Whole Transcriptome Analysis Identifies TNS4 as a Key Effector of Cetuximab and a Regulator of the Oncogenic Activity of KRAS Mutant Colorectal Cancer Cell Lines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Tumor Xenograft Mouse
2.2. RNA-seq Analysis
2.3. Cancer Cell Line Encyclopedia (CCLE) Data Analysis
2.4. The Cancer Genome Atlas (TCGA) Data Analysis
2.5. Gene Set Enrichment Analysis
2.6. Cell Culture and Reagents
2.7. Anchorage-Independent Growth Assay
2.8. Cell Growth Inhibition Assay
2.9. Western Blotting and Antibodies
2.10. RNAi Studies
2.11. Quantitative Real-Time PCR Analysis
3. Results
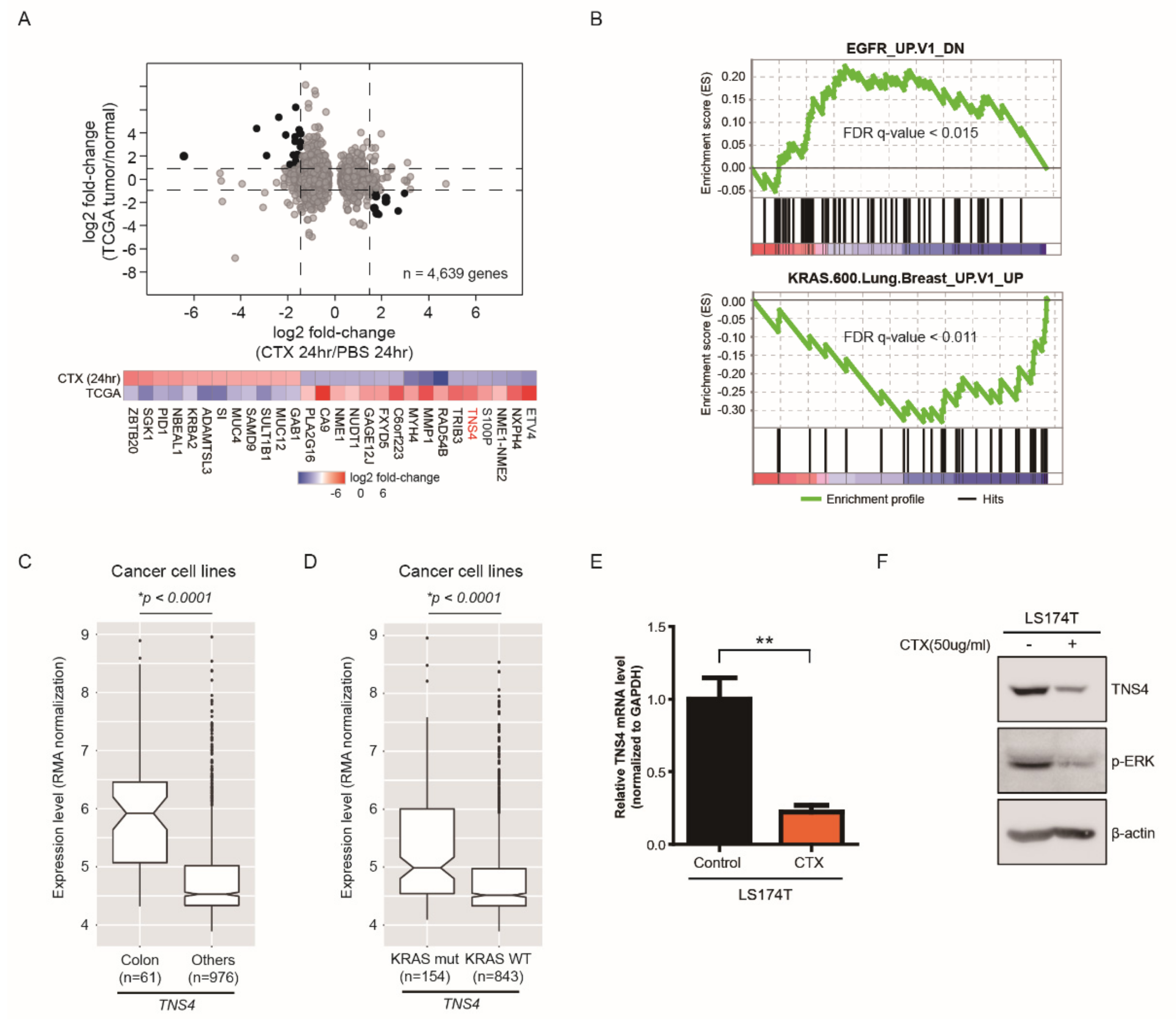
3.1. Whole Transcriptome Analysis Identified a Subset of Genes That Are Significantly Modulated by Cetuximab Treatment in Xenograft Mouse Tumors Induced by KRASG12D Mutation-Bearing LS174T CRC Cells
3.2. TNS4 is Significantly Downregulated by Cetuximab Treatment in a Subset of CRC Cell Lines
3.3. TNS4 Expression Is Associated with Oncogenic Potential of Colon Cancer Cell Lines
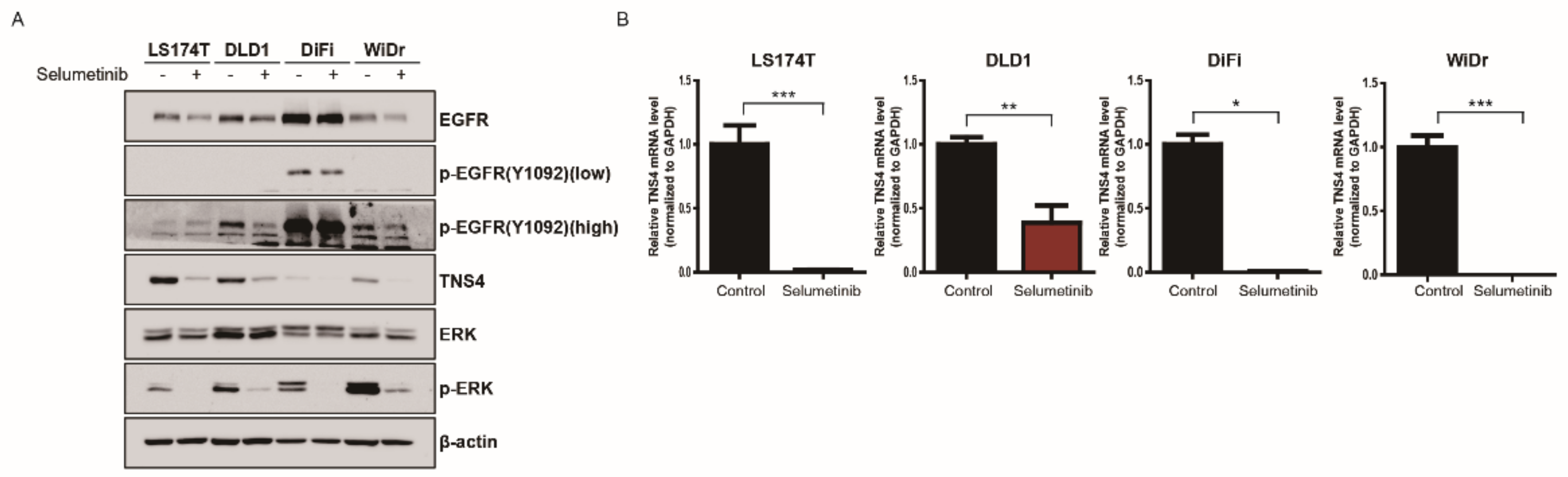
3.4. TNS4 Expression Is Regulated by MAP Kinase Pathway at a Transcriptional Level
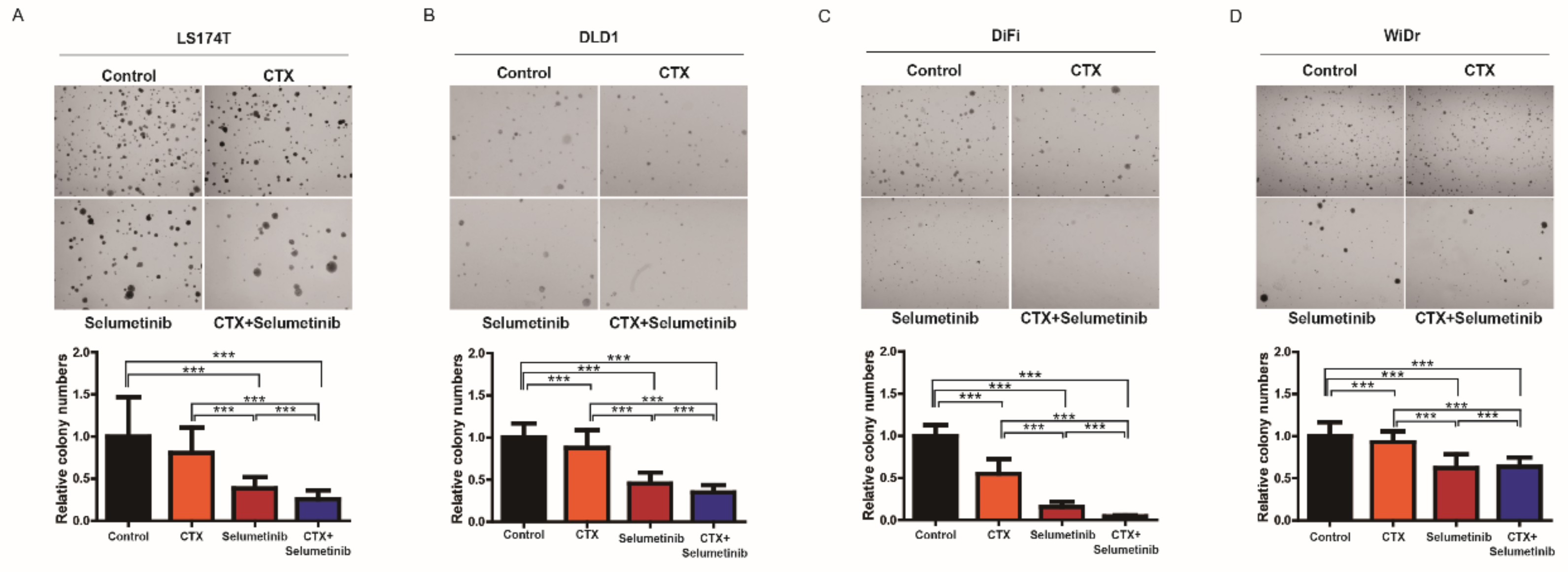
3.5. Combinatorial Treatment with Selumetinib Increases the Sensitivity of Cetuximab in a Subset of KRAS Mutation-Bearing Colorectal Cancer Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Ocvirk, J.; Brodowicz, T.; Wrba, F.; Ciuleanu, T.E.; Kurteva, G.; Beslija, S.; Koza, I.; Papai, Z.; Messinger, D.; Yilmaz, U.; et al. Cetuximab plus FOLFOX6 or FOLFIRI in metastatic colorectal cancer: CECOG trial. World J. Gastroenterol. 2010, 16, 3133–3143. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; Andre, T.; Chan, E.; Lordick, F.; Punt, C.J.; et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Siena, S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J. Clin. Oncol. 2010, 28, 1254–1261. [Google Scholar] [CrossRef]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Tam, I.Y.; Chung, L.P.; Suen, W.S.; Wang, E.; Wong, M.C.; Ho, K.K.; Lam, W.K.; Chiu, S.W.; Girard, L.; Minna, J.D.; et al. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin. Cancer Res. 2006, 12, 1647–1653. [Google Scholar] [CrossRef]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Kuwano, H.; Takahashi, T.; Mitsudomi, T. Mutations of the epidermal growth factor receptor gene in lung cancer: Biological and clinical implications. Cancer Res. 2004, 64, 8919–8923. [Google Scholar] [CrossRef]
- Marchetti, A.; Martella, C.; Felicioni, L.; Barassi, F.; Salvatore, S.; Chella, A.; Camplese, P.P.; Iarussi, T.; Mucilli, F.; Mezzetti, A.; et al. EGFR mutations in non-small-cell lung cancer: Analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J. Clin. Oncol. 2005, 23, 857–865. [Google Scholar] [CrossRef]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef]
- Irahara, N.; Baba, Y.; Nosho, K.; Shima, K.; Yan, L.; Dias-Santagata, D.; Iafrate, A.J.; Fuchs, C.S.; Haigis, K.M.; Ogino, S. NRAS mutations are rare in colorectal cancer. Diagn. Mol. Pathol. 2010, 19, 157–163. [Google Scholar] [CrossRef]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef]
- Lo Nigro, C.; Ricci, V.; Vivenza, D.; Granetto, C.; Fabozzi, T.; Miraglio, E.; Merlano, M.C. Prognostic and predictive biomarkers in metastatic colorectal cancer anti-EGFR therapy. World J. Gastroenterol. 2016, 22, 6944–6954. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Jonker, D.; Daneshmand, M.; Hanson, J.E.; O’Callaghan, C.J.; Marginean, C.; Zalcberg, J.R.; Simes, J.; Moore, M.J.; Tebbutt, N.C.; et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer--results from NCIC CTG/AGITG CO.17. Clin. Cancer Res. 2014, 20, 744–753. [Google Scholar] [CrossRef]
- Tian, S.; Simon, I.; Moreno, V.; Roepman, P.; Tabernero, J.; Snel, M.; van’t Veer, L.; Salazar, R.; Bernards, R.; Capella, G. A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut 2013, 62, 540–549. [Google Scholar] [CrossRef]
- Tan, C.; Du, X. KRAS mutation testing in metastatic colorectal cancer. World J. Gastroenterol. 2012, 18, 5171–5180. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Kruspig, B.; Monteverde, T.; Neidler, S.; Hock, A.; Kerr, E.; Nixon, C.; Clark, W.; Hedley, A.; Laing, S.; Coffelt, S.B.; et al. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, H.P.; Pranz, K.; Musteanu, M.; Grabner, B.; Hruschka, N.; Mohrherr, J.; Aigner, P.; Stiedl, P.; Brcic, L.; Laszlo, V.; et al. Afatinib restrains K-RAS-driven lung tumorigenesis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greulich, H.; Chen, T.H.; Feng, W.; Janne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef]
- Cho, J.; Kim, S.; Du, J.; Meyerson, M. Autophosphorylation of the carboxyl-terminal domain is not required for oncogenic transformation by lung-cancer derived EGFR mutants. Int. J. Cancer 2018, 143, 679–685. [Google Scholar] [CrossRef]
- Sawazaki, S.; Oshima, T.; Sakamaki, K.; Aoyama, T.; Sato, T.; Shiozawa, M.; Yoshikawa, T.; Rino, Y.; Imada, T.; Masuda, M. Clinical Significance of Tensin 4 Gene Expression in Patients with Gastric Cancer. In Vivo 2017, 31, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Cho, A.; Watanabe, H.; Choi, Y.L.; Aziz, M.; Kassner, M.; Joung, J.G.; Park, A.K.; Francis, J.M.; Bae, J.S.; et al. Integrated genomic approaches identify upregulation of SCRN1 as a novel mechanism associated with acquired resistance to erlotinib in PC9 cells harboring oncogenic EGFR mutation. Oncotarget 2016, 7, 13797–13809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ghamdi, S.; Albasri, A.; Cachat, J.; Ibrahem, S.; Muhammad, B.A.; Jackson, D.; Nateri, A.S.; Kindle, K.B.; Ilyas, M. Cten is targeted by Kras signalling to regulate cell motility in the colon and pancreas. PLoS ONE 2011, 6, e20919. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.H. Tensins. Curr. Biol. 2017, 27, R331–R332. [Google Scholar] [CrossRef] [PubMed]
- Albasri, A.; Al-Ghamdi, S.; Fadhil, W.; Aleskandarany, M.; Liao, Y.C.; Jackson, D.; Lobo, D.N.; Lo, S.H.; Kumari, R.; Durrant, L.; et al. Cten signals through integrin-linked kinase (ILK) and may promote metastasis in colorectal cancer. Oncogene 2011, 30, 2997–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.K.; Chiu, Y.T.; Sze, K.M.; Ng, I.O. Tensin4 is up-regulated by EGF-induced ERK1/2 activity and promotes cell proliferation and migration in hepatocellular carcinoma. Oncotarget 2015, 6, 20964–20976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, M.; Amit, I.; Citri, A.; Shay, T.; Carvalho, S.; Lavi, S.; Milanezi, F.; Lyass, L.; Amariglio, N.; Jacob-Hirsch, J.; et al. A reciprocal tensin-3-cten switch mediates EGF-driven mammary cell migration. Nat. Cell Biol. 2007, 9, 961–969. [Google Scholar] [CrossRef]
- Bennett, D.T.; Reece, T.B.; Foley, L.S.; Sjoberg, A.; Meng, X.; Fullerton, D.A.; Weyant, M.J. C-terminal tensin-like protein mediates invasion of human lung cancer cells and is regulated by signal transducer and activator of transcription 3. J. Thorac Cardiovasc. Surg. 2015, 149, 369–375. [Google Scholar] [CrossRef]
- Liao, Y.C.; Chen, N.T.; Shih, Y.P.; Dong, Y.; Lo, S.H. Up-regulation of C-terminal tensin-like molecule promotes the tumorigenicity of colon cancer through beta-catenin. Cancer Res. 2009, 69, 4563–4566. [Google Scholar] [CrossRef]
- Albasri, A.; Seth, R.; Jackson, D.; Benhasouna, A.; Crook, S.; Nateri, A.S.; Chapman, R.; Ilyas, M. C-terminal Tensin-like (CTEN) is an oncogene which alters cell motility possibly through repression of E-cadherin in colorectal cancer. J. Pathol. 2009, 218, 57–65. [Google Scholar] [CrossRef]
- Hung, S.Y.; Shih, Y.P.; Chen, M.; Lo, S.H. Up-regulated cten by FGF2 contributes to FGF2-mediated cell migration. Mol. Carcinog. 2014, 53, 787–792. [Google Scholar] [CrossRef]
- Hong, S.Y.; Shih, Y.P.; Li, T.; Carraway, K.L., 3rd; Lo, S.H. CTEN prolongs signaling by EGFR through reducing its ligand-induced degradation. Cancer Res. 2013, 73, 5266–5276. [Google Scholar] [CrossRef]
- Lamba, S.; Russo, M.; Sun, C.; Lazzari, L.; Cancelliere, C.; Grernrum, W.; Lieftink, C.; Bernards, R.; Di Nicolantonio, F.; Bardelli, A. RAF suppression synergizes with MEK inhibition in KRAS mutant cancer cells. Cell Rep. 2014, 8, 1475–1483. [Google Scholar] [CrossRef]
- Sun, C.; Hobor, S.; Bertotti, A.; Zecchin, D.; Huang, S.; Galimi, F.; Cottino, F.; Prahallad, A.; Grernrum, W.; Tzani, A.; et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014, 7, 86–93. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci. Transl. Med. 2014, 6, 224ra226. [Google Scholar] [CrossRef]
- Perera, D.; Venkitaraman, A.R. Oncogenic KRAS triggers MAPK-dependent errors in mitosis and MYC-dependent sensitivity to anti-mitotic agents. Sci. Rep. 2016, 6, 29741. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Kim, N.; Kang, K.; Kim, W.; Won, J.; Cho, J. Whole Transcriptome Analysis Identifies TNS4 as a Key Effector of Cetuximab and a Regulator of the Oncogenic Activity of KRAS Mutant Colorectal Cancer Cell Lines. Cells 2019, 8, 878. https://doi.org/10.3390/cells8080878
Kim S, Kim N, Kang K, Kim W, Won J, Cho J. Whole Transcriptome Analysis Identifies TNS4 as a Key Effector of Cetuximab and a Regulator of the Oncogenic Activity of KRAS Mutant Colorectal Cancer Cell Lines. Cells. 2019; 8(8):878. https://doi.org/10.3390/cells8080878
Chicago/Turabian StyleKim, Sujin, Nayoung Kim, Keunsoo Kang, Wonkyung Kim, Jonghwa Won, and Jeonghee Cho. 2019. "Whole Transcriptome Analysis Identifies TNS4 as a Key Effector of Cetuximab and a Regulator of the Oncogenic Activity of KRAS Mutant Colorectal Cancer Cell Lines" Cells 8, no. 8: 878. https://doi.org/10.3390/cells8080878