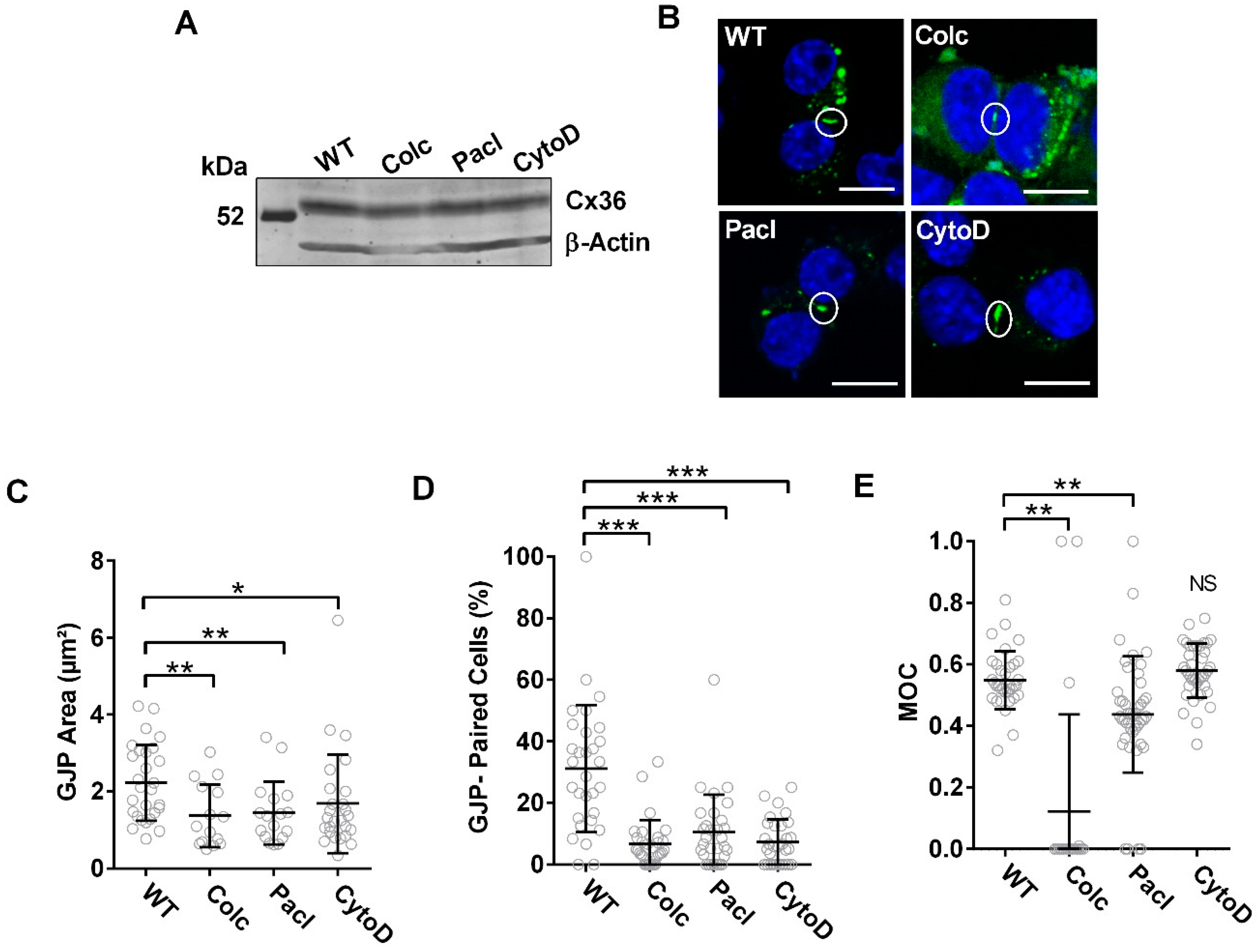
3.3. Cx36 Transport Correlates to the Modulation of Cytoskeletal Dynamics
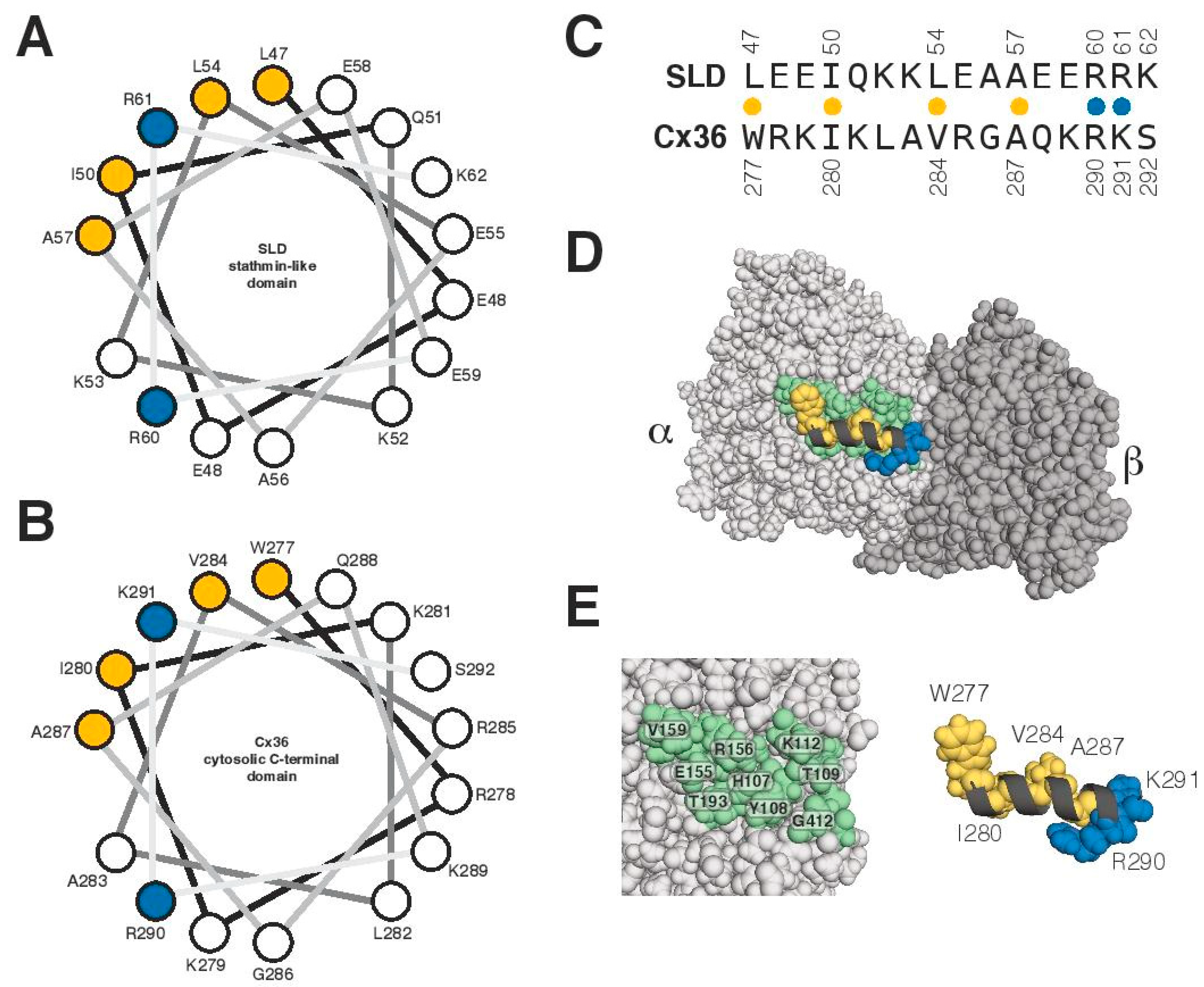
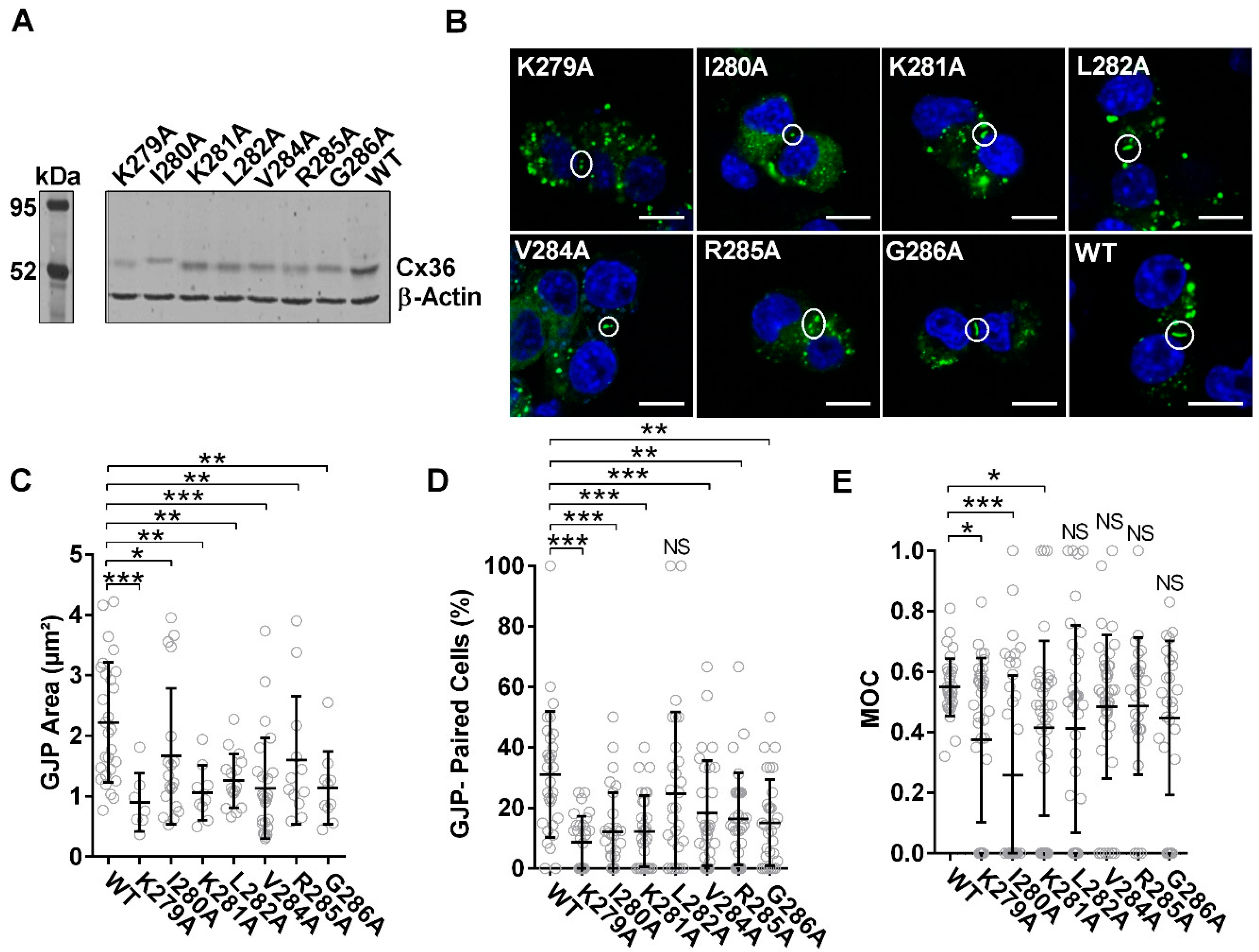
3.4. Characterization of the Tubulin-Binding Motif
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pereda, A.E. Electrical synapses and their functional interactions with chemical synapses. Nat. Rev. Neurosci. 2014, 15, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Pereda, A.E. Neurobiology: All synapses are created equal. Curr. Biol. 2015, 25, R38–R41. [Google Scholar] [CrossRef] [PubMed]
- Zoidl, G.; Meier, C.; Petrasch-Parwez, E.; Zoidl, C.; Habbes, H.W.; Kremer, M.; Srinivas, M.; Spray, D.C.; Dermietzel, R. Evidence for a role of the N-terminal domain in subcellular localization of the neuronal connexin36 (Cx36). J. Neurosci. Res. 2002, 69, 448–465. [Google Scholar] [CrossRef] [PubMed]
- Alev, C.; Urschel, S.; Sonntag, S.; Zoidl, G.; Fort, A.G.; Höher, T.; Matsubara, M.; Willecke, K.; Spray, D.C.; Dermietzel, R. The neuronal connexin36 interacts with and is phosphorylated by CaMKII in a way similar to CaMKII interaction with glutamate receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 20964–20969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Corsso, C.; Iglesias, R.; Zoidl, G.; Dermietzel, R.; Spray, D.C. Calmodulin dependent protein kinase increases conductance at gap junctions formed by the neuronal gap junction protein connexin36. Brain Res. 2012, 1487, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collingridge, G.; Isaac, J.; Wang, Y. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 2004, 5, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.; Wollmuth, L.; McBain, C.; Menniti, F.; Vance, K.; Ogden, K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Stoppini, L.; Miyamoto, E.; Muller, D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase. Biol Chem. 1993, 268, 7863–7867. [Google Scholar]
- Sheng, M.; Hoogenraad, C.C. The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu. Rev. Biochem. 2007, 76, 823–847. [Google Scholar] [CrossRef] [PubMed]
- Derkach, V.; Oh, M.; Guire, E.; Soderling, T. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 2007, 8, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza, M.; Fernandez-Villalobos, G.; Stein, I.; Kasumova, G.; Zhang, P.; Bayer, K.; Otmakhov, N.; Hell, J.W.; Lisman, J. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J. Neurosci. 2011, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Takahashi, E.; Li, W.; Halt, A.; Wiltgen, B.; Ehniger, D.; Li, G.D.; Hell, J.W.; Kennedy, M.B.; Silva, A.J. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J. Neurosci. 2007, 27, 13843–13853. [Google Scholar] [CrossRef] [PubMed]
- Dent, E.W.; Gupton, S.L.; Gertler, F.B. The Growth Cone Cytoskeleton in Axon Outgrowth and Guidance. Cold Spring Harb. Perspect. Biol. 2011, 3, a001800. [Google Scholar] [CrossRef] [PubMed]
- Hoogenraad, C.C.; Bradke, F. Control of neuronal polarity and plasticity—A renaissance for microtubules? Trends Cell Biol. 2009, 19, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Kneussel, M.; Wagner, W. Myosin motors at neuronal synapses: Drivers of membrane transport and actin dynamics. Nat. Rev. Neurosci. 2013, 14, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Yuen, E.Y.; Jiang, Q.; Feng, J.; Yan, Z. Microtubule Regulation of N-Methyl-D-aspartate Receptor Channels in Neurons. J. Biol. Chem. 2005, 280, 29420–29427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rossum, D.; Kuhse, J.; Betz, H. Dynamic interaction between soluble tubulin and C-terminal domains of N-methyl-D-aspartate receptor subunits. J. Neurochem. 1999, 72, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Maas, C.; Belgardt, D.; Lee, H.K.; Heisler, F.F.; Lappe-Siefke, C.; Magiera, M.M.; van Dijk, J.; Hausrat, T.J.; Janke, C.; Kneussel, M. Synaptic activation modifies microtubules underlying transport of postsynaptic cargo. Proc. Natl. Acad. Sci. USA 2009, 106, 8731–8736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alev, C.; Zoidl, G.; Dermietzel, R. The Cytoskeleton; Humana Press: Totowa, NJ, USA, 2013; pp. 135–149. ISBN 978-01-9850-970-7. [Google Scholar]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Giepmans, B.N.; Verlaan, I.; Moolenaar, W.H. Connexin-43 interactions with ZO-1 and alpha- and beta-tubulin. Cell Commun. Adhes. 2001, 8, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Siu, R.C.F.; Smirnova, E.; Brown, C.A.; Zoidl, C.; Spray, D.C.; Donaldson, L.W.; Zoidl, G. Structural and Functional Consequences of Connexin 36 (Cx36) Interaction with Calmodulin. Front. Mol. Neurosci. 2016, 9, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamzei-sichani, F.; Kamasawa, N.; Janssen, W.G.M.; Yasumura, T.; Davidoson, K.G.V.; Hof, P.R.; Wearne, S.L.; Stewart, M.G.; Young, S.R.; Whittington, M.A.; et al. Gap junctions on hippocampal mossy fiber axons demonstrated by thin-section electron microscopy and freeze – fracture replica immunogold labeling. PNAS 2007, 104, 12548–12553. [Google Scholar] [CrossRef] [PubMed]
- Bautisa, W.; Nagy, J. Connexin36 in gap junctions forming electrical synapses between motoneurons in sexually dimorphic motor nuclei in spinal cord of rat and mouse. Eur. J. Neurosci. 2014, 39, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Corsso, C.; Srinivas, M.; Urban-Maldonado, M.; Moreno, A.P.; Fort, A.G.; Fishman, G.I.; Spray, D.C. Transfection of mammalian cells with connexins and measurement of voltage sensitivity of their gap junctions. Nat. Protoc. 2006, 1, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Gardner, P.; Schrijver, I. The role of the cytoskeleton in the formation of gap junctions by Connexin 30. Exp. Cell Res. 2009, 215, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Lauf, U.; Giepmans, B.N.G.; Lopez, P.; Braconnot, S.; Chen, S.C.; Falk, M.M. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 10446–10451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Raveh, B.; London, N.; Schueler-Furman, O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins 2010, 78, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Burr, G.S.; Mitchell, C.K.; Keflemariam, Y.J.; Heidelberger, R.; O’Brien, J. Calcium-dependent binding of calmodulin to neuronal gap junction proteins. Biochem. Biophys. Res. Commun. 2005, 335, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.N.; Jan, L.Y. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Jordan, K.; Simek, J.; Shao, Q.; Jedeszko, C.; Walton, P.; Laird, D.W. Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J. Cell Sci. 2005, 118, 4451–4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, K.; Solan, J.L.; Dominguez, M.; Sia, M.; Hand, A.; Lampe, P.D.; Laird, D.W. Trafficking, assembly, and function of a connexin43-green fluorescent protein chimera in live mammalian cells. Mol. Biol. Cell 1999, 10, 2033–2050. [Google Scholar] [CrossRef] [PubMed]
- Finger, T.E. Cell types and lineages in taste buds. Chem. Senses 2005, 30 (Suppl. 1), 54–55. [Google Scholar] [CrossRef]
- Coles, C.H.; Bradke, F. Coordinating neuronal actin-microtubule dynamics. Curr. Biol. 2015, 25, R677–R691. [Google Scholar] [CrossRef] [PubMed]
- Epifantseva, I.; Shaw, R.M. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta 2018, 1860, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Saidi Brikci-Nigassa, A.; Clement, M.J.; Ha-Duong, T.; Adjadj, E.; Ziani, L.; Pastre, D.; Curmi, P.A.; Savarin, P. Phosphorylation controls the interaction of the connexin43 C-terminal domain with tubulin and microtubules. Biochemistry 2012, 51, 4331–4342. [Google Scholar] [CrossRef] [PubMed]
- Mignot, I.; Pecqueur, L.; Dorléans, A.; Karuppasamy, M.; Ravelli, R.B.G.; Dreier, B.; Plückthun, A.; Knossow, M.; Gigant, B. Design and Characterization of Modular Scaffolds for Tubulin Assembly. J. Biol. Chem. 2012, 287, 31085–31094. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ramani, A.; Soni, K.; Gottardo, M.; Zheng, S.; Ming Gooi, L.; Li, W.; Feng, S.; Mariappan, A.; Wason, A.; et al. Molecular basis for CPAP-tubulin interaction in controlling centriolar and ciliary length. Nat. Commun. 2016, 7, 11874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Yang, W.; Lurtz, M.M.; Chen, Y.; Jiang, J.; Huang, Y.; Louis, C.F.; Yang, J.J. Calmodulin mediates the Ca2+-dependent regulation of Cx44 gap junctions. Biophys. J. 2009, 96, 2832–2848. [Google Scholar] [CrossRef] [PubMed]
- Dodd, R.; Peracchia, C.; Stolady, D.; Török, K. Calmodulin Association with Connexin32-derived Peptides Suggests trans -Domain Interaction in Chemical Gating of Gap Junction Channels. J. Biol. Chem. 2008, 283, 26911–26920. [Google Scholar] [CrossRef] [PubMed]
- Myllykoski, M.; Kuczera, K.; Kursula, P. Complex formation between calmodulin and a peptide from the intracellular loop of the gap junction protein connexin43: Molecular conformation and energetics of binding. Biophys. Chem. 2009, 144, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Kopp, R.F.; Chen, Y.; Yang, J.J.; Roe, M.W.; Veenstra, R.D. Gating of connexin 43 gap junctions by a cytoplasmic loop calmodulin binding domain. Am. J. Physiol. Cell Physiol. 2012, 302, C1548–C1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mruk, K.; Farley, B.M.; Ritacco, A.W.; Kobertz, W.R. Calmodulation meta-analysis: Predicting calmodulin binding via canonical motif clustering. J. Gen. Physiol. 2014, 144, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witman, G.B. Axonemal dyneins. Curr. Opin. Cell Biol. 1992, 4, 74–79. [Google Scholar] [CrossRef]
- Wilkerson, C.G.; King, S.M.; Witman, G.B. Molecular analysis of the gamma heavy chain of Chlamydomonas flagellar outer-arm dynein. J. Cell Sci. 1994, 107, 497–506. [Google Scholar] [PubMed]
- Urschel, S.; Höher, T.; Schubert, T.; Alev, C.; Söhl, G.; Wörsdörfer, P.; Asahara, T.; Dermietzel, R.; Weiler, R.; Willecke, K.; et al. Protein Kinase A-mediated Phosphorylation of Connexin36 in Mouse Retina Results in Decreased Gap Junctional Communication between AII Amacrine Cells. J. Biol. Chem. 2006, 281, 33163–33171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, Z.; Blackburn, M.R.; Wang, S.W.; Ribelayga, C.P.; O’Brien, J. Adenosine and Dopamine Receptors Coregulate Photoreceptor Coupling via Gap Junction Phosphorylation in Mouse Retina. J. Neurosci. 2013, 33, 3135–3150. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Lin, Y.P.; Mitchell, C.K.; Ram, S.; O’Brien, J. Two-color fluorescent analysis of connexin 36 turnover: Relationship to functional plasticity. J. Cell Sci. 2015, 3888–3897. [Google Scholar] [CrossRef] [PubMed]
- Bazzigaluppi, P.; Isenia, S.C.; Haasdijk, E.D.; Elgersma, Y.; De Zeeuw, C.I.; van der Giessen, R.S.; de Jeu, M.T.G. Modulation of Murine Olivary Connexin 36 Gap Junctions by PKA and CaMKII. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Hendsch, Z.S.; Tidor, B. Do salt bridges stablize proteins—A continuum electrostatic analysis. Prot. Sci. 1994, 3, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Close-range electrostatic interactions in proteins. ChemBioChem 2002, 3, 604–617. [Google Scholar] [CrossRef]
- Sindelar, C.V.; Hendsch, Z.S.; Tidor, B. Effects of salt bridges on protein structure and design. Protein Sci. 1998, 7, 1898–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, A.; Summa, C.M.; Geremia, S.; Randaccio, L.; Pavone, V.; DeGrado, W.F. Retrostructural analysis of metalloproteins: Application to the design of a minimal model for diiron proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 6298–6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calhoun, J.R.; Nastri, F.; Maglio, O.; Pavone, V.; Lombardi, A.; DeGrado, W.F. Artificial diiron proteins: From structure to function. Biopolym. Peptide Sci. Sect. 2005, 80, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Zeroka, D.; Jensen, J.O.; Samuels, A.C. Rotation/Inversion Study of the Amino Group in Ethylamine. J. Phys. Chem. A 1998, 5639, 6571–6579. [Google Scholar] [CrossRef]
- Arpino, J.A.J.; Reddington, S.C.; Halliwell, L.M.; Rizkallah, P.J.; Jones, D.D. Random Single Amino Acid Deletion Sampling Unveils Structural Tolerance and the Benefits of Helical Registry Shift on GFP Folding and Structure. Structure 2014, 22, 889–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simm, A.M.; Baldwin, A.J.; Busse, K.; Jones, D.D. Investigating protein structural plasticity by surveying the consequence of an amino acid deletion from TEM-1 β-lactamase. FEBS Lett. 2007, 581, 3904–3908. [Google Scholar] [CrossRef] [PubMed]
- Simek, J.; Churko, J.; Shao, Q.; Laird, D.W. Cx43 has distinct mobility within plasma-membrane domains, indicative of progressive formation of gap-junction plaques. J. Cell Sci. 2009, 122, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Viesselmann, C.; Nam, S.; Merriam, E.; Dent, E.W. Activity-dependent dynamic microtubule invasion of dendritic spines. J. Neurosci. 2008, 28, 13094–13105. [Google Scholar] [CrossRef] [PubMed]
- Lynn, B.D.; Li, X.; Nagy, J.I. Under Construction: Building the Macromolecular Superstructure and Signaling Components of an Electrical Synapse. J. Membr. Biol. 2012, 245, 303–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligon, L.A.; Steward, O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000, 427, 351–361. [Google Scholar] [CrossRef]
- Wang, Z.; Edwards, J.G.; Riley, N.; Provance, D.W.; Karcher, R.; Li, X.; Davison, I.G.; Ikebe, M.; Mercer, J.A.; Kauer, J.A.; et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell 2008, 135, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Osterweil, E.; Wells, D.G.; Mooseker, M.S. A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J. Cell Biol. 2005, 168, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Bär, J.; Kobler, O.; van Bommel, B.; Mikhaylova, M. Periodic F-actin structures shape the neck of dendritic spines. Sci. Rep. 2016, 6, 37136. [Google Scholar] [CrossRef]
- Watanabe, H.; Washioka, H.; Tonosaki, A. Gap junction and its cytoskeletal undercoats as involved in invagination-endocytosis. Tohoku J. Exp. Med. 1988, 156, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Wayakanon, P.; Bhattacharjee, R.; Nakahama, K.I.; Morita, I. The role of the Cx43 C-terminus in GJ plaque formation and internalization. Biochem. Biophys. Res. Commun. 2012, 420, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Mohrmann, R.; Khr, G.; Hatt, H.; Sprengel, R.; Gottmann, K. Deletion of the C-terminal domain of the NR2B subunit alters channel properties and synaptic targeting of N-methyl-D-aspartate receptors in nascent neocortical synapses. J. Neurosci. Res. 2002, 68, 265–275. [Google Scholar] [CrossRef]
- Lei, S.; Czerwinska, E.; Czerwinski, W.; Walsh, M.P.; MacDonald, J.F. Regulation of NMDA Receptor Activity by F-Actin and Myosin Light Chain Kinase. J. Neurosci. 2001, 21, 8464–8472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, M.; Rozental, R.; Kojima, T.; Dermietzel, R.; Mehler, M.; Condorelli, D.F.; Kessler, J.A.; Spray, D.C. Functional properties of channels formed by the neuronal gap junction protein connexin36. J. Neurosci. 1999, 19, 9848–9855. [Google Scholar] [CrossRef]
- Rash, J.E.; Yasumura, T.; Davidson, K.G.; Furman, C.S.; Dudek, F.E.; Nagy, J.I. Identification of cells expressing Cx43, Cx30, Cx26, Cx32 and Cx36 in gap junctions of rat brain and spinal cord. Cell Commun. Adhes. 2001, 8, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E.; Olson, C.O.; Pouliot, W.A.; Davidson, K.G.V.; Yasumura, T.; Furman, C.S.; Royer, S.; Kamasawa, N.; Nagy, J.I.; Dudek, F.E. Connexin36 vs. connexin32, “miniature” neuronal gap junctions, and limited electrotonic coupling in rodent suprachiasmatic nucleus. Neuroscience 2007, 149, 350–371. [Google Scholar] [CrossRef] [PubMed]
- Kothmann, W.W.; Li, X.; Burr, G.S.; O’Brien, J. Connexin 35/36 is phosphorylated at regulatory sites in the retina. Vis. Neurosci. 2007, 24, 363–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, X.; Winbow, V.M.; Patel, L.S.; Burr, G.S.; Mitchell, C.K.; O’Brien, J. Protein kinase A mediates regulation of gap junctions containing connexin35 through a complex pathway. Brain Res. Mol. Brain Res. 2005, 135, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, K.M. GDNF: A novel factor with therapeutic potential for neurodegenerative disorders. Mol. Neurobiol. 1999, 19, 43–59. [Google Scholar] [CrossRef] [PubMed]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, C.A.; del Corsso, C.; Zoidl, C.; Donaldson, L.W.; Spray, D.C.; Zoidl, G. Tubulin-Dependent Transport of Connexin-36 Potentiates the Size and Strength of Electrical Synapses. Cells 2019, 8, 1146. https://doi.org/10.3390/cells8101146
Brown CA, del Corsso C, Zoidl C, Donaldson LW, Spray DC, Zoidl G. Tubulin-Dependent Transport of Connexin-36 Potentiates the Size and Strength of Electrical Synapses. Cells. 2019; 8(10):1146. https://doi.org/10.3390/cells8101146
Chicago/Turabian StyleBrown, Cherie A., Cristiane del Corsso, Christiane Zoidl, Logan W. Donaldson, David C. Spray, and Georg Zoidl. 2019. "Tubulin-Dependent Transport of Connexin-36 Potentiates the Size and Strength of Electrical Synapses" Cells 8, no. 10: 1146. https://doi.org/10.3390/cells8101146