Regulation Is in the Air: The Relationship between Hypoxia and Epigenetics in Cancer

 , , , , and
, , , , and
Abstract
:1. Microenvironment in Solid Tumors: Hypoxia
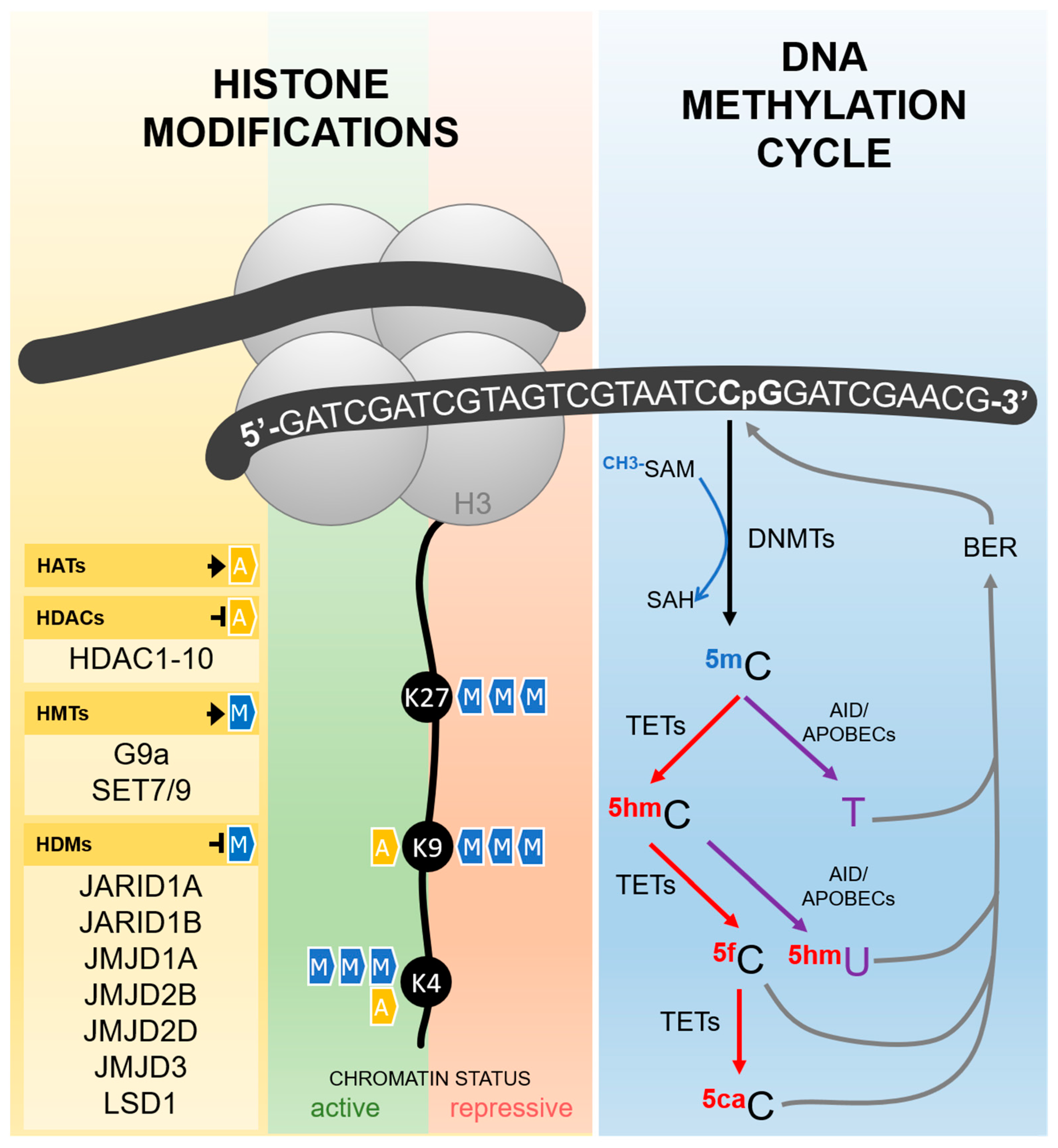
2. Hypoxia and DNA Methylation
3. Hypoxia and Histone Modifications
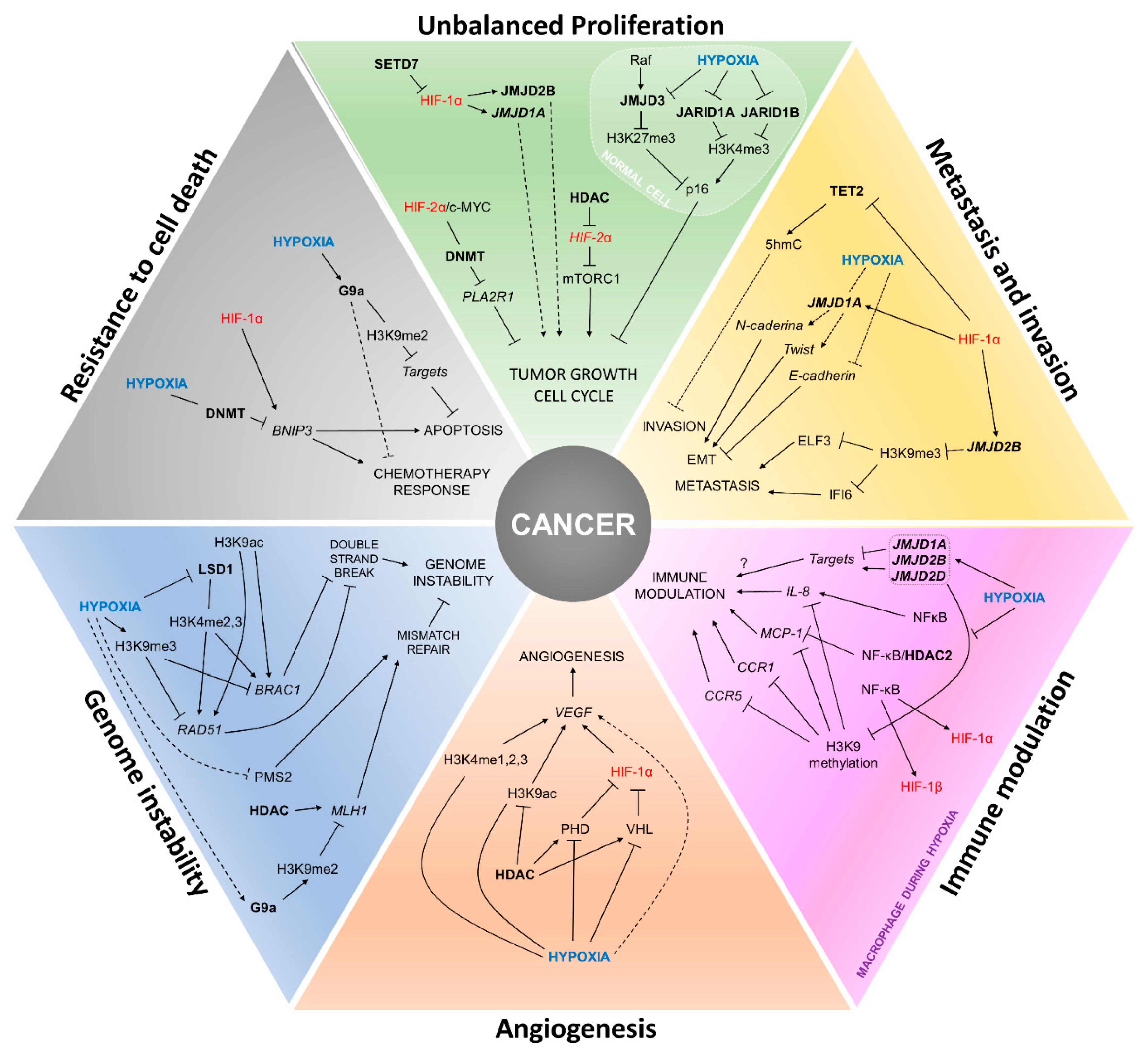
4. Painting the Cancer Hallmarks with Epigenetics
4.1. Unbalanced Proliferation
4.2. Angiogenesis
4.3. Metastasis and Invasion
4.4. Genomic Instability
4.5. Immune Modulation
4.6. Resistance to Cell Death
5. Final Considerations
Acknowledgments
Conflicts of Interest
References
- Evans, S.M.; Koch, C.J. Prognostic significance of tumor oxygenation in humans. Cancer Lett. 2003, 195, 1–16. [Google Scholar] [CrossRef]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9 (Suppl. 5), 4–9. [Google Scholar] [CrossRef]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar] [PubMed]
- Bersten, D.C.; Sullivan, A.E.; Peet, D.J.; Whitelaw, M.L. bHLH-PAS proteins in cancer. Nat. Rev. Cancer 2013, 13, 827–841. [Google Scholar] [CrossRef]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef]
- Jiang, B.H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef]
- O’Rourke, J.F.; Tian, Y.M.; Ratcliffe, P.J.; Pugh, C.W. Oxygen-regulated and transactivating domains in endothelial PAS protein 1: Comparison with hypoxia-inducible factor-1alpha. J. Biol. Chem. 1999, 274, 2060–2071. [Google Scholar] [CrossRef]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and characterization of hypoxia-inducible factor (HIF)-3alpha in human kidney: Suppression of HIF-mediated gene expression by HIF-3alpha. Biochem. Biophys Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef]
- Yang, S.L.; Wu, C.; Xiong, Z.F.; Fang, X. Progress on hypoxia-inducible factor-3: Its structure, gene regulation and biological function (Review). Mol. Med. Rep. 2015, 12, 2411–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, J.T.; Wang, Z.; Nuyten, D.S.; Rodriguez, E.H.; Schaner, M.E.; Salim, A.; Wang, Y.; Kristensen, G.B.; Helland, A.; Børresen-Dale, A.L.; et al. Gene expression programs in response to hypoxia: Cell type specificity and prognostic significance in human cancers. PLoS Med. 2006, 3, e47. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharm. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar]
- Martin, C.M.; Ferdous, A.; Gallardo, T.; Humphries, C.; Sadek, H.; Caprioli, A.; Garcia, J.A.; Szweda, L.I.; Garry, M.G.; Garry, D.J. Hypoxia-inducible factor-2alpha transactivates Abcg2 and promotes cytoprotection in cardiac side population cells. Circ. Res. 2008, 102, 1075–1081. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar]
- Slack, J.M. Conrad Hal Waddington: The last Renaissance biologist? Nat. Rev. Genet. 2002, 3, 889–895. [Google Scholar] [CrossRef]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, L.; Zhao, Y.; Zhang, J.; Wang, D.; Chen, J.; He, Y.; Wu, J.; Zhang, Z.; Liu, Z. Hypoxia induces genomic DNA demethylation through the activation of HIF-1α and transcriptional upregulation of MAT2A in hepatoma cells. Mol. Cancer Ther 2011, 10, 1113–1123. [Google Scholar] [CrossRef]
- Hermes, M.; Osswald, H.; Mattar, J.; Kloor, D. Influence of an altered methylation potential on mRNA methylation and gene expression in HepG2 cells. Exp. Cell Res. 2004, 294, 325–334. [Google Scholar] [CrossRef]
- Hermes, M.; von Hippel, S.; Osswald, H.; Kloor, D. S-adenosylhomocysteine metabolism in different cell lines: Effect of hypoxia and cell density. Cell Physiol Biochem. 2005, 15, 233–244. [Google Scholar] [CrossRef]
- Wu, X.; Sun, J.; Zhang, X.; Li, X.; Liu, Z.; Yang, Q.; Li, L. Epigenetic signature of chronic cerebral hypoperfusion and beneficial effects of S-adenosylmethionine in rats. Mol. Neurobiol 2014, 50, 839–851. [Google Scholar] [CrossRef]
- Skowronski, K.; Dubey, S.; Rodenhiser, D.; Coomber, B. Ischemia dysregulates DNA methyltransferases and p16INK4a methylation in human colorectal cancer cells. Epigenetics 2010, 5, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.H.; Bao, Y.; Wang, X.; Yan, F.; Guo, S.; Ma, Y.; Xu, D.; Jin, L.; Xu, J.; Wang, J. Hypoxic-stabilized EPAS1 proteins transactivate DNMT1 and cause promoter hypermethylation and transcription inhibition of EPAS1 in non-small cell lung cancer. FASEB J. 2018, 32. [Google Scholar] [CrossRef]
- Gao, X.; Hicks, K.C.; Neumann, P.; Patel, T.B. Hypoxia inducible factors regulate the transcription of the sprouty2 gene and expression of the sprouty2 protein. PLoS ONE 2017, 12, e0171616. [Google Scholar] [CrossRef]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef]
- Mariani, C.J.; Vasanthakumar, A.; Madzo, J.; Yesilkanal, A.; Bhagat, T.; Yu, Y.; Bhattacharyya, S.; Wenger, R.H.; Cohn, S.L.; Nanduri, J.; et al. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 2014, 7, 1343–1352. [Google Scholar] [CrossRef]
- Lin, G.; Sun, W.; Yang, Z.; Guo, J.; Liu, H.; Liang, J. Hypoxia induces the expression of TET enzymes in HepG2 cells. Oncol Lett 2017, 14, 6457–6462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.-Z.; Chen, S.-F.; Nieh, S.; Benner, C.; Ger, L.-P.; Jan, C.-I.; Ma, L.; Chien, C.-H.; Hishida, T.; Chang, H.-T. Hypoxia drives breast malignancy through a TET-TNFα-p38-MAPK signaling axis. Cancer Res. 2015, 75, 3912–3924. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.P.; Miles, S.L. Silencing HIF-1α induces TET2 expression and augments ascorbic acid induced 5-hydroxymethylation of DNA in human metastatic melanoma cells. Biochem. Biophys. Res. Commun. 2017, 490, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.P.; Chen, H.F.; Chen, S.Y.; Cheng, W.C.; Wang, H.W.; Shen, Z.J.; Song, C.; Teng, S.C.; He, C.; Wu, K.J. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014, 15, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vindrieux, D.; Devailly, G.; Augert, A.; Le Calvé, B.; Ferrand, M.; Pigny, P.; Payen, L.; Lambeau, G.; Perrais, M.; Aubert, S.; et al. Repression of PLA2R1 by c-MYC and HIF-2alpha promotes cancer growth. Oncotarget 2014, 5, 1004–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef]
- Seo, K.S.; Park, J.H.; Heo, J.Y.; Jing, K.; Han, J.; Min, K.N.; Kim, C.; Koh, G.Y.; Lim, K.; Kang, G.Y.; et al. SIRT2 regulates tumour hypoxia response by promoting HIF-1α hydroxylation. Oncogene 2015, 34, 1354–1362. [Google Scholar] [CrossRef]
- Kim, Y.; Nam, H.J.; Lee, J.; Park, D.Y.; Kim, C.; Yu, Y.S.; Kim, D.; Park, S.W.; Bhin, J.; Hwang, D.; et al. Methylation-dependent regulation of HIF-1α stability restricts retinal and tumour angiogenesis. Nat. Commun. 2016, 7, 10347. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.J.; Park, Y.S.; Cho, J.H.; Moon, B.; An, H.J.; Lee, J.Y.; Xie, Z.; Wang, Y.; Pocalyko, D.; Lee, D.C.; et al. Regulation of hypoxia responses by flavin adenine dinucleotide-dependent modulation of HIF-1α protein stability. EMBO J. 2017, 36, 1011–1028. [Google Scholar] [CrossRef]
- Fu, L.; Chen, L.; Yang, J.; Ye, T.; Chen, Y.; Fang, J. HIF-1α-induced histone demethylase JMJD2B contributes to the malignant phenotype of colorectal cancer cells via an epigenetic mechanism. Carcinogenesis 2012, 33, 1664–1673. [Google Scholar] [CrossRef]
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell Biol. 2010, 30, 344–353. [Google Scholar] [CrossRef]
- Wellmann, S.; Bettkober, M.; Zelmer, A.; Seeger, K.; Faigle, M.; Eltzschig, H.K.; Bührer, C. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem. Biophys. Res. Commun. 2008, 372, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.B.; Denko, N.; Barton, M.C. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res. 2008, 640, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Park, B.; Choi, K.; Moon, Y.; Lee, H.Y.; Park, H. Hypoxic reprograming of H3K27me3 and H3K4me3 at the INK4A locus. FEBS Lett. 2016, 590, 3407–3415. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.S.; Eisinger-Mathason, T.S.; Sadri, N.; Ochocki, J.D.; Gade, T.P.; Amin, R.K.; Simon, M.C. Epigenetic re-expression of HIF-2α suppresses soft tissue sarcoma growth. Nat. Commun. 2016, 7, 10539. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chu, A.; Turker, M.S.; Glazer, P.M. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol. Cell Biol. 2011, 31, 3339–3350. [Google Scholar] [CrossRef]
- Yamada, D.; Kobayashi, S.; Yamamoto, H.; Tomimaru, Y.; Noda, T.; Uemura, M.; Wada, H.; Marubashi, S.; Eguchi, H.; Tanemura, M.; et al. Role of the hypoxia-related gene, JMJD1A, in hepatocellular carcinoma: Clinical impact on recurrence after hepatic resection. Ann. Surg. Oncol. 2012, 19 (Suppl. 3), 355–364. [Google Scholar] [CrossRef]
- Chen, H.; Yan, Y.; Davidson, T.L.; Shinkai, Y.; Costa, M. Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res. 2006, 66, 9009–9016. [Google Scholar] [CrossRef]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol. Cell Biol. 2003, 23, 3265–3273. [Google Scholar] [CrossRef]
- Tausendschön, M.; Dehne, N.; Brüne, B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine 2011, 53, 256–262. [Google Scholar] [CrossRef]
- Safronova, O.; Pluemsampant, S.; Nakahama, K.; Morita, I. Regulation of chemokine gene expression by hypoxia via cooperative activation of NF-kappaB and histone deacetylase. Int. J. Biochem. Cell Biol. 2009, 41, 2270–2280. [Google Scholar] [CrossRef]
- Nabel, C.S.; Manning, S.A.; Kohli, R.M. The curious chemical biology of cytosine: Deamination, methylation, and oxidation as modulators of genomic potential. ACS Chem. Biol. 2012, 7, 20–30. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Santini, V.; Melnick, A.; Maciejewski, J.P.; Duprez, E.; Nervi, C.; Cocco, L.; Ford, K.G.; Mufti, G. Epigenetics in focus: Pathogenesis of myelodysplastic syndromes and the role of hypomethylating agents. Crit. Rev. Oncol. Hematol. 2013, 88, 231–245. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B.L. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics 2007, 2, 119–125. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Fritzsche, S.; Kenzelmann, M.; Hoffmann, M.J.; Müller, M.; Engers, R.; Gröne, H.J.; Schulz, W.A. Concomitant down-regulation of SPRY1 and SPRY2 in prostate carcinoma. Endocr Relat Cancer 2006, 13, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Sutterlüty, H.; Mayer, C.E.; Setinek, U.; Attems, J.; Ovtcharov, S.; Mikula, M.; Mikulits, W.; Micksche, M.; Berger, W. Down-regulation of Sprouty2 in non-small cell lung cancer contributes to tumor malignancy via extracellular signal-regulated kinase pathway-dependent and -independent mechanisms. Mol. Cancer Res. 2007, 5, 509–520. [Google Scholar] [CrossRef]
- Faratian, D.; Sims, A.H.; Mullen, P.; Kay, C.; Um, I.; Langdon, S.P.; Harrison, D.J. Sprouty 2 is an independent prognostic factor in breast cancer and may be useful in stratifying patients for trastuzumab therapy. PLoS ONE 2011, 6, e23772. [Google Scholar] [CrossRef]
- Song, K.; Gao, Q.; Zhou, J.; Qiu, S.J.; Huang, X.W.; Wang, X.Y.; Fan, J. Prognostic significance and clinical relevance of Sprouty 2 protein expression in human hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2012, 11, 177–184. [Google Scholar] [CrossRef]
- Chen, T.; Dent, S.Y. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef]
- Nakagawa, T.; Guarente, L. Sirtuins at a glance. J. Cell Sci. 2011, 124, 833–838. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Garofalo, O.; Cox, D.W.; Bachelard, H.S. Brain levels of NADH and NAD+ under hypoxic and hypoglycaemic conditions in vitro. J. Neurochem 1988, 51, 172–176. [Google Scholar] [CrossRef]
- Wang, R.H.; Zheng, Y.; Kim, H.S.; Xu, X.; Cao, L.; Luhasen, T.; Lee, M.H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol. Cell 2008, 32, 11–20. [Google Scholar] [CrossRef]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.; Kurimasa, A.; et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef]
- Peters, C.J.; Rees, J.R.; Hardwick, R.H.; Hardwick, J.S.; Vowler, S.L.; Ong, C.A.; Zhang, C.; Save, V.; O’Donovan, M.; Rassl, D.; et al. A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology 2010, 139, 1995–2004.e15. [Google Scholar] [CrossRef]
- Garber, M.E.; Troyanskaya, O.G.; Schluens, K.; Petersen, S.; Thaesler, Z.; Pacyna-Gengelbach, M.; van de Rijn, M.; Rosen, G.D.; Perou, C.M.; Whyte, R.I.; et al. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl. Acad. Sci. USA 2001, 98, 13784–13789. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.L.; Tsukasaki, K.; O’Neill, M.C.; Yamada, Y.; Onimaru, Y.; Matsumoto, K.; Ohashi, J.; Yamashita, Y.; Tsutsumi, S.; Kaneda, R.; et al. A genomic analysis of adult T-cell leukemia. Oncogene 2007, 26, 1245–1255. [Google Scholar] [CrossRef]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Lan, F.; Nottke, A.C.; Shi, Y. Mechanisms involved in the regulation of histone lysine demethylases. Curr. Opin. Cell Biol. 2008, 20, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef]
- Hosseini, A.; Minucci, S. A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 2017, 9, 1123–1142. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Wu, N.; Yang, M.; Gaur, U.; Xu, H.; Yao, Y.; Li, D. Alpha-Ketoglutarate: Physiological Functions and Applications. Biomol. Ther. (Seoul) 2016, 24, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- De Miguel, M.P.; Alcaina, Y.; de la Maza, D.S.; Lopez-Iglesias, P. Cell metabolism under microenvironmental low oxygen tension levels in stemness, proliferation and pluripotency. Curr. Mol. Med. 2015, 15, 343–359. [Google Scholar] [CrossRef]
- Zheng, L.; Kelly, C.J.; Colgan, S.P. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C350–C360. [Google Scholar] [CrossRef] [Green Version]
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [Green Version]
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009, 107, 1053–1062. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Watson, J.A.; Watson, C.J.; McCann, A.; Baugh, J. Epigenetics, the epicenter of the hypoxic response. Epigenetics 2010, 5, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R. Sensing hypoxia: Physiology, genetics and epigenetics. J. Physiol. 2013, 591, 2245–2257. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Naemura, M.; Murasaki, C.; Inoue, Y.; Okamoto, H. Transcriptional Regulation of the p16 Tumor Suppressor Gene. Anticancer Res. 2015, 35, 4397–4401. [Google Scholar]
- Romagosa, C.; Simonetti, S.; López-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Ramon y Cajal, S. p16(Ink4a) overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsagué, X.; Laporte, L.; Bosch, F.X.; de Sanjosé, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- Jin, Y.; Kato, T.; Furu, M.; Nasu, A.; Kajita, Y.; Mitsui, H.; Ueda, M.; Aoyama, T.; Nakayama, T.; Nakamura, T.; et al. Mesenchymal stem cells cultured under hypoxia escape from senescence via down-regulation of p16 and extracellular signal regulated kinase. Biochem. Biophys Res. Commun. 2010, 391, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Vindrieux, D. PLA2R1: Expression and function in cancer. Biochim. Biophys. Acta 2014, 1846, 40–44. [Google Scholar] [CrossRef]
- Choi, S.B.; Park, J.B.; Song, T.J.; Choi, S.Y. Molecular mechanism of HIF-1-independent VEGF expression in a hepatocellular carcinoma cell line. Int. J. Mol. Med. 2011, 28, 449–454. [Google Scholar] [CrossRef]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed. Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef]
- Qian, D.Z.; Wang, X.; Kachhap, S.K.; Kato, Y.; Wei, Y.; Zhang, L.; Atadja, P.; Pili, R. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004, 64, 6626–6634. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.Z.; Kato, Y.; Shabbeer, S.; Wei, Y.; Verheul, H.M.; Salumbides, B.; Sanni, T.; Atadja, P.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors: The hydroxamic acid derivative LBH589. Clin. Cancer Res. 2006, 12, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef]
- Uchiyama, R.; Uhara, H.; Uchiyama, A.; Ogawa, E.; Takazawa, Y.; Ashida, A.; Koga, H.; Hayashi, K.; Kiniwa, Y.; Okuyama, R. 5-Hydroxymethylcytosine as a useful marker to differentiate between malignant melanomas and benign melanocytic nevi. J. Dermatol. Sci. 2014, 73, 161–163. [Google Scholar] [CrossRef]
- Maia, L.L.; Peterle, G.T.; Dos Santos, M.; Trivilin, L.O.; Mendes, S.O.; de Oliveira, M.M.; Dos Santos, J.G.; Stur, E.; Agostini, L.P.; Couto, C.V.M.D.; et al. JMJD1A, H3K9me1, H3K9me2 and ADM expression as prognostic markers in oral and oropharyngeal squamous cell carcinoma. PLoS ONE 2018, 13, e0194884. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, Z.; Huo, S.; Chen, Z.; Ou, Z.; Mai, J.; Ding, S.; Zhang, J. Overexpression of ELF3 facilitates cell growth and metastasis through PI3K/Akt and ERK signaling pathways in non-small cell lung cancer. Int. J. Biochem. Cell Biol. 2018, 94, 98–106. [Google Scholar] [CrossRef]
- Nakarai, C.; Osawa, K.; Matsubara, N.; Ikeuchi, H.; Yamano, T.; Okamura, S.; Kamoshida, S.; Tsutou, A.; Takahashi, J.; Ejiri, K.; et al. Significance of ELF3 mRNA expression for detection of lymph node metastases of colorectal cancer. Anticancer Res. 2012, 32, 3753–3758. [Google Scholar]
- Cheriyath, V.; Kaur, J.; Davenport, A.; Khalel, A.; Chowdhury, N.; Gaddipati, L. G1P3 (IFI6), a mitochondrial localised antiapoptotic protein, promotes metastatic potential of breast cancer cells through mtROS. Br. J. Cancer 2018, 119, 52–64. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- King, M.C.; Marks, J.H.; Mandell, J.B.; Group, N.Y.B.C.S. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Alshareeda, A.T.; Negm, O.H.; Aleskandarany, M.A.; Green, A.R.; Nolan, C.; TigHhe, P.J.; Madhusudan, S.; Ellis, I.O.; Rakha, E.A. Clinical and biological significance of RAD51 expression in breast cancer: A key DNA damage response protein. Breast Cancer Res. Treat. 2016, 159, 41–53. [Google Scholar] [CrossRef]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [Green Version]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Webster, R.; Müller, H.A.; Mudie, S.; Rocha, S. Evolutionary conserved regulation of HIF-1β by NF-κB. PLoS Genet. 2011, 7, e1001285. [Google Scholar] [CrossRef]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.R.; Hsu, M.C.; Luo, C.W.; Chen, L.T.; Shan, Y.S.; Hung, W.C. The histone methyltransferase G9a as a therapeutic target to override gemcitabine resistance in pancreatic cancer. Oncotarget 2016, 7, 61136–61151. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Sun, W.; Hao, X.; Wei, M.; Su, X.; Zhang, Y.; Su, L.; Liu, X. EHMT2 inhibitor BIX-01294 induces apoptosis through PMAIP1-USP9X-MCL1 axis in human bladder cancer cells. Cancer Cell Int. 2015, 15, 4. [Google Scholar] [CrossRef]
- Ren, A.; Qiu, Y.; Cui, H.; Fu, G. Inhibition of H3K9 methyltransferase G9a induces autophagy and apoptosis in oral squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2015, 459, 10–17. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [Green Version]
- Murai, M.; Toyota, M.; Suzuki, H.; Satoh, A.; Sasaki, Y.; Akino, K.; Ueno, M.; Takahashi, F.; Kusano, M.; Mita, H.; et al. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin. Cancer Res. 2005, 11, 1021–1027. [Google Scholar]



{kind=link}
{kind=link}
| Hypoxia and DNA Methylation | |||||
| Hypoxic or Hypoxic-Like Condition | Epigenetic Modifier/Modification Involved | HIF Involved | Cancer/Cell Type | Functional Impact | Reference |
| 0.5% O2 for 24 h | 5hmC | ND | Eleven human and murine cell lines from different normal tissues and tumor types (HepG2, HT-1080, MCF10A, H358, MCF7, Hep3B, LLC, mESC WT, N2a, mES Tet1-/- and A549) | Decreased 5hmC levels following hypoxia. In MCF7, 5hmC was decreased near transcription start sites of NSD1, FOXA1 and CDKN2A | [31] |
| 1% O2 for 24 h | DNA methylation | HIF-1α | Human hepatoma cells (Hep3B) | MAT2A induction; decreased SAM levels; genomic DNA hypomethylation | [32] |
| ND | Human hepatoblastoma cells (HepG2) | Increased SAM levels | [33] | ||
| Human cervix adenocarcinoma cells (HeLa) | Decreased SAM levels | [34] | |||
| In vivo (rats) cerebral hypoperfusion (ischemia) for 90 days | DNMT3A | ND | Brain | Decreased SAM production; higher global methylation levels; higher DNMT3A expression levels | [35] |
| In vitro ischemia for 24 h | DNMTs | ND | Human colorectal carcinoma cells (HCT116) | Decreased DNMTs expression, which may contribute to the low DNA methylation observed in colorectal tumors | [36] |
| 1% O2 for 24 h | DNMT1 and DNMT3A | ND | Human hepatoma cells (Hep3B) | Increased DNMTs expression | [32] |
| 2% O2 for up to 96 h | DNMT1 | HIF-2α | Healthy human fetal lung fibroblasts (HFL1) and lung cancer | Increased DNMT1 expression; HIF-2α hypermethylation and decreased expression | [37] |
| 3% O2 for 24 h | HIF-1α and HIF-2α | Human hepatoma cells (HuH7 and Hep3B) | DNMT1 recruitment to SPRY2 promoter and its consequent decreased expression | [38] | |
| 1% O2 for up to 8 days | DNMT1 and DNMT3B | HIF-1α | Human primary cardiac fibroblasts (HCF) | Increased DNMTs expression | [39] |
| 1% O2 for 48 h | TET1 | Human neuroblastoma cells (SK-N-BE) | Increased TET1 expression; accumulation of 5-hydroxymethylcitosine in hypoxia-responsive genes | [40] | |
| 1% O2 for 24 h | TETs | Human hepatoma cells (HepG2) | Induced expression of TET enzymes | [41] | |
| 1% O2 for 24 h | TET1 and TET3 | Human breast cancer cell lines (MCF7 and MDA-MB-231) and primary breast cancer cells | Global hydroxymethylation; TNFα overexpression and activation of the TNFα-p38-MAPk signaling axis | [42] | |
| Normoxia | TET2 | Human metastatic melanoma cells (WM9) and human glioblastoma cells (T98G) | Reduced TET2 expression | [43] | |
| 1% O2 for 18 h | TET1 | HIF-1α and HIF-2α | Human hypopharynx carcinoma cells (FaDu) and human non-small cell lung cancer cells (derived from lymph node metastasis, H1299) | Increased TET1 expression; regulation of gene expression in response to hypoxia; INSIG1 induced expression; promotion of epithelial-mesenchymal transition | [44] |
| VHL deficiency + c-MYC amplification | DNA methylation | HIF-2α | Human kidney cancer cells (ACHN, RCC10 and 786-O) | HIF-2α stabilization and PLA2R1 repression by promoter hypermethylation | [45] |
| Hypoxia and Histone Modifications | |||||
| Hypoxic or Hypoxic-Like Condition | Epigenetic Modifier/Modification Involved | HIF Involved | Cancer/Cell Type | Functional Impact | Reference |
| In vitro 1% O2 for up to 24 h; in vivo (mice) 8% O2 | SIRT1 | HIF-1α | Human fibrosarcoma cells (HT1080), human colon cancer cells (HCT116), human embryonic kidney cells (HEK293 and HEK293T) and mice | In normoxia, SIRT1 deacetylates HIF-1α, blocking p300 recruitment, and represses HIF-1α targets. This is reversed in hypoxia, when SIRT1 levels decrease | [46] |
| In vitro 1% O2; in vivo (Sirt1+/– mice) 6% O2 | HIF-2α | Human hepatoma cells (Hep3B) and mice | Stimulation of HIF-2α activity | [47] | |
| 1% O2 for 16 h | SIRT2 | HIF-1α | Human cervix adenocarcinoma cells (HeLa) | Increased affinity of HIF-1α by PHD2, after deacetylation by SIRT2; HIF-1α degradation by proteasome | [48] |
| In vitro 1% O2 for up to 30 h; in vivo (mice) 10% O2 for up to 14 days | SET7/9 and LSD1 | Human cervix adenocarcinoma cells (HeLa) and mice | Regulation of HIF-1α stability | [49] | |
| 1% O2 for up to 8 h | LSD1 | Human embryonic kidney cells (HEK293T), human lung adenosquamous carcinoma cells (NCI-H596), human colon adenocarcinoma cells (Colo-205) and human clear cell renal cell carcinoma cells (RCC4) | Increased HIF-1α stability; glycolysis upregulation | [50] | |
| 1% O2 for up to 24 h | Human lung adenosquamous carcinoma cells (NCI-H596) | Decreased RFK expression, reduced FAD+ levels; HIF-1α degradation. | [50]. | ||
| 1% O2 for 24 h | JMJD2B | Human colorectal cancer cells (SW480 and HCT116) | Increased JMJD expression and proliferation induction; reduced H3K9me3 levels in ELF3 and IFI6 | [51] | |
| 0.5% O2 for 16 h | JMJD1A | Human clear cell renal cell carcinoma cells (RCC4) and human colon cancer cells (HCT116) | Increased JMJD expression and regulation of hypoxia-inducible genes | [52] | |
| In vitro 0.5% O2 or chemical hypoxia for up to 18 h; in vivo (rats) 8% O2 for up to 12 h | Human embryonic kidney cells (HEK293); brain, heart, kidney and liver | Increased JMJD1A expression | [53] | ||
| 0.5% O2 for 16 h | JMJD1A and JMJD2B | Human prostate cancer cells (LNCaP), human cervix adenocarcinoma cells (HeLa) and human renal adenocarcinoma cells (786–0 RCC) | Increased JMJD expression | [54] | |
| Human osteosarcoma cells (U2OS), human breast cancer cells (MCF-7), human cervix adenocarcinoma cells (HeLa), human neuroblastoma cells (IMR32) and human promyelocytic leukaemia cells (HL60) | [55] | ||||
| 0.1–0.5% O2 for up to 72 h; 0.2% O2 for 48 h | HATs? TETs? | Human Burkitt’s lymphoma cells (P493-6), mouse embryonic fibroblasts (MEF) and mouse hepatoma cells (Hepa 1–6) | PDK1 activation; blockage of PDH activity; reduced acetyl-CoA synthesis; widespread repression of RNA and mRNA synthesis | [56,57] | |
| 0.5% O2 for up to 48 h | KDM4C | ND | Human glioblastoma cells (SF188), human embryonic kidney cells (HEK293T), mouse embryonic fibroblasts (MEF), human neuroblastoma cells (SH-SY5Y), mouse bone marrow cells (32D) and mice fetal liver cells (FL5.12) | Increased 2-hydroxyglutarate (2HG) production; KDM4C inhibition, increased H3K9me3 levels | [58] |
| <0.5% O2 for up to 4 days | JMJD3, JARID1A and JARID1B | ND | Human lung fibroblasts (IMR-90) | JMJD3 activity is reduced, resulting in increased H3K27me3 levels in p16 promoter; JARID1A and JARID1B activity is also reduced, leading to increased H3K4me3 | [59] |
| 0.5% O2 for 16 h | H3ac | HIF-2α | Primary mice undifferentiated pleomorphic sarcoma cells | Reduced H3ac levels in HIF-2α promoter leads to decreased expression | [60] |
| 0.01% O2 for 48 h | H3K4me and H3K9ac | ND | Breast carcinoma cells (MCF-7) | Increased H3K4me1,2,3 and H4K9ac in VEGF promoter leads to transcriptional activation | [61] |
| H3K4me2,3, H3K9me3 and H3K9ac, LSD1 | HIF-independent | Reduced H3K4me2,3, increased H3K9me3 and decreased H3K9ac levels in BRCA1 and RAD51 promoters with a consequent decreased expression | |||
| 1% O2 for up to 72 h | JMJD1A | ND | Hepatocellular carcinoma cells (PLC, HuH7, and HepG2) | JMJD1A silencing during hypoxia leads to reduced N-cadherin and Twist levels and increased E-cadherin levels | [62] |
| 0.5% O2 for up to 24 h | G9a | HIF-1α-independent | Human lung carcinoma cells (A549), HEK293 and mouse embryonic stem cells (MES) | Higher H3K9me2 levels mediated in part by G9a in MLH1 promoter region, decreasing its expression | [63] |
| <10 ppm O2 or desferrioxamine mesylate treatment for up to 48 h | Histone acethylation | ND | Mouse fibroblasts (3340) and human cervix adenocarcinoma cells (HeLa) | Decreased MLH1 and PMS2 levels; increased mutation frequency | [64] |
| 1% O2 for 24 h | JMJD1A, JMJD2B and JMJD2D | ND | Murine macrophages (RAW264.7) | Increased H3K9me in MCP-1, CCR1 and CCR5, reducing their expression | [65] |
| 0.5% O2 for 24 h | HDAC2 | ND | Human cervix adenocarcinoma cells (HeLa) | HDAC2 recruitment by NF-κB leads to MCP-1 downregulation | [66] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camuzi, D.; de Amorim, Í.S.S.; Ribeiro Pinto, L.F.; Oliveira Trivilin, L.; Mencalha, A.L.; Soares Lima, S.C. Regulation Is in the Air: The Relationship between Hypoxia and Epigenetics in Cancer. Cells 2019, 8, 300. https://doi.org/10.3390/cells8040300
Camuzi D, de Amorim ÍSS, Ribeiro Pinto LF, Oliveira Trivilin L, Mencalha AL, Soares Lima SC. Regulation Is in the Air: The Relationship between Hypoxia and Epigenetics in Cancer. Cells. 2019; 8(4):300. https://doi.org/10.3390/cells8040300
Chicago/Turabian StyleCamuzi, Diego, Ísis Salviano Soares de Amorim, Luis Felipe Ribeiro Pinto, Leonardo Oliveira Trivilin, André Luiz Mencalha, and Sheila Coelho Soares Lima. 2019. "Regulation Is in the Air: The Relationship between Hypoxia and Epigenetics in Cancer" Cells 8, no. 4: 300. https://doi.org/10.3390/cells8040300