Lasting DNA Damage and Aberrant DNA Repair Gene Expression Profile Are Associated with Post-Chronic Cadmium Exposure in Human Bronchial Epithelial Cells

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Establishment of Chronic and Post-Chronic Cd-Exposed Human Bronchial Epithelial BEAS-2B Cell Models
2.2. Determination of Cell Proliferation and Cell Viability
2.3. Detection of DNA Damage and DNA Repair Capacity
2.4. Determination of Colony Formation Ability
2.5. Detection of Histone H2AFX and γH2AFX Expression
2.6. Assessment of Global DNA Repair-Related Gene Expression Profile
2.7. Analysis of Clinical Data Obtained from Online Databases
2.8. Statistical Analysis
3. Results
3.1. Establishment of a Post-Chronic Cd-Exposed BEAS-2B Cell Line (the “CR0” Cells)
3.2. Cd-Resistant Cells Retain Cd Tolerance Feature for an Extended Period of Time
3.3. Exposure to Cd Resulted in Cells Having Lasting DNA Damage and Reduced DNA Repair Capacity
3.4. Aberrant Expression of DNA Repair Gene Profile Is Associated with Post-Chronic Cd Exposure
3.5. Short-Term Cd Exposure Reveals Different DNA Repair Transcriptomic Landscapes in Normal and Cd-Exposed Cells
3.6. Clinical Database Analysis of Lung Cancer Samples with Different Smoking Histories
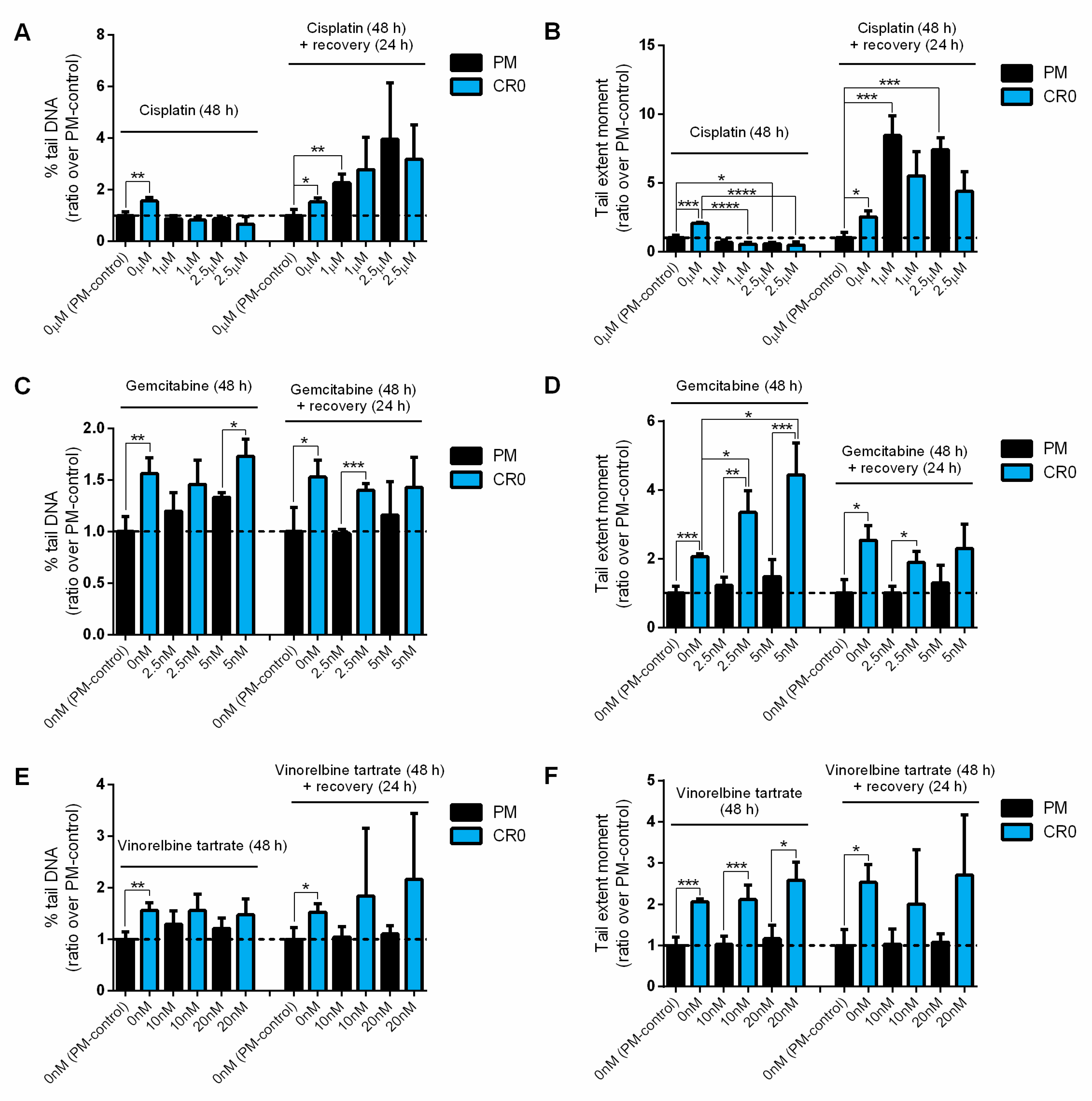
3.7. Post-Chronic Cd Exposure Resulted in Cells Having Reduced DNA Repair Capacity and More Susceptible to Chemotherapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hossain, M.B.; Vahter, M.; Concha, G.; Broberg, K. Low-level environmental cadmium exposure is associated with DNA hypomethylation in Argentinean women. Environ. Health Perspect. 2012, 120, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P. Mechanisms of cadmium carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.; Xu, Y.M.; Wu, D.D.; Lau, A.T.Y. Recent insights into human bronchial proteomics – how are we progressing and what is next? Expert. Rev. Proteomics 2018, 15, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Bertin, G.; Averbeck, D. Cadmium: Cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochimie 2006, 88, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Kumar, A.; Lal, A.; Pant, M. Cellular mechanisms of cadmium-induced toxicity: A review. Int. J. Environ. Health Res. 2014, 24, 378–399. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gu, X.; Zhang, X.; Kong, J.; Ding, N.; Qi, Y.; Zhang, Y.; Wang, J.; Huang, D. Cadmium delays non-homologous end joining (NHEJ) repair via inhibition of DNA-PKcs phosphorylation and downregulation of XRCC4 and Ligase IV. Mutat. Res. 2015, 779, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; Lei, Y.X.; Wang, C.X. Analysis of aberrant methylation in DNA repair genes during malignant transformation of human bronchial epithelial cells induced by cadmium. Toxicol. Sci. 2012, 125, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Youn, C.K.; Kim, S.H.; Lee, D.Y.; Song, S.H.; Chang, I.Y.; Hyun, J.W.; Chung, M.H.; You, H.J. Cadmium down-regulates human OGG1 through suppression of Sp1 activity. J. Biol. Chem. 2005, 280, 25185–25195. [Google Scholar] [CrossRef]
- Giaginis, C.; Gatzidou, E.; Theocharis, S. DNA repair systems as targets of cadmium toxicity. Toxicol. Appl. Pharmacol. 2006, 213, 282–290. [Google Scholar] [CrossRef]
- Andrew, A.S.; Warren, A.J.; Barchowsky, A.; Temple, K.A.; Klei, L.; Soucy, N.V.; O’Hara, K.A.; Hamilton, J.W. Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environ. Health Perspect. 2003, 111, 825–835. [Google Scholar] [CrossRef]
- Bartosiewicz, M.; Penn, S.; Buckpitt, A. Applications of gene arrays in environmental toxicology: Fingerprints of gene regulation associated with cadmium chloride, benzo(a)pyrene, and trichloroethylene. Environ. Health Perspect. 2001, 109, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Viau, M.; Gastaldo, J.; Bencokova, Z.; Joubert, A.; Foray, N. Cadmium inhibits non-homologous end-joining and over-activates the MRE11-dependent repair pathway. Mutat. Res. 2008, 654, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.X.; Lu, Q.; Shao, C.; He, C.C.; Lei, Z.N.; Lian, Y.Y. Expression profiles of DNA repair-related genes in rat target organs under subchronic cadmium exposure. Genet. Mol. Res. 2015, 14, 515–524. [Google Scholar] [CrossRef]
- Potts, R.J.; Bespalov, I.A.; Wallace, S.S.; Melamede, R.J.; Hart, B.A. Inhibition of oxidative DNA repair in cadmium-adapted alveolar epithelial cells and the potential involvement of metallothionein. Toxicology 2001, 161, 25–38. [Google Scholar] [CrossRef]
- Singh, K.P.; Kumari, R.; Pevey, C.; Jackson, D.; DuMond, J.W. Long duration exposure to cadmium leads to increased cell survival, decreased DNA repair capacity, and genomic instability in mouse testicular Leydig cells. Cancer Lett. 2009, 279, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Anetor, J.I. Rising environmental cadmium levels in developing countries: Threat to genome stability and health. Niger. J. Physiol. Sci. 2012, 27, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, T.S.; Martens, D.S.; Hara, A.; Plusquin, M.; Vangronsveld, J.; Roels, H.A.; Staessen, J.A. Association of total cancer and lung cancer with environmental exposure to cadmium: The meta-analytical evidence. Cancer Causes Control. 2015, 26, 1281–1288. [Google Scholar] [CrossRef]
- Cowley, M.; Skaar, D.A.; Jima, D.D.; Maguire, R.L.; Hudson, K.M.; Park, S.S.; Sorrow, P.; Hoyo, C. Effects of Cadmium Exposure on DNA Methylation at Imprinting Control Regions and Genome-Wide in Mothers and Newborn Children. Environ. Health Perspect. 2018, 126, 037003. [Google Scholar] [CrossRef]
- Liang, Z.-L.; Wu, D.-D.; Yao, Y.; Yu, F.-Y.; Yang, L.; Tan, H.W.; Hylkema, M.N.; Rots, M.G.; Xu, Y.M.; Lau, A.T.Y. Epiproteome profiling of cadmium-transformed human bronchial epithelial cells by quantitative histone post-translational modification-enzyme-linked immunosorbent assay. J. Appl. Toxicol. 2018, 38, 888–895. [Google Scholar] [CrossRef]
- Sanders, A.P.; Smeester, L.; Rojas, D.; DeBussycher, T.; Wu, M.C.; Wright, F.A.; Zhou, Y.H.; Laine, J.E.; Rager, J.E.; Swamy, G.K.; et al. Cadmium exposure and the epigenome: Exposure-associated patterns of DNA methylation in leukocytes from mother-baby pairs. Epigenetics 2014, 9, 212–221. [Google Scholar] [CrossRef]
- Xu, Y.M.; Wu, D.D.; Zheng, W.; Yu, F.Y.; Yang, F.; Yao, Y.; Zhou, Y.; Ching, Y.P.; Lau, A.T. Proteome profiling of cadmium-induced apoptosis by antibody array analyses in human bronchial epithelial cells. Oncotarget 2016, 7, 6146–6158. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Barisone, G.A.; Diaz, E. Focus formation: A cell-based assay to determine the oncogenic potential of a gene. J. Vis. Exp. 2014, 94, e51742. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lefever, S.; Vandesompele, J.; Speleman, F.; Pattyn, F. RTPrimerDB: The portal for real-time PCR primers and probes. Nucleic Acids Res. 2009, 37, D942–D945. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Firsanov, D.V.; Solovjeva, L.V.; Svetlova, M.P. H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clin. Epigenetics 2011, 2, 283–297. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yang, Y.S.; Francis, D.; Rogers, H.J.; Li, P.; Zhang, Q. Cadmium stress alters gene expression of DNA mismatch repair related genes in Arabidopsis seedlings. Chemosphere 2008, 73, 1138–1144. [Google Scholar] [CrossRef]
- Zhou, T.; Jia, X.; Chapin, R.E.; Maronpot, R.R.; Harris, M.W.; Liu, J.; Waalkes, M.P.; Eddy, E.M. Cadmium at a non-toxic dose alters gene expression in mouse testes. Toxicol. Lett. 2004, 154, 191–200. [Google Scholar] [CrossRef]
- Lin, Y.S.; Caffrey, J.L.; Chang, M.H.; Dowling, N.; Lin, J.W. Cigarette smoking, cadmium exposure, and zinc intake on obstructive lung disorder. Respir. Res. 2010, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, L.M.; dos Santos, G.C.; dos Santos, R.A.; Takahashi, C.S.; Bianchi Mde, L.; Antunes, L.M. Evaluation of curcumin and cisplatin-induced DNA damage in PC12 cells by the alkaline comet assay. Hum. Exp. Toxicol. 2010, 29, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Jones, N.J. Assessment of DNA interstrand crosslinks using the modified alkaline comet assay. Methods Mol. Biol. 2012, 817, 165–181. [Google Scholar] [PubMed]
- Faroon, O.; Ashizawa, A.; Wright, F.A.; Tucker, P.; Jenkins, K.; Ingerman, L.; Rudisill, C. Toxicological Profile for Cadmium; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2012.
- Hartwig, A. Mechanisms in cadmium-induced carcinogenicity: Recent insights. Biometals 2010, 23, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.; Brodeur, J. Toxic interactions among environmental pollutants: Corroborating laboratory observations with human experience. Environ. Health Perspect. 1994, 102, 11–17. [Google Scholar] [CrossRef]
- Adams, S.V.; Quraishi, S.M.; Shafer, M.M.; Passarelli, M.N.; Freney, E.P.; Chlebowski, R.T.; Luo, J.; Meliker, J.R.; Mu, L.; Neuhouser, M.L.; et al. Dietary cadmium exposure and risk of breast, endometrial, and ovarian cancer in the Women’s Health Initiative. Environ. Health Perspect. 2014, 122, 594–600. [Google Scholar] [CrossRef]
- Yang, X.F.; Ge, Y.M.; Zhang, H.T.; Ning, H.M.; Jiang, J.Q.; Qi, Y.H.; Wang, Z.L. Damaging effects of water-borne cadmium chloride on DNA of lung cells of immature mice. Genet. Mol. Res. 2012, 11, 4323–4329. [Google Scholar] [CrossRef]
- Chen, D.J.; Xu, Y.M.; Du, J.Y.; Huang, D.Y.; Lau, A.T. Cadmium induces cytotoxicity in human bronchial epithelial cells through upregulation of eIF5A1 and NF-kappaB. Biochem. Biophys. Res. Commun. 2014, 445, 95–99. [Google Scholar] [CrossRef]
- Waalkes, M.P. Cadmium carcinogenesis. Mutat. Res. 2003, 533, 107–120. [Google Scholar] [CrossRef]
- IARC Meeting of the IARC working group on beryllium, cadmium, mercury and exposures in the glass manufacturing industry. Scand. J. Work Environ. Health 1993, 19, 360–363. [CrossRef]
- Schmittgen, T.D.; Zakrajsek, B.A. Effect of experimental treatment on housekeeping gene expression: Validation by real-time, quantitative RT-PCR. J. Biochem. Biophys. Methods 2000, 46, 69–81. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.J.; Du, Y.W.; Hao, Y.Q.; Su, Z.Z.; Zhang, L.; Zhao, L.J.; Zhang, J. RNA-binding motif protein 5 inhibits the proliferation of cigarette smoke-transformed BEAS-2B cells through cell cycle arrest and apoptosis. Oncol. Rep. 2016, 35, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ji, Y.; Zhang, J.; Su, Z.; Liu, M.; Tong, J.; Ge, C.; Chen, T.; Li, J. Aberrant DNA methylation in radon and/or cigarette smoke-induced malignant transformation in BEAS-2B human lung cell line. J. Toxicol. Environ. Health Part A 2017, 80, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.J.; Xu, Y.M.; Lau, A.T.Y. Electronic cigarette: A recent update of its toxic effects on humans. J. Cell. Physiol. 2018, 233, 4466–4478. [Google Scholar] [CrossRef] [PubMed]
- Hart, B.A.; Potts, R.J.; Watkin, R.D. Cadmium adaptation in the lung—A double-edged sword? Toxicology 2001, 160, 65–70. [Google Scholar] [CrossRef]
- Lau, A.T.Y.; Tan, H.W.; Xu, Y.M. Epigenetic effects of dietary trace elements. Curr. Pharmacol. Rep. 2017, 3, 232–241. [Google Scholar] [CrossRef]
- Everson, T.M.; Punshon, T.; Jackson, B.P.; Hao, K.; Lambertini, L.; Chen, J.; Karagas, M.R.; Marsit, C.J. Cadmium-associated differential methylation throughout the placental genome: Epigenome-wide association study of two U.S. birth cohorts. Environ. Health Perspect. 2018, 126, 017010. [Google Scholar] [CrossRef]
- Reese, S.E.; Zhao, S.; Wu, M.C.; Joubert, B.R.; Parr, C.L.; Haberg, S.E.; Ueland, P.M.; Nilsen, R.M.; Midttun, O.; Vollset, S.E.; et al. DNA methylation score as a biomarker in newborns for sustained maternal smoking during pregnancy. Environ. Health Perspect. 2017, 125, 760–766. [Google Scholar] [CrossRef]
- Tellez-Plaza, M.; Tang, W.Y.; Shang, Y.; Umans, J.G.; Francesconi, K.A.; Goessler, W.; Ledesma, M.; Leon, M.; Laclaustra, M.; Pollak, J.; et al. Association of global DNA methylation and global DNA hydroxymethylation with metals and other exposures in human blood DNA samples. Environ. Health Perspect. 2014, 122, 946–954. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym | DNA Repair Pathways | No. of Genes 1 | Gene Name (UniProtKB accession ID) |
|---|---|---|---|
| Major DNA Repair Pathways | |||
| BER | Base excision repair | 38 | APEX1 (P27695); CCNO (P22674); ERCC1 (P07992); ERCC4 (Q92889); ERCC5 (P28715); ERCC6 (Q03468); FEN1 (P39748); GADD45A (P24522); HUS1 (O60921); LIG1 (P18858); LIG3 (P49916); MBD4 (O95243); MPG (P29372); MUTYH (Q9UIF7); NEIL1 (Q96FI4); NEIL2 (Q969S2); NEIL3 (Q8TAT5); NTHL1 (P78549); OGG1 (O15527); PARP1 (P09874); PARP2 (Q9UGN5); PARP3 (Q9Y6F1); PCNA (P12004); POLB (P06746); POLD3 (Q15054); RAD23A (P54725); RAD23B (P54727); RAD52 (P43351); RPA1 (P27694); RPA2 (P15927); RPA3 (P35244); SIRT1 (Q96EB6); SMUG1 (Q53HV7); TDG (Q13569); TP53 (P04637); UNG (P13051); WRN (Q14191); XRCC1 (P18887) |
| NER | Nucleotide excision repair | 37 | ATR (Q13535); ATXN3 (P54252); CCNH (P51946); CDK7 (P50613); CETN2 (P41208); DDB1 (Q16531); DDB2 (Q92466); ERCC1 (P07992); ERCC2 (P18074); ERCC3 (P19447); ERCC4 (Q92889); ERCC5 (P28715); ERCC6 (Q03468); ERCC8 (Q13216); GADD45A (P24522); GTF2H1 (P32780); GTF2H2 (Q13888); GTF2H3 (Q13889); GTF2H4 (Q92759); LIG1 (P18858); LIG3 (P49916); MMS19 (Q96T76); NTHL1 (P78549); OGG1 (O15527); PCNA (P12004); POLD3 (Q15054); RAD23A (P54725); RAD23B (P54727); RFC1 (P35251); RPA1 (P27694); RPA2 (P15927); RPA3 (P35244); SIRT1 (Q96EB6); TP53 (P04637); XAB2 (Q9HCS7); XPA (P23025); XRCC1 (P18887) |
| MMR | Mismatch repair | 19 | ABL1 (P00519); EXO1 (Q9UQ84); MLH1 (P40692); MLH3 (Q9UHC1); MSH2 (P43246); MSH3 (P20585); MSH4 (O15457); MSH5 (O43196); MSH6 (P52701); PCNA (P12004); PMS1 (P54277); PMS2 (P54278); POLD3 (Q15054); RFC1 (P35251); RPA1 (P27694); RPA2 (P15927); RPA3 (P35244); TDG (Q13569); TREX1 (Q9NSU2) |
| HRR | Homologous recombinational repair | 49 | ATM (Q13315); ATR (Q13535); BLM (P54132); BRCA1 (P38398); BRCA2 (P51587); BRIP1 (Q9BX63); CHEK1 (O14757); DMC1 (Q14565); ERCC1 (P07992); ERCC4 (Q92889); ERCC6 (Q03468); FANCA (O15360); FANCB (Q8NB91); FANCF (Q9NPI8); FANCG (O15287); FEN1 (P39748); GADD45A (P24522); H2AFX (P16104); HUS1 (O60921); LIG3 (P49916); MDC1 (Q14676); MRE11 (P49959); MSH2 (P43246); MSH4 (O15457); MSH5 (O43196); MSH6 (P52701); NBN (O60934); PARP1 (P09874); PARP2 (Q9UGN5); PARP3 (Q9Y6F1); RAD17 (O75943); RAD21 (O60216); RAD50 (Q92878); RAD51 (Q06609); RAD51B (O15315); RAD51C (O43502); RAD51D (O75771); RAD52 (P43351); RAD54B (Q9Y620); RAD54L (Q92698); REV1 (Q9UBZ9); RPA1 (P27694); RPA2 (P15927); RPA3 (P35244); SIRT1 (Q96EB6); TP53BP1 (Q12888); WRN (Q14191); XRCC1 (P18887); XRCC2 (O43543) |
| NHEJ | Non-homologous end-joining | 22 | ATM (Q13315); ATP23 (Q9Y6H3); BRCA1 (P38398); ERCC1 (P07992); ERCC4 (Q92889); LIG4 (P49917); MDC1 (Q14676); MLH1 (P40692); MRE11 (P49959); NBN (O60934); PARP1 (P09874); PARP2 (Q9UGN5); PARP3 (Q9Y6F1); PRKDC (P78527); RAD50 (Q92878); SIRT1 (Q96EB6); TP53BP1 (Q12888); WRN (Q14191); XRCC1 (P18887); XRCC4 (Q13426); XRCC5 (P13010); XRCC6 (P12956) |
| Other DNA Repair Pathways | |||
| POL | Polymerases | 4 | PCNA (P12004); POLB (P06746); POLD3 (Q15054); REV1 (Q9UBZ9) |
| DRD | Direct reversal of damage | 3 | ALKBH1 (Q13686); ALKBH3 (Q96Q83); MGMT (P16455) |
| SMNP | Sanitization/modulation of nucleotide pools | 3 | DUT (P33316); NUDT1 (P36639); RRM2B (Q7LG56) |
| FA | Fanconi anemia | 9 | BRCA1 (P38398); BRCA2 (P51587); BRIP1 (Q9BX63); FANCA (O15360); FANCB (Q8NB91); FANCF (Q9NPI8); FANCG (O15287); RAD51 (Q06609); RAD51C (O43502) |
| TLM | Telomere maintenance | 26 | ATR (Q13535); BRCA1 (P38398); BRCA2 (P51587); ERCC1 (P07992); ERCC4 (Q92889); FEN1 (P39748); MRE11 (P49959); NBN (O60934); PARP1 (P09874); PARP3 (Q9Y6F1); PCNA (P12004); POLD3 (Q15054); RAD50 (Q92878); RAD51 (Q06609); RAD51C (O43502); RAD51D (O75771); RFC1 (P35251) RPA1 (P27694); RPA2 (P15927); RPA3 (P35244); SIRT1 (Q96EB6); TP53 (P04637); WRN (Q14191); XRCC1 (P18887); XRCC5 (P13010); XRCC6 (P12956) |
| TLS | Translesion synthesis | 3 | PCNA (P12004); POLD3 (Q15054); REV1 (Q9UBZ9) |
| CSM | Chromatin structure and modification | 16 | CHEK1 (O14757); ERCC6 (Q03468); FEN1 (P39748); H2AFX (P16104); LIG3 (P49916); PCNA (P12004); RAD17 (O75943); RAD21 (O60216); RAD51 (Q06609); RAD51C (O43502); RPA1 (P27694); RPA2 (P15927); RPA3 (P35244); SIRT1 (Q96EB6); TP53 (P04637); XRCC1 (P18887) |
| MMEJR | Microhomology-mediated end-joining repair | 6 | FEN1 (P39748); LIG3 (P49916); MRE11 (P49959); NBN (O60934); PARP1 (P09874); XRCC1 (P18887) |
| SDA | Genes defective in diseases associated with sensitivity to DNA-damaging agents | 7 | ATM (Q13315); ATXN3 (P54252); BLM (P54132); ERCC6 (Q03468); ERCC8 (Q13216); NBN (O60934); WRN (Q14191) |
| UBM | Ubiquitination and modification (Rad6 pathways) | 6 | RAD18 (Q9NS91); SHPRH (Q149N8); UBE2A (P49459); UBE2B (P63146); UBE2N (P61088); UBE2V2 (Q15819) |
| OTHER | Other DNA damage-related | 4 | CRY1 (Q16526); SUMO1 (P63165); TOP3A (Q13472); TOP3B (O95985) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, H.W.; Liang, Z.-L.; Yao, Y.; Wu, D.-D.; Mo, H.-Y.; Gu, J.; Chiu, J.-F.; Xu, Y.-M.; Lau, A.T.Y. Lasting DNA Damage and Aberrant DNA Repair Gene Expression Profile Are Associated with Post-Chronic Cadmium Exposure in Human Bronchial Epithelial Cells. Cells 2019, 8, 842. https://doi.org/10.3390/cells8080842
Tan HW, Liang Z-L, Yao Y, Wu D-D, Mo H-Y, Gu J, Chiu J-F, Xu Y-M, Lau ATY. Lasting DNA Damage and Aberrant DNA Repair Gene Expression Profile Are Associated with Post-Chronic Cadmium Exposure in Human Bronchial Epithelial Cells. Cells. 2019; 8(8):842. https://doi.org/10.3390/cells8080842
Chicago/Turabian StyleTan, Heng Wee, Zhan-Ling Liang, Yue Yao, Dan-Dan Wu, Hai-Ying Mo, Jiang Gu, Jen-Fu Chiu, Yan-Ming Xu, and Andy T. Y. Lau. 2019. "Lasting DNA Damage and Aberrant DNA Repair Gene Expression Profile Are Associated with Post-Chronic Cadmium Exposure in Human Bronchial Epithelial Cells" Cells 8, no. 8: 842. https://doi.org/10.3390/cells8080842