Advances in Targeting the Epidermal Growth Factor Receptor Pathway by Synthetic Products and Its Regulation by Epigenetic Modulators as a Therapy for Glioblastoma

 and
and 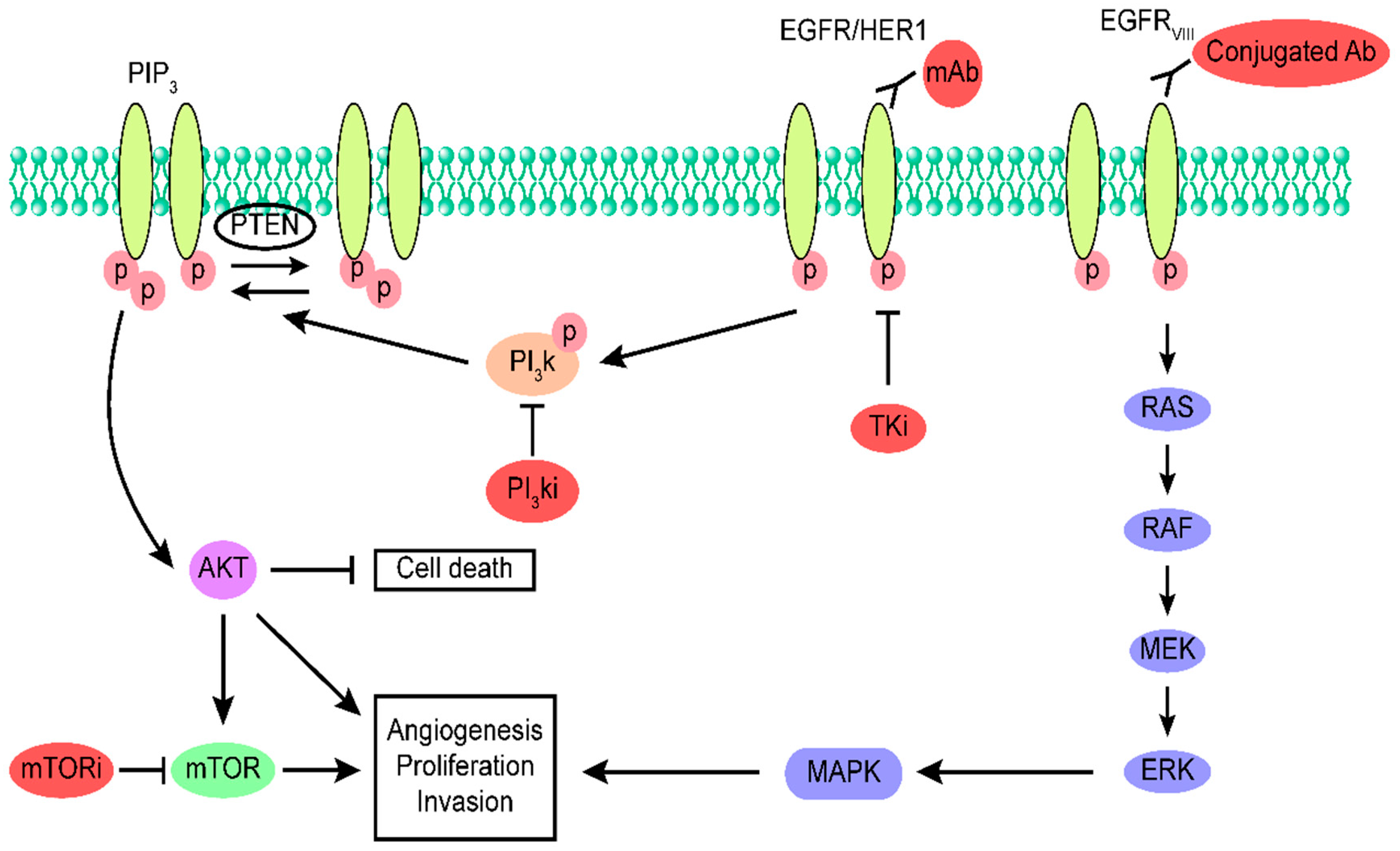{kind=link}
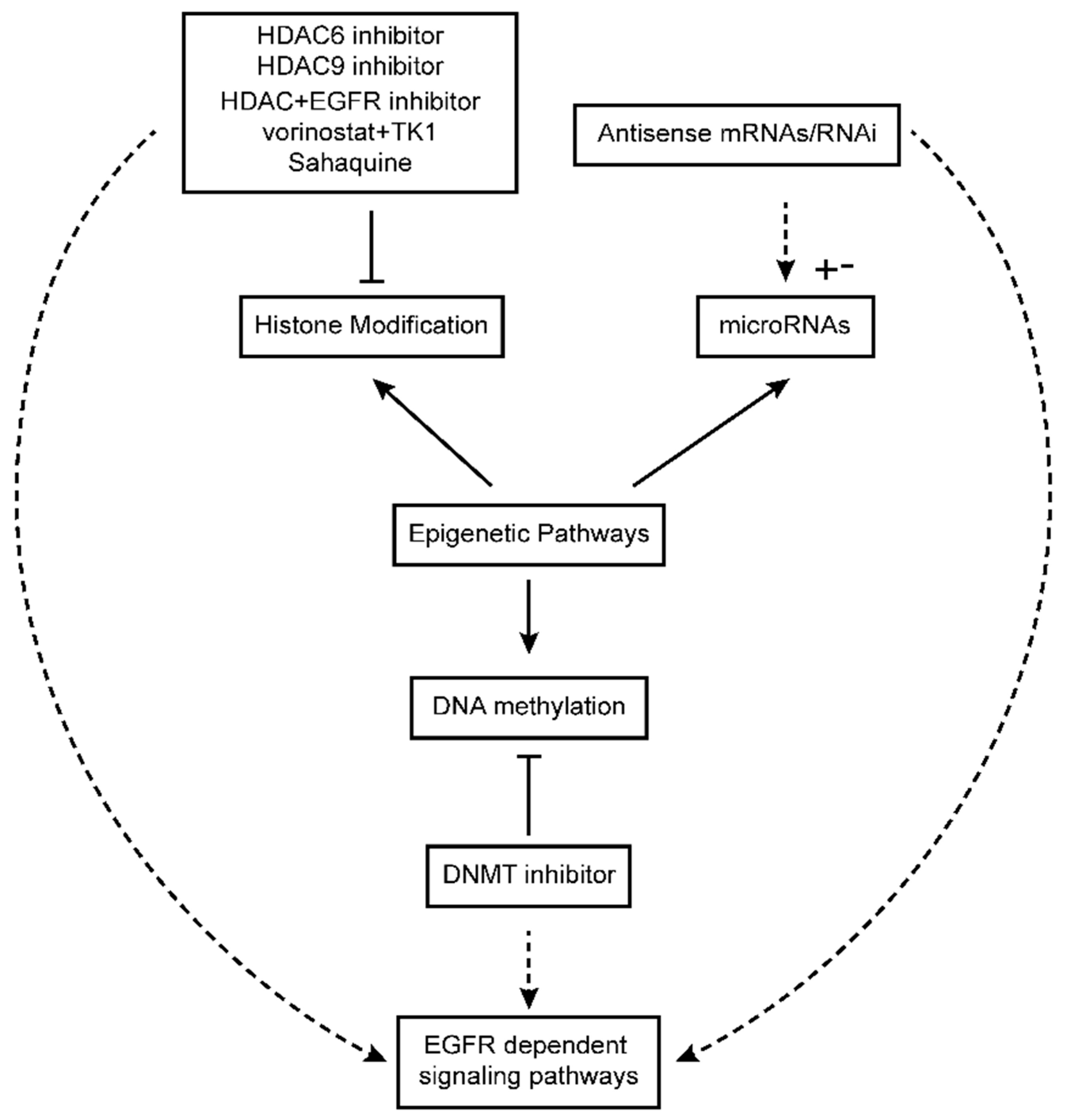{kind=link}
Abstract
:1. Introduction
2. Epidermal Growth Factor Receptor Family and Their Ligands
3. Epidermal Growth Factor Receptor Amplification
4. Alteration of EGFR and Other Contributing Factors in Glioblastoma
5. Targeting EGFR in Glioblastoma Using Drugs
5.1. Monoclonal Antibodies as Therapeutic Agents for Glioblastoma
5.2. Tyrosine Kinase Inhibitors as Therapeutic Agents for Glioblastoma
5.3. mTOR Inhibitors as Therapeutic Agents for Glioblastoma
5.4. PI3K Inhibitors as Therapeutic Agents for Glioblastoma
6. Epigenetics in Glioblastoma
6.1. Modulation of EGFR-Dependent Signaling Pathways in the Context of Epigenetic Drugs in Glioblastoma
6.2. DNA Methylation Inhibitors in Glioblastoma Treatment and Their Impact on EGFR-Dependent Pathways
6.3. Histone Deacetylase Inhibitors in Glioblastoma Therapy and Their Influence on EGFR Signaling Pathway
6.4. MicroRNAs in Glioblastoma Therapy and Their Impact on EGFR Signaling Pathway
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Russell, D.S.; Rubinstein, L.J. Pathology of Tumors of the Central Nervous System; Arnold: London, UK, 1989. [Google Scholar]
- Chen, Y.; Hu, X.; Li, Y.; Zhang, H.; Fu, R.; Liu, Y.; Hu, J.; Deng, Q.; Luo, Q.; Zhang, D.; et al. Ars2 promotes cell proliferation and tumorigenicity in glioblastoma through regulating miR-6798-3p. Sci. Rep. 2018, 8, 15602. [Google Scholar] [CrossRef] [PubMed]
- Kusaczuk, M.; Krętowski, R.; Stypułkowska, A.; Cechowska-Pasko, M. Molecular and cellular effects of a novel hydroxamate-based HDAC inhibitor–belinostat in glioblastoma cell lines: A preliminary report. Invest. New Drugs 2016, 34, 552–564. [Google Scholar] [PubMed]
- Xuan, F.; Huang, M.; Zhao, E.; Cui, H. MINA53 deficiency leads to glioblastoma cell apoptosis via inducing DNA replication stress and diminishing DNA damage response. Cell Death Dis. 2018, 17, 1062. [Google Scholar]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nature Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, P.; Aldape, K.D. Insights from molecular profiling of adult Glioma. J. Clin. Oncol. 2017, 35, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Louis, N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhao, Y.; Zhao, E.; Wang, X.; Dong, Z.; Chen, Y.; Yang, L.; Cui, H. Cancer-testis specific gene OIP5: A downstream gene of E2F1 that promotes tumorigenesis and metastasis in glioblastoma by stabilizing E2F1 signaling. Neuro. Oncol. 2018, 20, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Lomas, M.F.; De Vito, S.; Lachance, J.F.B.; Houde, J.; Nilson, L.A. Determination of EGFR signaling output by opposing gradients of BMP and JAK/STAT activity. Curr. Biol. 2016, 26, 2572–2582. [Google Scholar]
- Abbas, M.N.; Kausar, S.; Sun, Y.X.; Tian, J.W.; Zhu, B.J.; Liu, C.L. Suppressor of cytokine signaling 6 can enhance epidermal growth factor receptor signaling pathway in Bombyx mori (Dazao). Develop. Comp. Immunol. 2018, 81, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Lei, Q.; Yang, R.; Zhu, S.; Ke, X.X.; Yang, L.; Cui, H.; Yi, L. Inhibition of neurotensin receptor 1 induces intrinsic apoptosis via let-7a-3p/Bcl-w axis in glioblastoma. Br. J. Cancer. 2017, 116, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Schmidt, U.; Behnke-Mursch, J.; Unterberg, A.; Wirtz, C. Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat. Rev. 2006, 32, 74–89. [Google Scholar] [CrossRef]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1, 1–14. [Google Scholar] [CrossRef]
- Koutsopoulos, A.V.; Mavroudis, D.; Dambaki, K.I.; Souglakos, J.; Tzortzaki, E.G.; Drositis, J.; Delides, G.S.; Georgoulias, V.; Stathopoulos, E.N. Simultaneous expression of c-erbB-1, c-erbB-2, c-erbB-3 and c-erbB-4 receptors in non-small-cell lung carcinomas: Correlation with clinical outcome. Lung Cancer 2007, 57, 193–200. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010, 12, 675–684. [Google Scholar] [CrossRef]
- Nandakumar, V.; Singh, T.; Katiyar, S.K. Multi-targeted prevention and therapy of cancer by proanthocyanidins. Cancer Lett. 2008, 269, 378–387. [Google Scholar] [CrossRef]
- Benetou, V.; Orfanos, P.; Lagiou, P.; Trichopoulos, D.; Boffetta, P.; Trichopoulos, A. Vegetables and fruits in relation to cancer risk: Evidence from the Greek EPIC cohort study. Cancer Epidemiol. Biomark. Prev. 2008, 17, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.D.; Park, Y.; Subar, A.F.; Hollenbeck, A.R.; Leitzmann, M.F.; Schatzkin, A.; Abnet, C.C. Fruit and vegetable intake and head and neck cancer risk in a large United States prospective cohort study. Int. J. Cancer 2008, 122, 2330–2336. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; Bromberg, J.E.C.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.; Ferreyra, V.S.; Kling, T.; Olsson, B.T.; Jakola, A.S.; Carén, H. Intra-Tumor DNA Methylation Heterogeneity in Glioblastoma; Implications for DNA Methylation-Based Classification. Neuro. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.H.; Barve, A.; Yu, S.; Huang, M.T.; Kong, A.N.T. Cancer chemoprevention by phytochemicals: Potential molecular targets, biomarkers and animal models. Acta Pharmacol. Sin. 2007, 28, 1409–1421. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [PubMed]
- Nair, P. Epidermal growth factor receptor family and its role in cancer progression. Cur. Sci. 2005, 88, 890–895. [Google Scholar]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Cho, H.S.; Leahy, D.J. Structure of the Extracellular Region of HER3 Reveals an Interdomain Tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000 Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.A.; Luwor, R.B.; Burgess, A.W. The Epidermal Growth Factor Receptor: Structure Function Informing the Design of Anticancer Therapeutics. Exp. Cell Res. 2018, 371, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Hung, M.C. EGFR signaling pathway in breast cancers: From traditional signal transduction to direct nuclear translocalization. Breast Cancer Res. Treat. 2006, 95, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Paul, I.; Bhattacharya, S.; Chatterjee, A.; Ghosh, M.K. Current Understanding on EGFR and Wnt/β-Catenin Signaling in Glioma and Their Possible Crosstalk. Genes Cancer 2013, 4, 427–446. [Google Scholar] [CrossRef]
- Abbas, M.N.; Kausar, S.; Cui, H. The biological functions of peroxiredoxins in innate immune responses of aquatic invertebrates. Fish Shellfish Immunol. 2019, 89, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Yarden, Y. Structure and function of epigen, the last EGFR ligand. Semin. Cell Dev. Biol. 2014, 28, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, I.M.; Bechmann, T.; Brandslund, I.; Madsen, J.S. Prognostic and predictive value of EGFR and EGFR-ligands in blood of breast cancer patients: A systematic review. Clin. Chem. Lab. Med. 2018, 56, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac Dis. 2010, 2, 48–51. [Google Scholar]
- Carlsson, J.; Wester, K.; De La Torre, M.; Malmström, P.U.; Gårdmark, T. EGFR-expression in primary urinary bladder cancer and corresponding metastases and the relation to HER2-expression. On the possibility to target these receptors with radionuclides. Radiol. Oncol. 2015, 49, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar]
- Senhaji, N.; Louati, S.; Chbani, L.; ElFatemi, H.; Hammas, N.; Mikou, K.; Maaroufi, M.; Benzagmout, M.; Boujraf, S.; El Bardai, S.; et al. EGFR Amplification and IDH Mutations in Glioblastoma Patients of the Northeast of Morocco. BioMed. Res. Int. 2017, 2017. Article ID 8045859, 7p. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.S.; Lee, M.C.; Kang, S.S.; Kim, J.H.; Jung, S.; Kim, Y.J.; Lee, J.H.; Ahn, K.Y.; Lee, J.S.; Cheon, J.Y. p53 Mutation and Epidermal Growth Factor Receptor Overexpression in Glioblastoma. J. Korean Med. Sci. 2001, 16, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; LeBrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Invest. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef]
- Thorne, A.H.; Zanca, C.; Furnari, F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro. Oncol. 2016, 18, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Chekhonin, I.V.; Chekhonina, V.P. The EGFR variant III mutant as a target for immunotherapy of glioblastoma multiforme. Eur. J. Pharmcol. 2017, 810, 70–82. [Google Scholar] [CrossRef]
- Guo, G.; Gong, K.; Wohlfeld, B.; Hatanpaa, K.J.; Zhao, D.; Habib, A.A. Ligand independent EGFR signaling. Cancer Res. 2015, 75, 3436–3441. [Google Scholar] [CrossRef]
- Cominelli, M.; Grisanti, S.; Mazzoleni, S.; Branca, C.; Buttolo, L.; Furlan, D.; Liserre, B.; Bonetti, M.F.; Medicina, D.; Buglione, V.P.M.; et al. EGFR amplified and over expressing glioblastoma and association with better response to adjuvant metronomic temozolomide. J. Nat. Cancer Inst. 2015, 107, 5. [Google Scholar] [CrossRef]
- Faulkner, C.; Palmer, A.; Williams, H.; Wragg, C.; Haynes, H.R.; White, P.; DeSouza, R.M.; Williams, M.; Hopkins, K.; Kurian, K.M. EGFR and EGFRvIII analysis in glioblastoma as therapeutic biomarkers. Br. J. Neurosurg. 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.L.; Lamborn, K.R.; Takahashi, M.; Chen, P.; Israel, M.A.; Berger, M.S.; Godfrey, T.; Nigro, J.; Prados, M.; Chang, S.; et al. Analysis of Complex Relationships between Age, p53, Epidermal Growth Factor Receptor, and Survival in Glioblastoma Patients. Cancer Res. 2001, 61, 1122–1128. [Google Scholar] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef]
- Kuga, D.; Mizoguchi, M.; Guan, Y.; Hata, N.; Yoshimoto, K.; Shono, T.; Suzuki, S.O.; Kukita, Y.; Tahira, T.; Nagata, S.; et al. Prevalence of copy-number neutral LOH in glioblastomas revealed by genomewide analysis of laser-microdissected tissues. Neuro. Oncol. 2008, 10, 995–1003. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. BRIM-3 Study Group. (). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Robins, H.I.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N.; et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Lamba, S.; Zanon, C.; Molenaar, R.J.; Hulsebos, T.J.M.; Troost, D.; van Tilborg, A.A.; Vandertop, W.P.; Leenstra, S.; van Noorden, C.J.F. Mutational profiling of kinases in gliblastoma. BMC Cancer 2014, 14, 718. [Google Scholar] [CrossRef]
- Quant, E.C.; Nutt, C.L.; Wang, D.; Batchelor, T.T. Targeting the Epidermal Growth Factor Pathway as Therapy for Glioblastoma. Curr. Cancer Ther. Rev. 2011, 7, 65–77. [Google Scholar] [CrossRef]
- Micallef, J.; Taccone, M.; Mukherjee, J.; Croul, S.; Busby, J.; Moran, M.F.; Guha, A. Epidermal growth factor receptor variant III-induced glioma invasion is mediated through myristoylated alanine-rich protein kinase C substrate overexpression. Cancer Res. 2009, 69, 19. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Pedro, N.Y.; Rangel-López, E.; Félix, G.V.; Pineda, B.; Sotelo, J. An Update in the Use of Antibodies to Treat Glioblastoma Multiforme. Autoimmune Dis. 2013. Article ID716813, 14p. [Google Scholar]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Berger, M.S.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Greer, K.; Herndon, J.E.; Kunwar, S.; et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro. Oncol. 2008, 10, 320–329. [Google Scholar] [CrossRef]
- Patel, D.; Lahiji, A.; Patel, S.; Franklin, M.; Jimenez, X.; Hicklin, D.J.; Kang, X. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res. 2007, 27, 3355–3366. [Google Scholar]
- Neyns, B.; Sadones, J.; Joosens, E.; Bouttens, F.; Verbeke, L.; Baurain, J.F.; D’Hondt, L.; Strauven, T.; Chaskis, C.; In’t Veld, P.; et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 2009, 20, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Combs, S.E.; Heeger, S.; Haselmann, R.; Edler, L.; Debus, J.; Schulz, E.D. Treatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (GERT)–phase I/II trial: Study protocol. BMC Cancer 2006, 18. [Google Scholar] [CrossRef]
- Martens, T.; Laabs, Y.; Gunther, H.S.; Kemming, D.; Zhu, Z.; Witte, L.; Hagel, C.; Westphal, M.; Lamszus, K. Inhibition of glioblastoma growth in a highly invasive nude mouse model can be achieved by targeting epidermal growth factor receptor but not vascular endothelial growth factor receptor-2. Clin. Cancer Res. 2008, 14, 5447–5458. [Google Scholar] [CrossRef]
- Hicks, M.J.; Chiuchiolo, M.J.; Ballon, D.; Dyke, J.P.; Aronowitz, E.; Funato, K.; Tabar, V.; Havlicek, D.; Fan, F.; Sondhi, D.; et al. Anti-epidermal growth factor receptor gene therapy for glioblastoma. PLoS ONE 2016, 11, e0162978. [Google Scholar] [CrossRef]
- Chakraborty, S.; Filippi, C.G.; Wong, T.; Ray, A.; Fralin, S.; Tsiouris, A.J.; Praminick, B.; Demopoulos, A.; McCrea, H.J.; Bodhinayake, I.; et al. Superselective intraarterial cerebral infusion of cetuximab after osmotic blood/brain barrier disruption for recurrent malignant glioma: Phase I study. J. Neuro. Oncol. 2016, 128, 405–415. [Google Scholar] [CrossRef]
- Wu, G.; Yang, W.; Barth, R.F.; Kawabata, S.; Swindall, M.; Bandyopadhyaya, A.K.; Tjarks, W.; Khorsandi, B.; Blue, T.E.; Ferketich, A.K.; et al. Molecular targeting and treatment of an epidermal growth factor receptor-positive glioma using boronated cetuximab. Clin. Cancer Res. 2007, 13, 1260–1268. [Google Scholar] [CrossRef]
- Barth, R.F.; Wu, G.; Meisen, W.H.; Nakkula, R.J.; Yang, W.; Huo, T.A.; Kellough, D.; Kaumaya, P.; Turro, C.; Agius, L.M.; et al. Design, synthesis, and evaluation of cisplatin-containing EGFR targeting bioconjugates as potential therapeutic agents for brain tumors. Onco. Targets Ther. 2016, 9, 2769–2781. [Google Scholar] [CrossRef]
- Eller, J.L.; Longo, S.L.; Kyle, M.M.; Bassano, D.; Hicklin, D.J.; Canute, G.W. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery 2005, 56, 155–162. [Google Scholar] [CrossRef]
- Li, L.; Quang, T.S.; Gracely, E.J.; Kim, J.H.; Emrich, J.G.; Yaeger, T.E.; Jenrette, J.M.; Cohen, S.C.; Black, P.; Brady, L.W. A Phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J. Neurosurg. 2010, 113, 192–198. [Google Scholar] [CrossRef]
- Talavera, A.; Friemann, R.; Gomez-Puerta, S.; Martinez-Fleites, C.; Garrido, G.; Rabasa, A.; Lopez-Requena, A.; Pupo, A.; Johansen, R.F.; Sanchez, O.; et al. Nimotuzumab, an antitumor antibody that targets the epidermal growth factor receptor, blocks ligand binding while permitting the active receptor conformation. Cancer Res. 2009, 69, 5851–5859. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.T.; Selva, J.C.; Figueredo, J.; Vaquer, J.; Toledo, C.; Quintanal, N.; Salva, S.; Domíngez, R.; Alert, J.; Marinello, J.J.; et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high-grade glioma patients: Results from a randomized, double blind trial. BMC Cancer 2013, 13, 299. [Google Scholar] [CrossRef]
- Bode, U.; Massimino, M.; Bach, F.; Zimmermann, M.; Khuhlaeva, E.; Westphal, M.; Fleischhack, G. Nimotuzumab treatment of malignant gliomas. Expert. Opin. Biol. Ther. 2012, 12, 1649–1659. [Google Scholar] [CrossRef]
- Ozdemir-Kaynak, E.; Qutub, A.A.; Yesil-Celiktas, O. Advances in Glioblastoma Multiforme Treatment: New Models for Nanoparticle Therapy. Front. Physiol. 2018, 9, 170. [Google Scholar] [CrossRef]
- Westphal, M.; Heese, O.; Steinbach, J.P.; Schnell, O.; Schackert, G.; Mehdorn, M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur. J. Cancer 2015, 51, 522–532. [Google Scholar] [CrossRef]
- Massimino, M.; Bode, U.; Biassoni, V.; Fleischhack, G. Nimotuzumab for pediatric diffuse intrinsic pontine gliomas. Expert. Opin. Biol. Ther. 2011, 11, 247–256. [Google Scholar] [CrossRef]
- Nicholas, M.K.; Lukas, R.V.; Jafri, N.F.; Faoro, L.; Salgia, R. Epidermal growth factor receptor - mediated signal transduction in the development and therapy of gliomas. Clin. Cancer Res. 2006, 12, 726170. [Google Scholar] [CrossRef]
- Lal, A.; Glazer, C.; Martinson, H.; Friedman, H.; Archer, G.; Sampson, J.; Riggins, G. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res. 2002, 62, 3335–3339. [Google Scholar]
- Struve, N.; Riedel, M.; Schulte, A.; Rieckmann, T.; Grob, T.J.; Gal, A.; Rothkamm, K.; Lamszus, K.; Petersen, C.; Dikomey, E.; et al. EGFRvIII does not affect radiosensitivity with or without gefitinib treatment in glioblastoma cells. Oncotarget 2015, 1–11. [Google Scholar] [CrossRef]
- Halatsch, M.E.; Gehrke, E.; Vougioukas, V.; Bötefür, I.; Efferth, T.; Gebhardt, E.; Domhof, S.; Schmidt, U.; Buchfelder, M. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J. Neurosurg. 2004, 100, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.; Abrey, L.; Lassman, A.; Chang, S.; Lamborn, K.; Kuhn, J.; Yung, W.; Gilbert, M.; Aldape, K.; Wen, P.; et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro. Oncol. 2010, 12, 95–103. [Google Scholar] [CrossRef]
- Yung, W.; Vredenburgh, J.; Cloughesy, T.; Nghiemphu, P.; Klencke, B.; Gilbert, M.; Reardon, D.; Prados, M. Safety and efficacy of erlotinib in first-relapse glioblastoma: A phase II open-label study. Neuro. Oncol. 2010, 12, 1061–1070. [Google Scholar] [CrossRef]
- Van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II Trial of Gefitinib in Recurrent Glioblastoma. J. Clin. Oncol. 2004, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Knobbe, C.B.; Merlo, A.; Reifenberger, G. Pten signaling in gliomas. Neuro. Oncol. 2002, 4, 196–211. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Labagnara, M.; Friedman, M.; Kwasnicki, A.; Murali, R. Glioblastoma: Molecular pathways, stem cells and therapeutic targets. Cancers 2015, 7, 538–555. [Google Scholar] [CrossRef] [PubMed]
- Duzgun, Z.; Eroglu, Z.; Avci, C.B. 2016. Role of mTOR in glioblastoma. Gene 2016, 575, 187–190. [Google Scholar] [CrossRef]
- Vignot, S.; Faivre, S.; Aguirre, D.; Raymond, E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann. Oncol. 2005, 16, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 67188. [Google Scholar] [CrossRef]
- Kuhn, J.G.; Chang, S.M.; Wen, P.Y.; Cloughesy, T.F.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.L.; Robins, H.I.; Mehta, M.; et al. Pharmacokinetic and tumor distribution characteristics of temsirolimus in patients with recurrent malignant glioma. Clin. Cancer Res. 2007, 13, 7401–7406. [Google Scholar] [CrossRef]
- Wang, M.Y.; Lu, K.V.; Zhu, S.; Dia, E.Q.; Vivanco, I.; Shackleford, G.M.; Sawyers, C.L. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006, 66, 7864–7869. [Google Scholar] [CrossRef]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Masui, K.; Kohmura, E. NT-39 glutaminase-mediated metabolic pathway involves glioblastoma resistance to mTOR-targeted therapies. Neuro. Oncol. 2014, 16, v167. [Google Scholar] [CrossRef]
- Weiss, W.A.; Wong, R.; Fan, Q.W.; Ilkhanizadeh, S.; Lu, M.; Beyer, J.; Xu, T. mTOR kinaseinhibitors and apoptosis in glioblastoma. Neuro. Oncol. 2014, 16, iii31. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Bader, A.G.; Kang, S.; Zhao, L.; Vogt, P.K. Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 2005, 5, 921–929. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J. Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Cheng, C.K.; Fan, Q.W.; Weiss, W.A. PI3K signaling in glioma--animal models and therapeutic challenges. Brain Pathol. 2009, 19, 112–120. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Endersby, R.; Baker, S.J. PTEN signaling in brain: Neuropathology and tumorigenesis. Oncogene 2008, 27, 5416–5430. [Google Scholar] [CrossRef]
- Garcia-Echeverria, C.; Sellers, W.R. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 2008, 27, 5511–5526. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Warmoes, M.O.; Shen, X.; Locasale, J.W. Epigenetics and cancer metabolism. Cancer Lett. 2015, 356, 309–314. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Alekseeva, E.; Tanas, A.; Prozorenko, E.; Zaytsev, A.; Kirsanova, O.; Strelnikov, V.; Zaletayev, D. Molecular pathology of the 10q23.3-26.3 chromosome region in glioblastoma. Ann. Oncol. 2016, 27. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Löffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Carén, H.; Pollard, S.M.; Beck, S. The good, the bad and the ugly: Epigenetic mechanisms in glioblastoma. Mol. Aspects Med. 2013, 34, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Llinàs-Arias, P.; Esteller, M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: An. update. Open Biol. 2017, 7, 170152. [Google Scholar] [CrossRef]
- Nebbioso, A.; Carafa, V.; Benedetti, R.; Altucci, L. Trials with ‘epigenetic’ drugs: An update. Mol. Oncol. 2012, 6, 657–682. [Google Scholar] [CrossRef]
- Jin, X.; Kim, L.J.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [CrossRef]
- Clarke, J.; Penas, C.; Pastori, C.; Komotar, R.J.; Bregy, A.; Shah, A.H.; Wahlestedt, C.; Ayad, N.G. Epigenetic pathways and glioblastoma treatment. Epigenetics 2013, 8, 785–795. [Google Scholar] [CrossRef]
- Kelly, A.D.; Issa, J.J. The promise of epigenetic therapy: Reprogramming the cancer epigenome. Curr. Opin. Genet. Dev. 2017, 42, 68–77. [Google Scholar] [CrossRef]
- Yuan, J.; Llamas, L.N.; Sander, B.; Golas, M.M. Synergistic anti-cancer effects of epigenetic drugs on medulloblastoma cells. Cell Oncol. 2017, 40, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Lovkvist, C.; Dodd, I.B.; Sneppen, K.; Haerter, J.O. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016, 44, 5123–5132. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Chédin, F. The DNMT3 family of mammalian de novo DNA methyltransferases. Prog. Mol. Biol. Transl. Sci. 2011, 101, 255–285. [Google Scholar] [PubMed]
- Kaiser, S.; Jurkowski, T.P.; Kellner, S.; Schneider, D.; Jeltsch, A.; Helm, M. The RNA methyltransferase Dnmt2 methylates DNA in the structural context of a tRNA. RNA Biol. 2017, 14, 1241–1251. [Google Scholar] [CrossRef]
- Hashimshony, T.; Zhang, J.; Keshet, I.; Bustin, M.; Cedar, H. Therole of DNA methylation in setting up chromatin structure during development. Nat. Genet. 2003, 34, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C.; Sandoval, J.; Sayols, S.; Chianese, C.; Giachini, C.; Heyn, H.; Esteller, M. Novel insights into DNA methylation features in spermatozoa: Stability and peculiarities. PLoS ONE 2012, 7, e44479. [Google Scholar] [CrossRef] [PubMed]
- Shimooka, Y.; Nishikawa, J.; Ohyama, T. Most methylation susceptible DNA sequences in human embryonic stem cells undergo a change in conformation or flexibility upon methylation. Biochemistry 2013, 52, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Wang, G.; Ding, H.; Chen, Y.; Yu, G.; Wang, J. Downregulation of metastasis suppressor 1 (MTSS1) is associated with nodal metastasis and poor outcome in Chinese patients with gastric cancer. BMC Cancer 2010, 10. [Google Scholar] [CrossRef]
- Lee, Y.G.; Macoska, J.A.; Korenchuk, S.; Pienta, K.J.M.I.M. A potential metastasis suppressor gene in bladder cancer. Neoplasia 2002, 4, 291–294. [Google Scholar] [CrossRef]
- Parr, C.; Jiang, W.G. Metastasis suppressor 1 (MTSS1) demonstrates prognostic value and anti-metastatic properties in breast cancer. Eur. J. Cancer 2009, 45, 1673–1683. [Google Scholar] [CrossRef]
- Wang, D.M.R.X.; Wang, T.; Li, T.; Zhu, J.W. MTSS1 Overexpression Correlates with Poor Prognosis in Colorectal Cancer. J. Gastrointest. Surg. 2011, 15, 1205–1212. [Google Scholar] [CrossRef]
- Callahan, C.A.; Ofstad, T.; Horng, L.; Wang, J.K.; Zhen, H.H.; Coulombe, P.A.; Oro, A.E. MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 2004, 18, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M.; Kusaczuk, M.; Kotyńska, J.; Gál, M.; Krętowski, R.; Cechowska-Pasko, M.; Naumowicz, M. The effect of quercetin on the electrical properties of model lipid membranes and human glioblastoma cells. Bioelectrochemistry 2018, 124, 133–141. [Google Scholar] [CrossRef]
- Utikal, J.; Gratchev, A.; Muller-Molinet, I.; Oerther, S.; Kzhyshkowska, J.; Arens, N.; Grobholz, R.; Kannookadan, S.; Goerdt, S. Theexpressionof metastasis suppressor MIM/MTSS1 is regulated by DNA methylation. Int. J. Cancer 2006, 119, 2287–2293. [Google Scholar]
- Fan, H.; Chen, L.; Zhang, F.; Quan, Y.; Su, X.; Qiu, X.; Zhao, Z.; Kong, K.L.; Dong, S.; Song, Y.; et al. MTSS1, a novel target of DNA methyltransferase 3B, functions as a tumor suppressor in hepatocellular carcinoma. Oncogene 2012, 31, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Schemionek, M.; Herrmann, O.; Reher, M.M.; Chatain, N.; Schubert, C.; Costa, I.G.; Hänzelmann, S.; Gusmao, E.G.; Kintsler, S.; Braunschweig, T.; et al. Mtss1 is a critical epigenetically regulated tumor suppressor in CML. Leukemia 2015, 30, 823–832. [Google Scholar] [CrossRef]
- Luxen, D.; Gielen, G.H.; Waha, A.; Isselstein, L.; Müller, T.; Koch, P.; Hammes, J.; Becker, A.; Simon, M.; Wurst, P.; et al. MTSS1 is epigenetically regulated in glioma cells and inhibits glioma cell motility. Trans. Oncol. 2017, 10, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, G.; Bhat, K.P.L.; Aldape. EGFR and EGFRvIII in glioblastoma: partners in crime. Cancer Cell 2013, 24, 403–404. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Giacomini, C.P.; Voge, H.; Jensen, K.C.; Florio, T.; Merlo, A.; Pollack, J.R.; Wong, A.J. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013, 32, 2670–2681. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Yelton, C.J.; Ray, S.K. Histone deacetylase enzymes and selective histone deacetylase inhibitors for antitumor effects and enhancement of antitumor immunity in glioblastoma. Neuroimmunol. Neuroinflamm. 2018, 5, 46. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Metivier, R.; Lin, C.Y.; Denger, S.; Ibberson, D.; Ivacevic, T.; Brand, H.; Benes, V.; Liu, E.T.; Gannon, F. Multiple mechanisms induce transcriptional silencing of a subset of genes, including oestrogen receptor alpha, in response to deacetylase inhibition by valproic acid and trichostatin A. Oncogene 2005, 24, 4894–4907. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Greer, C.B.; Cecchini, K.R.; Harris, L.N.; Tuck, D.P.; Kim, T.H. HDAC inhibitors induce transcriptional repression of high copy number genes in breast cancer through elongation blockade. Oncogene 2013, 32, 2828–2835. [Google Scholar] [CrossRef]
- Robert, C.; Rassool, F.V. Chapter Three - HDAC Inhibitors: Roles of DNA Damage and Repair. Adv. Cancer Res. 2012, 116, 87–129. [Google Scholar]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 875824. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; He, X.; Liu, W.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef]
- Sakuma, T.; Uzawa, K.; Onda, T.; Shiiba, M.; Yokoe, H.; Shibahara, T.; Tanzawa, H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 2006, 29, 117–124. [Google Scholar] [CrossRef]
- Inoue, A.; Yoshida, N.; Omoto, Y.; Oguchi, S.; Yamori, T.; Kiyama, R.; Hayashi, S. Development of cDNA microarray for expression profiling of estrogen-responsive genes. J. Mol. Endocrinol. 2002, 29, 175–192. [Google Scholar] [CrossRef]
- Bazzaro, M.; Lin, Z.; Santillan, A.; Lee, M.K.; Wang, M.C.; Chan, K.C.; Bristow, R.E.; Mazitschek, R.; Bradner, J.; Roden, R.B. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin. Cancer Res. 2008, 14, 73407347. [Google Scholar] [CrossRef]
- Ray, A.; Das, D.S.; Song, Y.; Hideshima, T.; Tai, Y.T.; Chauhan, D.; Anderson, K.C. Combination of a novel HDAC6 inhibitor ACY-241 and anti-PD-L1 antibody enhances anti-tumor immunity and cytotoxicity in multiple myeloma. Leukemia 2018, 32, 843–846. [Google Scholar] [CrossRef]
- Kausar, S.; Abbas, M.N.; Qian, C.; Zhu, B.J.; Sun, Y.; Sun, Y.X.; Wang, L.; Wei, G.Q.; Maqsood, I.; Liu, C.L. Serpin-14 negatively regulates prophenoloxidase activation and expression of antimicrobial peptides in Chinese oak silkworm Antheraea pernyi. Dev. Comp. Immunol. 2017, 76, 45–55. [Google Scholar] [CrossRef]
- Yang, R.; Wu, Y.; Wang, M.; Sun, Z.; Zou, J.; Zhang, Y.; Cui, H. HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation. Oncotarget 2015, 6, 7644–7656. [Google Scholar] [CrossRef]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Bae, S.M.; Kim, Y.W.; Lee, H.N.; Kim, Y.W.; Park, T.C.; Ro, D.Y.; Shin, J.C.; Shin, S.J.; Seo, J.S.; et al. Gene expression profiles in squamous cell cervical carcinoma using array-based comparative genomic hybridization analysis. Int. J. Gynecol. Cancer 2007, 17, 687–696. [Google Scholar] [CrossRef]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jian, W.; Zhao, J.; Wang, G. Overexpression of HDAC9 is associated with poor prognosis and tumor progression of breast cancer in Chinese females. Oncotarget Ther. 2018, 11, 2177–2184. [Google Scholar] [CrossRef]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.-G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef]
- Duque, M.B.; Pinheiro, K.D.V.; Thomaz, A.; Da Silva, C.A.; Freire, N.H.; Brunetto, A.T.; Schwartsmann, G.; Jaeger, M.; De Farias, C.B.; Roesler, R. Combined Inhibition of HDAC and EGFR Reduces Viability and Proliferation and Enhances STAT3 mRNA Expression in Glioblastoma Cells. J. Mol. Neurosci. 2019, 1–9. [Google Scholar]
- Liffers, K.; Kolbe, K.; Westphal, M.; Lamszus, K.; Schulte, A. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Targ. Oncol. 2015. [Google Scholar] [CrossRef]
- Jane, E.P.; Premkumar, D.R.; Addo-Yobo, S.O.; Pollack, I.F. Abrogation of mitogen-activated protein kinase and Akt signalling by vandetanib synergistically potentiates histone deacetylase inhibitor-induced apoptosis in human glioma cells. J. Pharmacol. Exp. Ther. 2009, 331, 327–337. [Google Scholar] [CrossRef]
- Marino, A.M.; Sofiadis, A.; Baryawno, N.; Johnsen, J.I.; Larsson, C.; Vukojević, V.; Ekström, T.J. Enhanced effects by 4-phenylbutyrate in combination with RTK inhibitors on proliferation in brain tumor cell models. Biochem. Biophys. Res. Commun. 2011, 411, 208–212. [Google Scholar] [CrossRef]
- Bezecny, P.; Wong, F.; Sang, N.J.P.X.; Pieri, C.; Mulholland, P.J.; Sheer, D. Addition of erlotinib changes gene expression in glioblastoma cell lines treated with vorinostat. Eur. J. Cancer 2011, 47, S578. [Google Scholar] [CrossRef]
- Chen, M.C.; Chen, C.H.; Wang, J.C.; Tsai, A.C.; Liou, J.P.; Pan, S.L.; Teng, C.M. The HDAC inhibitor, MPT0E028, enhances erlotinib-induced cell death in EGFR-TKI-resistant NSCLC cells. Cell Death Dis. 2013, 4, e810. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, C.; Gao, J.; Abbas, M.N.; Kausar, S.; Qian, C.; Wang, L.; Wei, G.; Zhu, B.J.; Liu, C.L. Comparative mitochondrial genome analysis of Daphnis nerii and other lepidopteran insects reveals conserved mitochondrial genome organization and phylogenetic relationships. PLoS ONE 2017, 12, e0178773. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Tong, L.; Luo, Y.; Su, M.; Zang, Y.; Li, J.; Lu, W.; Chen, Y. The discovery of colchicine-SAHA hybrids as a new class of antitumor agents. Bioorganic Med. Chem. 2013, 21, 3240–3244. [Google Scholar] [CrossRef]
- Zhang, I.; Beus, M.; Stochaj, U.; Le, P.U.; Zorc, B.; Rajić, Z.; Petrecca, K.; Maysinger, D. Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule. Cell Death Discov. 2019, 5, 41. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Pileggi, A.; Klein, D.; Fotino, C.; Bravo-Egana, V.; Rosero, S.; Doni, M.; Podetta, M.; Ricordi, C.; Molano, R.D.; Pastori, R.L. MicroRNAs in islet immunobiology and transplantation. Immunol. Res. 2013, 57, 185–196. [Google Scholar] [CrossRef]
- Moller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A systematic review of microRNA in glioblastoma multiforme: Micro modulators in the mesenchymal mode of migration and invasion. Mol. Neurobiol. 2013, 47, 131–144. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Ohetal, H.J. Let-7microRNAinhibitsthe proliferation of human glioblastoma cells. J. NeuroOncol. 2011, 102, 19–24. [Google Scholar] [CrossRef]
- Papagiannakopoulos, T.; Friedmann-Morvinski, D.; Neveu, P.; Dugas, J.C.; Gill, R.M.; Huillard, E.; Liu, C.; Zong, H.; Rowitch, D.H.; Barres, B.A.; et al. Pro-neuralmiR-128isagliomatumor suppressor that targets mitogenic kinases. Oncogene 2012, 31, 1884–1895. [Google Scholar] [CrossRef]
- Zhang, Y.; Chao, T.; Li, R.; Liu, W.; Chen, Y.; Yan, X.; Gong, Y.; Yin, B.; Liu, W.; Qiang, B.; et al. MicroRNA-128 inhibits glioma cells proliferation by targeting transcription factor E2F3a. J. Mol. Med. 2009, 87, 43–51. [Google Scholar] [CrossRef]
- Wuchty, S.; Arjona, D.; Li, A.; Kotliarov, Y.; Walling, J.; Ahn, S.; Zhang, A.; Marić, D.; Anolik, R.; Zenklusen, J.C.; et al. Prediction of associations between microRNAs and gene expression in glioma biology. PLoS ONE 2011, 6, e14681. [Google Scholar] [CrossRef]
- Zhou, X.; Ren, Y.; Moore, L.; Mei, M.; You, Y.; Xu, P.; Wang, B.; Wang, G.; Jia, Z.; Pu, P.; et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab. Invest. 2010, 90, 144–155. [Google Scholar] [CrossRef]
- Peng, Y.; He, X.; Chen, H.; Duan, H.; Shao, B.; Yang, F.; Li, H.; Yang, P.; Zeng, Y.; Zheng, J.; et al. Inhibition of microRNA-299-5p sensitizes glioblastoma cells to temozolomide via the MAPK/ERK signaling pathway. Biosci. Rep. 2018, 38, BSR20181051. [Google Scholar] [CrossRef]
- Zubieta, D.M.G.; Hamood, M.A.; Beydoun, R.; Pall, A.E.; Kondapalli, K.C. MicroRNA-135a regulates NHE9 to inhibit proliferation and migration of glioblastoma cells. Cell Commun. Signal. 2017, 15, 55. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Z.; Huang, J.; Huang, S.; Li, Y.; Yu, S.; Yu, S.; Liu, X. miR-7 inhibits glioblastoma growth by simultaneously interfering with the PI3K/ATK and Raf/MEK/ERK pathways. Intl. J. Oncol. 2014, 44, 1571–1580. [Google Scholar] [CrossRef]
- Wang, W.; Dai, L.X.; Zhang, S.; Yang, Y.; Yan, N.; Fan, P.; Dai, L.; Tian, H.W.; Cheng, L.; Zhang, X.M.; et al. Regulation of epidermal growth factor receptor signaling by plasmid-based microRNA-7 inhibits human malignant gliomas growth and metastasis in vivo. Neoplasma 2013, 60, 274–283. [Google Scholar] [CrossRef]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. MicroRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef]
- Liu, X.; Li, G.; Su, Z.; Jiang, Z.; Chen, L.; Wang, J.; Yu, S.; Liu, Z. Poly (amido amine) is an ideal carrier of miR-7 for enhancing gene silencing effects on the EGFR pathway in U251 glioma cells. Oncol. Rep. 2013, 29, 1387–1394. [Google Scholar] [CrossRef]
- Abbas, M.N.; Zhu, B.J.; Kausar, S.; Dai, L.S.; Sun, Y.X.; Tian, J.W.; Liu, C.L. Suppressor of cytokine signalling 2-12 regulates antimicrobial peptides and ecdysteroid signaling pathways in B. mori (Dazao). J. Insect Physiol. 2017, 103, 47–56. [Google Scholar] [CrossRef]
- Zhang, K.L.; Zhou, X.; Han, L.; Chen, L.Y.; Chen, L.C.; Shi, Z.D.; Yang, M.; Ren, Y.; Yang, J.X.; Frank, T.S.; et al. MicroRNA-566 activates EGFR signaling and its inhibition sensitizes glioblastoma cells to nimotuzumab. Mol. Cancer 2014, 13, 63. [Google Scholar] [CrossRef]
- Serna, E.; Lopez-Gines, C.; Monleon, D.; Muñoz-Hidalgo, L.; Callaghan, R.C.; Gil-Benso, R.; Martinetto, H.; Gregori-Romero, A.; Gonzalez-Darder, J.; Cerda-Nicolas, M. Correlation between EGFR Amplification and the Expression of MicroRNA-200c in Primary Glioblastoma Multiforme. PLoS ONE 2014, 9, e102927. [Google Scholar] [CrossRef]
- Vidal, L.; Blagden, S.; Attard, G.; de Bono, J. Making sense of antisense. Eur. J. Cancer. 2005, 41, 28128. [Google Scholar] [CrossRef]
- Mur, P.; De Lope, Á.R.; Díaz-Crespo, F.J.; Hernández-Iglesias, T.; Ribalta, T.; Fiano, C.; García, J.F.; Rey, J.A.; Mollejo, M.; Meléndez, B. Impact on prognosis of the regional distribution of MGMT methylation with respect to the CpG island methylator phenotype and age in glioma patients. J. Neurooncol. 2015, 122, 441–450. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, J.; Hao, J.; Shi, Z.; Wang, Y.; Han, L.; Yu, S.; You, Y.; Jiang, T.; Wang, J.; et al. High level of miR-221/222 confers increased cell invasion and poor prognosis in glioma. J. Transl. Med. 2012, 10, 119. [Google Scholar] [CrossRef]
- Sharma, V.; Koul, N.; Joseph, C.; Dixit, D.; Ghosh, S.; Sen, E. HDAC inhibitor, scriptaid, induces glioma cell apoptosis through JNK activation and inhibits telomerase activity. J. Cell. Mol. Med. 2010, 14, 2151–2161. [Google Scholar] [CrossRef]
- Vargas, J.E.; Filippi-Chiela, E.C.; Suhre, T.; Kipper, F.C.; Bonatto, D.; Lenz, G. Inhibition of HDAC increases the senescence induced by natural polyphenols in glioma cells. Biochem. Cell Biol. 2014, 92, 297–304. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadeem Abbas, M.; Kausar, S.; Wang, F.; Zhao, Y.; Cui, H. Advances in Targeting the Epidermal Growth Factor Receptor Pathway by Synthetic Products and Its Regulation by Epigenetic Modulators as a Therapy for Glioblastoma. Cells 2019, 8, 350. https://doi.org/10.3390/cells8040350
Nadeem Abbas M, Kausar S, Wang F, Zhao Y, Cui H. Advances in Targeting the Epidermal Growth Factor Receptor Pathway by Synthetic Products and Its Regulation by Epigenetic Modulators as a Therapy for Glioblastoma. Cells. 2019; 8(4):350. https://doi.org/10.3390/cells8040350
Chicago/Turabian StyleNadeem Abbas, Muhammad, Saima Kausar, Feng Wang, Yongju Zhao, and Hongjuan Cui. 2019. "Advances in Targeting the Epidermal Growth Factor Receptor Pathway by Synthetic Products and Its Regulation by Epigenetic Modulators as a Therapy for Glioblastoma" Cells 8, no. 4: 350. https://doi.org/10.3390/cells8040350