Polyhexamethyleneguanidine Phosphate-Induced Cytotoxicity in Liver Cells Is Alleviated by Tauroursodeoxycholic Acid (TUDCA) via a Reduction in Endoplasmic Reticulum Stress

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
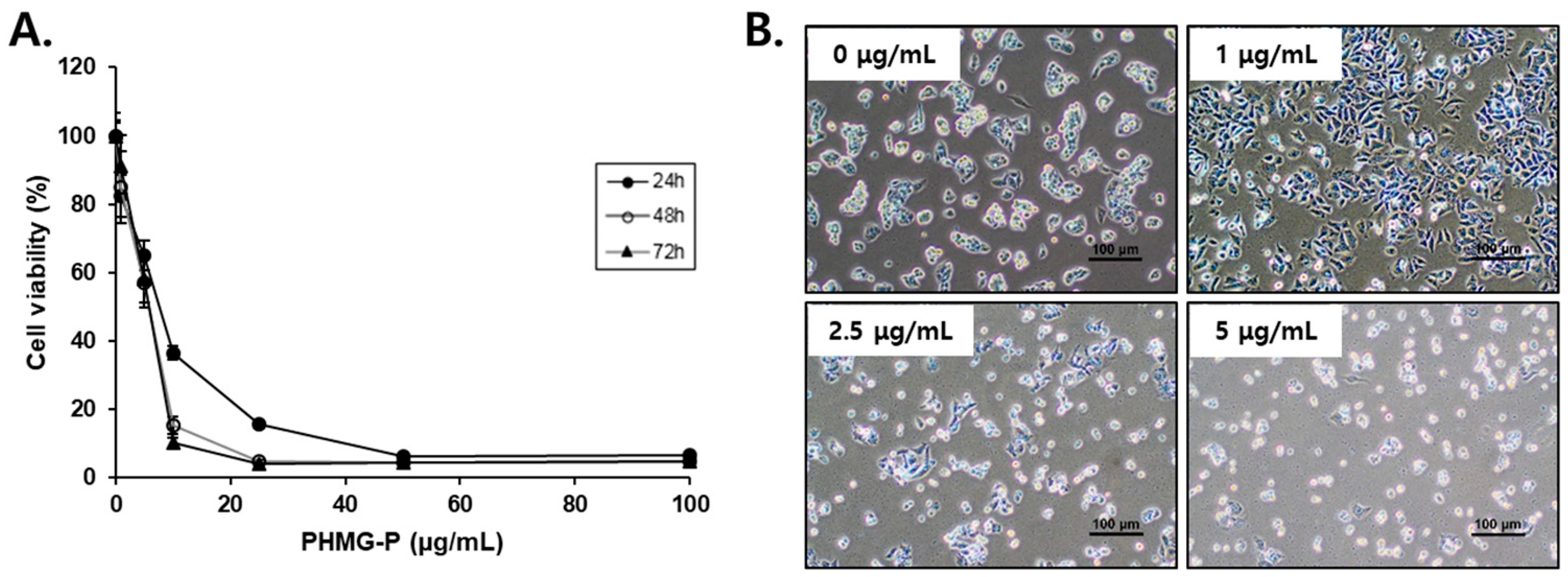
2.2. Cell Viability Assays
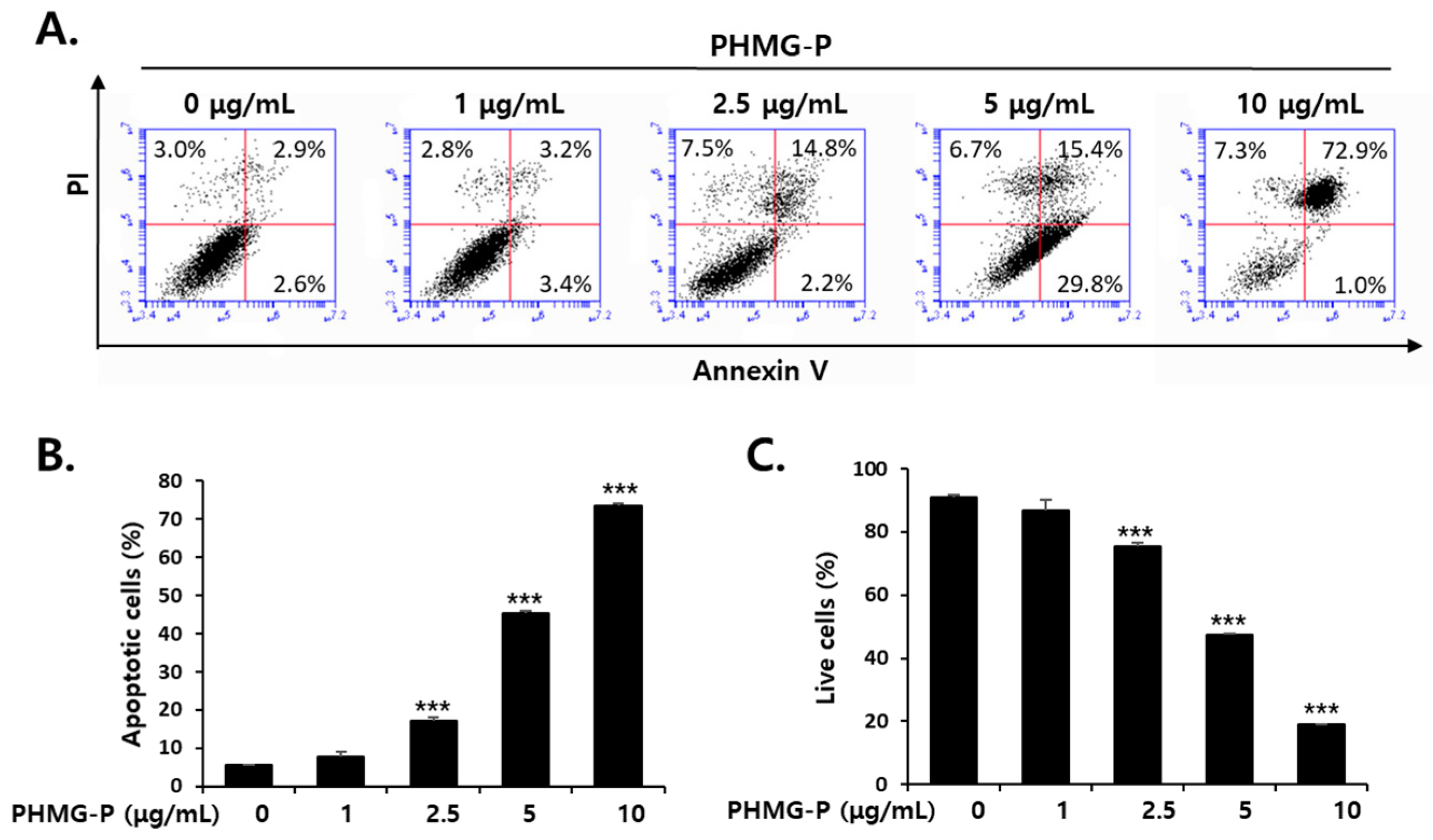
2.3. Fluorescence-Activated Cell Sorting Analysis of Apoptosis
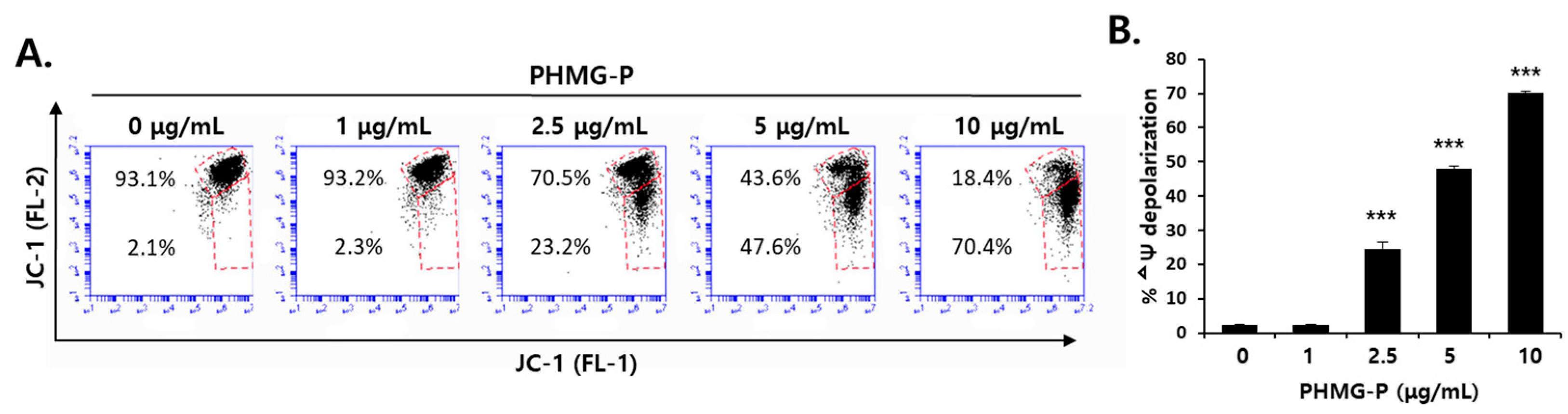
2.4. FACS Analysis of Mitochondrial Membrane Potential
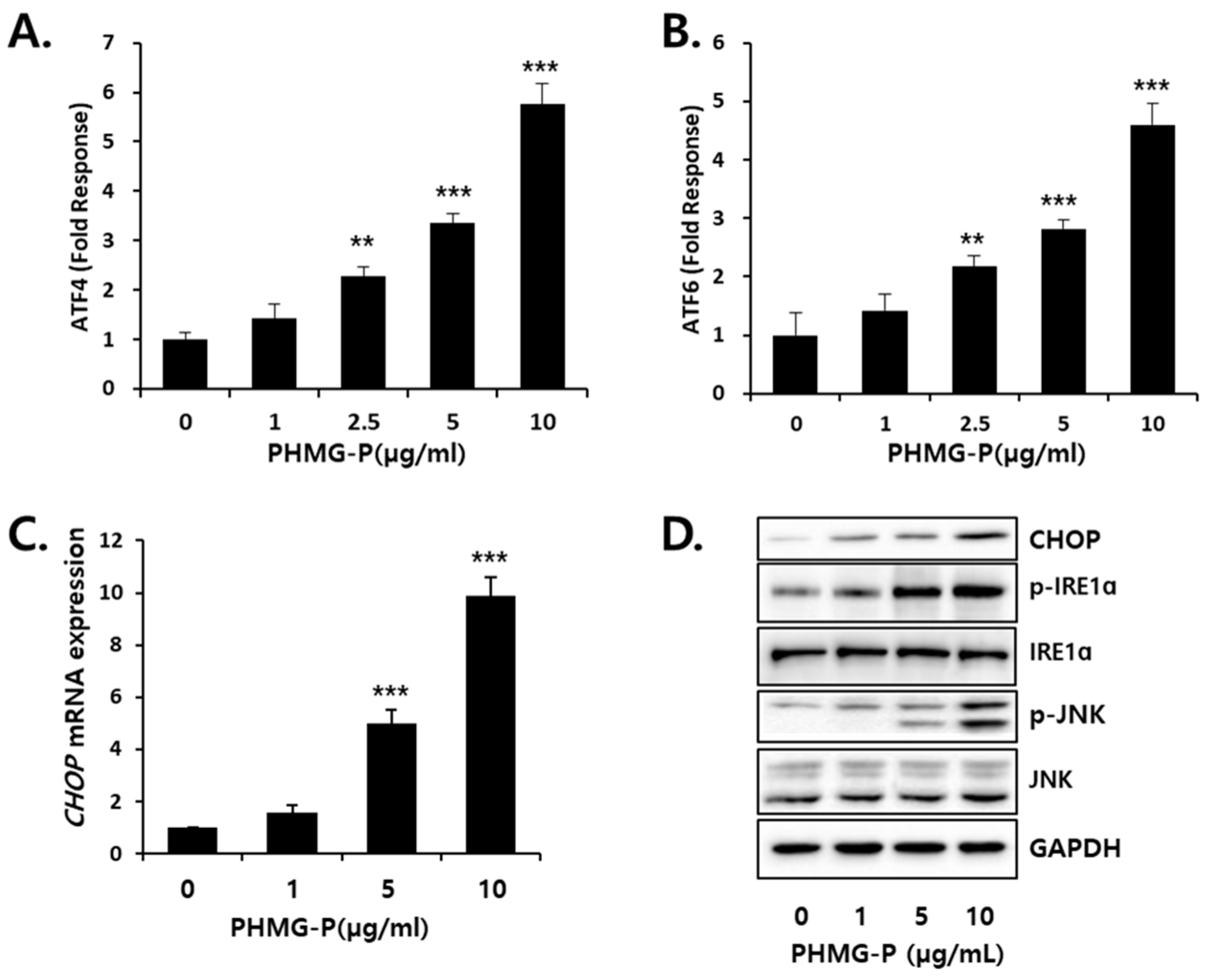
2.5. Luciferase Reporter Assay for ER Stress Response
2.6. Western Blotting Analysis
2.7. Caspase-3 Activity
2.8. Real-Time Reverse Transcription-Polymerase Chain Reaction
2.9. Statistical Analysis
3. Results
3.1. PHMG-P Cytotoxicity in Liver Cells
3.2. Apoptosis Induced by PHMG-P in HepG2 Cells
3.3. ER Stress Induced by PHMG-P in HepG2 Cells
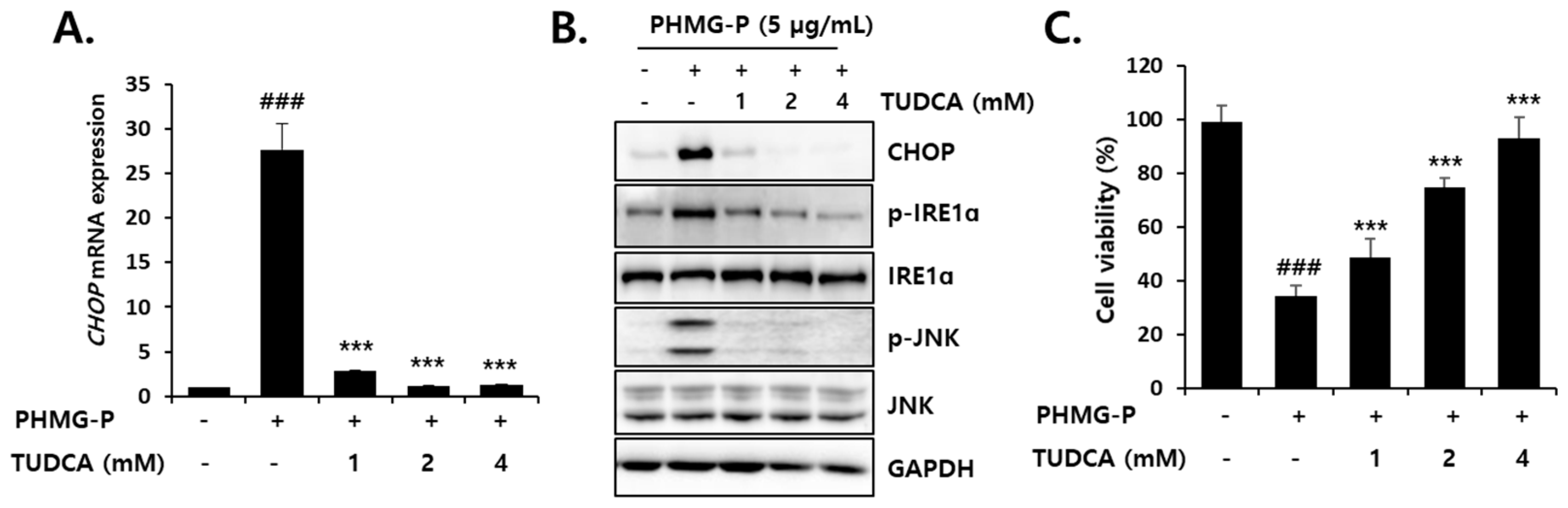
3.4. Inhibition of PHMG-P-Induced ER Stress by TUDCA
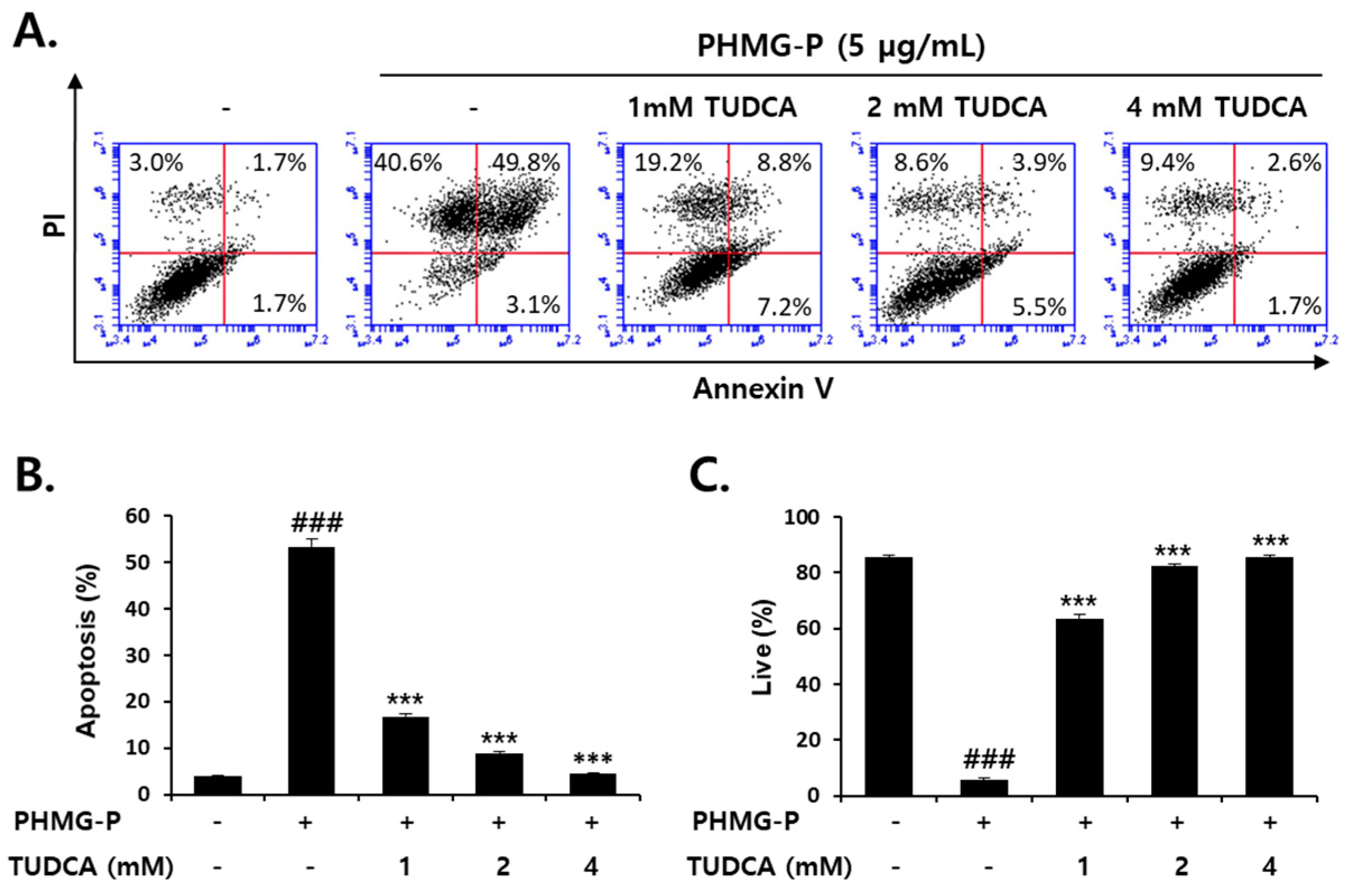
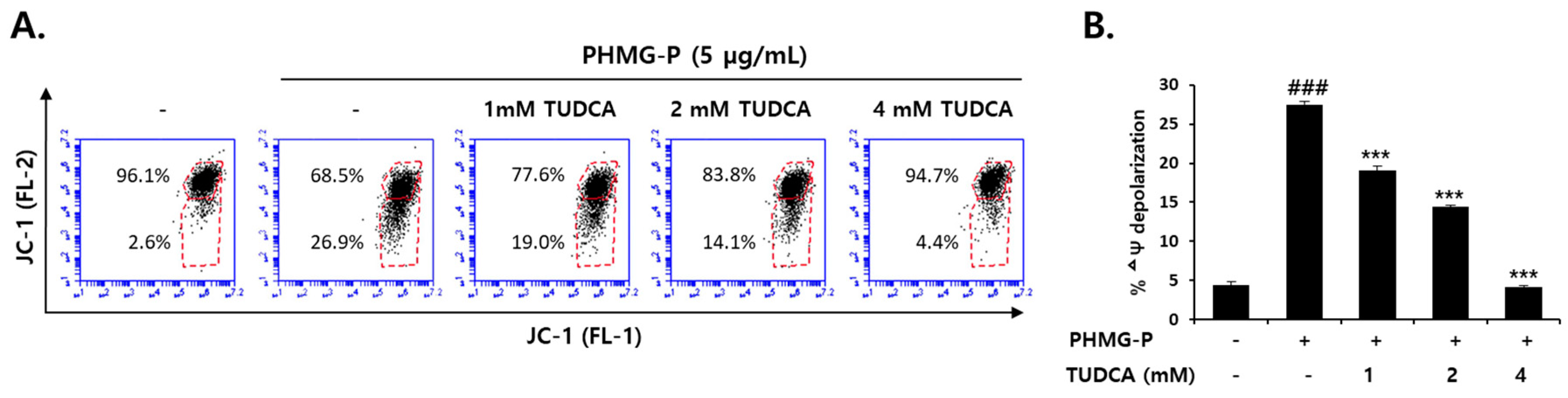
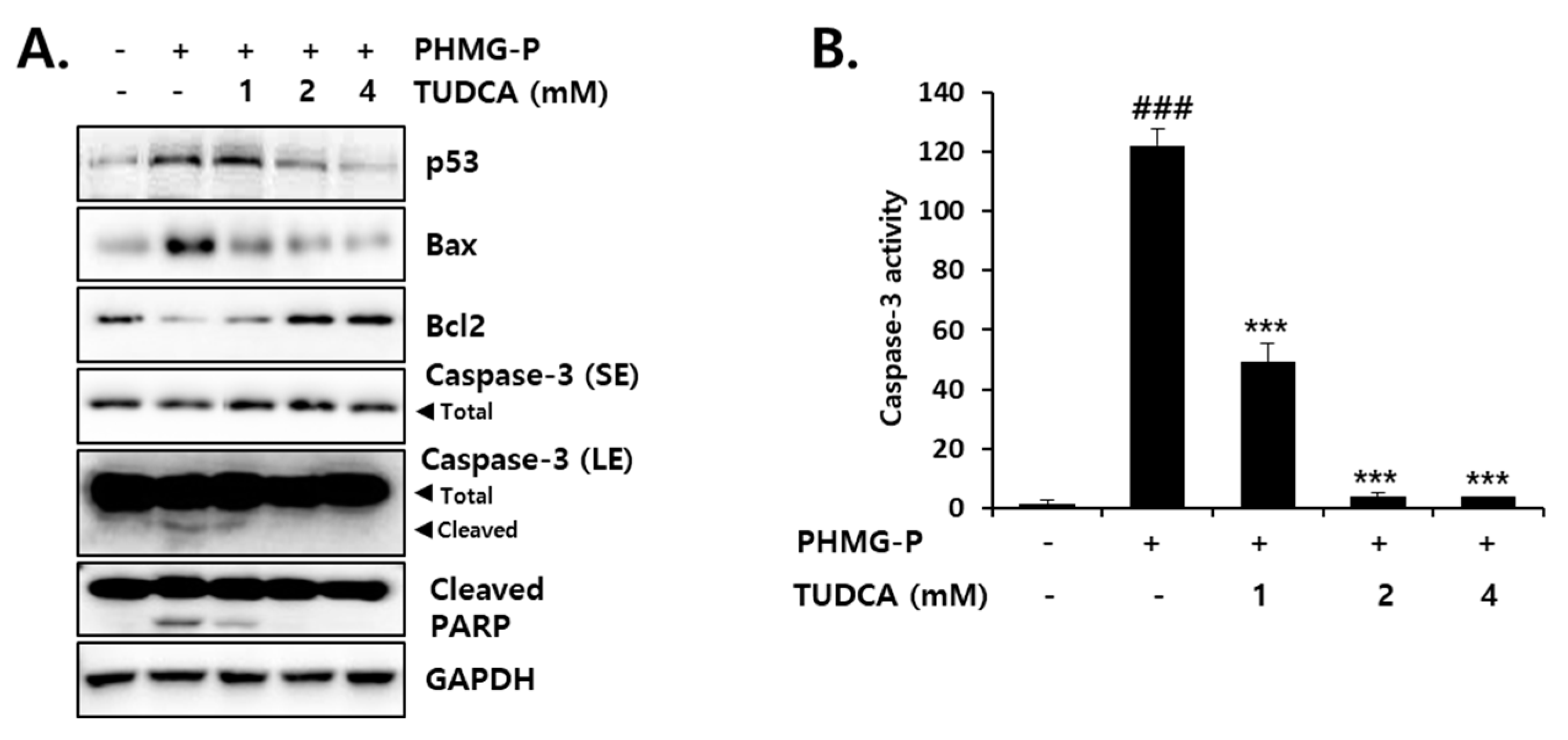
3.5. Inhibition of PHMG-P-Induced Apoptosis by TUDCA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kim, H.R.; Hwang, G.W.; Naganuma, A.; Chung, K.H. Adverse health effects of humidifier disinfectants in Korea: lung toxicity of polyhexamethylene guanidine phosphate. J. Toxicol. Sci. 2016, 41, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Park, S.J.; Kim, S.; Lee, K.; Chang, J. Lung fibroblasts may play an important role in clearing apoptotic bodies of bronchial epithelial cells generated by exposure to PHMG-P-containing solution. Toxicol. Lett. 2018, 286, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Jung, K.J.; Yoon, S.J.; Lee, K.; Kim, B. Polyhexamethyleneguanidine phosphate induces cytotoxicity through disruption of membrane integrity. Toxicology 2019, 414, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Lee, K.; Park, C.W.; Song, J.A.; Shin, D.Y.; Park, Y.J.; Chung, K.H. Polyhexamethylene guanidine phosphate aerosol particles induce pulmonary inflammatory and fibrotic responses. Arch. Toxicol. 2016, 90, 617–632. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Misawa, Y.; Miyamoto, H.; Makino, M.; Nagai, K.; Shiraishi, T.; Nakagawa, Y.; Yamato, S.; Tachikawa, E.; Zenda, H. A comparative study of characteristics of current-type and conventional-type cationic bactericides. Biol. Pharm. Bull. 2001, 24, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Oule, M.K.; Azinwi, R.; Bernier, A.M.; Kablan, T.; Maupertuis, A.M.; Mauler, S.; Nevry, R.K.; Dembele, K.; Forbes, L.; Diop, L. Polyhexamethylene guanidine hydrochloride-based disinfectant: A novel tool to fight meticillin-resistant Staphylococcus aureus and nosocomial infections. J. Med. Microbiol. 2008, 57, 1523–1528. [Google Scholar] [CrossRef]
- Oule, M.K.; Quinn, K.; Dickman, M.; Bernier, A.M.; Rondeau, S.; DeMoissac, D.B.; Boisvert, A.; Diop, L. Akwaton, polyhexamethylene-guanidine hydrochloride-based sporicidal disinfectant: A novel tool to fight bacterial spores and nosocomial infections. J. Med. Microbiol. 2012, 61, 1421–1427. [Google Scholar] [CrossRef]
- Vitt, A.; Sofrata, A.; Slizen, V.; Sugars, R.V.; Gustafsson, A.; Gudkova, E.I.; Kazeko, L.A.; Ramberg, P.; Buhlin, K. Antimicrobial activity of polyhexamethylene guanidine phosphate in comparison to chlorhexidine using the quantitative suspension method. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 36. [Google Scholar] [CrossRef]
- Broxton, P.; Woodcock, P.M.; Heatley, F.; Gilbert, P. Interaction of some polyhexamethylene biguanides and membrane phospholipids in Escherichia coli. J. Appl. Bacteriol. 1984, 57, 115–124. [Google Scholar] [CrossRef]
- Ikeda, T.; Ledwith, A.; Bamford, C.H.; Hann, R.A. Interaction of a polymeric biguanide biocide with phospholipid membranes. Biochim. Biophys. Acta. 1984, 769, 57–66. [Google Scholar] [CrossRef]
- Ikeda, T.; Tazuke, S.; Watanabe, M. Interaction of biologically active molecules with phospholipid membranes. I. Fluorescence depolarization studies on the effect of polymeric biocide bearing biguanide groups in the main chain. Biochim. Biophys. Acta. 1983, 735, 380–386. [Google Scholar] [CrossRef]
- Carmona-ibeiro, A.M.; deMeloCarrasco, L.D. Cationic antimicrobial polymers and their assemblies. Int. J. Mol. Sci. 2013, 14, 9906–9946. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.E.; Lee, J.Y.; Lee, C.H.; Mushtaq, S.; Song, H.Y.; Song, L.; Choi, S.J.; Lee, K.; Jeon, J. Quantification of inhaled aerosol particles composed of toxic household disinfectant using radioanalytical method. Chemosphere 2018, 207, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, W.; Kim, Y.B.; Kim, B.; Lee, K. Time course of polyhexamethyleneguanidine phosphate-induced lung inflammation and fibrosis in mice. Toxicol. Appl. Pharmacol. 2018, 345, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Song, J.A.; Park, H.J.; Yang, M.J.; Jung, K.J.; Yang, H.S.; Song, C.W.; Lee, K. Polyhexamethyleneguanidine phosphate induces severe lung inflammation, fibrosis, and thymic atrophy. Food. Chem. Toxicol. 2014, 69, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Lachenmeier, D.W.; Monakhova, Y.B.; Rehm, J. Influence of unrecorded alcohol consumption on liver cirrhosis mortality. World. J. Gastroenterol. 2014, 20, 7217–7222. [Google Scholar] [CrossRef] [PubMed]
- Ostapenko, Y.N.; Brusin, K.M.; Zobnin, Y.V.; Shchupak, A.Y.; Vishnevetskiy, M.K.; Sentsov, V.G.; Novikova, O.V.; Alekseenko, S.A.; Lebed’ko, O.A.; Puchkov, Y.B. Acute cholestatic liver injury caused by polyhexamethyleneguanidine hydrochloride admixed to ethyl alcohol. Clin. Toxicol. 2011, 49, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Asiedu-Gyekye, I.J.; Mahmood, S.A.; Awortwe, C.; Nyarko, A.K. A preliminary safety evaluation of polyhexamethylene guanidine hydrochloride. Int. J. Toxicol. 2014, 33, 523–531. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, H.H.; Cho, K.H. Acute cardiovascular toxicity of sterilizers, PHMG, and PGH: severe inflammation in human cells and heart failure in zebrafish. Cardiovasc. Toxicol. 2013, 13, 148–160. [Google Scholar] [CrossRef]
- Jung, H.N.; Zerin, T.; Podder, B.; Song, H.Y.; Kim, Y.S. Cytotoxicity and gene expression profiling of polyhexamethylene guanidine hydrochloride in human alveolar A549 cells. Toxicol. In Vitro 2014, 28, 684–692. [Google Scholar] [CrossRef]
- Shin, D.Y.; Jeong, M.H.; Bang, I.J.; Kim, H.R.; Chung, K.H. MicroRNA regulatory networks reflective of polyhexamethylene guanidine phosphate-induced fibrosis in A549 human alveolar adenocarcinoma cells. Toxicol. Lett. 2018, 287, 49–58. [Google Scholar] [CrossRef]
- Kim, H.R.; Shin, D.Y.; Chung, K.H. The role of NF-kappaB signaling pathway in polyhexamethylene guanidine phosphate induced inflammatory response in mouse macrophage RAW264.7 cells. Toxicol. Lett. 2015, 233, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Frasch, S.C.; Warner, M.L.; Henson, P.M. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell. Death. Differ. 1998, 5, 551–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, G.; Kroemer, G. Mechanisms of apoptotic phosphatidylserine exposure. Cell. Res. 2013, 23, 1247–1248. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.E.; Gabai, V.L. Cell Death and Survival Assays. Methods. Mol. Biol. 2018, 1709, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell. Death. Dis. 2012, 3, e430. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell. Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS. J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bras, M.; Queenan, B.; Susin, S.A. Programmed cell death via mitochondria: different modes of dying. Biochemistry 2005, 70, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Tolkovsky, A.M.; Zakeri, Z. Elan vital, elan letal: one life but multiple deaths. Cell. Death. Differ. 2008, 15, 1089–1090. [Google Scholar] [CrossRef] [PubMed]
- Savitskaya, M.A.; Onishchenko, G.E. Mechanisms of Apoptosis. Biochemistry 2015, 80, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein. Cell. 2014, 5, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foufelle, F.; Fromenty, B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol. Res. Perspect. 2016, 4, e00211. [Google Scholar] [CrossRef] [PubMed]
- Kleizen, B.; Braakman, I. Protein folding and quality control in the endoplasmic reticulum. Curr. Opin. Cell. Biol. 2004, 16, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold. Spring. Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Deniaud, A.; Sharafeldein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. ER stress: Can the liver cope? J. Hepatol. 2006, 45, 321–333. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell. Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell. Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Ma, Y.; Brewer, J.W.; Diehl, J.A.; Hendershot, L.M. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Okada, T.; Yoshida, H.; Akazawa, R.; Negishi, M.; Mori, K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366, 585–594. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Mak, B.C.; Wang, Q.; Laschinger, C.; Lee, W.; Ron, D.; Harding, H.P.; Kaufman, R.J.; Scheuner, D.; Austin, R.C.; McCulloch, C.A. Novel function of PERK as a mediator of force-induced apoptosis. J. Biol. Chem. 2008, 283, 23462–23472. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Oyadomari, S.; Mori, K.; Mori, M. Nitric oxide-induced apoptosis in RAW264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J. Biol. Chem. 2002, 277, 12343–12350. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Nutt, L.K.; Pataer, A.; Pahler, J.; Fang, B.; Roth, J.; McConkey, D.J.; Swisher, S.G. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 2002, 277, 9219–9225. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Gavin, J.; Quilty, F.; Majer, F.; Gilsenan, G.; Byrne, A.M.; Long, A.; Radics, G.; Gilmer, J.F. A fluorescent analogue of tauroursodeoxycholic acid reduces ER stress and is cytoprotective. Bioorg. Med. Chem. Lett. 2016, 26, 5369–5372. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 2002, 36, 592–601. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhong, J.J.; Jin, J.F.; Yin, X.M.; Miao, H. Tauroursodeoxycholate, a chemical chaperone, prevents palmitate-induced apoptosis in pancreatic beta-cells by reducing ER stress. Exp. Clin. Endocrinol. Diabetes. 2013, 121, 43–47. [Google Scholar] [CrossRef]
- Hou, Y.; Yang, H.; Cui, Z.; Tai, X.; Chu, Y.; Guo, X. Tauroursodeoxycholic acid attenuates endoplasmic reticulum stress and protects the liver from chronic intermittent hypoxia induced injury. Exp. Ther. Med. 2017, 14, 2461–2468. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xia, Y.; Li, B.; Xu, H.; Wang, C.; Liu, Y.; Li, Y.; Li, C.; Gao, N.; Li, L. Induction of ER stress-mediated apoptosis by ceramide via disruption of ER Ca(2+) homeostasis in human adenoid cystic carcinoma cells. Cell. Biosci. 2014, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Gani, A.R.; Uppala, J.K.; Ramaiah, K.V. Tauroursodeoxycholic acid prevents stress induced aggregation of proteins in vitro and promotes PERK activation in HepG2 cells. Arch. Biochem. Biophys. 2015, 568, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Uppala, J.K.; Gani, A.R.; Ramaiah, K.V.A. Chemical chaperone, TUDCA unlike PBA, mitigates protein aggregation efficiently and resists ER and non-ER stress induced HepG2 cell death. Sci. Rep. 2017, 7, 3831. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequences | |
|---|---|---|
| CHOP | F: CAGAACCAGCAGAGGTCACA | R: AGCTGTGCCACTTTCCTTTC |
| 18S | F: CAGCCACCCGAGATTGAGCA | R: TAGTAGCGACGGGCGGTGTG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Kwon, D.; Lee, S.; Ki, S.H.; Jeong, H.G.; Hong, J.T.; Lee, Y.-H.; Jung, Y.-S. Polyhexamethyleneguanidine Phosphate-Induced Cytotoxicity in Liver Cells Is Alleviated by Tauroursodeoxycholic Acid (TUDCA) via a Reduction in Endoplasmic Reticulum Stress. Cells 2019, 8, 1023. https://doi.org/10.3390/cells8091023
Kim SH, Kwon D, Lee S, Ki SH, Jeong HG, Hong JT, Lee Y-H, Jung Y-S. Polyhexamethyleneguanidine Phosphate-Induced Cytotoxicity in Liver Cells Is Alleviated by Tauroursodeoxycholic Acid (TUDCA) via a Reduction in Endoplasmic Reticulum Stress. Cells. 2019; 8(9):1023. https://doi.org/10.3390/cells8091023
Chicago/Turabian StyleKim, Sou Hyun, Doyoung Kwon, Seunghyun Lee, Sung Hwan Ki, Hye Gwang Jeong, Jin Tae Hong, Yun-Hee Lee, and Young-Suk Jung. 2019. "Polyhexamethyleneguanidine Phosphate-Induced Cytotoxicity in Liver Cells Is Alleviated by Tauroursodeoxycholic Acid (TUDCA) via a Reduction in Endoplasmic Reticulum Stress" Cells 8, no. 9: 1023. https://doi.org/10.3390/cells8091023