Evolutionary Conservation and Divergence of Genes Encoding 3-Hydroxy-3-methylglutaryl Coenzyme A Synthase in the Allotetraploid Cotton Species Gossypium hirsutum

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of HMGS Proteins in Gossypium and Other Species
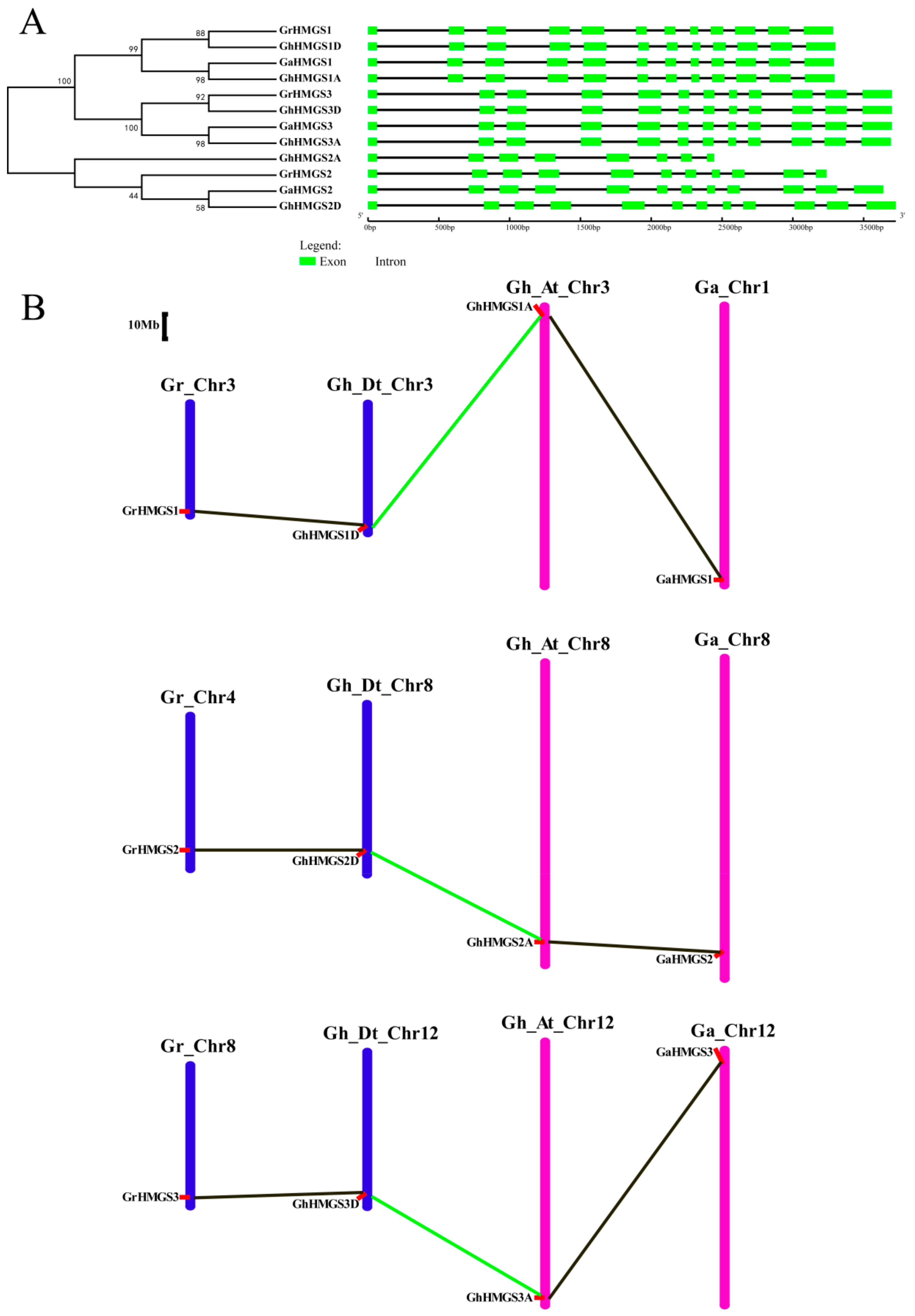
2.2. Phylogenetic Analysis, Gene Structure and Chromosomal Mapping
2.3. Plant Materials and Treatments
2.4. RNA Isolation, cDNA Synthesis, and Primer Design
2.5. Reverse Transcription PCR (RT-PCR) and Quantitative Real-Time RT-PCR (qRT-PCR)
3. Results
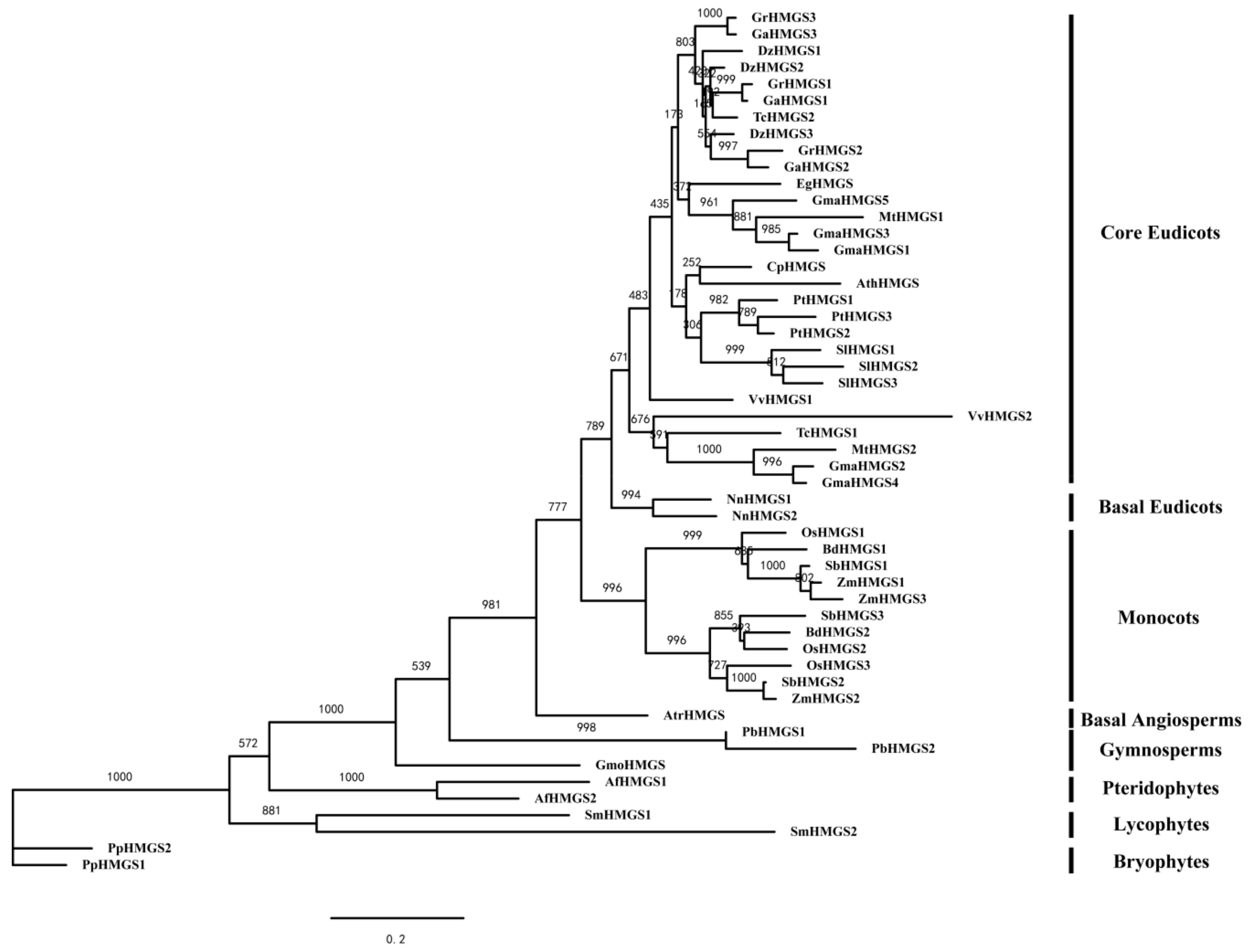
3.1. Identification and Phylogenetic Analysis of HMGS Genes in Green Plants
3.2. Phylogenetic Analysis of Gossypium HMGS Genes
3.3. Expression Profiles of HMGS Genes in Various G. hirsutum Tissues
3.4. Expression Analysis of HMGS Genes in Response to Phytohormone Treatments
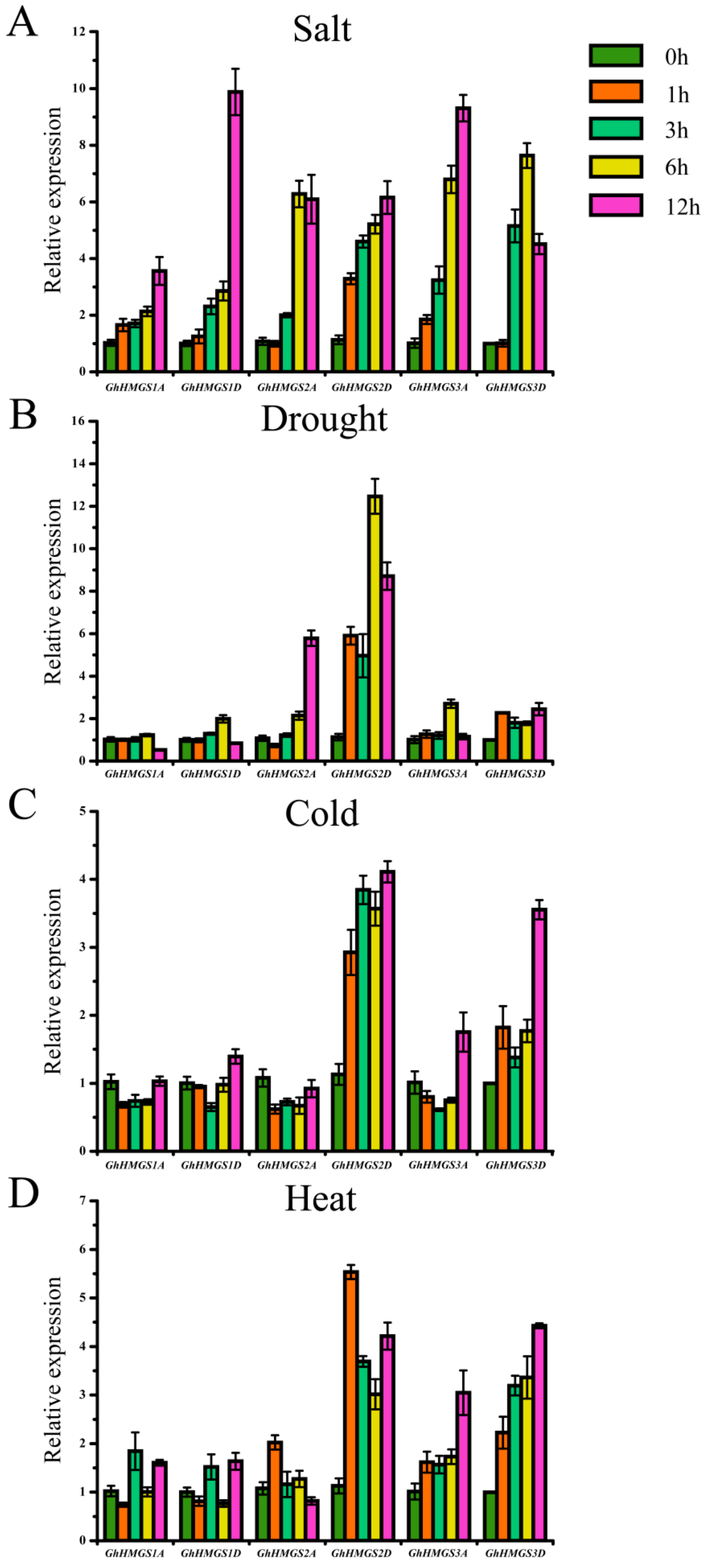
3.5. Expression Analysis of HMGS Genes in Response to Abiotic Stresses
4. Discussion
4.1. Comparative Genomic Analysis of the HMGS Gene Family in Green Plants
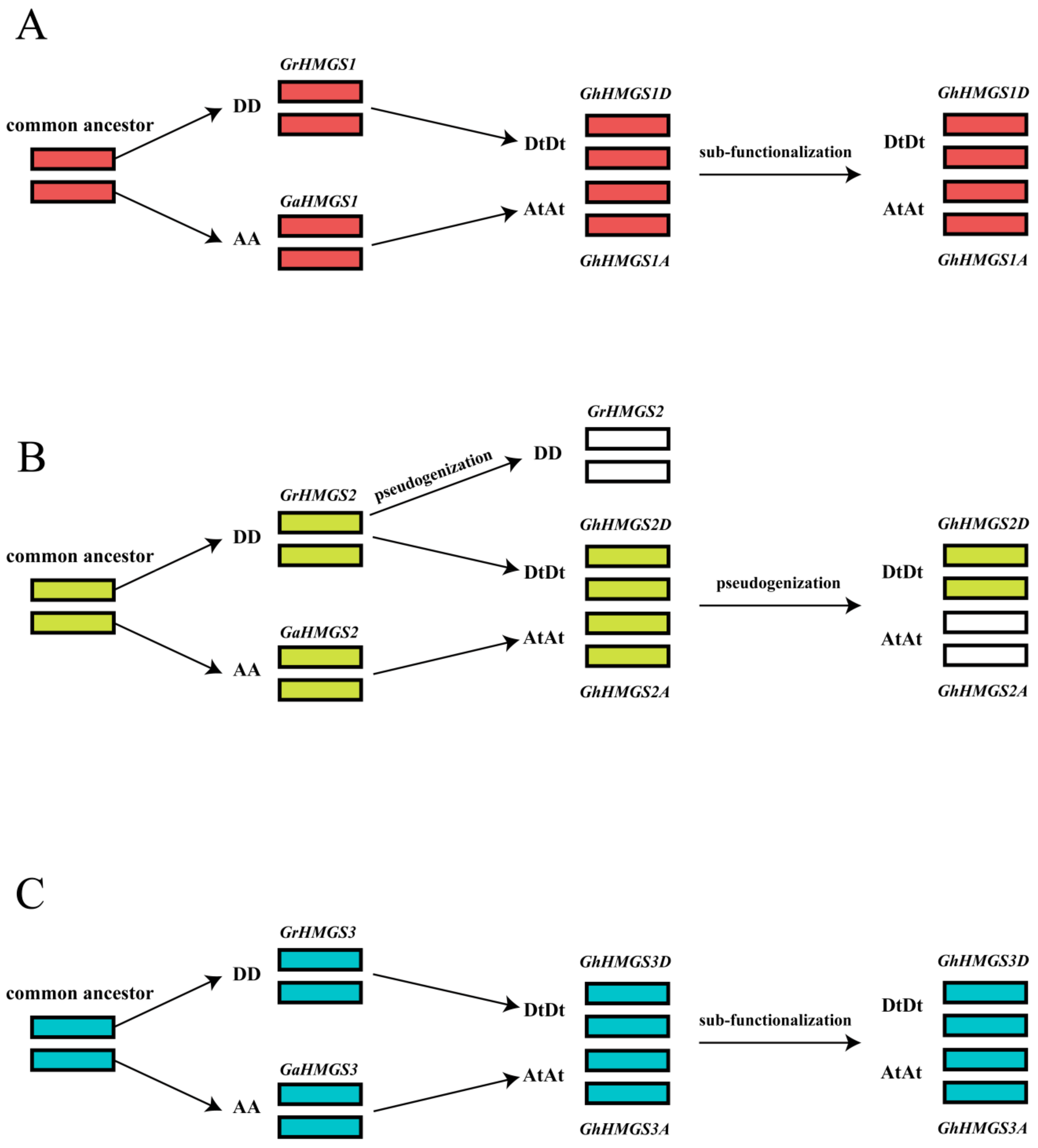
4.2. Evolutionary Conservation of the Cotton HMGS Gene Family
4.3. Evolutionary Divergence of the Cotton HMGS Gene Family
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Doyle, J.J.; Egan, A.N. Dating the origins of polyploidy events. New Phytol. 2010, 186, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.S.; Liu, H.Y.; Chiang, Y.C.; Chiou, W.L. Polyploidy and speciation in Pteris (Pteridaceae). J. Bot. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Schneider, H.; Liu, H.M.; Chang, Y.F.; Ohlsen, D.; Perrie, L.R.; Shepherd, L.; Kessler, M.; Karger, D.N.; Hennequin, S.; Marquardt, J.; et al. Neo- and Paleopolyploidy contribute to the species diversity of Asplenium-the most species-rich genus of ferns. J. Syst. Evol. 2017, 55, 353–364. [Google Scholar] [CrossRef]
- Del Pozo, J.C.; Ramirez-Parra, E. Whole genome duplications in plants: An overview from Arabidopsis. J. Exp. Bot. 2015, 66, 6991–7003. [Google Scholar] [CrossRef] [PubMed]
- Vision, T.J.; Brown, D.G.; Tanksley, S.D. The origins of genomic duplications in Arabidopsis. Science 2000, 290, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Yang, J.; Gu, X. Expression divergence between duplicate genes. Trends Genet. 2005, 21, 602–607. [Google Scholar] [CrossRef]
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Guan, X.; Liang, W.; Chen, J.; Fang, L.; Hu, Y.; Guo, W.; Rong, J.; Xu, G.; Zhang, T. Divergence and evolution of cotton bHLH proteins from diploid to allotetraploid. BMC Genomics 2018, 19, 162. [Google Scholar] [CrossRef] [PubMed]
- Magadum, S.; Banerjee, U.; Murugan, P.; Gangapur, D.; Ravikesavan, R. Gene duplication as a major force in evolution. J. Genet. 2013, 92, 155–161. [Google Scholar] [CrossRef]
- Wendel, J.F. Genome evolution in polyploids. Plant Mol. Biol. 2000, 42, 225–249. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef]
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.; Guo, W.; Chen, X.; Stelly, D.M.; Rabinowicz, P.D.; Town, C.D.; et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef]
- Wendel, J.F.; Albert, V.A. Phylogenetics of the cotton genus (Gossypium): Character-state weighted parsimony analysis of chloroplast-DNA restriction site data and its systematic and biogeographic implications. Syst. Bot. 1992, 17, 115–143. [Google Scholar] [CrossRef]
- Wendel, J.F.; Cronn, R.C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 2003, 78, 139–186. [Google Scholar] [Green Version]
- Wendel, J.F. New World tetraploid cottons contain Old World cytoplasm. Proc. Natl. Acad. Sci. USA 1989, 86, 4132–4136. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef]
- Du, X.; Huang, G.; He, S.; Yang, Z.; Sun, G.; Ma, X.; Li, N.; Zhang, X.; Sun, J.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, J.; Zuo, D.; Wang, Q.; Malik, W.; Zhang, Y.; Abid, M.A.; Cheng, H.; Yang, Q.; Song, G. Recent insights into cotton functional genomics: Progress and future perspectives. Plant Biotechnol. J. 2018, 16, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Thulasiram, H.V.; Erickson, H.K.; Poulter, C.D. Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis. Science 2007, 316, 73–76. [Google Scholar] [CrossRef] [PubMed]
- McGarvey, D.J.; Croteau, R. Terpenoid metabolism. Plant Cell 1995, 7, 1015–1026. [Google Scholar] [CrossRef]
- Lange, B.M.; Rujan, T.; Martin, W.; Croteau, R. Isoprenoid biosynthesis: The evolution of two ancient and distinct pathways across genomes. Proc. Natl. Acad. Sci. USA 2000, 97, 13172–13177. [Google Scholar] [CrossRef] [Green Version]
- Laule, O.; Furholz, A.; Chang, H.S.; Zhu, T.; Wang, X.; Heifetz, P.B.; Gruissem, W.; Lange, M. Crosstalk between cytosolic and plastidial pathways of isoprenoid biosynthesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2003, 100, 6866–6871. [Google Scholar] [CrossRef]
- Bick, J.A.; Lange, B.M. Metabolic cross talk between cytosolic and plastidial pathways of isoprenoid biosynthesis: Unidirectional transport of intermediates across the chloroplast envelope membrane. Arch. Biochem. Biophys. 2003, 415, 146–154. [Google Scholar] [CrossRef]
- Liao, P.; Wang, H.; Hemmerlin, A.; Nagegowda, D.A.; Bach, T.J.; Wang, M.; Chye, M.L. Past achievements, current status and future perspectives of studies on 3-hydroxy-3-methylglutaryl-CoA synthase (HMGS) in the mevalonate (MVA) pathway. Plant Cell Rep. 2014, 33, 1005–1022. [Google Scholar] [CrossRef] [PubMed]
- Vranova, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef]
- Hemmerlin, A.; Harwood, J.L.; Bach, T.J. A raison d’etre for two distinct pathways in the early steps of plant isoprenoid biosynthesis? Prog. Lipid Res. 2012, 51, 95–148. [Google Scholar] [CrossRef]
- Chang, J.; Ning, Y.; Xu, F.; Cheng, S.; Li, X. Research advance of 3-hydroxy-3-methylglutaryl-coenzyme a synthase in plant isoprenoid biosynthesis. J. Anim. Plant Sci. 2015, 25, 1441–1450. [Google Scholar]
- Montamat, F.; Guilloton, M.; Karst, F.; Delrot, S. Isolation and characterization of a cDNA encoding Arabidopsis thaliana 3-hydroxy-3-methylglutaryl-coenzyme A synthase. Gene 1995, 167, 197–201. [Google Scholar] [CrossRef]
- Alex, D.; Bach, T.J.; Chye, M.L. Expression of Brassica juncea 3-hydroxy-3-methylglutaryl CoA synthase is developmentally regulated and stress-responsive. Plant J. 2000, 22, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Song, Q.; Ye, J.; Wang, L.; Xu, F. Characterization, function, and transcriptional profiling analysis of 3-hydroxy-3-methylglutaryl-CoA synthase gene (GbHMGS1) towards stresses and exogenous hormone treatments in Ginkgo biloba. Molecules 2017, 22, 1706. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, Q.; Wang, C.; Chen, L.; Sun, Z.; Zhu, X.; Tang, Y.; Shao, J.; Wu, Y. Characterization of genes involved in isoprenoid diphosphate biosynthesis in maize. J. Plant Growth Regul. 2015, 34, 294–308. [Google Scholar] [CrossRef]
- Ren, A.; Ouyang, X.; Shi, L.; Jiang, A.L.; Mu, D.S.; Li, M.J.; Han, Q.; Zhao, M.W. Molecular characterization and expression analysis of GlHMGS, a gene encoding hydroxymethylglutaryl-CoA synthase from Ganoderma lucidum (Ling-zhi) in ganoderic acid biosynthesis pathway. World J. Microb. Biot. 2013, 29, 523–531. [Google Scholar] [CrossRef]
- Tholl, D.; Lee, S. Terpene Specialized Metabolism in Arabidopsis thaliana. Arabidopsis Book 2011, 9, e0143. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New developments in the family and domain prediction database. Nucleic Acids Res. 2012, 40, D306–D312. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, W.; He, Q.; Daud, M.K.; Chen, J.; Zhu, S. Characterization of 19 genes encoding membrane-bound fatty acid desaturases and their expression profiles in Gossypium raimondii under low temperature. PLoS ONE 2015, 10, e0123281. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, W. OLIGO 7 primer analysis software. Methods Mol. Biol. 2007, 402, 35–60. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Li, W.; Ren, Z.; Wang, Z.; Sun, K.; Pei, X.; Liu, Y.; He, K.; Zhang, F.; Song, C.; Zhou, X.; et al. Evolution and stress responses of Gossypium hirsutum SWEET genes. Int. J. Mol. Sci. 2018, 19, 769. [Google Scholar] [CrossRef]
- Yao, H.; Guo, L.; Fu, Y.; Borsuk, L.A.; Wen, T.J.; Skibbe, D.S.; Cui, X.; Scheffler, B.E.; Cao, J.; Emrich, S.J.; et al. Evaluation of five ab initio gene prediction programs for the discovery of maize genes. Plant Mol. Biol. 2005, 57, 445–460. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Wang, X.; Xu, F.; Chen, Q.; Tao, T.; Lei, J.; Zhang, W.; Liao, Y.; Chang, J.; Li, X. Cloning, expression profiling and functional analysis of CnHMGS, a gene encoding 3-hydroxy-3-methylglutaryl coenzyme A synthase from Chamaemelum nobile. Molecules 2016, 21, 316. [Google Scholar] [CrossRef]
- Lombard, J.; Moreira, D. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef]
- Schwender, J.; Gemunden, C.; Lichtenthaler, H.K. Chlorophyta exclusively use the 1-deoxyxylulose 5-phosphate/2-C-methylerythritol 4-phosphate pathway for the biosynthesis of isoprenoids. Planta 2001, 212, 416–423. [Google Scholar] [CrossRef]
- Li, W.; Liu, W.; Wei, H.; He, Q.; Chen, J.; Zhang, B.; Zhu, S. Species-specific expansion and molecular evolution of the 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) gene family in plants. PLoS ONE 2014, 9, e94172. [Google Scholar] [CrossRef]
- Lecharny, A.; Boudet, N.; Gy, I.; Aubourg, S.; Kreis, M. Introns in, introns out in plant gene families: A genomic approach of the dynamics of gene structure. J. Struct. Funct. Genomics 2003, 3, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Sunilkumar, G.; Campbell, L.M.; Puckhaber, L.; Stipanovic, R.D.; Rathore, K.S. Engineering cottonseed for use in human nutrition by tissue-specific reduction of toxic gossypol. Proc. Natl. Acad. Sci. USA 2006, 103, 18054–18059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellon, J.E.; Dowd, M.K.; Beltz, S.B.; Moore, G.G. Growth inhibitory effects of gossypol and related compounds on fungal cotton root pathogens. Lett. Appl. Microbiol. 2014, 59, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Percifield, R.; Wendel, J.F. Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid. Genetics 2004, 168, 2217–2226. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Cronn, R.; Percifield, R.; Wendel, J.F. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA 2003, 100, 4649–4654. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Adams, K.L. Expression partitioning between genes duplicated by polyploidy under abiotic stress and during organ development. Curr. Biol. 2007, 17, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Force, A. The probability of duplicate gene preservation by subfunctionalization. Genetics 2000, 154, 459–473. [Google Scholar]
- Xiao, J.; Sekhwal, M.K.; Li, P.; Ragupathy, R.; Cloutier, S.; Wang, X.; You, F.M. Pseudogenes and their genome-wide prediction in plants. Int. J. Mol. Sci. 2016, 17, 1991. [Google Scholar] [CrossRef] [PubMed]
- Thibaud-Nissen, F.; Ouyang, S.; Buell, C.R. Identification and characterization of pseudogenes in the rice gene complement. BMC Genomics 2009, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Z.; Li, W.; Zhu, W.; Ren, Z.; Wang, Z.; Li, L.; Jia, L.; Zhu, S.; Ma, Z. Genome-wide identification and comparative analysis of the 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) gene family in Gossypium. Molecules 2018, 23, 193. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Zhang, Z.; Zhu, W.; Ren, Z.; Jia, L.; Li, W.; Ma, Z. Evolutionary Conservation and Divergence of Genes Encoding 3-Hydroxy-3-methylglutaryl Coenzyme A Synthase in the Allotetraploid Cotton Species Gossypium hirsutum. Cells 2019, 8, 412. https://doi.org/10.3390/cells8050412
Liu W, Zhang Z, Zhu W, Ren Z, Jia L, Li W, Ma Z. Evolutionary Conservation and Divergence of Genes Encoding 3-Hydroxy-3-methylglutaryl Coenzyme A Synthase in the Allotetraploid Cotton Species Gossypium hirsutum. Cells. 2019; 8(5):412. https://doi.org/10.3390/cells8050412
Chicago/Turabian StyleLiu, Wei, Zhiqiang Zhang, Wei Zhu, Zhongying Ren, Lin Jia, Wei Li, and Zongbin Ma. 2019. "Evolutionary Conservation and Divergence of Genes Encoding 3-Hydroxy-3-methylglutaryl Coenzyme A Synthase in the Allotetraploid Cotton Species Gossypium hirsutum" Cells 8, no. 5: 412. https://doi.org/10.3390/cells8050412