Glycosylation in the Tumor Microenvironment: Implications for Tumor Angiogenesis and Metastasis


1
Center for Biomedical Mass Spectrometry, Department of Biochemistry, Boston University School of Medicine, Boston, MA 02118, USA
2
Department of Pathology and Laboratory Medicine, Boston University School of Medicine, Boston, MA 02118, USA
*
Authors to whom correspondence should be addressed.
Cells 2019, 8(6), 544; https://doi.org/10.3390/cells8060544
Submission received: 14 May 2019
/
Revised: 31 May 2019
/
Accepted: 1 June 2019
/
Published: 5 June 2019
Abstract
:Just as oncogene activation and tumor suppressor loss are hallmarks of tumor development, emerging evidence indicates that tumor microenvironment-mediated changes in glycosylation play a crucial functional role in tumor progression and metastasis. Hypoxia and inflammatory events regulate protein glycosylation in tumor cells and associated stromal cells in the tumor microenvironment, which facilitates tumor progression and also modulates a patient’s response to anti-cancer therapeutics. In this review, we highlight the impact of altered glycosylation on angiogenic signaling and endothelial cell adhesion, and the critical consequences of these changes in tumor behavior.
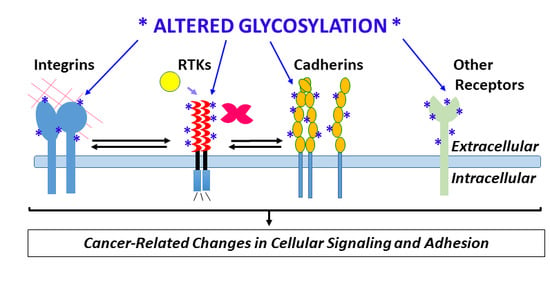
1. Introduction
A thin layer of endothelial cells lines the interior surfaces of blood and lymphatic vessels, releases signals that control vascular relaxation and contraction, secretes factors that regulate blood clotting, and plays an important role in immune function and platelet adhesion. The relationship between tumor cells and endothelial cells is complex. To sustain rapid cellular proliferation and a high metabolic rate, solid tumors develop a vascular network that fulfills tumors’ need for nutrients and oxygen and also aids in the removal of metabolic waste products [1]. In rapidly growing tumors, an angiogenic switch, often triggered by hypoxia-induced expression of vascular endothelial growth factor (VEGF) and other angiogenesis-inducing molecules, causes normally quiescent endothelial cells to proliferate and sprout [2,3,4,5,6]. In the tumor microenvironment (TME), dysregulation of angiogenic signals contributes to the development of hyper-permeable and highly heterogeneous blood vessels, and also aids in entry of tumor cells into (intravasation) and out of (extravasation) the blood stream via trans-endothelial migration [7]. Tumor-associated endothelial cells often exhibit decreased adhesion between neighboring cells and with the extracellular matrix, with profound consequences relevant to the development and treatment of cancer. Consequently, abnormally organized and leaky tumor blood vessels contribute to tumor angiogenesis, inflammatory cell infiltration, metastasis, and the development of resistance to chemotherapeutic agents in tumors of diverse origin [2,7,8,9,10,11,12,13].
Key instigators of angiogenic signaling, e.g., vascular endothelial growth factor receptor-2 (VEGFR-2), fibroblast growth factor receptor-1 (FGFR-1), Notch and Tie receptors, and critical endothelial adhesion molecules including vascular endothelial cadherin (VE-cadherin, also called cadherin-5), integrins, and immunoglobulin-like cell adhesion molecules (Ig-CAMs), are highly glycosylated. Global changes in glycosylation of these receptors and others could have wide-ranging consequences for their biological activity and interactions with other molecules [14]. The discovery that galectins, which bind β-galactoside sugars on glycoproteins, bind specific glycosylated forms of VEGFR-2, trigger receptor activation, and mediate resistance to anti-VEGF therapies used to treat cancer, represents a major shift in understanding and targeting VEGFR-2 signaling in cancer and other angiogenesis-associated diseases [15,16]. In addition, enzymatic removal or mutation of specific VEGFR-2 glycosylation sites amplifies ligand-dependent VEGFR-2 activation and signaling [17], indicating that angiogenic signaling of VEGFR-2 also is affected by changes in glycosylation independently of galectins.
Changes in glycosylation promoted by hypoxia, the shunting of glycolytic intermediates into glycan synthesis via the hexosamine biosynthesis pathway (HBP), and pro-inflammatory cytokines, have the potential to alter core physiologic characteristics of endothelial cells and contribute to dysregulation of endothelial signaling (Figure 1) [18,19,20,21]. There remains a critical need to understand how tumor microenvironment-induced changes in endothelial cell glycosylation alter angiogenic signaling, dysregulate adhesion, contribute to the formation of abnormal tumor vasculature, and promote tumor metastasis. The goal of this review is to highlight recent developments that have advanced our understanding of tumor microenvironment-directed changes in glycosylation that alter vascular endothelial cell signaling and adhesion and thereby contribute to tumorigenesis.
2. Glycoprotein and Glycosaminoglycan Synthesis and Recognition by Lectins
The luminal surface of endothelial cells contains an extensive network of membrane-bound glycoproteins and proteoglycans, called the endothelial glycocalyx. There is an increasing awareness that the endothelial glycocalyx plays a critical role in vascular physiology and pathology, especially with relation to tumor angiogenesis and interactions between endothelial cells and tumor cells that mediate trans-endothelial migration. Protein N- and O-glycosylation, as well as glycosaminoglycan (GAG) synthesis, involve multiple enzymatic steps that occur co- and/or post-translationally, are influenced by enzyme and substrate levels, and result in considerable structural diversity [22,23,24,25]. Tumor-associated endothelial cells are exposed to a hypoxic, hyper-glycolytic, and pro-inflammatory milieu [26,27,28]. Endothelial cell glycosylation is supremely sensitive to hypoxia and inflammation [29,30,31,32]. Tumor-associated endothelial cells adopt a hyper-glycolytic metabolic state [27,33,34]. The enzyme fructose-6-phosphate-amidotransferase (GFAT) converts fructose-6-phosphate into glucosamine-6-phosphate, and in so doing shunts glycolytic intermediates into the HBP, linking metabolism and glycosylation [35,36,37]. Glucosamine-6-phosphate is the common precursor to all amino sugars used in glycoprotein synthesis [38,39]. Ultimately, changes in endothelial cell glycosylation alter protein interactions and function at the plasma membrane [36,37,40,41,42,43].
Protein glycosylation changes dramatically in cancer, and has been studied extensively in tumor epithelial cells, where it regulates cellular adhesion, cell-matrix interactions, and signaling via receptor tyrosine kinases (RTKs) [40,44,45,46,47,48,49]. In fact, ST6Gal-I, responsible for the attachment of sialic acid to glycoproteins via 2,6-linkage, regulates transcription factors involved in stem cell maintenance [50]. However, until recently there has been little understanding of how changes in endothelial cell glycosylation in the tumor microenvironment influence endothelial barrier function, adhesion, cell-matrix interactions, and cell signaling.
2.1. N-Glycosylation
N-glycosylation occurs on asparagine (N) residues within the NXS/T motif, where any amino acid X except for proline (X ≠ P) follows asparagine, and serine or threonine (S/T) occupy the third position. N-glycosylation is a complex, multi-step co- and/or post-translational process that is initiated by the transfer of N-acetyl-glucosamine-1-phosphate (GlcNAc-1-P) to a dolichol-phosphate on the cytoplasmic face of the endoplasmic reticulum (ER) membrane by GlcNAc-1-phosphotransferase (encoded by the human DPAGT1 gene, yeast ALG7) [23}. Notably, tunicamycin, an analog of uridine diphosphate-N-acetylglucosamine (UDP-HexNAc), inhibits this step and has been used widely to study N-glycosylation. After this initial step, an additional N-acetylglucosamine (GlcNAc) and five mannose (Man) residues are added sequentially. Then, the entire dolichol-linked glycan is flipped into the ER lumen, where four additional- Man residues and three glucose residues are added. This precursor, assembled from 14 monosaccharides, is then transferred by multi-subunit enzyme, oligosaccharyltransferase (OST), to an asparagine residue within the NXS/T motif. The nascent glycoprotein next undergoes interaction with chaperones to ensure quality control. Glycoproteins that ‘pass’ this quality control step proceed through multiple steps and are trimmed during protein folding to remove glucose. Further trimming and processing occurs in the ER and Golgi and produces a heterogeneous set of N-linked glycans.
2.2. Mucin-Type O-Glycosylation
Mucin-type O-glycans, also called O-GalNAc glycans, are initiated by the transfer of N-acetylgalactosamine (GalNAc) by polypeptide GalNAc-transferases (ppGalNAcTs) to specific Ser and Thr residues on O-glycosylated proteins. There are 20 human polypeptide GalNAc-transferase genes. This process occurs in the Golgi apparatus. O-glycosylated regions of proteins are frequently rich in serine, threonine and proline residues. O-glycans are commonly found on mucins, a class of glycoproteins that may each contain hundreds of such O-glycans, but other proteins can also be O-glycosylated, including membrane-associated glycoproteins such as P-selectin glycoprotein ligand 1 (PSGL-1). While all mucin-type O-glycans start with O-GalNAc there is considerable structural variability. There are four common O-glycan core structures, and additional rare core structures have also been elucidated [22].
2.3. O-GlcNAc
In contrast to the complex glycans on cell surface glycoproteins, O-linked β-N-acetylglucosamine (O-GlcNAc) modification of Ser and Thr residues occurs on intracellular proteins and is involved in signaling and the regulation of enzyme activity [51]. Two key enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), catalyze the addition and removal of O-GlcNAc, respectively, from intracellular proteins. O-GlcNAc modification of endothelial nitric oxide synthase (eNOS) results in inactivation of the phosphorylated enzyme in the context of diabetes [52,53]. In addition, elevated flux through the HBP leads to increased protein modification by O-GlcNAc and impairs angiogenesis, potentially by inhibiting Akt signaling in endothelial cells [54]. Decreasing levels of OGT in prostate cancer cells diminished expression of VEGF and reduced endothelial tube formation in vitro, and regulation of this process involved FOXM1 [55].
2.4. Glycosaminoglycans
Glycosaminoglycans (GAGs) are long unbranched polysaccharides with repeating disaccharide units that are a major component of the extracellular matrix (ECM). They undergo sulfation at distinct positions and also undergo epimerization of uronic acid, resulting in the generation of a diverse set of molecules with distinct physical and biological properties. With the exception of hyaluronan, GAGs are covalently linked via serine residues to GAG-bearing proteins (proteoglycans) that reside on the cell surface and within the ECM. Six classes of GAGs exist, including chondroitin sulfate, dermatan sulfate, heparan sulfate, heparin, hyaluronan, and keratan sulfate. Hyaluronan (HA), a high-molecular-weight, non-sulfated glycosaminoglycan, is synthesized at the cell surface and is subsequently incorporated into the extracellular matrix [24,45].
2.5. Glycan-Binding Proteins
Lectins are a class of glycan-binding proteins that recognize carbohydrate substructures within larger branched carbohydrates. Lectins are notable for their low-affinity interactions, which mediate “rolling” in of leukocytes and cancer cells when they interact with glycans on the endothelial cell surface. In this review, we will discuss two major classes of lectins, which are categorized by the substructures they bind. The first, galectins, recognize glycans with exposed galactose residues [56]. The second, selectins, are a family of calcium-dependent cell adhesion molecules that recognize sialylated, fucosylated carbohydrate ligands with low affinity [57]. Selectins are upregulated in inflammatory conditions to recruit platelets and leukocytes to sites of injury or infection, but may also be co-opted in the context of cancer to facilitate tumor cell adhesion to endothelial cells. In addition to lectins, a broad array of molecules, including many growth factors, have the ability to bind with carbohydrate moieties on glycoproteins and glycosaminoglycans, and in so doing they mediate cell–cell and cell–matrix interactions.
The modification of membrane glycoproteins by N- and O-glycans, cytoplasmic O-glycosylation, the production and deposition of glycosaminoglycans, and the recognition of motifs on glycoconjugates by lectins, have been characterized extensively in the epithelial context. Next, we will discuss specific glycoproteins that are involved in endothelial cell adhesion, and how carbohydrate modifications may impact the function of these molecules.
3. Endothelial Cell Adhesion Molecules
Much of what is known about the impact of altered glycosylation on cell–cell and cell–matrix adhesion is derived from studies of aberrant glycosylation in tumor cells [58,59,60]. For example, increased β-1,6 branching and increased sialylation on N-linked glycans that occurs during tumorigenesis lessens cell–cell adhesion [58,61]. In contrast, knowledge of the impact of altered glycosylation on endothelial adhesion molecules is primarily based on the interaction of endothelial cell adhesion molecules with immune cells in the context acute inflammatory conditions. Glycans on the surface of leukocytes, and to a lesser extent, glycans on the surface of endothelial cells, play a crucial role in leukocyte recruitment. Glycosyltransferases, including α1,3 fucosyltransferases, α2,3 sialyltransferases, core 2 N-acetylglucosaminlytransferases, β1,4 galactosyltransferases, and polypeptide N-acetylgalactosaminyltransferases are involved in the synthesis of selectin ligands that mediate leukocyte rolling by binding to selectins [62]. Major glycoconjugates and lectins involved in endothelial cell adhesion and signaling are shown in Figure 1.
3.1. ICAM-1
Intercellular adhesion molecule 1 (ICAM-1/CD54) is involved in trans-endothelial migration of leukocytes and serves as a ligand for integrins on leukocytes. Scott et al. (2013) showed that activated endothelial cells expressed two forms of ICAM-1, the more abundant of which displayed N-glycans modified with α2,6-linked sialic acids, while the less abundant form displayed primarily high-mannose type glycans. Inhibition of α-mannosidase to force expression of high-mannose N-glycans led to increased monocyte rolling and adhesion, as compared with ICAM-1 displaying more processed N-glycans, suggesting that the high-mannose glycans could serve as leukocyte ligands. However, in cells with ICAM-1 displaying high-mannose glycans, interactions with the actin cytoskeleton were lost, suggesting that the glycosylation status and adhesion properties of ICAM-1 are modulated by inflammation [63].
3.2. Endothelial Selectins: E-Selectin (ELAM) and P-Selectin
E-selectin is an endothelial-specific lectin that recognizes glycans containing the sialyl-Lewis x substructure (SLex; NeuAc α2,3Gal β1,4(Fuc α1,3)-GlcNAc, its expression is activated by cytokines, and it is involved in recruitment of neutrophils to sites of inflammation [64,65]. Aberrant expression of glycans bearing the SLex motif in multiple types of cancer, including colon cancer [66,67] and prostate cancer [68,69,70], has been implicated in facilitating tumor cell adhesion to the endothelial cells, and facilitating tumor cell metastasis via interaction with selectins [71,72]. P-selectin is expressed in both platelets and activated endothelial cells. In endothelial cells, P-selectin is stored in Weibel–Palade bodies and is rapidly released and translocated to the cell surface in response to inflammation. P-selectin glycoprotein ligand-1 (PGSL-1/CD162) is a ligand of P-selectin that is expressed on leukocytes and contains mucin-type O-glycans. Interestingly, P-selectin deficient mice show a decreased rate of tumor growth and decreased metastasis compared to wild-type mice [73]. This can be explained in part by the fact that tumors frequently express glycosylated ligands with sialyl-Lewis x structures, bind platelets and leukocytes via P-selectin, and use these interactions to initiate contact with endothelial cells at distant sites and extravasate [74]. Some tumor cells express P-selectin and initiate this process in a platelet-independent manner [75].
3.3. VCAM-1
Vascular cell adhesion molecule 1 (VCAM-1/CD106) is an endothelial glycoprotein, its expression is upregulated in response to TNF-α, IL-1 and IL-4, and it is involved in leukocyte adhesion to endothelial cells, an interaction mediated by VCAM-1 binding to α4 integrins (i.e., α4β1 and α4β7) on leukocytes. In response to IL-1 and IL-4, α2,6-sialyltransferase (ST6Gal-I) expression is enhanced. A decrease in α2,6-linked sialic acids increased VCAM-1-dependent adhesion, while α2,3-linked sialic acids did not impact adhesion [76].
3.4. PECAM (CD31)
Platelet endothelial cell adhesion molecule (PECAM) is involved in cell adhesion, mechanical stress sensing, angiogenic signaling, and also has an anti-apoptotic role [77]. It is a major component of intercellular junctions in endothelial cells. In addition, it has been shown to have lectin-like properties and recognize α2,6-sialic acid, and this property is involved in regulation of hemophilic interactions [78,79]. PECAM glycans bearing α2,6-sialic acid are essential for endothelial tube formation, and removal of these sialic acid residues disrupts endothelial tube formation [80,81]. Several N-glycans are located at the hemophilic binding interface [82], suggesting that α2,6-sialylated glycans modulate homophilic PECAM-dependent interactions. A decrease in α2,6-sialylation reduces the levels of PECAM at the cell surface and increases its role in apoptosis, and may regulate interactions between PECAM, VEGFR2, and integrin-β3 [77,83]. Therefore, α2,6-sialylated glycans appear to be critical for endothelial cell survival, as they stabilize membrane proteins, leading to their retention at the cell surface and thereby impact pro-angiogenic signaling.
3.5. IGPR-1
The Ig-containing and proline-rich receptor-1(IGPR-1) is a newly identified Ig-CAM that is uniquely expressed in human and other higher mammalians, but not in rodents [84]. IGPR-1 is expressed in endothelial cells and regulates endothelial barrier function and angiogenesis [85]. More importantly, IGPR-1 expression is elevated in various tumors including, colon cancer [86]. Although it is heavily glycosylated [84], the role of glycosylation in IGPR-1 function has not been studied.
3.6. VE-Cadherin
Vascular endothelial cadherin (VE-cadherin/CD144) is an endothelial-specific adhesion molecule that is an essential player in the formation of cell–cell endothelial adherens junctions and controls vascular permeability. Analyses of VE-cadherin N-glycans indicate that it bears predominantly sialylated biantennary and hybrid-type glycans, and it may also be O-manosylated [87,88]. Sialic acid-bearing glycans on VE-cadherin are likely important for maintenance of endothelial cell adherens junctions [89].
3.7. Endomucin
Endomucin is a highly sialylated, type I O-glycosylated protein that is endothelial specific. Endomucin is involved in angiogenesis [90], and recent evidence suggests that the α1,3-fucosyltransferase FUT7, upregulated by IL-1β, induced monocyte-endothelial adhesion via fucosylation of endomucin [91].
As we have noted here, several of the glycoproteins and glycan-binding proteins discussed above, including ICAM-1, E-Selectin, P-Selectin, VCAM-1, and PECAM, are known to initiate specific adhesive interactions only when modified (or binding to) specific glycan substructures. As a result, changes in glycosylation alter the functions of these proteins. Below, we will discuss factors that influence endothelial glycosylation and in so doing alter endothelial cell adhesion.
4. Factors that Influence Endothelial Glycosylation
Endothelial glycosylation is evolutionarily conserved in both developmental and inflammatory processes. Yano et al. (2007) examined the endothelium of hagfish to understand evolutionarily-conserved features of the endothelium using lectins LCA (Lens culinaris agglutinin) and HP (Helix pomatia) that bind carbohydrate structures containing α-linked mannose and α-N-acetylgalactosamine respectively, to characterize differences in glycosylation between endothelial cells in different vascular beds. Their analyses revealed that vascular bed-associated differences in glycosylation facilitated histamine-induced adhesion of leukocytes in capillaries and post-capillary venules but not in the aortic endothelium or arterioles, suggesting a link between inflammation and altered glycosylation [92]. Using similar methods, Jilani et al. (2003) demonstrated that lectin affinities differed between the vasculature of chicken embryos at early and late stages of development, suggesting that endothelial glycosylation plays a role in embryonic development [93]. These patterns are likely relevant to human to the biology of human cells as well.
Inflammatory cytokines TNF-alpha and interleukin-1, and bacterial lipopolysaccharide increase expression of ST6Gal-I and also increase the binding of lectins with affinity for sialic acid to the endothelium. E-selectin, ICAM-1, and VCAM-1 were reported as glycoprotein substrates for ST6Gal-I [94]. DW Scott et al. demonstrated that inflammatory stimuli including TNF-α, LPS, and IL-1β induce changes in expression of specific endothelial glycoproteins involved in monocyte adhesion including ICAM-1 and VCAM-1, as well as expression of enzymes involved in N-glycan processing including α-mannosidase, which catalyzed the removal of two mannose residues from GlcNAcMan5GlcNAc2, the committed step in the synthesis of complex N-linked glycans [95]. These investigators showed that endothelial responses to inflammatory stimuli vary between vascular beds [96].
Within the tumor microenvironment, inflammatory stimuli, hypoxia, and tumor-secreted signaling factors alter expression of endothelial cell surface carbohydrates by impacting the underlying expression of enzymes involved in carbohydrate synthesis [16,95,96,97]. Table 1 shows the reported impact of various cytokines and hypoxia on endothelial glycosylation. Pro-inflammatory signals including IFN-γ and IL-17 increase the expression of α2,6-linked sialic acid-containing carbohydrate epitopes on the endothelial cell surface glycoproteins. In contrast, immunosuppressive cytokines IL-10 and TGF-β1 reduce α2,6-linked sialic acid-containing carbohydrate epitopes on N-linked glycans [16]. In addition, tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) alter endothelial surface N-glycosylation and this correlates with increased monocyte adhesion [76,95]. Critically, immune-mediated mechanisms that alter glycosylation and the expression of glycan-binding proteins have been shown to lead to acquired resistance to anti-angiogenic therapies via changes in the interaction with glycan-binding proteins [98].
The impact of hypoxia on endothelial cells in the tumor microenvironment has been extensively studied [99,100,101]. Hypoxia-inducible factor (HIF-1) is a heterodimeric transcription factor composed of subunits HIF-1β/aryl hydrocarbon receptor nuclear translocator (ARNT) and either HIF-1α or HIF-2α. Under normoxic conditions, prolyl-hydroxylase (PHD) enzymes including PHD2 hydroxylate HIFα, leading to HIF-1 inactivation, followed by its ubiquitination by the von Hippel–Lindau tumor suppressor (pVHL), an E3 ubiquitin ligase, and subsequent degradation [102,103,104]. However, under hypoxic conditions such as those in the tumor microenvironment, PHD2, unable to bind oxygen, no longer hydroxylates HIFα, and this results in its accumulation [105]. HIF-1α and HIF-2α regulate different and, in some cases, opposing, sets of genes [106,107]. While there is some evidence that HIF-1 signaling alters glycosylation, the extent of its influence on endothelial glycosylation, the potential differential roles of HIF-1α and HIF-2α, and the physiological impact of the resulting changes in glycosylation are unclear. In addition to hypoxia, the role of metabolism in endothelial cell glycosylation is an intriguing, though unexplored, subject. It has been reported that the glycolytic activator PFKFB3 regulates endothelial cell rearrangement during vessel sprouting, in part by reducing intercellular adhesion [108]. The role of glycosylation in reducing intercellular adhesion should be further investigated in this context.
Abnormal endothelial cell glycosylation and increased expression of lectins, which bind glycan epitopes, aid the development of resistance to anti-angiogenic cancer therapeutics [14,109]. Further exploration should be done to understand the impact of these changes in both acute and chronic inflammation. Hypoxia, a common feature of the tumor microenvironment, also appears to alter endothelial cell glycosylation, leading to the production of glycoproteins bearing carbohydrate structures with less α2,6-linked sialic acid, greater branching of β1,6 N-glycan structures, and elongation with poly-LacNAc residues [16]. Culturing endothelial cells in a tumor conditioned medium from colon carcinoma cell line HT29 induced increased β1,6-GlcNAc branching of endothelial cell glycans, suggesting that factors secreted by tumor cells also influence glycosylation in their environment [32]. Inflammatory cues, hypoxia, and tumor-secreted factors, by triggering changes in endothelial surface carbohydrate structures, may alter angiogenic signaling by modifying the properties of endothelial glycoproteins that are key mediators of signaling and adhesion. To improve our understanding of the role these changes play in tumor progression, metastasis, and treatment, further study is required in animal models and human tissue.
5. Glycosylation and VEGFR2 Pro-Angiogenic Signaling
Vascular endothelial growth factors (VEGFs), first identified based on their role in vascular permeability, bind to extracellular matrix proteoglycans (specifically, heparan sulfate proteoglycans, HSPGs), resulting in their sequestration and controlled release from the extracellular matrix in cases of tissue damage or remodeling by matrix-metalloproteinases. Upon release, they are available to promote angiogenesis to repair tissue, although this process is dysregulated in the tumor microenvironment. Additional factors, including fibroblast growth factors, and angiogenic inhibitors such as thrombospondin and platelet factor 4, also interact with and are in some instances stabilized by HSPGs [110]. Using proximity ligation assays in primary brain endothelial cells, Xu et al. (2011) demonstrated that heparan sulfate and VEGFR2 interact directly, and that the number of heparan sulfate-VEGFR2 complexes increased in response to stimulation with VEGF165 and VEGF121 [111]. HSPGs also bind gremlin (Drm), and alter its activation of VEGFR2 [112].
Most endothelial surface proteins bear N- and/or O-linked glycans. Multiple adhesion molecules bind glycoconjugates expressed on the surfaces of endothelial cells [113]. The cell-surface receptor tyrosine kinase VEGFR2 is involved in pro-angiogenic signaling in endothelial cells and plays a critical role in tumor angiogenesis. The extracellular domain of VEGFR2 is highly modified by N-linked glycans [114], and glycans, especially α2,6-linked N-glycans at site N247 on Ig-like domain 3 near the ligand binding pocket, influence ligand-dependent signaling [17]. Immune-mediated mechanisms that alter glycosylation and influence endothelial cell signaling are implicated in acquired resistance to anti-angiogenic therapies, highlighting the convergence of immunosuppressive and pro-angiogenic signaling in the tumor microenvironment. Chiodelli et al. (2017) also found that VEGFR2-associated NeuAc plays an important role in modulating VEGF/VEGFR2 interaction, pro-angiogenic activation of endothelial cells and neovascularization [14].
Galectin-3 (Gal-3) is able to induce angiogenesis in a glycan-dependent manner by binding to glycoproteins on the surface of endothelial cells [15]. VEGFR-2 N-glycans are involved in retention of the receptor at the endothelial cell surface via interaction with Gal-3 [115]. Rabinovich et al. studied anti-VEGF refractory tumors and found that glycans on the endothelial surface glycoproteins, including VEGFR2, were remodeled to selectively bind galectin-1 (Gal-1) expressed by the tumor cells. Endothelial cells displayed high levels of β1,6-GlcNAc-branched N-glycans and low levels of α2,6-linked sialic acid in anti-VEGF refractory tumors compared to tumors that were sensitive to anti-VEGF treatment. Binding of Gal-1 to VEGFR2 resulted in VEGF-independent activation of the receptor [16]. The group also found that hypoxia upregulates expression of galectin-1 (Gal-1) via HIF-1-dependent and -independent mechanisms. In Kaposi’s sarcoma, activation of the transcription factor nuclear factor κB (NF-κB) by reactive oxygen species resulted in higher levels of Gal-1 expression that promoted angiogenesis and tumorigenesis [116]. In another study by the same group, HIF-1α was found to increase Gal-1 expression in colorectal cancer (CRC) cells, and the group identified two hypoxia-responsive elements upstream to the transcriptional start site of the Gal-1 gene that are essential for HIF-1-mediated galectin-1 expression [16]. Tumor microenvironment-dependent changes in endothelial cell glycosylation are summarized in Figure 2.
6. Glycosaminoglycans in Tumor Angiogenesis and Metastasis
Within the ECM, GAGS play a role in regulating migration of endothelial cells, providing a scaffold that guides endothelial cell tube formation, and stabilizes neovasculature. An excellent review by Oliveira-Ferrer, et al. describes the varied roles of GAGs in metastasis [117]. Here, we will primarily discuss the role of GAGs as they relate to endothelial cell function (or dysfunction) in cancer.
6.1. Heparan Sulfate Proteoglycans (HSPGs)
HSPGs are a well-studied group of proteins that bear long heparan sulfate chains consisting of 50–200 glucuronic acid disaccharide repeats with variable patterns of sulfation, and reside both on the endothelial cell surface and within the extracellular matrix. HSPG modifications including sulfation create binding sites for various ligands, including adhesive proteins, chemokines, growth factors and growth factor-binding proteins, proteases and protease inhibitors, and morphogens [118,119,120,121,122]. Critically, these interactions are sensitive to the position and linkage of sulfate modifications. Transmembrane HSPGs including syndecans, glycpicans, and perlecan reside on the cell surface and are involved in extracellular matrix assembly and maintenance. Both VEGFR2 and VEGF (including VEGF165 but not VEGF121) interact with heparan sulfate, and ligand-stimulation has been reported to increase heparan sulfate-VEGFR2 complex formation and vascular permeability [111]. VEGF HS-binding domains encoded by exons 6 and 7 are responsible for the interaction of VEGF ligands with HS, and result in the sequestration of VEGF in the extracellular matrix that may subsequently be released by proteases and heparanase during ECM degradation by proteases associated with angiogenesis [123,124,125]. The ability of VEGF165 to bind HS is partially controlled by its interaction with endothelial transglutaminase-2 [126].
Additional growth factors, including PDGF-B, contain HS-interacting domains [127,128]. TGF-β isoforms also bind HS, and HS plays a role in gradient formation of cytokines [129,130]. By regulating heparan sulfate modifications on endothelial cells, heparan sulfatases affect tumor angiogenesis in a number of contexts, including ovarian and breast cancer. Downregulation of endosulfatases responsible for removal of 6-O sulfate from HS in response to hypoxia, as well as downregulation in tumor cells, results in the presence of more highly sulfated forms of HS, thus increasing growth factor binding and downstream signaling [131].
6.2. Chondroitin Sulfate (CS)
Chondroitin sulfate (CS), composed of repeating units of the disaccharide GalNAc-GlcA, is also variably-sulfated in a tissue-specific manner by carbohydrate sulfotransferases. Expression of specific sulfated forms of CS on the surface of tumor cells facilitates their interaction with platelets and endothelial cells by creating ligands that bind P-selectin, e.g., in breast cancer [132]. Moreover, the sulfation pattern of CS on versican appears to be critical for interaction with L-selectin, P-selectin, and CD44, molecules involved in endothelial cell adhesion and/or tumor angiogenesis [133]. However, the full role of such modifications in tumor angiogenesis remains to be determined.
6.3. Hyaluronan (HA)
Hyaluronan (HA) is a negatively charged, nonsulfated GAG. Unlike other GAGs, hyaluronan (HA) is not covalently linked to a core protein. Rather, it is deposited in the extracellular matrix, where it may interact with ECM proteins and other GAGs. In healthy tissue, the coordinated expression and activity of HA synthases and hyaluronidases maintain a homeostasis. In tumors, higher expression of low-molecular weight HA is often present and is associated with inflammatory conditions [134], and contributes to tumor angiogenesis by impairing cellular adhesion [135,136]. HA also seems to play a role in tumor-associated macrophage trafficking to tumor stroma [137].
7. Endothelial Glycosylation Regulates Tumor Cell Trans-Endothelial Migration
The binding to glycosylated epitopes on tumors by selectins (E-selectin, P-selectin) and galectins expressed on endothelial cells, and of tumor-expressed lectins to endothelial glycans, mediates a process of rolling followed by stable heterotypic adhesion. This process mirrors the process through which platelets and leukocytes interact with the endothelium. The glycan-binding proteins on endothelial cells recognize glycan substructures on platelets, leukocytes, and circulating tumor cells. Conversely, L-selectin expressed on leukocytes (specifically, T cells) also recognizes glycan structures on endothelial cells, allowing leukocytes to attach to specific endothelial beds, based purely on the glycans expressed on the endothelial surface [138]. Sulfated glycans also play a role in this process in lymphatic endothelium. There is evidence that these interactions are regulated by the spatial and temporal expression of glycosyltransferases and sulfotransferases in endothelial cells in a bed-specific manner, and by inflammatory signals.
Galectin-3 (Gal-3) expressed on endothelial cells is a major actor in tumor metastasis. Gal-3 is the only human lectin of the ‘chimera’ galectin subtype. It can exist as a monomer, or form multivalent complexes of up to five Gal-3 molecules via its non-lectin domain, allowing it to facilitate the interaction of multiple glycoproteins. By binding T antigen on MUC-1, Gal-3 promotes adhesion of tumor cells to the endothelium in breast and prostate cancer [139,140,141]. Circulating Gal-3 can also increase tumor cell adhesion to and migration across the endothelium by interacting with MUC1 on tumor cells, leading to exposure of additional glycosylated ligands including CD44 that bind E-selectin on endothelial cells [142]. Under flow conditions, highly metastatic MDA-MB-435 human breast carcinoma cells that express high levels of T antigen and Gal-3 showed increased adhesion to endothelial cells compared to similar non-metastatic cells [143].
Glycan-mediated intravasation, rolling, and extravasation of tumor cells contribute to tumor metastasis (Figure 3). For example, in colon and prostate cancers, glycans with the SLex motif (a tetra-saccharide containing both sialic acid and α1,3 linked fucose) are involved in tumor metastasis [66,67]. Forced reduction in the expression of α1,3 fucosyltransferases reduced incidence of prostate cancer in mice [68,69,70]. As previously noted, this can be explained by tumor cell adhesion to the endothelial cells via interaction with selectins [71,72]. In patients with multiple myeloma, high expression of ST3Gal6, which catalyzes the 2,3-linked attachment of sialic acid residues to glycoproteins, correlates with lower overall survival. Knockdown of ST3GAL6 in multiple myeloma cells diminished the cells’ ability to undergo trans-endothelial migration and reduced ability to roll on P-selectin in vitro [144].
8. Toward Therapeutic Strategies that Target Endothelial Glycosylation
Several anti-cancer therapeutic strategies that target tumor vasculature have been proposed, and include (a) the inhibition of tumor angiogenesis and (b) treatments that promote blood vessel normalization to enhance delivery of chemotherapeutic agents and reduce metastasis [2,145,146,147,148]. In clinical trials, anti-angiogenic therapies have shown promise in patients with colorectal, lung, breast, and other cancers, but resistance to these therapies often develops rapidly [98,145,149,150,151]. Additional drug targets that aid in vascular normalization are being investigated [146]. There remain gaps in our understanding of tumor-associated endothelial cell pathobiology, including how tumor microenvironment-induced changes in the glycosylation of endothelial adhesion and signaling molecules contribute to altered angiogenesis. Addressing this gap in knowledge could lead to the design and delivery of pharmacological agents that aid in normalizing blood vessels, prevent metastasis and increase responsiveness to targeted chemotherapeutics.
A number of approaches that target protein glycosylation attempt to address this gap. Therapeutic targeting of glycan-mediated processes has been explored, including the use of glycomimetics [152]. Partial inhibition of OST, the enzyme involved in the initiation of N-linked glycosylation, is an approach pioneered by Contessa et al. [153,154]. Among the molecular targets of this strategy are receptor tyrosine kinases such as EGFR, which are highly N-glycosylated. The approach is currently being tested in a number of pre-clinical models [155]. While it has not been tested in the context of angiogenesis, it is notable that VEGFR2 and additional RTKs involved in pro-angiogenic signaling are highly N-glycosylated, and therefore might also be susceptible to targeting by this drug, potentially in combination with other approaches. Another breakthrough involves the development of fucosyltransferases inhibitor 2-fluorofucose (2-FF) by Okeley et al. (2013) [156]. Many selectin ligands are fucosylated, and administration of 2-FF could potentially block these interactions and attenuate trans-endothelial migration of tumor cells. In pre-clinical models, 2-FF inhibited leukocyte-endothelium interactions [157], inhibits liver cancer HepG2 proliferation, migration, and tumor formation [158], and reduced fucosylated E-selectin ligand expression in human invasive ductal carcinoma [159]. Multiple fucosyltransferases in humans catalyze the attachment of fucose via specific linkages to glycans. It is likely that the development of fucosyltransferase-specific inhibitors will ultimately be the most successful strategy, as this will enable targeting of specific fucose linkages involved in metastasis while minimizing off-target effects. Thioglycosides are a class of compounds that are currently being tested as glycosylated decoys to reduce selectin-dependent leukocyte adhesion [160]. It remains to be seen whether a similar approach might be applied in the context of cancer treatment. Additionally, targeting selectin-mediated cell adhesion to endothelial cells may represent an opportunity to control tumor immunity [161]. As discussed previously, heparanase is elevated in multiple types of cancer and promotes tumor invasion, angiogenesis, and metastasis. Heparanase inhibitors that prevent the release of heparan sulfate side chains have been tested in pre-clinical and clinical settings, and reduce tumor metastasis by maintaining ECM integrity and partially restoring vascular function [162,163,164,165].
9. Conclusions
Tumor-associated endothelial cells are significantly influenced by signals from nearby tumor cells, stromal cells and infiltrating immune cells. Glycans on endothelial adhesion molecules including ICAM-1, VCAM-1, and PECAM, and glycan-binding proteins (lectins) expressed on the surfaces of endothelial, immune, and cancer cells, alter the adhesive properties of endothelial cells and facilitate (or disfavor) immune and tumor cell infiltration. In addition, altered endothelial cell glycosylation in the tumor microenvironment has been shown to impact VEGFR2-mediated angiogenic signaling. Further investigation will be needed to understand how changes in tumor-associated endothelial cell glycosylation machinery, with cues from the tumor microenvironment, dysregulate endothelial cell signaling and adhesion, and contribute to the formation of abnormal and leaky tumor blood vessels. Since glycosylation is not template based, different sites within the same protein may be occupied by different glycan structures, and a single protein may have many glycoforms with different biological functions. Major barriers to progress in this field have included (a) the technical challenge of analyzing glycan heterogeneity, (b) the low abundance of plasma membrane receptors and adhesion molecules, and (c) the complexity of linking non-template-based protein glycosylation status to biological function. Despite these challenges, significant progress has been made towards elucidating the roles of normal and aberrant glycosylation in endothelial processes, and we further expect that advances will be made in these areas in the years ahead. We predict that recent advances in mass spectrometry-based methods for the characterization of glycoconjugates, in combination with gene expression analyses in model systems and tissue, CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 gene editing, and the application of fluorophore-conjugated lectins for live cell and tissue imaging, among others, will enable the establishment of a clear relationship between changes in glycan structures on the cell surface and altered endothelial function in tumor-associated endothelial cells. The knowledge gained in this exciting and emerging field of biology can lead to development of a new class of therapeutics to combat cancer and other diseases.
Author Contributions
Conceptualization, K.B.C. and N.R.; resources, K.B.C., N.R. and C.E.C.; writing—original draft preparation, K.B.C.; writing—review and editing, N.R. and C.E.C.; visualization, K.B.C.; supervision, N.R. and C.E.C.; funding sources, K.B.C., N.R. and C.E.C.
Funding
The authors acknowledge the support of NIH grants F32 CA196157 (to KBC), R21CA191970 and R21CA193958 (to NR), and P41 GM104603 (to CEC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Chen, H.; Davis-Smyth, T.; Gerber, H.P.; Nguyen, T.N.; Peers, D.; Chisholm, V.; Hillan, K.J.; Schwall, R.H. Vascular endothelial growth factor is essential for corpus luteum angiogenesis. Nat. Med. 1998, 4, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Alitalo, K.; Carmeliet, P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell 2002, 1, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Vaisman, N.; Gospodarowicz, D.; Neufeld, G. Characterization of the receptors for vascular endothelial growth factor. J. Biol. Chem. 1990, 265, 19461–19466. [Google Scholar]
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef]
- Maishi, N.; Hida, K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 2017, 108, 1921–1926. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar] [PubMed]
- Liang, W.; Ferrara, N. The Complex Role of Neutrophils in Tumor Angiogenesis and Metastasis. Cancer Immunol. Res. 2016, 4, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franses, J.W.; Drosu, N.C.; Gibson, W.J.; Chitalia, V.C.; Edelman, E.R. Dysfunctional endothelial cells directly stimulate cancer inflammation and metastasis. Int. J. Cancer 2013, 133, 1334–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiodelli, P.; Rezzola, S.; Urbinati, C.; Federici Signori, F.; Monti, E.; Ronca, R.; Presta, M.; Rusnati, M. Contribution of vascular endothelial growth factor receptor-2 sialylation to the process of angiogenesis. Oncogene 2017, 36, 6531–6541. [Google Scholar] [CrossRef] [PubMed]
- Nangia-Makker, P.; Honjo, Y.; Sarvis, R.; Akahani, S.; Hogan, V.; Pienta, K.J.; Raz, A. Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am. J. Pathol. 2000, 156, 899–909. [Google Scholar] [CrossRef]
- Croci, D.O.; Cerliani, J.P.; Dalotto-Moreno, T.; Mendez-Huergo, S.P.; Mascanfroni, I.D.; Dergan-Dylon, S.; Toscano, M.A.; Caramelo, J.J.; Garcia-Vallejo, J.J.; Ouyang, J.; et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef]
- Chandler, K.B.; Leon, D.R.; Kuang, J.; Meyer, R.D.; Rahimi, N.; Costello, C.E. N-glycosylation regulates VEGFR2 ligand-dependent activation and signaling. Manuscript submitted for publication. 2019. [Google Scholar]
- Dewald, J.H.; Colomb, F.; Bobowski-Gerard, M.; Groux-Degroote, S.; Delannoy, P. Role of Cytokine-Induced Glycosylation Changes in Regulating Cell Interactions and Cell Signaling in Inflammatory Diseases and Cancer. Cells 2016, 5, 43. [Google Scholar] [CrossRef]
- Delmotte, P.; Degroote, S.; Lafitte, J.J.; Lamblin, G.; Perini, J.M.; Roussel, P. Tumor necrosis factor alpha increases the expression of glycosyltransferases and sulfotransferases responsible for the biosynthesis of sialylated and/or sulfated Lewis x epitopes in the human bronchial mucosa. J. Biol. Chem. 2002, 277, 424–431. [Google Scholar] [CrossRef]
- Colomb, F.; Krzewinski-Recchi, M.A.; El Machhour, F.; Mensier, E.; Jaillard, S.; Steenackers, A.; Harduin-Lepers, A.; Lafitte, J.J.; Delannoy, P.; Groux-Degroote, S. TNF regulates sialyl-Lewisx and 6-sulfo-sialyl-Lewisx expression in human lung through up-regulation of ST3GAL4 transcript isoform BX. Biochimie 2012, 94, 2045–2053. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; Krzewinski-Recchi, M.A.; Cazet, A.; Vincent, A.; Lehoux, S.; Lafitte, J.J.; Van Seuningen, I.; Delannoy, P. IL-6 and IL-8 increase the expression of glycosyltransferases and sulfotransferases involved in the biosynthesis of sialylated and/or sulfated Lewisx epitopes in the human bronchial mucosa. Biochem. J. 2008, 410, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockhausen, I.; Schachter, H.; Stanley, P. O-GalNAc Glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Stanley, P.; Taniguchi, N.; Aebi, M. N-Glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Hascall, V.; Esko, J.D. Hyaluronan. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Harjes, U.; Bensaad, K.; Harris, A.L. Endothelial cell metabolism and implications for cancer therapy. Br. J. Cancer 2012, 107, 1207–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Vallejo, J.J.; van Dijk, W.; van Die, I.; Gringhuis, S.I. Tumor necrosis factor-alpha up-regulates the expression of beta1,4-galactosyltransferase I in primary human endothelial cells by mRNA stabilization. J. Biol. Chem. 2005, 280, 12676–12682. [Google Scholar] [CrossRef]
- Garcia-Vallejo, J.J.; van Dijk, W.; van Het Hof, B.; van Die, I.; Engelse, M.A.; van Hinsbergh, V.W.; Gringhuis, S.I. Activation of human endothelial cells by tumor necrosis factor-alpha results in profound changes in the expression of glycosylation-related genes. J. Cell Physiol. 2006, 206, 203–210. [Google Scholar] [CrossRef]
- Chacko, B.K.; Scott, D.W.; Chandler, R.T.; Patel, R.P. Endothelial surface N-glycans mediate monocyte adhesion and are targets for anti-inflammatory effects of peroxisome proliferator-activated receptor gamma ligands. J. Biol. Chem. 2011, 286, 38738–38747. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Li, J.; Geng, M. The glycan profile of endothelial cells in the present of tumor-conditioned medium and potential roles of beta-1,6-GlcNAc branching on HUVEC conformation. Mol. Cell Biochem. 2010, 340, 143–152. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef]
- Lucena, M.C.; Carvalho-Cruz, P.; Donadio, J.L.; Oliveira, I.A.; de Queiroz, R.M.; Marinho-Carvalho, M.M.; Sola-Penna, M.; de Paula, I.F.; Gondim, K.C.; McComb, M.E.; et al. Epithelial Mesenchymal Transition Induces Aberrant Glycosylation through Hexosamine Biosynthetic Pathway Activation. J. Biol. Chem. 2016, 291, 12917–12929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel Rahman, A.M.; Ryczko, M.; Nakano, M.; Pawling, J.; Rodrigues, T.; Johswich, A.; Taniguchi, N.; Dennis, J.W. Golgi N-glycan branching N-acetylglucosaminyltransferases I, V and VI promote nutrient uptake and metabolism. Glycobiology 2015, 25, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Ryczko, M.C.; Pawling, J.; Chen, R.; Abdel Rahman, A.M.; Yau, K.; Copeland, J.K.; Zhang, C.; Surendra, A.; Guttman, D.S.; Figeys, D.; et al. Metabolic Reprogramming by Hexosamine Biosynthetic and Golgi N-Glycan Branching Pathways. Sci. Rep. 2016, 6, 23043. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.N.; Vidal, N.; Berthezene, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, R.; Hawkins, M.; Barzilai, N.; Rossetti, L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature 1998, 393, 684–688. [Google Scholar] [CrossRef]
- Dennis, J.W.; Lau, K.S.; Demetriou, M.; Nabi, I.R. Adaptive regulation at the cell surface by N-glycosylation. Traffic 2009, 10, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, M.; Granovsky, M.; Quaggin, S.; Dennis, J.W. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 2001, 409, 733–739. [Google Scholar] [CrossRef]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef]
- Dennis, J.W.; Nabi, I.R.; Demetriou, M. Metabolism, cell surface organization, and disease. Cell 2009, 139, 1229–1241. [Google Scholar] [CrossRef]
- Lopez-Sambrooks, C.; Shrimal, S.; Khodier, C.; Flaherty, D.P.; Rinis, N.; Charest, J.C.; Gao, N.; Zhao, P.; Wells, L.; Lewis, T.A.; et al. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat. Chem. Biol. 2016, 12, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Varki, A.; Kannagi, R.; Toole, B.; Stanley, P. Glycosylation Changes in Cancer. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Lau, K.S.; Dennis, J.W. N-Glycans in cancer progression. Glycobiology 2008, 18, 750–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contessa, J.N.; Bhojani, M.S.; Freeze, H.H.; Rehemtulla, A.; Lawrence, T.S. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008, 68, 3803–3809. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Bouchie, M.P.; Kukuruzinska, M.A. Protein N-glycosylation in oral cancer: Dysregulated cellular networks among DPAGT1, E-cadherin adhesion and canonical Wnt signaling. Glycobiology 2014, 24, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, H.; Cohen, S.; Bishayee, S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: A study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J. Biol. Chem. 2001, 276, 5375–5383. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.J.; Holdbrooks, A.T.; Chakraborty, A.; Grizzle, W.E.; Landen, C.N.; Buchsbaum, D.J.; Conner, M.G.; Arend, R.C.; Yoon, K.J.; Klug, C.A.; et al. The Tumor-Associated Glycosyltransferase ST6Gal-I Regulates Stem Cell Transcription Factors and Confers a Cancer Stem Cell Phenotype. Cancer Res. 2016, 76, 3978–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federici, M.; Menghini, R.; Mauriello, A.; Hribal, M.L.; Ferrelli, F.; Lauro, D.; Sbraccia, P.; Spagnoli, L.G.; Sesti, G.; Lauro, R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation 2002, 106, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Musicki, B.; Kramer, M.F.; Becker, R.E.; Burnett, A.L. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc. Natl. Acad. Sci. USA 2005, 102, 11870–11875. [Google Scholar] [CrossRef]
- Luo, B.; Soesanto, Y.; McClain, D.A. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting Akt activity in endothelial cells. Arterioscler Thromb. Vasc. Biol. 2008, 28, 651–657. [Google Scholar] [CrossRef]
- Lynch, T.P.; Ferrer, C.M.; Jackson, S.R.; Shahriari, K.S.; Vosseller, K.; Reginato, M.J. Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J. Biol. Chem. 2012, 287, 11070–11081. [Google Scholar] [CrossRef]
- Cummings, R.D.; Liu, F.T. Galectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Cummings, R.D.; McEver, R.P. C-type Lectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Guo, H.B.; Lee, I.; Kamar, M.; Pierce, M. N-acetylglucosaminyltransferase V expression levels regulate cadherin-associated homotypic cell-cell adhesion and intracellular signaling pathways. J. Biol. Chem. 2003, 278, 52412–52424. [Google Scholar] [CrossRef] [PubMed]
- Jamal, B.T.; Nita-Lazar, M.; Gao, Z.; Amin, B.; Walker, J.; Kukuruzinska, M.A. N-glycosylation status of E-cadherin controls cytoskeletal dynamics through the organization of distinct beta-catenin- and gamma-catenin-containing AJs. Cell Health Cytoskeleton 2009, 2009, 67–80. [Google Scholar] [PubMed]
- Nita-Lazar, M.; Rebustini, I.; Walker, J.; Kukuruzinska, M.A. Hypoglycosylated E-cadherin promotes the assembly of tight junctions through the recruitment of PP2A to adherens junctions. Exp. Cell Res. 2010, 316, 1871–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cheng, L.; Wang, L.J.; Liu, H.C.; Li, L.; Wang, X.L.; Geng, M.Y. Cell surface sialic acid inhibits Cx43 gap junction functions in constructed Hela cancer cells involving in sialylated N-cadherin. Mol. Cell Biochem. 2010, 344, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, M.; Gleissner, C.A.; Ley, K. Glycosylation in immune cell trafficking. Immunol. Rev. 2009, 230, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.W.; Dunn, T.S.; Ballestas, M.E.; Litovsky, S.H.; Patel, R.P. Identification of a high-mannose ICAM-1 glycoform: Effects of ICAM-1 hypoglycosylation on monocyte adhesion and outside in signaling. Am. J. Physiol. Cell Physiol. 2013, 305, C228–C237. [Google Scholar] [CrossRef] [PubMed]
- Tiemeyer, M.; Swiedler, S.J.; Ishihara, M.; Moreland, M.; Schweingruber, H.; Hirtzer, P.; Brandley, B.K. Carbohydrate ligands for endothelial-leukocyte adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1991, 88, 1138–1142. [Google Scholar] [CrossRef]
- Phillips, M.L.; Nudelman, E.; Gaeta, F.C.; Perez, M.; Singhal, A.K.; Hakomori, S.; Paulson, J.C. ELAM-1 mediates cell adhesion by recognition of a carbohydrate ligand, sialyl-Lex. Science 1990, 250, 1130–1132. [Google Scholar] [CrossRef]
- Nakamori, S.; Kameyama, M.; Imaoka, S.; Furukawa, H.; Ishikawa, O.; Sasaki, Y.; Izumi, Y.; Irimura, T. Involvement of carbohydrate antigen sialyl Lewis(x) in colorectal cancer metastasis. Dis. Colon Rectum 1997, 40, 420–431. [Google Scholar] [CrossRef]
- Gout, S.; Morin, C.; Houle, F.; Huot, J. Death receptor-3, a new E-Selectin counter-receptor that confers migration and survival advantages to colon carcinoma cells by triggering p38 and ERK MAPK activation. Cancer Res. 2006, 66, 9117–9124. [Google Scholar] [CrossRef]
- Barthel, S.R.; Gavino, J.D.; Wiese, G.K.; Jaynes, J.M.; Siddiqui, J.; Dimitroff, C.J. Analysis of glycosyltransferase expression in metastatic prostate cancer cells capable of rolling activity on microvascular endothelial (E)-selectin. Glycobiology 2008, 18, 806–817. [Google Scholar] [CrossRef] [PubMed]
- Barthel, S.R.; Hays, D.L.; Yazawa, E.M.; Opperman, M.; Walley, K.C.; Nimrichter, L.; Burdick, M.M.; Gillard, B.M.; Moser, M.T.; Pantel, K.; et al. Definition of molecular determinants of prostate cancer cell bone extravasation. Cancer Res. 2013, 73, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Barthel, S.R.; Wiese, G.K.; Cho, J.; Opperman, M.J.; Hays, D.L.; Siddiqui, J.; Pienta, K.J.; Furie, B.; Dimitroff, C.J. Alpha 1,3 fucosyltransferases are master regulators of prostate cancer cell trafficking. Proc. Natl. Acad. Sci. USA 2009, 106, 19491–19496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St Hill, C.A. Interactions between endothelial selectins and cancer cells regulate metastasis. Front. Biosci. (Landmark Ed.) 2011, 16, 3233–3251. [Google Scholar] [CrossRef] [PubMed]
- Witz, I.P. Tumor-microenvironment interactions: The selectin-selectin ligand axis in tumor-endothelium cross talk. Cancer Treat. Res. 2006, 130, 125–140. [Google Scholar]
- Kim, Y.J.; Borsig, L.; Varki, N.M.; Varki, A. P-selectin deficiency attenuates tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 1998, 95, 9325–9330. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Borsig, L.; Han, H.L.; Varki, N.M.; Varki, A. Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium. Am. J. Pathol. 1999, 155, 461–472. [Google Scholar] [CrossRef]
- Reyes-Reyes, M.E.; George, M.D.; Roberts, J.D.; Akiyama, S.K. P-selectin activates integrin-mediated colon carcinoma cell adhesion to fibronectin. Exp. Cell Res. 2006, 312, 4056–4069. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Smith, C.W.; Katkin, J.P.; Thurmon, L.M.; Xu, X.; Mendoza, L.H.; Ballantyne, C.M. Endothelial alpha 2,6-linked sialic acid inhibits VCAM-1-dependent adhesion under flow conditions. J. Immunol. 1999, 163, 2867–2876. [Google Scholar]
- Kitazume, S.; Imamaki, R.; Kurimoto, A.; Ogawa, K.; Kato, M.; Yamaguchi, Y.; Tanaka, K.; Ishida, H.; Ando, H.; Kiso, M.; et al. Interaction of platelet endothelial cell adhesion molecule (PECAM) with alpha2,6-sialylated glycan regulates its cell surface residency and anti-apoptotic role. J. Biol. Chem. 2014, 289, 27604–27613. [Google Scholar] [CrossRef]
- Kitazume, S.; Imamaki, R.; Ogawa, K.; Taniguchi, N. Sweet role of platelet endothelial cell adhesion molecule in understanding angiogenesis. Glycobiology 2014, 24, 1260–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazume, S.; Imamaki, R.; Ogawa, K.; Komi, Y.; Futakawa, S.; Kojima, S.; Hashimoto, Y.; Marth, J.D.; Paulson, J.C.; Taniguchi, N. Alpha2,6-sialic acid on platelet endothelial cell adhesion molecule (PECAM) regulates its homophilic interactions and downstream antiapoptotic signaling. J. Biol. Chem. 2010, 285, 6515–6521. [Google Scholar] [CrossRef] [PubMed]
- Lertkiatmongkol, P.; Paddock, C.; Newman, D.K.; Zhu, J.; Thomas, M.J.; Newman, P.J. The Role of Sialylated Glycans in Human Platelet Endothelial Cell Adhesion Molecule 1 (PECAM-1)-mediated Trans Homophilic Interactions and Endothelial Cell Barrier Function. J. Biol. Chem. 2016, 291, 26216–26225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Liu, A.; Miranda-Ribera, A.; Hyun, S.W.; Lillehoj, E.P.; Cross, A.S.; Passaniti, A.; Grimm, P.R.; Kim, B.Y.; Welling, P.A.; et al. NEU1 sialidase regulates the sialylation state of CD31 and disrupts CD31-driven capillary-like tube formation in human lung microvascular endothelia. J. Biol. Chem. 2014, 289, 9121–9135. [Google Scholar] [CrossRef] [PubMed]
- Paddock, C.; Zhou, D.; Lertkiatmongkol, P.; Newman, P.J.; Zhu, J. Structural basis for PECAM-1 homophilic binding. Blood 2016, 127, 1052–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamaki, R.; Ogawa, K.; Kizuka, Y.; Komi, Y.; Kojima, S.; Kotani, N.; Honke, K.; Honda, T.; Taniguchi, N.; Kitazume, S. Glycosylation controls cooperative PECAM-VEGFR2-beta3 integrin functions at the endothelial surface for tumor angiogenesis. Oncogene 2018, 37, 4287–4299. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N.; Rezazadeh, K.; Mahoney, J.E.; Hartsough, E.; Meyer, R.D. Identification of IGPR-1 as a novel adhesion molecule involved in angiogenesis. Mol. Biol. Cell 2012, 23, 1646–1656. [Google Scholar] [CrossRef]
- Wang, Y.H.W.; Meyer, R.D.; Bondzie, P.A.; Jiang, Y.; Rahimi, I.; Rezazadeh, K.; Mehta, M.; Laver, N.M.V.; Costello, C.E.; Rahimi, N. IGPR-1 Is Required for Endothelial Cell-Cell Adhesion and Barrier Function. J. Mol. Biol. 2016, 428, 5019–5033. [Google Scholar] [CrossRef]
- Woolf, N.; Pearson, B.E.; Bondzie, P.A.; Meyer, R.D.; Lavaei, M.; Belkina, A.C.; Chitalia, V.; Rahimi, N. Targeting tumor multicellular aggregation through IGPR-1 inhibits colon cancer growth and improves chemotherapy. Oncogenesis 2017, 6, e378. [Google Scholar] [CrossRef]
- Geyer, H.; Geyer, R.; Odenthal-Schnittler, M.; Schnittler, H.J. Characterization of human vascular endothelial cadherin glycans. Glycobiology 1999, 9, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Larsen, I.S.B.; Narimatsu, Y.; Joshi, H.J.; Siukstaite, L.; Harrison, O.J.; Brasch, J.; Goodman, K.M.; Hansen, L.; Shapiro, L.; Honig, B.; et al. Discovery of an O-mannosylation pathway selectively serving cadherins and protocadherins. Proc. Natl. Acad. Sci. USA 2017, 114, 11163–11168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindberg, G. Resialylation of sialic acid deficit vascular endothelium, circulating cells and macromolecules may counteract the development of atherosclerosis: A hypothesis. Atherosclerosis 2007, 192, 243–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park-Windhol, C.; Ng, Y.S.; Yang, J.; Primo, V.; Saint-Geniez, M.; D’Amore, P.A. Endomucin inhibits VEGF-induced endothelial cell migration, growth, and morphogenesis by modulating VEGFR2 signaling. Sci. Rep. 2017, 7, 17138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ju, N.; Yang, X.; Chen, L.; Yu, C. The alpha1,3-fucosyltransferase FUT7 regulates IL-1beta-induced monocyte-endothelial adhesion via fucosylation of endomucin. Life Sci. 2018, 192, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Gale, D.; Massberg, S.; Cheruvu, P.K.; Monahan-Earley, R.; Morgan, E.S.; Haig, D.; von Andrian, U.H.; Dvorak, A.M.; Aird, W.C. Phenotypic heterogeneity is an evolutionarily conserved feature of the endothelium. Blood 2007, 109, 613–615. [Google Scholar] [CrossRef] [PubMed]
- Jilani, S.M.; Murphy, T.J.; Thai, S.N.; Eichmann, A.; Alva, J.A.; Iruela-Arispe, M.L. Selective binding of lectins to embryonic chicken vasculature. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2003, 51, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Hanasaki, K.; Varki, A.; Stamenkovic, I.; Bevilacqua, M.P. Cytokine-induced beta-galactoside alpha-2,6-sialyltransferase in human endothelial cells mediates alpha 2,6-sialylation of adhesion molecules and CD22 ligands. J. Biol. Chem. 1994, 269, 10637–10643. [Google Scholar]
- Scott, D.W.; Vallejo, M.O.; Patel, R.P. Heterogenic endothelial responses to inflammation: Role for differential N-glycosylation and vascular bed of origin. J. Am. Heart Assoc. 2013, 2, e000263. [Google Scholar] [CrossRef]
- Scott, D.W.; Patel, R.P. Endothelial heterogeneity and adhesion molecules N-glycosylation: Implications in leukocyte trafficking in inflammation. Glycobiology 2013, 23, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Croci, D.O.; Mendez-Huergo, S.P.; Cerliani, J.P.; Rabinovich, G.A. Immune-Mediated and Hypoxia-Regulated Programs: Accomplices in Resistance to Anti-angiogenic Therapies. Handb. Exp. Pharmacol. 2018, 249, 31–61. [Google Scholar] [PubMed]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Rehman, J.; Tang, H.; Wary, K.; Mittal, M.; Chaturvedi, P.; Zhao, Y.Y.; Komarova, Y.A.; Vogel, S.M.; Malik, A.B. HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Investig. 2015, 125, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Fong, G.H. Prolyl hydroxylase domain 2 protein suppresses hypoxia-induced endothelial cell proliferation. Hypertension (Dallas, Tex. 1979) 2007, 49, 178–184. [Google Scholar] [CrossRef]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Leite de Oliveira, R.; Deschoemaeker, S.; Henze, A.T.; Debackere, K.; Finisguerra, V.; Takeda, Y.; Roncal, C.; Dettori, D.; Tack, E.; Jonsson, Y.; et al. Gene-targeting of Phd2 improves tumor response to chemotherapy and prevents side-toxicity. Cancer Cell 2012, 22, 263–277. [Google Scholar] [CrossRef]
- Kuchnio, A.; Moens, S.; Bruning, U.; Kuchnio, K.; Cruys, B.; Thienpont, B.; Broux, M.; Ungureanu, A.A.; Leite de Oliveira, R.; Bruyere, F.; et al. The Cancer Cell Oxygen Sensor PHD2 Promotes Metastasis via Activation of Cancer-Associated Fibroblasts. Cell Rep. 2015, 12, 992–1005. [Google Scholar] [CrossRef] [Green Version]
- Branco-Price, C.; Zhang, N.; Schnelle, M.; Evans, C.; Katschinski, D.M.; Liao, D.; Ellies, L.; Johnson, R.S. Endothelial cell HIF-1alpha and HIF-2alpha differentially regulate metastatic success. Cancer Cell 2012, 21, 52–65. [Google Scholar] [CrossRef]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2alpha regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443. [Google Scholar] [CrossRef]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.C.; Vandekeere, S.; Bouche, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7, 12240. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Cerliani, J.P.; Pinto, N.A.; Morosi, L.G.; Rabinovich, G.A. Regulatory role of glycans in the control of hypoxia-driven angiogenesis and sensitivity to anti-angiogenic treatment. Glycobiology 2014, 24, 1283–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J.; Shing, Y. Control of angiogenesis by heparin and other sulfated polysaccharides. Adv. Exp. Med. Biol. 1992, 313, 355–364. [Google Scholar] [PubMed]
- Xu, D.; Fuster, M.M.; Lawrence, R.; Esko, J.D. Heparan sulfate regulates VEGF165- and VEGF121-mediated vascular hyperpermeability. J. Biol. Chem. 2011, 286, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Chiodelli, P.; Mitola, S.; Ravelli, C.; Oreste, P.; Rusnati, M.; Presta, M. Heparan sulfate proteoglycans mediate the angiogenic activity of the vascular endothelial growth factor receptor-2 agonist gremlin. Arterioscler Thromb. Vasc. Biol. 2011, 31, e116–e127. [Google Scholar] [CrossRef] [PubMed]
- Nangia-Makker, P.; Baccarini, S.; Raz, A. Carbohydrate-recognition and angiogenesis. Cancer Metastasis Rev. 2000, 19, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Chandler, K.B.; Leon, D.R.; Meyer, R.D.; Rahimi, N.; Costello, C.E. Site-Specific N-Glycosylation of Endothelial Cell Receptor Tyrosine Kinase VEGFR-2. J. Proteome Res. 2017, 16, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.I.; Jefferies, K.C.; Panjwani, N. Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J. Biol. Chem. 2011, 286, 29913–29921. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Salatino, M.; Rubinstein, N.; Cerliani, J.P.; Cavallin, L.E.; Leung, H.J.; Ouyang, J.; Ilarregui, J.M.; Toscano, M.A.; Domaica, C.I.; et al. Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. J. Exp. Med. 2012, 209, 1985–2000. [Google Scholar] [CrossRef]
- Oliveira-Ferrer, L.; Legler, K.; Milde-Langosch, K. Role of protein glycosylation in cancer metastasis. Semin. Cancer Biol. 2017, 44, 141–152. [Google Scholar] [CrossRef]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Selleck, S.B. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002, 71, 435–471. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.; Kjellen, L. Heparan sulfate: Lessons from knockout mice. J. Clin. Investig. 2001, 108, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Rapraeger, A.C. Heparan sulfate-growth factor interactions. Methods Cell Biol. 2002, 69, 83–109. [Google Scholar] [PubMed]
- Carmeliet, P.; Ng, Y.S.; Nuyens, D.; Theilmeier, G.; Brusselmans, K.; Cornelissen, I.; Ehler, E.; Kakkar, V.V.; Stalmans, I.; Mattot, V.; et al. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat. Med. 1999, 5, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Ruhrberg, C.; Gerhardt, H.; Golding, M.; Watson, R.; Ioannidou, S.; Fujisawa, H.; Betsholtz, C.; Shima, D.T. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002, 16, 2684–2698. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Beckouche, N.; Bignon, M.; Lelarge, V.; Mathivet, T.; Pichol-Thievend, C.; Berndt, S.; Hardouin, J.; Garand, M.; Ardidie-Robouant, C.; Barret, A.; et al. The interaction of heparan sulfate proteoglycans with endothelial transglutaminase-2 limits VEGF165-induced angiogenesis. Sci. Signal. 2015, 8, ra70. [Google Scholar] [CrossRef]
- LaRochelle, W.J.; May-Siroff, M.; Robbins, K.C.; Aaronson, S.A. A novel mechanism regulating growth factor association with the cell surface: Identification of a PDGF retention domain. Genes Dev. 1991, 5, 1191–1199. [Google Scholar] [CrossRef]
- Ostman, A.; Andersson, M.; Betsholtz, C.; Westermark, B.; Heldin, C.H. Identification of a cell retention signal in the B-chain of platelet-derived growth factor and in the long splice version of the A-chain. Cell Regul. 1991, 2, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Rider, C.C. Heparin/heparan sulphate binding in the TGF-beta cytokine superfamily. Biochem. Soc. Trans. 2006, 34, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.; Rushton, G.; Gallagher, J.T. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J. Biol. Chem. 1997, 272, 18000–18006. [Google Scholar] [CrossRef] [PubMed]
- Khurana, A.; Beleford, D.; He, X.; Chien, J.; Shridhar, V. Role of heparan sulfatases in ovarian and breast cancer. Am. J. Cancer Res. 2013, 3, 34–45. [Google Scholar] [PubMed]
- Monzavi-Karbassi, B.; Stanley, J.S.; Hennings, L.; Jousheghany, F.; Artaud, C.; Shaaf, S.; Kieber-Emmons, T. Chondroitin sulfate glycosaminoglycans as major P-selectin ligands on metastatic breast cancer cell lines. Int. J. Cancer 2007, 120, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Hirose, M.; Hirose, J.; Nagakubo, D.; Plaas, A.H.; Miyasaka, M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J. Biol. Chem. 2000, 275, 35448–35456. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, F.M.; Vitale, D.L.; Demarchi, G.; Cristina, C.; Alaniz, L. The immunological effect of hyaluronan in tumor angiogenesis. Clin. Transl. Immunol. 2015, 4, e52. [Google Scholar] [CrossRef] [PubMed]
- Koyama, H.; Hibi, T.; Isogai, Z.; Yoneda, M.; Fujimori, M.; Amano, J.; Kawakubo, M.; Kannagi, R.; Kimata, K.; Taniguchi, S.; et al. Hyperproduction of hyaluronan in neu-induced mammary tumor accelerates angiogenesis through stromal cell recruitment: Possible involvement of versican/PG-M. Am. J. Pathol. 2007, 170, 1086–1099. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Miyoshi, S.; Mikami, T.; Koyama, H.; Kitazawa, M.; Takeoka, M.; Sano, K.; Amano, J.; Isogai, Z.; Niida, S.; et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. 2010, 70, 7073–7083. [Google Scholar] [CrossRef]
- Renkonen, J.; Tynninen, O.; Hayry, P.; Paavonen, T.; Renkonen, R. Glycosylation might provide endothelial zip codes for organ-specific leukocyte traffic into inflammatory sites. Am. J. Pathol. 2002, 161, 543–550. [Google Scholar] [CrossRef]
- Glinsky, V.V.; Huflejt, M.E.; Glinsky, G.V.; Deutscher, S.L.; Quinn, T.P. Effects of Thomsen-Friedenreich antigen-specific peptide P-30 on beta-galactoside-mediated homotypic aggregation and adhesion to the endothelium of MDA-MB-435 human breast carcinoma cells. Cancer Res. 2000, 60, 2584–2588. [Google Scholar] [PubMed]
- Glinsky, V.V.; Glinsky, G.V.; Rittenhouse-Olson, K.; Huflejt, M.E.; Glinskii, O.V.; Deutscher, S.L.; Quinn, T.P. The role of Thomsen-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res. 2001, 61, 4851–4857. [Google Scholar] [PubMed]
- Yu, L.G.; Andrews, N.; Zhao, Q.; McKean, D.; Williams, J.F.; Connor, L.J.; Gerasimenko, O.V.; Hilkens, J.; Hirabayashi, J.; Kasai, K.; et al. Galectin-3 interaction with Thomsen-Friedenreich disaccharide on cancer-associated MUC1 causes increased cancer cell endothelial adhesion. J. Biol. Chem. 2007, 282, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Guo, X.; Nash, G.B.; Stone, P.C.; Hilkens, J.; Rhodes, J.M.; Yu, L.G. Circulating galectin-3 promotes metastasis by modifying MUC1 localization on cancer cell surface. Cancer Res. 2009, 69, 6799–6806. [Google Scholar] [CrossRef]
- Khaldoyanidi, S.K.; Glinsky, V.V.; Sikora, L.; Glinskii, A.B.; Mossine, V.V.; Quinn, T.P.; Glinsky, G.V.; Sriramarao, P. MDA-MB-435 human breast carcinoma cell homo- and heterotypic adhesion under flow conditions is mediated in part by Thomsen-Friedenreich antigen-galectin-3 interactions. J. Biol. Chem. 2003, 278, 4127–4134. [Google Scholar] [CrossRef] [PubMed]
- Glavey, S.V.; Manier, S.; Natoni, A.; Sacco, A.; Moschetta, M.; Reagan, M.R.; Murillo, L.S.; Sahin, I.; Wu, P.; Mishima, Y.; et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood 2014, 124, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K.; Duda, D.G.; Clark, J.W.; Loeffler, J.S. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 2006, 3, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arao, T.; Matsumoto, K.; Furuta, K.; Kudo, K.; Kaneda, H.; Nagai, T.; Sakai, K.; Fujita, Y.; Tamura, D.; Aomatsu, K.; et al. Acquired drug resistance to vascular endothelial growth factor receptor 2 tyrosine kinase inhibitor in human vascular endothelial cells. Anticancer Res. 2011, 31, 2787–2796. [Google Scholar] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Rinis, N.; Golden, J.E.; Marceau, C.D.; Carette, J.E.; Van Zandt, M.C.; Gilmore, R.; Contessa, J.N. Editing N-Glycan Site Occupancy with Small-Molecule Oligosaccharyltransferase Inhibitors. Cell Chem. Biol. 2018, 25, 1231–1241.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baro, M.; Lopez Sambrooks, C.; Quijano, A.; Saltzman, W.M.; Contessa, J. Oligosaccharyltransferase Inhibition Reduces Receptor Tyrosine Kinase Activation and Enhances Glioma Radiosensitivity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Lopez Sambrooks, C.; Baro, M.; Quijano, A.; Narayan, A.; Cui, W.; Greninger, P.; Egan, R.; Patel, A.; Benes, C.H.; Saltzman, W.M.; et al. Oligosaccharyltransferase Inhibition Overcomes Therapeutic Resistance to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2018, 78, 5094–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeley, N.M.; Alley, S.C.; Anderson, M.E.; Boursalian, T.E.; Burke, P.J.; Emmerton, K.M.; Jeffrey, S.C.; Klussman, K.; Law, C.L.; Sussman, D.; et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl. Acad. Sci. USA 2013, 110, 5404–5409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Nguyen, P.; Nguyen, M.; Okeley, N.M.; Benjamin, D.R.; Senter, P.D.; Vercellotti, G.M. The fucosylation inhibitor, 2-fluorofucose, inhibits vaso-occlusion, leukocyte-endothelium interactions and NF-kB activation in transgenic sickle mice. PLoS ONE 2015, 10, e0117772. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fukuda, T.; Hang, Q.; Hou, S.; Isaji, T.; Kameyama, A.; Gu, J. Inhibition of fucosylation by 2-fluorofucose suppresses human liver cancer HepG2 cell proliferation and migration as well as tumor formation. Sci. Rep. 2017, 7, 11563. [Google Scholar] [CrossRef] [PubMed]
- Carrascal, M.A.; Silva, M.; Ramalho, J.S.; Pen, C.; Martins, M.; Pascoal, C.; Amaral, C.; Serrano, I.; Oliveira, M.J.; Sackstein, R.; et al. Inhibition of fucosylation in human invasive ductal carcinoma reduces E-selectin ligand expression, cell proliferation, and ERK1/2 and p38 MAPK activation. Mol. Oncol. 2018, 12, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Gao, X.; Solar, V.D.; Yu, X.; Antonopoulos, A.; Friedman, A.E.; Matich, E.K.; Atilla-Gokcumen, G.E.; Nasirikenari, M.; Lau, J.T.; et al. Thioglycosides Are Efficient Metabolic Decoys of Glycosylation that Reduce Selectin Dependent Leukocyte Adhesion. Cell Chem. Biol. 2018, 25, 1519–1532.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsig, L. Selectins in cancer immunity. Glycobiology 2018, 28, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundin, L.; Larsson, H.; Kreuger, J.; Kanda, S.; Lindahl, U.; Salmivirta, M.; Claesson-Welsh, L. Selectively desulfated heparin inhibits fibroblast growth factor-induced mitogenicity and angiogenesis. J. Biol. Chem. 2000, 275, 24653–24660. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, K.D.; Karoli, T.; Liu, L.; Dredge, K.; Copeman, E.; Li, C.P.; Davis, K.; Hammond, E.; Bytheway, I.; Kostewicz, E.; et al. Synthesis and biological evaluation of polysulfated oligosaccharide glycosides as inhibitors of angiogenesis and tumor growth. J. Med. Chem. 2010, 53, 1686–1699. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Ilan, N.; Naggi, A.; Casu, B. Heparanase: Structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr. Pharm. Des. 2007, 13, 2057–2073. [Google Scholar] [CrossRef]
- Heyman, B.; Yang, Y. Mechanisms of heparanase inhibitors in cancer therapy. Exp. Hematol. 2016, 44, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Major classes of glycans and glycosaminoglycans involved in endothelial cell signaling and adhesion. Representative glycan-binding lectins (Gal-1, Gal-3, E-selectin, P-selectin, and L-selectin), growth factors (such as vascular endothelial growth factor, VEGF), and glycoconjugates including N- and O-linked glycans, and glycosaminoglycans, are shown. Abbreviations: HS, heparan sulfate; CS, chondroitin sulfate; HA, hyaluronan; GalNAc, N-acetylgalactosamine; GlcNAc, N-acetylglucosamine; Man, mannose; Gal, galactose; Fuc, fucose; Sialic acid, N-acetylneuraminic acid; GlcA, glucuronic acid; IdoA, iduronic acid; S, sulfate.
Figure 1.
Major classes of glycans and glycosaminoglycans involved in endothelial cell signaling and adhesion. Representative glycan-binding lectins (Gal-1, Gal-3, E-selectin, P-selectin, and L-selectin), growth factors (such as vascular endothelial growth factor, VEGF), and glycoconjugates including N- and O-linked glycans, and glycosaminoglycans, are shown. Abbreviations: HS, heparan sulfate; CS, chondroitin sulfate; HA, hyaluronan; GalNAc, N-acetylgalactosamine; GlcNAc, N-acetylglucosamine; Man, mannose; Gal, galactose; Fuc, fucose; Sialic acid, N-acetylneuraminic acid; GlcA, glucuronic acid; IdoA, iduronic acid; S, sulfate.

Figure 2.
Tumor microenvironment-mediated changes in endothelial cell glycosylation. Endothelial glycoproteins are shown, including integrins, receptor tyrosine kinases (RTKs), VE-cadherin, and Ig-like cell adhesion molecules (IgCAMs). Glycans synthesized in the endoplasmic reticulum (ER) and Golgi have the potential to alter signaling and adhesion.
Figure 2.
Tumor microenvironment-mediated changes in endothelial cell glycosylation. Endothelial glycoproteins are shown, including integrins, receptor tyrosine kinases (RTKs), VE-cadherin, and Ig-like cell adhesion molecules (IgCAMs). Glycans synthesized in the endoplasmic reticulum (ER) and Golgi have the potential to alter signaling and adhesion.

Figure 3.
Glycan-mediated intravasation, rolling, and extravasation. Glycan modifications on endothelial cells (red) alter endothelial adhesion and may contribute to endothelial permeability and tumor cells (blue) intravasation. Circulating lectins such as Gal-3 and endothelial lectins including E-selectin initiate rolling, adhesion, and extravasation of tumor cells to the endothelium, frequently at distant sites. Integrins also assist in this process.
Figure 3.
Glycan-mediated intravasation, rolling, and extravasation. Glycan modifications on endothelial cells (red) alter endothelial adhesion and may contribute to endothelial permeability and tumor cells (blue) intravasation. Circulating lectins such as Gal-3 and endothelial lectins including E-selectin initiate rolling, adhesion, and extravasation of tumor cells to the endothelium, frequently at distant sites. Integrins also assist in this process.


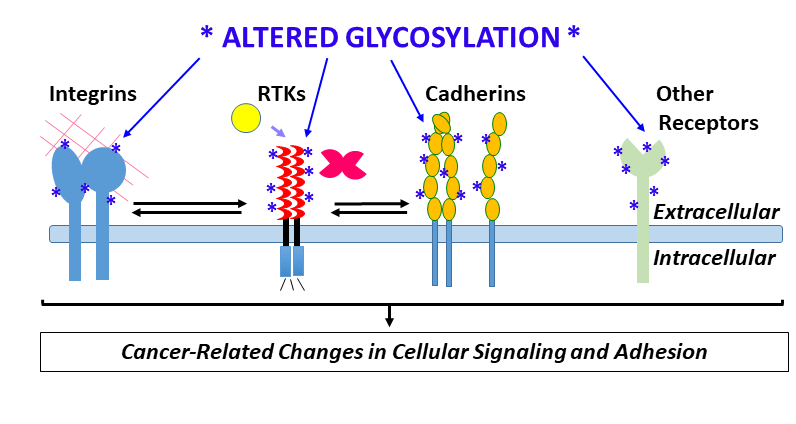{kind=link}
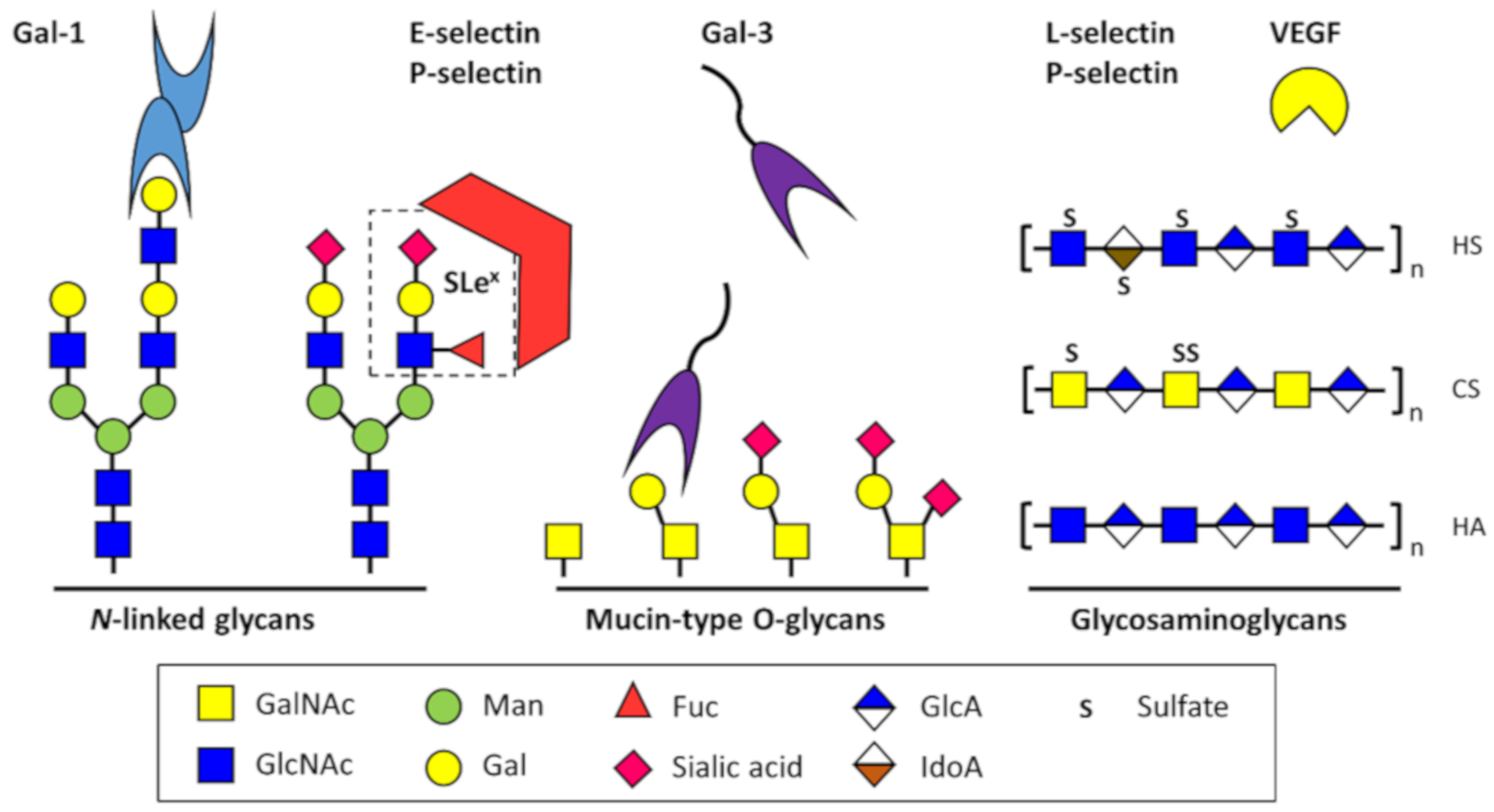{kind=link}
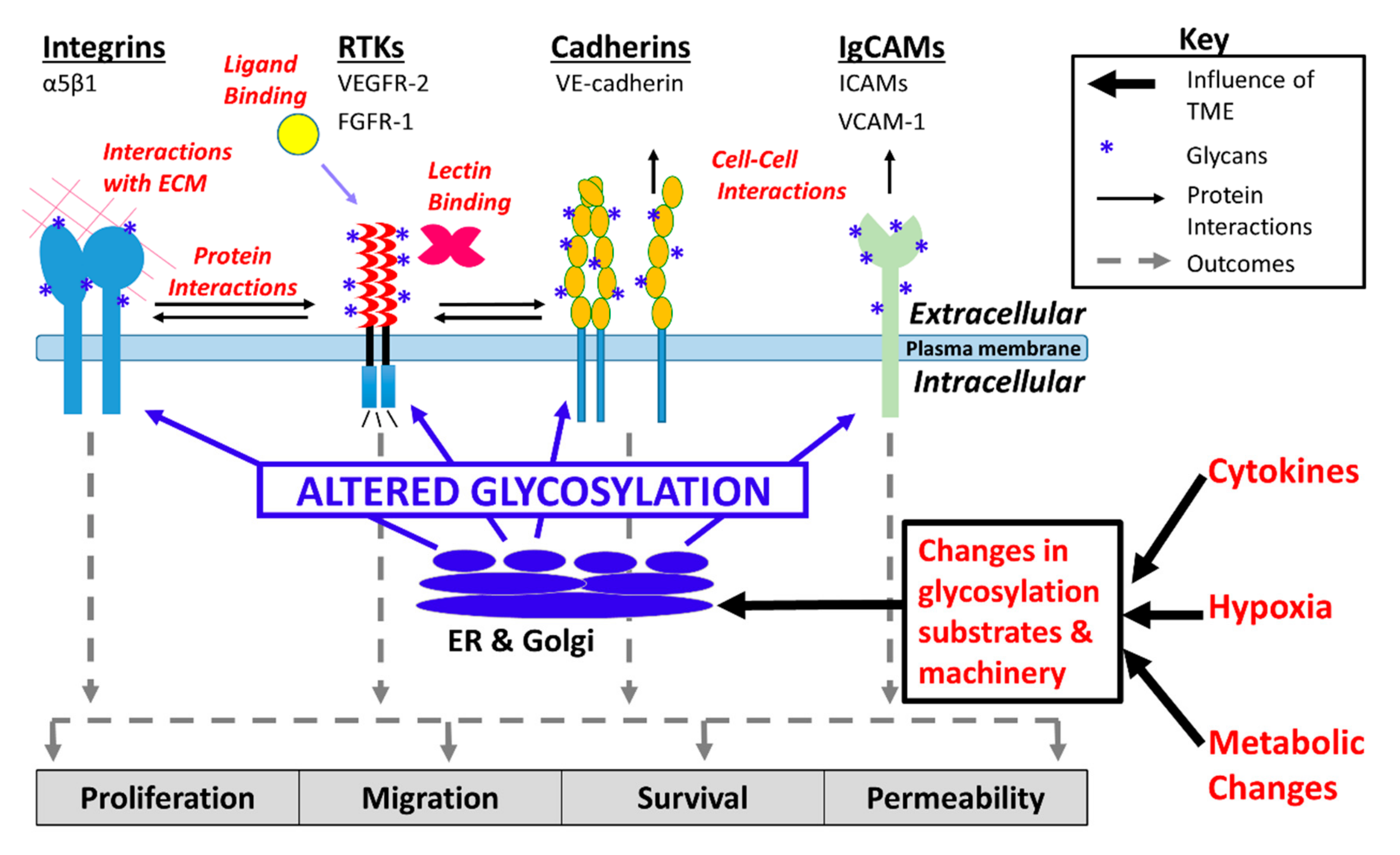{kind=link}
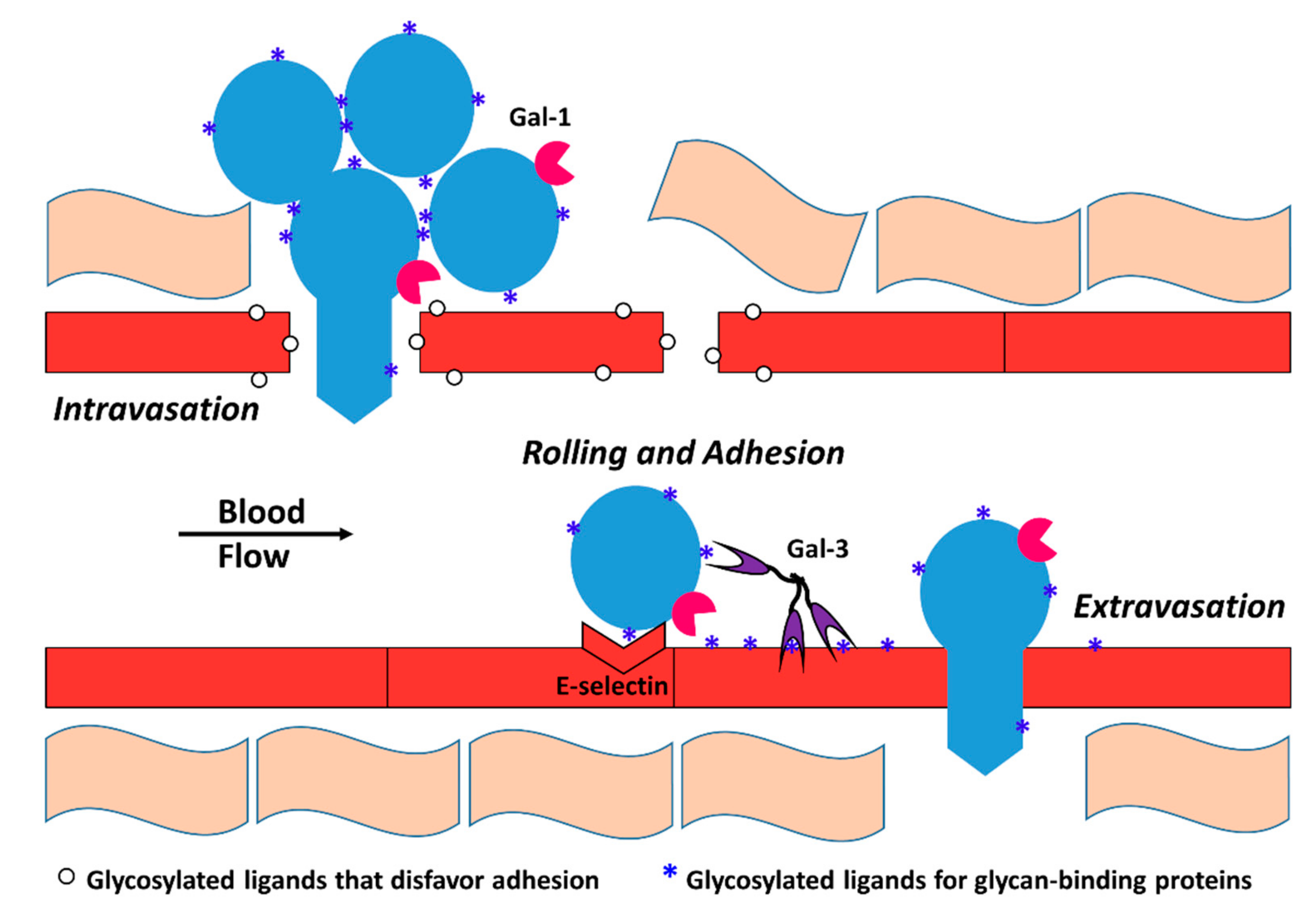{kind=link}
Table 1.
Factors that influence endothelial cell glycosylation. Pro- and anti-inflammatory actors that have been reported to impact endothelial cell glycosylation are shown.
Table 1.
Factors that influence endothelial cell glycosylation. Pro- and anti-inflammatory actors that have been reported to impact endothelial cell glycosylation are shown.
| Factor | Inflam. | Source(s) | Impact on Endothelial Glycosylation |
|---|---|---|---|
| TNF-α | Pro | macrophages, CD4+ lympho-cytes, NK cells, neutrophils | ↑ ST6Gal-I and α-mannosidase expression [94,95] |
| IL-1* | Pro | macrophages, monocytes, fibroblasts, and dendritic cells | ↑ ST6Gal-I expression [94] |
| IL-1β | Pro | macrophages, dendritic cells | ↑ α-mannosidase expression [95] |
| IFN-γ | Pro | NK, NKT cells, and CD4+ Th1 and CD8+ CTL effector T cells | ↑ α2,6-linked sialic acids [16] (presumably via ↑ ST6Gal-I expr.) |
| IL-17 | Pro | Th (CD4+) cells | ↑ α2,6-linked sialic acids [16] (presumably via ↑ ST6Gal-I expr.) |
| IL-10 | Anti | Th2, mast cells, CD4+ CD25+ Foxp3+ Treg | ↓ α2,6-linked sialic acids [16] (presumably via ↓ ST6Gal-I expr.) |
| TGF-β1 | Anti | Platelets, most leukocytes | ↓ α2,6-linked sialic acids [16] (presumably via ↓ ST6Gal-I expr.) |
| Hypoxia | N/A | N/A | ↓ α2,6-linked sialic acids, ↑ β1,6 branching, elongation of poly-LacNAc chains [16] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Brown Chandler, K.; E. Costello, C.; Rahimi, N. Glycosylation in the Tumor Microenvironment: Implications for Tumor Angiogenesis and Metastasis. Cells 2019, 8, 544. https://doi.org/10.3390/cells8060544
AMA Style
Brown Chandler K, E. Costello C, Rahimi N. Glycosylation in the Tumor Microenvironment: Implications for Tumor Angiogenesis and Metastasis. Cells. 2019; 8(6):544. https://doi.org/10.3390/cells8060544
Chicago/Turabian StyleBrown Chandler, Kevin, Catherine E. Costello, and Nader Rahimi. 2019. "Glycosylation in the Tumor Microenvironment: Implications for Tumor Angiogenesis and Metastasis" Cells 8, no. 6: 544. https://doi.org/10.3390/cells8060544
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.