Presenilins and γ-Secretase in Membrane Proteostasis



Department of Neurology, University of Bonn, 53127 Bonn, Germany
*
Author to whom correspondence should be addressed.
Cells 2019, 8(3), 209; https://doi.org/10.3390/cells8030209
Submission received: 1 February 2019
/
Revised: 26 February 2019
/
Accepted: 27 February 2019
/
Published: 1 March 2019
(This article belongs to the Special Issue Proteostasis and Autophagy)
{kind=link}
{kind=link}
Abstract
:The presenilin (PS) proteins exert a crucial role in the pathogenesis of Alzheimer disease (AD) by mediating the intramembranous cleavage of amyloid precursor protein (APP) and the generation of amyloid β-protein (Aβ). The two homologous proteins PS1 and PS2 represent the catalytic subunits of distinct γ-secretase complexes that mediate a variety of cellular processes, including membrane protein metabolism, signal transduction, and cell differentiation. While the intramembrane cleavage of select proteins by γ-secretase is critical in the regulation of intracellular signaling pathways, the plethora of identified protein substrates could also indicate an important role of these enzyme complexes in membrane protein homeostasis. In line with this notion, PS proteins and/or γ-secretase has also been implicated in autophagy, a fundamental process for the maintenance of cellular functions and homeostasis. Dysfunction in the clearance of proteins in the lysosome and during autophagy has been shown to contribute to neurodegeneration. This review summarizes the recent knowledge about the role of PS proteins and γ-secretase in membrane protein metabolism and trafficking, and the functional relation to lysosomal activity and autophagy.
1. Introduction
Since mutations causative of early-onset familial Alzheimer disease (FAD) have been identified in the PSEN1 and PSEN2 genes [1,2,3], pathophysiological functions of the encoded proteins presenilin 1 (PS1) and PS2 have been extensively studied. PS1 and 2 are multifunctional proteins involved in fundamental cellular events, such as cell differentiation, intracellular signaling, and membrane trafficking [4]. The best-characterized function of PS proteins is their role in intramembrane proteolysis as the catalytic component of the γ-secretase complex [5,6,7]. A fundamental role of γ-secretase in cell differentiation and development has been attributed to the regulated intramembrane cleavage of Notch, and PSEN1/PSEN2 double knockout (PSsDKO) mice that completely lack γ-secretase activity closely resemble the developmental phenotypes of Notch-deficient mice [8,9]. PS proteins also exert critical roles in the maintenance of cellular homeostasis and function by modulating membrane protein degradation [10] and intracellular vesicle/protein trafficking [4,11].
Lysosomal activity and autophagy are critical for cellular function and viability by maintaining cellular homeostasis of lipids, sugars, and proteins [12]. Impairment of metabolite degradation in lysosomes and during autophagy could promote neurodegeneration and, thus, could also be implicated in the pathogenesis of AD [13,14,15]. Noticeably, it has been reported that FAD-related clinical PS mutants could impair autophagy and lysosomal activity, and disturb membrane protein metabolism.
This review provides an overview of the present knowledge on the role of PS proteins in the homeostasis of membrane protein metabolism that might be dependent or independent on the catalytic activity of γ-secretase.
2. Presenilins
In humans, there are two homologous genes, PSEN1 and PSEN2, located on chromosome 14 and 1, respectively. The encoded proteins PS1 and PS2 are multi-pass transmembrane proteins synthesized into the membrane system of the endoplasmic reticulum (ER) [16,17,18]. After synthesis and during transport from the ER to Golgi compartments, full-length PSs undergo endoproteolysis and assembly with the additional proteins nicastrin (NCT), anterior-pharynx defective-1 (APH1), and presenilin-enhancer-2 (PEN2) to form catalytically active γ-secretase complexes [19,20,21,22,23,24,25,26,27,28,29] (Figure 1). Besides the localization in the ER and Golgi [30], PS proteins are also found in additional subcellular membrane systems, including the plasma membrane (PM) [31], endosomes [32], lysosomes [33], nuclear envelope [34], and mitochondria [35]. However, while PS1 mainly localizes in the Golgi compartment, at the PM, and in endosomes, PS2 is predominantly localized in endosomes and lysosomes [36,37]. The localization of PS2 in the endolysosomal compartment depends on the phosphorylation at serine residue 19 in its intracellular N-terminal domain [37]. PS2 can be phosphorylated at additional sites in the N-terminal domain and within the hydrophilic loop domain by several protein kinases, including aurora A, casein kinase-1 (CK1) and CK2 [17,37,38,39]. PS1 can also be phosphorylated within the hydrophilic loop domain at several sites upon generation of C-terminal fragments (CTFs) by endoproteolytic cleavage [20,38,40,41,42,43,44,45]. However, the effects of phosphorylation in the CTFs on the activity and localization of γ-secretase are not clear. Rather, the phosphorylation of PS1 and PS2 CTFs inhibits their degradation by caspases, retards the progression of apoptosis, and regulates the interaction with β-catenin and related signaling pathways [41,42,43,44]. The phosphorylation of PS1-CTFs also affects autophagy (see below) [46,47]. PSs can also undergo ubiquitination in vitro [48], but its effect on proteasomal degradation of PSs is not clear. Due to efficient endoproteolysis, the cellular level of full-length PSs (PSs-FL) is very low, while substantial amounts of PSs-N-terminal fragments (NTFs) and -CTFs are detected [19,49,50,51,52]. PSs are expressed not only in neurons and glial cells [53,54,55,56,57,58,59] but also in tissues outside the brain [8].
3. Functions of Presenilins in Membrane Protein Metabolism
3.1. γ-Secretase as an Intramembrane Cleaving Protease
The most extensively characterized function of PS proteins is that as the catalytic components of γ-secretase complexes in assembly with additional proteins NCT, APH1, and PEN2 [22,23,24] (Figure 1). γ-Secretase is an aspartyl protease responsible for proteolysis of type I membrane proteins [5,6,7]. It has been proposed that the substrate proteins—which generally have short ectodomains [60,61,62] and are recognized by γ-secretase, possibly by NCT [63,64,65,66]—initially bind to a substrate-docking site, and are subsequently passed to the active site of γ-secretase where the α-helical conformation of the substrate’s transmembrane domain is focally changed to a β-sheet conformation to enable hydrolytic cleavage [67,68]. Details on the molecular features of γ-secretase and substrate processing have been described in recent excellent reviews [5,69,70].
More than 90 membrane proteins have been identified as γ-secretase substrates [71,72]. An important characteristic is that the protease usually does not cleave substrates with large ectodomains, although cleavage of full-length amyloid-precursor-like protein 1 (APLP1) and E-Cadherin has been described [31,73]. Rather, γ-secretase-dependent cleavage occurs after precedent shedding of the respective ectodomains of the individual substrate proteins. Ectodomain shedding of type I membrane proteins can be exerted by many different proteases, including members of the A Disintegrin And Metalloproteinase (ADAM) family, many additional metalloproteases, and several aspartyl-, serine, and cysteine proteases [74]. Since shedding usually occurs close to the transmembrane domain, the remaining membrane-tethered CTFs have relatively short ectodomains. It has been shown that the length of the ectodomain can affect the efficacy of the subsequent cleavage of these CTFs by γ-secretase [60,61,62]. Thus, γ-secretase cleavage usually generates two different products: short extracellular fragments that can be secreted from cells and intracellular domains that are liberated into the cytosol. While it is reported that the extracellular fragment of a γ-secretase substrate, the B-cell maturation antigen (BCMA), mediates survival of activated B cells [62], fundamental physiological functions of extracellular fragments of other substrate proteins of γ-secretase remain to be established. On the other hand, the intracellular domains of select substrates can regulate signal transduction pathways and gene transcription. The best-understood example is the intracellular domain (ICD) of Notch that, when liberated from cellular membranes by γ-secretase cleavage, can translocate to the nucleus to regulate transcription and determine cell fate decisions [75,76]. Nuclear translocation, control of gene transcription, and modulation of intracellular signaling pathways have also been shown for the ICDs of the amyloid precursor protein (APP) [77,78], ErbB4 [79,80,81,82,83,84,85], CD44 [86,87,88,89,90,91,92], and ephrinB2 [93,94,95]. However, for most of the γ-secretase substrates, a physiological role for the cleavage products has not been identified, and it has been speculated that γ-secretase activity might also be important for the degradation of CTFs generated by ectodomain shedding that otherwise would remain in cellular membranes and potentially interfere with membrane dynamics [60,96].
Notably, the conditional deletion of PS proteins or other components of the γ-secretase complex in neurons causes age-dependent neurodegeneration, and it has been speculated that this phenotype could involve the accumulation of membrane-tethered γ-secretase protein substrates [97,98,99]. Indeed, transgenic overexpression of APP CTFs in mouse brain neurons impaired synaptic function and caused neurodegeneration [100,101]. Evidence from cellular and mouse models indicates that increased levels of APP CTFs could interfere with mitochondrial function by changing membrane contacts with the ER [102], endocytosis [103,104], axonal trafficking of neurotrophic factors [105,106], neurite outgrowth [107], and synaptic plasticity [108]. While the exact mechanisms underlying these effects remain to be further characterized, it has been proposed that accumulated APP CTFs might interfere with intracellular signaling and subcellular trafficking by increased interaction with endocytic adaptor proteins, FE65 and ARH [109], heterotrimeric Go-proteins [107], or the Ca2+ sensor synaptotagmin 7 [108]. Impaired signaling upon inhibition of γ-secretase has also been observed for the triggering receptor expressed on myeloid cells 2 (TREM2) [110,111]. This immune receptor mediates signaling in macrophages and microglia via its co-receptor Dap12 [112,113]. Since accumulated TREM2 CTFs can also interact with Dap12, it was proposed that the TREM2 CTF lacking the ligand binding ectodomain upon ectodomain shedding competes with functional full-length TREM2 for coupling to limited amounts of Dap12.
In the future, it will be important to further understand the involvement of substrate accumulation in cellular dysfunction observed upon genetic and pharmacological inhibition of γ-secretase in different cell types. Here, it is also interesting to note that certain mutations in the genes encoding PS1 or the γ-secretase components NCT and PEN2 are associated with hidradenitis suppurativa [114,115]. Although impaired cleavage of Notch might be implicated [116], additional mechanisms could also contribute to pathogenesis [117].
3.2. Presenilins as Modulators of Vesicle/Protein Trafficking
In addition to the catalytic function within the γ-secretase complex, PS proteins also interact with several proteins to modulate their biological activity, such as β-catenin and glycogen synthase kinase 3β (GSK3β) for Wnt/β-catenin signaling and inositol 1,4,5-trisphosphate receptor (InsP3R), ryanodine receptors, and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump for cellular calcium homeostasis [71]. Presenilins also interact with proteins involved in intracellular vesicular trafficking, including Rab11 [118], Rab guanosine diphosphate dissociation inhibitor (RabGDI) [119], phospholipase D1 (PLD1) [120], syntaxin 1A [121], syntaxin 5 [122], X11α/β [123], and Annexin A2 (featured in the next paragraph) [47]. Interestingly, PS1 deficiency accelerates while FAD-linked PS1 mutants impair anterograde transport of APP-containing vesicles [124,125]. Additionally, PS1 deficiency negatively affects bulk or receptor-mediated endocytosis and transcytosis [57,109,126,127,128]. In line with a role of PS in membrane trafficking, the localization and transport of several membrane proteins, including APP [124,129,130], APLP1 [129], C-terminal fragment of APP (APP-CTF) [131,132], TrkB [129], N-Cadherin [133], intracellular adhesion molecule 5 (ICAM5) [134], NMDA (N-methyl-D-aspartate) receptor [135], transferrin receptor [136], tyrosinase [137], epidermal growth factor receptor (EGFR) [138], integrin β1 [139], low-density lipoprotein (LDL) receptor [109], vATPase V0a1 subunit [140], EphB [141], LDL receptor-related protein 1 (LRP1) [127], and the triggering receptor expressed on myeloid cells 2 (TREM2), are altered in PS-deficient or PS-mutant cells [128]. Interestingly, PS-dependent vATPase V0a1 transport and maturation can have an impact on lysosomal acidification and subsequent lysosomal/autophagic function (see below).
4. Presenilin in Autophagy
Autophagy is important for neural viability, indicated by the age-dependent neurodegeneration and intraneuronal accumulation of protein aggregates observed in mouse brains upon conditional deletion of key autophagy genes, ATG5 or ATG7, in neuronal progenitor cells [142,143]. Autophagy also contributes to neurogenesis and differentiation [144], neuronal maturation [145], and synaptic refinement [146]. Due to the importance of autophagy in the maintenance of neural cells, impaired autophagy has been suspected to contribute to the development of neurodegenerative diseases, such as Parkinson disease, Huntington disease, and Alzheimer disease [13,14,15].
Autophagy describes a process in which cellular material is targeted to the lysosome/vacuole for degradation and recycling of macromolecular constituents [147]. Three types of autophagy have been described in mammals: macroautophagy, chaperone-mediated autophagy, and microautophagy. Macroautophagy (hereafter called autophagy) involves several steps: phagophore formation by vesicle nucleation, vesicle elongation and sequestration of cytoplasmic material, autophagosome formation by phagophore maturation, autolysosome formation by fusion of autophagosome and lysosome, and eventually degradation of autolysosomal contents [12,14]. Each of these steps is coordinated by a panel of proteins [12,14].
The first indication on a relation of PS proteins and autophagy resulted from the observation that PSEN1-null mouse fibroblasts and neurons showed enlarged lysosomal organelles with an accumulation of α- and β-synucleins [148]. The accumulation of synucleins appeared to be caused by increased production and decreased degradation rather than from impairment of axonal transport and could be rescued by the expression of wild-type or clinical mutant PS1 or by modulation of cellular calcium homeostasis. Enlarged lysosomal organelles and mislocalization of synucleins could not be induced by pharmacological inhibition of γ-secretase in PS1-expressing neurons. Essenlens et al. also reported a possible defect of autophagy by showing an accumulation of telencephalin (ICAM5) in autophagic vacuoles in PSEN1-null mouse neurons [134]. The increase in ICAM5 appeared to result from decreased degradation, not from the impaired transport of newly synthesized ICAM5 in the early secretory pathway. Accumulation of ICAM5 was detected exclusively in autophagic vacuoles, which are not acidified in PSEN1-null neurons, suggesting impairment in the fusion of autophagosomes with lysosomes. Noticeably, ICAM5 accumulation in PSEN1-null cells could be rescued by the expression of wild-type or dysfunctional PS1, but not by pharmacological inhibition of γ-secretase in wild-type neurons. Cataldo et al. also reported an increase of lysosomal proteins cathepsin D (CatD) and the cation-independent 215-kDa-form mannose-6-phosphate receptor (MPR215), which targets lysosomal proteins to lysosomes, in human and mouse brains harboring clinical PSEN1 or PSEN2 mutations [149]. Together, these studies indicate the implication of PS1 and/or PS2 in lysosomal function and autophagy, probably independent of the catalytic activity of γ-secretase.
In line with this notion, Lee et al. reported decreased turnover of long-lived proteins in PSEN1- KO blastocysts associated with increased formation of autophagosomes [140], suggesting impaired autophagy. Ultrastructural analysis revealed the accumulation of enlarged autophagosomes and early autolysosomes containing undigested materials, suggesting impaired clearance of autophagic vacuoles after fusion with lysosomes. In support of dysfunctional vesicle fusion and lysosomal impairment, PSEN1-KO cells showed decreased maturation of CatD and increased lysosomal pH. However, the impairments in lysosomal acidification, cathepsin B (CatB) activity, and CatD processing were not mimicked by pharmacological or genetic inhibition of γ-secretase activity. Thus, the acidification defect in PSEN1-KO cells occurred independently of the catalytic activity of γ-secretase and was attributed to impaired interaction of v-ATPase V0a1 subunit with full-length PS1 that resulted in reduced maturation and targeting to the lysosome. Defective autophagosome accumulation and acidification were also observed in brains of PS1-deficient mice, as well as in human fibroblasts with FAD mutant PS1, in which impaired autophagy could be rescued by restoring lysosomal pH using cyclic adenosine monophosphate (cAMP) [150]. Neely et al. also reported impaired proteolysis of long-lived protein together with alterations in other markers of autophagy in PSsDKO and FAD mutant cells [151]. However, the acidification of lysosomes was not impaired, but rather enhanced in PSsDKO cells. Noticeably, pharmacological inhibition of γ-secretase activity did not induce similar phenotypes, while the expression of catalytically inactive or FAD mutant PS1 in PSEN1-null cells normalized levels of the microtubule-associated protein 1 light chain 3 (LC3) II that accumulated in PS-deficient cells.
Together, these studies indicate that alterations in the endolysosomal system and autophagy in PS-deficient cells could be independent of the catalytic activity of γ-secretase, and suggest a role of PS proteins in the fusion of autophagosomes with lysosomes. However, there is controversy in the field regarding the molecular mechanism by which PS regulates autophagy. For example, Zhang et al. did not observe an impairment of autophagic flux, cellular vesicular acidification, CatD maturation and expression, and v-ATPase V0a1 subunit glycosylation with showing no physical interaction with full-length PS1 [152]. Instead, the PS-dependent regulation of the CLEAR (coordinated lysosomal expression and regulation) gene network encoding components for lysosomal biogenesis was revealed. Coen et al. also reported normal lysosomal acidification but a significant alteration in lysosomal calcium storage/release in PSsDKO cells [153]. Decreased lysosomal calcium levels were found in PSsDKO mouse embryonic fibroblasts (MEFs) and PSEN1-KO neurons; noticeably, this defect was rescued by expression of wild-type or γ-secretase-dysfunctional PS1 in PSsDKO MEFs. Neely Kayala et al. also reported lower lysosomal calcium levels in PSsDKO MEFs, and altered expression of lysosomal calcium efflux channels, known as two-pore channels (TPC). These alterations were not only observed in PSsDKO, but also in PSEN1 and PSEN2 single-KO MEFs [154]. Decreased lysosomal Ca2+ levels were also observed in PSEN1-KO blastocysts, and was rescued by inhibition of the endolysosomal transient receptor potential cation channel mucolipin subfamily member 1 (TRPML1), indicating the involvement of TRPML1 in the PS-dependent regulation of lysosomal calcium [155]. Interestingly, impaired autophagy in the PSEN1-KO blastocysts could be restored by pharmacological normalization of lysosomal pH but not by modulation of lysosomal calcium level. Additionally, the decrease in the lysosomal calcium level could be induced by pharmacological inhibition of vATPase and a subsequent increase in lysosomal pH, suggesting that the impairment in lysosomal acidification in PS-deficient cells results in a disturbance of lysosomal calcium homeostasis and autophagy.
Impairment in autophagy in PS mutant cells has also been attributed to altered expression and activity of acid sphingomyelinase (ASM) [156]. ASM levels in blood plasma and in fibroblasts from individuals carrying FAD-associated PSEN1 mutations were increased together with an accumulation of LC3II and long-lived proteins under serum-starved conditions. Interestingly, the increase of LC3II could be induced by the addition of ASM to wild-type fibroblasts and neurons.
Another PS-dependent mechanism related to the regulation of autophagy could involve the mechanistic target of rapamycin complex 1 (mTORC1) and transcription factor EB (TFEB) [157]. PSsDKO and PSEN1-KO MEFs showed dysregulated lysosomal amino acid sensing by TORC1, which inhibits the activation and translocation of TFEB to the nucleus and subsequent CLEAR gene network expression. Noticeably, the impairment of CLEAR gene network activity could be rescued by the expression of wild-type PS1, PS2, or the γ-secretase-dysfunctional form of PS1, but not by the expression of clinical PS1 mutants. Additionally, pharmacological γ-secretase inhibition did not impair CLEAR gene network activity, suggesting that the impairment is independent of γ-secretase activity. It has been proposed that PS-dependent alterations in mTORC1 and CLEAR gene expression involve the calcium/calmodulin-dependent protein kinase (CaMK)-cAMP response element binding protein (CREB) and related sestrin pathway. Impairment of CLEAR gene network activity has also been observed in induced pluripotent stem cell (iPSC)-derived human neurons carrying clinical PSEN1 mutations, and was associated with increased intracellular amyloid β-protein (Aβ), phosphorylated tau, cleaved caspase 3, and degenerated microtubules. Involvement of TFEB and CREB-mediated nuclear signaling pathway in autophagy impairment have also been observed in other iPSC-derived human neural cells carrying clinical PSEN1 mutation or lacking PSEN1 [158,159]. Regarding the CREB-mediated nuclear signaling pathway, reduced ERK activity initiates the activation and translocation of GSK3β to the nucleus, which decreases the expression of CREB and autophagy-related genes in PSEN1-KO neural stem cells [159].
The effect of PS proteins in autophagy might also depend on their phosphorylation state [46,47]. Inhibition of PS1 phosphorylation at Ser367 by mutagenesis of this site (PS1-S367A) led to increased LC3II and p62 levels in mouse brain and cultured primary neurons [46]. Levels of APP-CTF cleaved by β-secretase (APP-CTFβ) and of Aβ40 and Aβ42 were also increased in the PS1-S367A mouse brain homogenate. These effects were attributed to the phosphorylation-dependent interaction of PS1 with Annexin A2 and N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) Vamp8, which, in turn, modulates the fusion of autophagosomes with lysosomes [47].
In contrast to most other reports, PS deficiency has also been found to be associated with the activation of autophagy. Száraz et al. reported activation of autophagy upon knockdown of PS1-, but not of PS2, in hepatocytes, as indicated by increased LC3 protein expression and inactivation of mTOR [160]. In the same study, it was suggested that the alteration in autophagy involved calcium homeostasis-related ER stress response. Autophagy activation has also been reported in bone marrow-derived mesenchymal stem cells (BM-MSCs) upon γ-secretase inhibition [161]. The pharmacological inhibition of γ-secretase activity resulted in increased expression of PTEN, which can stimulate autophagy [162], and in decreased phosphorylation of PI3K, Akt, and mTOR, suggesting that the PTEN-PI3K/Akt/mTOR pathway could activate autophagy upon γ-secretase inhibition in BM-MSCs.
5. Concluding Remarks
Extensive research has significantly improved our understandings of the role of PS/γ-secretase in membrane protein degradation, lysosomal activity, and in autophagy (Figure 2), but the underlying mechanisms remain controversial. Thus, it will be important to further dissect the molecular mechanisms and pathways related to PS-dependent membrane protein homeostasis, and to understand the physiological and pathophysiological implications not only in the nervous system but also in the periphery. It would be interesting to investigate whether PS1 and PS2 differentially affect membrane protein metabolism since both proteins show differential subcellular distribution [36,37] and could exert individual functions [163,164]. Another important question to clarified in the future is whether and how clinical PS mutants affect membrane protein homeostasis and contribute to AD development. Deletion of PSs or other γ-secretase components in neurons results in age-dependent neurodegeneration [97,98,99,135], suggesting that PS deficiency or clinical PS mutants can contribute to AD development in addition to known alterations on Aβ production. Cell bioengineering technology, such as iPSC generation and genome editing, can provide powerful experimental systems to identify the molecular effects caused by disease-related mutations in more authentic, isogenic human neural cells. Thus, an examination of membrane protein homeostasis in human neural cells carrying FAD-related clinical PSEN mutations could provide important clues to more comprehensively understand how PSEN mutations cause AD development.
Funding
The laboratory of Molecular Cell Biology is supported by grants of the German Research Foundation (DFG), the Alzheimer Research Initiative (AFI), and by the IMI2 Program of the European Union (EU).
Acknowledgments
We thank the members of the laboratory for their excellent work and stimulating discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.P.; McCarthy, J.V. Beyond γ-secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cell Signal. 2016, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.W. The γ-secretase complex: From structure to function. Front. Cell. Neurosci. 2014, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.J.; Coleman-Vaughan, C.; McCarthy, J.V. Regulated intramembrane proteolysis: Emergent role in cell signalling pathways. Biochem. Soc. Trans. 2017, 45, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Herreman, A.; Hartmann, D.; Annaert, W.; Saftig, P.; Craessaerts, K.; Serneels, L.; Umans, L.; Schrijvers, V.; Checler, F.; Vanderstichele, H.; et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. USA 1999, 96, 11872–11877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoviel, D.B.; Hadjantonakis, A.K.; Ikeda, M.; Zheng, H.; Hyslop, P.S.; Bernstein, A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999, 13, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Jurisch-Yaksi, N.; Sannerud, R.; Annaert, W. A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta 2013, 1828, 2815–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenough, M.A. The Role of Presenilin in Protein Trafficking and Degradation-Implications for Metal Homeostasis. J. Mol. Neurosci. 2016, 60, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in Age-Associated Neurodegeneration. Cells 2018, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Kovacs, D.M.; Fausett, H.J.; Page, K.J.; Kim, T.W.; Moir, R.D.; Merriam, D.E.; Hollister, R.D.; Hallmark, O.G.; Mancini, R.; Felsenstein, K.M.; et al. Alzheimer-associated presenilins 1 and 2: Neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat. Med. 1996, 2, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Capell, A.; Grünberg, J.; Pesold, B.; Schindzielorz, A.; Prior, R.; Podlisny, M.B.; Fraser, P.; Hyslop, P.S.; Selkoe, D.J.; et al. The Alzheimer’s disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol. Med. 1996, 2, 673–691. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Beullens, M.; Contreras, B.; Levesque, L.; Craessaerts, K.; Cordell, B.; Moechars, D.; Bollen, M.; Fraser, P.; George-Hyslop, P.S.; et al. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer’s disease-associated presenilins. J. Biol. Chem. 1997, 272, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Spitzer, L.; Kim, G.; Ratovitsky, T.; Davenport, F.; Nordstedt, C.; Seeger, M.; et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 1996, 17, 181–190. [Google Scholar] [CrossRef]
- Walter, J.; Grünberg, J.; Capell, A.; Pesold, B.; Schindzielorz, A.; Citron, M.; Mendla, K.; George-Hyslop, P.S.; Multhaup, G.; Selkoe, D.J.; et al. Proteolytic processing of the Alzheimer disease-associated presenilin-1 generates an in vivo substrate for protein kinase C. Proc. Natl. Acad. Sci. USA 1997, 94, 5349–5354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef] [PubMed]
- LaVoie, M.J.; Fraering, P.C.; Ostaszewski, B.L.; Ye, W.; Kimberly, W.T.; Wolfe, M.S.; Selkoe, D.J. Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J. Biol. Chem. 2003, 278, 37213–37222. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, F.; Sanjo, N.; Kawarai, T.; Hasegawa, H.; Duthie, M.; Li, W.; Ruan, X.; Luthra, A.; Mount, H.T.; et al. APH-1 interacts with mature and immature forms of presenilins and nicastrin and may play a role in maturation of presenilin.nicastrin complexes. J. Biol. Chem. 2003, 278, 7374–7380. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.J.; Wang, H.; Li, H.; Kim, B.S.; Shah, S.; Lee, H.J.; Thinakaran, G.; Kim, T.W.; Yu, G.; Xu, H. PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem. 2003, 278, 7850–7854. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Shirotani, K.; Edbauer, D.; Haass, C.; Steiner, H. Requirement of PEN-2 for stabilization of the presenilin N-/C-terminal fragment heterodimer within the gamma-secretase complex. J. Biol. Chem. 2004, 279, 23255–23261. [Google Scholar] [CrossRef] [PubMed]
- Spasic, D.; Tolia, A.; Dillen, K.; Baert, V.; De Strooper, B.; Vrijens, S.; Annaert, W. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J. Biol. Chem. 2006, 281, 26569–26577. [Google Scholar] [CrossRef] [PubMed]
- Annaert, W.G.; Levesque, L.; Craessaerts, K.; Dierinck, I.; Snellings, G.; Westaway, D.; George-Hyslop, P.S.; Cordell, B.; Fraser, P.; De Strooper, B. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J. Cell Biol. 1999, 147, 277–294. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Shioi, J.; Serban, G.; Georgakopoulos, A.; Sarner, S.; Nagy, V.; Baki, L.; Wen, P.; Efthimiopoulos, S.; Shao, Z.; et al. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002, 21, 1948–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, S.H.; Bagshaw, R.D.; Guiral, M.; Zhang, S.; Ackerley, C.A.; Pak, B.J.; Callahan, J.W.; Mahuran, D.J. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 2003, 278, 26687–26694. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Nakamura, S.I.; Honda, T.; Takashima, A.; Nakayama, H.; Ono, F.; Sakakibara, I.; Doi, K.; Kawamura, S.; Yoshikawa, Y. Age-related changes in the localization of presenilin-1 in cynomolgus monkey brain. Brain Res. 2001, 922, 30–41. [Google Scholar] [CrossRef]
- Ankarcrona, M.; Hultenby, K. Presenilin-1 is located in rat mitochondria. Biochem. Biophys. Res. Commun. 2002, 295, 766–770. [Google Scholar] [CrossRef]
- Meckler, X.; Checler, F. Presenilin 1 and Presenilin 2 Target γ-Secretase Complexes to Distinct Cellular Compartments. J. Biol. Chem. 2016, 291, 12821–12837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; De Baets, G.; De Wever, V.; Habets, R.; Baert, V.; et al. Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Aβ Pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Grünberg, J.; Schindzielorz, A.; Haass, C. Proteolytic fragments of the Alzheimer’s disease associated presenilins-1 and -2 are phosphorylated in vivo by distinct cellular mechanisms. Biochemistry 1998, 37, 5961–5967. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Schindzielorz, A.; Grünberg, J.; Haass, C. Phosphorylation of presenilin-2 regulates its cleavage by caspases and retards progression of apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 1391–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, M.; Nordstedt, C.; Petanceska, S.; Kovacs, D.M.; Gouras, G.K.; Hahne, S.; Fraser, P.; Levesque, L.; Czernik, A.J.; George-Hyslop, P.S.; et al. Evidence for phosphorylation and oligomeric assembly of presenilin 1. Proc. Natl. Acad. Sci. USA 1997, 94, 5090–5094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschenbaum, F.; Hsu, S.C.; Cordell, B.; McCarthy, J.V. Substitution of a glycogen synthase kinase-3beta phosphorylation site in presenilin 1 separates presenilin function from beta-catenin signaling. J. Biol. Chem. 2001, 276, 7366–7375. [Google Scholar] [CrossRef] [PubMed]
- Kirschenbaum, F.; Hsu, S.C.; Cordell, B.; McCarthy, J.V. Glycogen synthase kinase-3beta regulates presenilin 1 C-terminal fragment levels. J. Biol. Chem. 2001, 276, 30701–30707. [Google Scholar] [CrossRef] [PubMed]
- Fluhrer, R.; Friedlein, A.; Haass, C.; Walter, J. Phosphorylation of presenilin 1 at the caspase recognition site regulates its proteolytic processing and the progression of apoptosis. J. Biol. Chem. 2004, 279, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Prager, K.; Wang-Eckhardt, L.; Fluhrer, R.; Killick, R.; Barth, E.; Hampel, H.; Haass, C.; Walter, J. A structural switch of presenilin 1 by glycogen synthase kinase 3beta-mediated phosphorylation regulates the interaction with beta-catenin and its nuclear signaling. J. Biol. Chem. 2007, 282, 14083–14093. [Google Scholar] [CrossRef] [PubMed]
- Matz, A.; Halamoda-Kenzaoui, B.; Hamelin, R.; Mosser, S.; Alattia, J.R.; Dimitrov, M.; Moniatte, M.; Fraering, P.C. Identification of new Presenilin-1 phosphosites: Implication for γ-secretase activity and Aβ production. J. Neurochem. 2015, 133, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Bustos, V.; Pulina, M.V.; Kelahmetoglu, Y.; Sinha, S.C.; Gorelick, F.S.; Flajolet, M.; Greengard, P. Bidirectional regulation of Aβ levels by Presenilin 1. Proc. Natl. Acad. Sci. USA 2017, 114, 7142–7147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos, V.; Pulina, M.V.; Bispo, A.; Lam, A.; Flajolet, M.; Gorelick, F.S.; Greengard, P. Phosphorylated Presenilin 1 decreases β-amyloid by facilitating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 7148–7153. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.P.; Yan, R.; McCarthy, J.V. A ubiquitin-binding CUE domain in presenilin-1 enables interaction with K63-linked polyubiquitin chains. FEBS Lett. 2015, 589, 1001–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercken, M.; Takahashi, H.; Honda, T.; Sato, K.; Murayama, M.; Nakazato, Y.; Noguchi, K.; Imahori, K.; Takashima, A. Characterization of human presenilin 1 using N-terminal specific monoclonal antibodies: Evidence that Alzheimer mutations affect proteolytic processing. FEBS Lett. 1996, 389, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.V.; Davis, J.B.; Gray, C.W.; Barton, A.J.; Bresciani, L.G.; Caivano, M.; Murphy, V.F.; Duff, K.; Hutton, M.; Hardy, J.; et al. Presenilin-1 is processed into two major cleavage products in neuronal cell lines. Neurodegeneration 1996, 5, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Podlisny, M.B.; Citron, M.; Amarante, P.; Sherrington, R.; Xia, W.; Zhang, J.; Diehl, T.; Levesque, G.; Fraser, P.; Haass, C.; et al. Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol. Dis. 1997, 3, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Ancolio, K.; Lopez-Perez, E.; Checler, F. Proteasome inhibitors prevent the degradation of familial Alzheimer’s disease-linked presenilin 1 and potentiate A beta 42 recovery from human cells. Mol. Med. 1998, 4, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Huynh, D.P.; Vinters, H.V.; Ho, D.H.; Ho, V.V.; Pulst, S.M. Neuronal expression and intracellular localization of presenilins in normal and Alzheimer disease brains. J. Neuropathol. Exp. Neurol. 1997, 56, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Weggen, S.; Diehlmann, A.; Buslei, R.; Beyreuther, K.; Bayer, T.A. Prominent expression of presenilin-1 in senile plaques and reactive astrocytes in Alzheimer’s disease brain. Neuroreport 1998, 9, 3279–3283. [Google Scholar] [PubMed]
- Diehlmann, A.; Ida, N.; Weggen, S.; Grünberg, J.; Haass, C.; Masters, C.L.; Bayer, T.A.; Beyreuther, K. Analysis of presenilin 1 and presenilin 2 expression and processing by newly developed monoclonal antibodies. J. Neurosci. Res. 1999, 56, 405–419. [Google Scholar] [CrossRef]
- Nadler, Y.; Alexandrovich, A.; Grigoriadis, N.; Hartmann, T.; Rao, K.S.; Shohami, E.; Stein, R. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 2008, 56, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Farfara, D.; Trudler, D.; Segev-Amzaleg, N.; Galron, R.; Stein, R.; Frenkel, D. γ-Secretase component presenilin is important for microglia β-amyloid clearance. Ann. Neurol. 2011, 69, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Case, A.; Eastman, A.J.; Nguyen, H.; Pollak, J.; Wiley, J.C.; Möller, T.; Morrison, R.S.; Garden, G.A. Presenilin 2 is the predominant γ-secretase in microglia and modulates cytokine release. PLoS ONE 2010, 5, e15743. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Case, A.; Alajajian, B.; Eastman, A.J.; Möller, T.; Garden, G.A. Presenilin 2 influences miR146 level and activity in microglia. J. Neurochem. 2013, 127, 592–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struhl, G.; Adachi, A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol. Cell 2000, 6, 625–636. [Google Scholar] [CrossRef]
- Funamoto, S.; Sasaki, T.; Ishihara, S.; Nobuhara, M.; Nakano, M.; Watanabe-Takahashi, M.; Saito, T.; Kakuda, N.; Miyasaka, T.; Nishikawa, K.; et al. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun. 2013, 4, 2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Lee, S.F.; Tabuchi, K.; Hao, Y.H.; Yu, C.; LaPlant, Q.; Ball, H.; Dann, C.E.; Südhof, T.; Yu, G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell 2005, 122, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F.; Haass, C.; Steiner, H. Regulated intramembrane proteolysis--lessons from amyloid precursor protein processing. J. Neurochem. 2011, 117, 779–796. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Bai, X.C.; Ma, D.; Xie, T.; Yan, C.; Sun, L.; Yang, G.; Zhao, Y.; Zhou, R.; Scheres, S.H.W.; et al. Three-dimensional structure of human γ-secretase. Nature 2014, 512, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolduc, D.M.; Montagna, D.R.; Gu, Y.; Selkoe, D.J.; Wolfe, M.S. Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc. Natl. Acad. Sci. USA 2016, 113, E509–E518. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Toward the structure of presenilin/γ-secretase and presenilin homologs. Biochim. Biophys. Acta 2013, 1828, 2886–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishima-Kawashima, M. Molecular mechanism of the intramembrane cleavage of the β-carboxyl terminal fragment of amyloid precursor protein by γ-secretase. Front. Physiol. 2014, 5, 463. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.V.; Twomey, C.; Wujek, P. Presenilin-dependent regulated intramembrane proteolysis and gamma-secretase activity. Cell. Mol. Life Sci. 2009, 66, 1534–1555. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, A.; Kovacs, D.M. The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 2011, 25, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Schauenburg, L.; Liebsch, F.; Eravci, M.; Mayer, M.C.; Weise, C.; Multhaup, G. APLP1 is endoproteolytically cleaved by γ-secretase without previous ectodomain shedding. Sci. Rep. 2018, 8, 1916. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018, 37, e99456. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim. Biophys. Acta. 2016, 1863, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, S.J.; Gomez-Lamarca, M. Notch after cleavage. Curr. Opin. Cell Biol. 2018, 51, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Pardossi-Piquard, R.; Checler, F. The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 2012, 120 (Suppl. 1), 109–124. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.Y.; Murphy, M.P.; Golde, T.E.; Carpenter, G. gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 2001, 294, 2179–2181. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jung, K.M.; Huang, Y.Z.; Bennett, L.B.; Lee, J.S.; Mei, L.; Kim, T.W. Presenilin-dependent gamma-secretase-like intramembrane cleavage of ErbB4. J. Biol. Chem. 2002, 277, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Feng, L. Implication of gamma-secretase in neuregulin-induced maturation of oligodendrocytes. Biochem. Biophys. Res. Commun. 2004, 314, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.C.; Allison, J.G.; Vidal, G.A.; Burow, M.E.; Beckman, B.S.; Marrero, L.; Jones, F.E. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J. Cell Biol. 2004, 167, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, G.A.; Naresh, A.; Marrero, L.; Jones, F.E. Presenilin-dependent gamma-secretase processing regulates multiple ERBB4/HER4 activities. J. Biol. Chem. 2005, 280, 19777–19783. [Google Scholar] [CrossRef] [PubMed]
- Arasada, R.R.; Carpenter, G. Secretase-dependent tyrosine phosphorylation of Mdm2 by the ErbB-4 intracellular domain fragment. J. Biol. Chem. 2005, 280, 30783–30787. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Murtie, J.; Koirala, S.; Patten, B.A.; Corfas, G. Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell 2006, 127, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Okochi, M.; Takeda, M.; Kaether, C.; Capell, A.; Zimmer, A.K.; Edbauer, D.; Walter, J.; Steiner, H.; Haass, C. Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Abeta-like peptide. J. Biol. Chem. 2002, 277, 44754–44759. [Google Scholar] [CrossRef] [PubMed]
- Murakami, D.; Okamoto, I.; Nagano, O.; Kawano, Y.; Tomita, T.; Iwatsubo, T.; De Strooper, B.; Yumoto, E.; Saya, H. Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene 2003, 22, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Ke, J.Z.; Zhang, Q.; Ke, H.Z.; Chalouni, C.; Vignery, A. The intracellular domain of CD44 promotes the fusion of macrophages. Blood 2006, 107, 796–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, L.; Guillaumot, P.; Frêche, B.; Luquain, C.; Christiansen, D.; Brugière, S.; Garin, J.; Manié, S.N. Gamma-secretase-dependent proteolysis of CD44 promotes neoplastic transformation of rat fibroblastic cells. Cancer Res. 2006, 66, 3681–3687. [Google Scholar] [CrossRef] [PubMed]
- De Falco, V.; Tamburrino, A.; Ventre, S.; Castellone, M.D.; Malek, M.; Manié, S.N.; Santoro, M. CD44 proteolysis increases CREB phosphorylation and sustains proliferation of thyroid cancer cells. Cancer Res. 2012, 72, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lee, H.W.; Kang, H.G.; Kim, H.Y.; Kim, S.J.; Chun, K.H. Cleaved CD44 intracellular domain supports activation of stemness factors and promotes tumorigenesis of breast cancer. Oncotarget 2015, 6, 8709–8721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, K.; Grieger Lindner, C.; Li, Y.; Urbánek, P.; Ruschel, A.; Minnich, K.; Bruder, D.; Gereke, M.; Sechi, A.; Herrlich, P. Gamma secretase dependent release of the CD44 cytoplasmic tail upregulates IFI16 in cd44-/- tumor cells, MEFs and macrophages. PLoS ONE 2018, 13, e0207358. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, A.; Litterst, C.; Ghersi, E.; Baki, L.; Xu, C.; Serban, G.; Robakis, N.K. Metalloproteinase/Presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J. 2006, 25, 1242–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waschbüsch, D.; Born, S.; Niediek, V.; Kirchgessner, N.; Tamboli, I.Y.; Walter, J.; Merkel, R.; Hoffmann, B. Presenilin 1 affects focal adhesion site formation and cell force generation via c-Src transcriptional and posttranslational regulation. J. Biol. Chem. 2009, 284, 10138–10149. [Google Scholar] [CrossRef] [PubMed]
- Kemmerling, N.; Wunderlich, P.; Theil, S.; Linnartz-Gerlach, B.; Hersch, N.; Hoffmann, B.; Heneka, M.T.; de Strooper, B.; Neumann, H.; Walter, J. Intramembranous processing by γ-secretase regulates reverse signaling of ephrin-B2 in migration of microglia. Glia 2017, 65, 1103–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopan, R.; Ilagan, M.X. Gamma-secretase: Proteasome of the membrane. Nat. Rev. Mol. Cell Biol. 2004, 5, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Chen, G.; Südhof, T.C.; Shen, J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J. Neurosci. 2009, 29, 7290–7301. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Iqbal, M.; Zheng, J.; Wines-Samuelson, M.; Shen, J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J. Neurosci. 2014, 34, 15912–15922. [Google Scholar] [CrossRef] [PubMed]
- Acx, H.; Serneels, L.; Radaelli, E.; Muyldermans, S.; Vincke, C.; Pepermans, E.; Müller, U.; Chávez-Gutiérrez, L.; De Strooper, B. Inactivation of γ-secretases leads to accumulation of substrates and non-Alzheimer neurodegeneration. EMBO Mol. Med. 2017, 9, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Oster-Granite, M.L.; McPhie, D.L.; Greenan, J.; Neve, R.L. Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J. Neurosci. 1996, 16, 6732–6741. [Google Scholar] [CrossRef] [PubMed]
- Nalbantoglu, J.; Tirado-Santiago, G.; Lahsaïni, A.; Poirier, J.; Goncalves, O.; Verge, G.; Momoli, F.; Welner, S.A.; Massicotte, G.; Julien, J.P.; et al. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 1997, 387, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Ginsberg, S.D.; Cataldo, A.M.; Mathews, P.M.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sato, Y.; Mohan, P.S.; Peterhoff, C.; Pensalfini, A.; Rigoglioso, A.; Jiang, Y.; Nixon, R.A. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 2016, 21, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Weissmiller, A.M.; Natera-Naranjo, O.; Reyna, S.M.; Pearn, M.L.; Zhao, X.; Nguyen, P.; Cheng, S.; Goldstein, L.S.; Tanzi, R.E.; Wagner, S.L.; et al. A γ-secretase inhibitor, but not a γ-secretase modulator, induced defects in BDNF axonal trafficking and signaling: Evidence for a role for APP. PLoS ONE 2015, 10, e0118379. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Weissmiller, A.M.; White, J.A.; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef] [PubMed]
- Deyts, C.; Clutter, M.; Herrera, S.; Jovanovic, N.; Goddi, A.; Parent, A.T. Loss of presenilin function is associated with a selective gain of APP function. Elife 2016, 5, e15645. [Google Scholar] [CrossRef] [PubMed]
- Barthet, G.; Jordà-Siquier, T.; Rumi-Masante, J.; Bernadou, F.; Müller, U.; Mulle, C. Presenilin-mediated cleavage of APP regulates synaptotagmin-7 and presynaptic plasticity. Nat. Commun. 2018, 9, 4780. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, I.Y.; Prager, K.; Thal, D.R.; Thelen, K.M.; Dewachter, I.; Pietrzik, C.U.; St George-Hyslop, P.; Sisodia, S.S.; De Strooper, B.; Heneka, M.T.; et al. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J. Neurosci. 2008, 28, 12097–12106. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef] [PubMed]
- Glebov, K.; Wunderlich, P.; Karaca, I.; Walter, J. Functional involvement of γ-secretase in signaling of the triggering receptor expressed on myeloid cells-2 (TREM2). J. Neuroinflamm. 2016, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef] [PubMed]
- Walter, J. The Triggering Receptor Expressed on Myeloid Cells 2: A Molecular Link of Neuroinflammation and Neurodegenerative Diseases. J. Biol. Chem. 2016, 291, 4334–4341. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. Gamma-secretase gene mutations in familial acne inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Mattei, P.; You, J.; Sobreira, N.L.; Hinds, G.A. γ-Secretase Mutation in an African American Family with Hidradenitis Suppurativa. JAMA Dermatol. 2015, 151, 668–670. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A. Concurrent hidradenitis suppurativa and Dowling-Degos disease taken down a ‘Notch’. Br. J. Dermatol. 2018, 178, 328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sisodia, S.S. Acne inversa caused by missense mutations in NCSTN is not fully compatible with impairments in Notch signaling. J. Investig. Dermatol. 2015, 135, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Dumanchin, C.; Czech, C.; Campion, D.; Cuif, M.H.; Poyot, T.; Martin, C.; Charbonnier, F.; Goud, B.; Pradier, L.; Frebourg, T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum. Mol. Genet. 1999, 8, 1263–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheper, W.; Zwart, R.; van der Sluijs, P.; Annaert, W.; Gool, W.A.; Baas, F. Alzheimer’s presenilin 1 is a putative membrane receptor for rab GDP dissociation inhibitor. Hum. Mol. Genet. 2000, 9, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Netzer, W.J.; Zhong, M.; Lin, Y.; Du, G.; Frohman, M.; Foster, D.A.; Sisodia, S.S.; Xu, H.; Gorelick, F.S.; et al. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc. Natl. Acad. Sci. USA 2006, 103, 1941–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.K.; Anderson, H.A.; Yu, G.; Robertson, A.G.; Allen, S.J.; Tyler, S.J.; Naylor, R.L.; Mason, G.; Wilcock, G.W.; Roche, P.A.; et al. Identification of syntaxin 1A as a novel binding protein for presenilin-1. Brain Res. Mol. Brain Res. 2000, 78, 100–107. [Google Scholar] [CrossRef]
- Suga, K.; Tomiyama, T.; Mori, H.; Akagawa, K. Syntaxin 5 interacts with presenilin holoproteins, but not with their N- or C-terminal fragments, and affects beta-amyloid peptide production. Biochem. J. 2004, 381, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.F.; McLoughlin, D.M.; Standen, C.; Miller, C.C. X11 alpha and x11 beta interact with presenilin-1 via their PDZ domains. Mol. Cell. Neurosci. 2000, 16, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Leem, J.Y.; Greenfield, J.P.; Wang, P.; Kim, B.S.; Wang, R.; Lopes, K.O.; Kim, S.H.; Zheng, H.; Greengard, P.; et al. Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J. Biol. Chem. 2003, 278, 3446–3454. [Google Scholar] [CrossRef] [PubMed]
- Lazarov, O.; Morfini, G.A.; Pigino, G.; Gadadhar, A.; Chen, X.; Robinson, J.; Ho, H.; Brady, S.T.; Sisodia, S.S. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer’s disease-linked mutant presenilin 1. J. Neurosci. 2007, 27, 7011–7020. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.R.; Nye, J.S.; Lamb, N.J.; Fernandez, A.; Kitzmann, M. Intracellular retention of caveolin 1 in presenilin-deficient cells. J. Biol. Chem. 2005, 280, 6663–6668. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; Van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, X.; Huang, T.; Jiang, L.L.; Tan, Z.; Zhang, M.; Cheng, I.H.; Wang, X.; Bu, G.; Zhang, Y.W.; et al. Intracellular trafficking of TREM2 is regulated by presenilin 1. Exp. Mol. Med. 2017, 49, e405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naruse, S.; Thinakaran, G.; Luo, J.J.; Kusiak, J.W.; Tomita, T.; Iwatsubo, T.; Qian, X.; Ginty, D.D.; Price, D.L.; Borchelt, D.R.; et al. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 1998, 21, 1213–1221. [Google Scholar] [CrossRef]
- Kaether, C.; Lammich, S.; Edbauer, D.; Ertl, M.; Rietdorf, J.; Capell, A.; Steiner, H.; Haass, C. Presenilin-1 affects trafficking and processing of betaAPP and is targeted in a complex with nicastrin to the plasma membrane. J. Cell Biol. 2002, 158, 551–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Yang, D.S.; Petanceska, S.; Yang, A.; Tandon, A.; Yu, G.; Rozmahel, R.; Ghiso, J.; Nishimura, M.; Zhang, D.M.; et al. Carboxyl-terminal fragments of Alzheimer beta-amyloid precursor protein accumulate in restricted and unpredicted intracellular compartments in presenilin 1-deficient cells. J. Biol. Chem. 2000, 275, 36794–36802. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; Zhang, Y.W.; Ikin, A.; Schmidt, S.D.; Bogush, A.; Levy, E.; Sheffield, R.; Nixon, R.A.; Liao, F.F.; Mathews, P.M.; et al. Alzheimer’s presenilin 1 modulates sorting of APP and its carboxyl-terminal fragments in cerebral neurons in vivo. J. Neurochem. 2007, 102, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Uemura, K.; Kitagawa, N.; Kohno, R.; Kuzuya, A.; Kageyama, T.; Chonabayashi, K.; Shibasaki, H.; Shimohama, S. Presenilin 1 is involved in maturation and trafficking of N-cadherin to the plasma membrane. J. Neurosci. Res. 2003, 74, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Esselens, C.; Oorschot, V.; Baert, V.; Raemaekers, T.; Spittaels, K.; Serneels, L.; Zheng, H.; Saftig, P.; De Strooper, B.; Klumperman, J.; et al. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J. Cell Biol. 2004, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.A.; Choi, S.Y.; Beglopoulos, V.; Malkani, S.; Zhang, D.; Shankaranarayana Rao, B.S.; Chattarji, S.; Kelleher, R.J.; Kandel, E.R.; Duff, K.; et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 2004, 42, 23–36. [Google Scholar] [CrossRef]
- Zhang, M.; Haapasalo, A.; Kim, D.Y.; Ingano, L.A.; Pettingell, W.H.; Kovacs, D.M. Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006, 20, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tang, P.; Wang, P.; Boissy, R.E.; Zheng, H. Regulation of tyrosinase trafficking and processing by presenilins: Partial loss of function by familial Alzheimer’s disease mutation. Proc. Natl. Acad. Sci. USA 2006, 103, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Repetto, E.; Yoon, I.S.; Zheng, H.; Kang, D.E. Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J. Biol. Chem. 2007, 282, 31504–31516. [Google Scholar] [CrossRef] [PubMed]
- Zou, K.; Hosono, T.; Nakamura, T.; Shiraishi, H.; Maeda, T.; Komano, H.; Yanagisawa, K.; Michikawa, M. Novel role of presenilins in maturation and transport of integrin beta 1. Biochemistry 2008, 47, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Barthet, G.; Dunys, J.; Shao, Z.; Xuan, Z.; Ren, Y.; Xu, J.; Arbez, N.; Mauger, G.; Bruban, J.; Georgakopoulos, A.; et al. Presenilin mediates neuroprotective functions of ephrinB and brain-derived neurotrophic factor and regulates ligand-induced internalization and metabolism of EphB2 and TrkB receptors. Neurobiol. Aging 2013, 34, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Dhaliwal, J.S.; Ceizar, M.; Vaculik, M.; Kumar, K.L.; Lagace, D.C. Knockout of Atg5 delays the maturation and reduces the survival of adult-generated neurons in the hippocampus. Cell Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Codogno, P.; Cuervo, A.M.; Deretic, V.; Elazar, Z.; Fueyo-Margareto, J.; Gewirtz, D.A.; Kroemer, G.; Levine, B.; Mizushima, N.; et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010, 6, 438–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.A.; Murphy, D.D.; Giasson, B.I.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Degradative organelles containing mislocalized alpha-and beta-synuclein proliferate in presenilin-1 null neurons. J. Cell Biol. 2004, 165, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Schmidt, S.D.; Terio, N.B.; Duff, K.; Beard, M.; Mathews, P.M.; Nixon, R.A. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.E.; Beckel, J.M.; Laties, A.M.; Mitchell, C.H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neely, K.M.; Green, K.N.; LaFerla, F.M. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a γ-secretase-independent manner. J. Neurosci. 2011, 31, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Garbett, K.; Veeraraghavalu, K.; Wilburn, B.; Gilmore, R.; Mirnics, K.; Sisodia, S.S. A role for presenilins in autophagy revisited: Normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J. Neurosci. 2012, 32, 8633–8648. [Google Scholar] [CrossRef] [PubMed]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neely Kayala, K.M.; Dickinson, G.D.; Minassian, A.; Walls, K.C.; Green, K.N.; Laferla, F.M. Presenilin-null cells have altered two-pore calcium channel expression and lysosomal calcium: Implications for lysosomal function. Brain Res. 2012, 1489, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Park, M.H.; Kim, B.R.; Lee, P.H.; Nakauchi, H.; Carter, J.E.; He, X.; Schuchman, E.H.; Bae, J.S. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J. Exp. Med. 2014, 211, 1551–1570. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Maestro, P.; Gargini, R.; Sproul, A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy Failure in Fibroblasts and iPSC-Derived Neurons of Alzheimer’s Disease-Associated Presenilin 1 Mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.M.; Ke, M.; Tan, Y.; Huang, Z.; Zhang, K.; Ai, N.; Ge, W.; Qin, D.; Lu, J.H.; Su, H. Presenilin 1 deficiency suppresses autophagy in human neural stem cells through reducing γ-secretase-independent ERK/CREB signaling. Cell Death Dis. 2018, 9, 879. [Google Scholar] [CrossRef] [PubMed]
- Száraz, P.; Bánhegyi, G.; Marcolongo, P.; Benedetti, A. Transient knockdown of presenilin-1 provokes endoplasmic reticulum stress related formation of autophagosomes in HepG2 cells. Arch. Biochem. Biophys. 2013, 538, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Song, B.Q.; Chi, Y.; Li, X.; Du, W.J.; Han, Z.B.; Tian, J.J.; Li, J.J.; Chen, F.; Wu, H.H.; Han, L.X.; et al. Inhibition of Notch Signaling Promotes the Adipogenic Differentiation of Mesenchymal Stem Cells Through Autophagy Activation and PTEN-PI3K/AKT/mTOR Pathway. Cell. Physiol. Biochem. 2015, 36, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contino, S.; Porporato, P.E.; Bird, M.; Marinangeli, C.; Opsomer, R.; Sonveaux, P.; Bontemps, F.; Dewachter, I.; Octave, J.N.; Bertrand, L.; et al. Presenilin 2-Dependent Maintenance of Mitochondrial Oxidative Capacity and Morphology. Front. Physiol. 2017, 8, 796. [Google Scholar] [CrossRef] [PubMed]
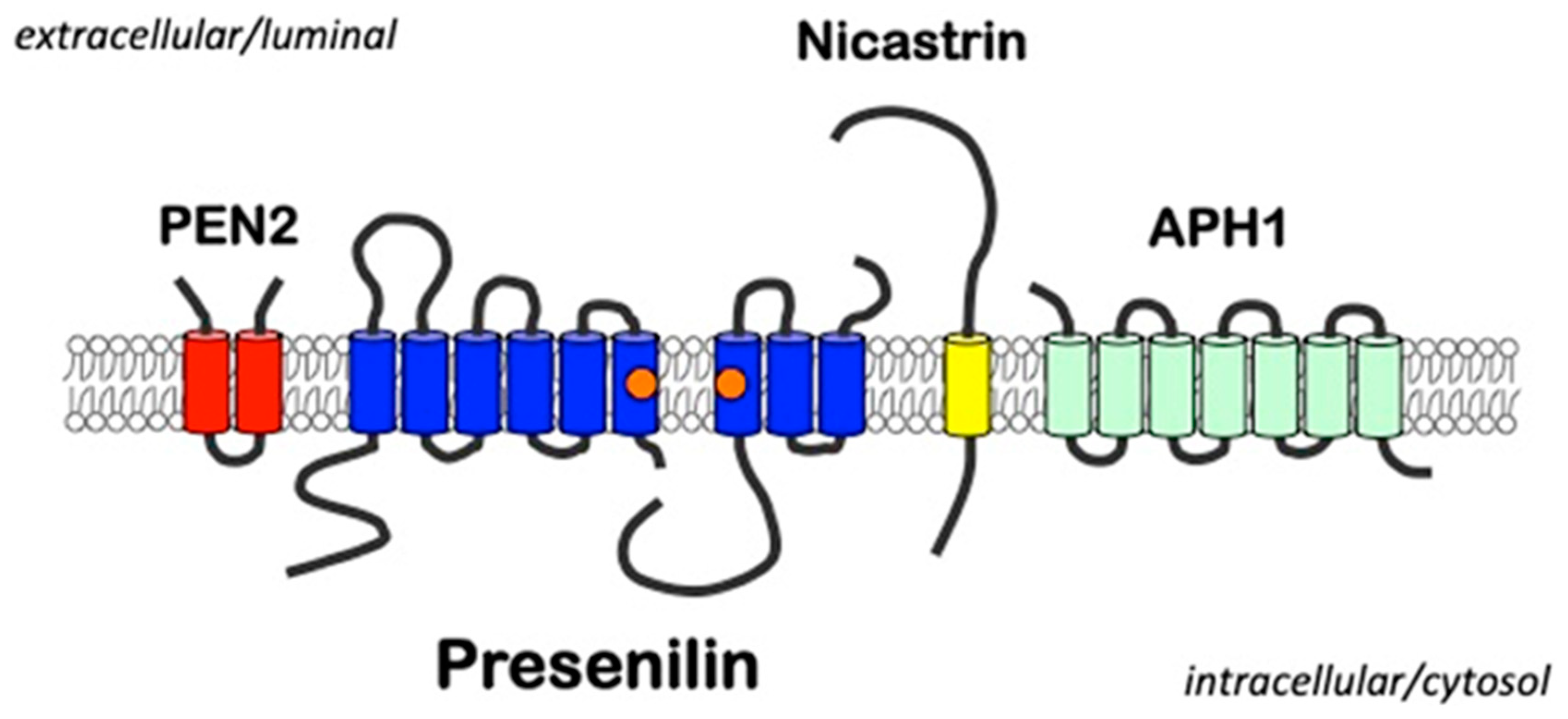
Figure 1.
A schematic of the γ-secretase complex. Presenilins represent the catalytic components of γ-secretase complexes that contain three additional proteins presenilin-enhancer-2 (PEN2), nicastrin, and anterior-pharynx defective-1 (APH1). The aspartyl residues in transmembrane domains 6 and 7 required for catalytic activity are indicated by orange circles.
Figure 1.
A schematic of the γ-secretase complex. Presenilins represent the catalytic components of γ-secretase complexes that contain three additional proteins presenilin-enhancer-2 (PEN2), nicastrin, and anterior-pharynx defective-1 (APH1). The aspartyl residues in transmembrane domains 6 and 7 required for catalytic activity are indicated by orange circles.

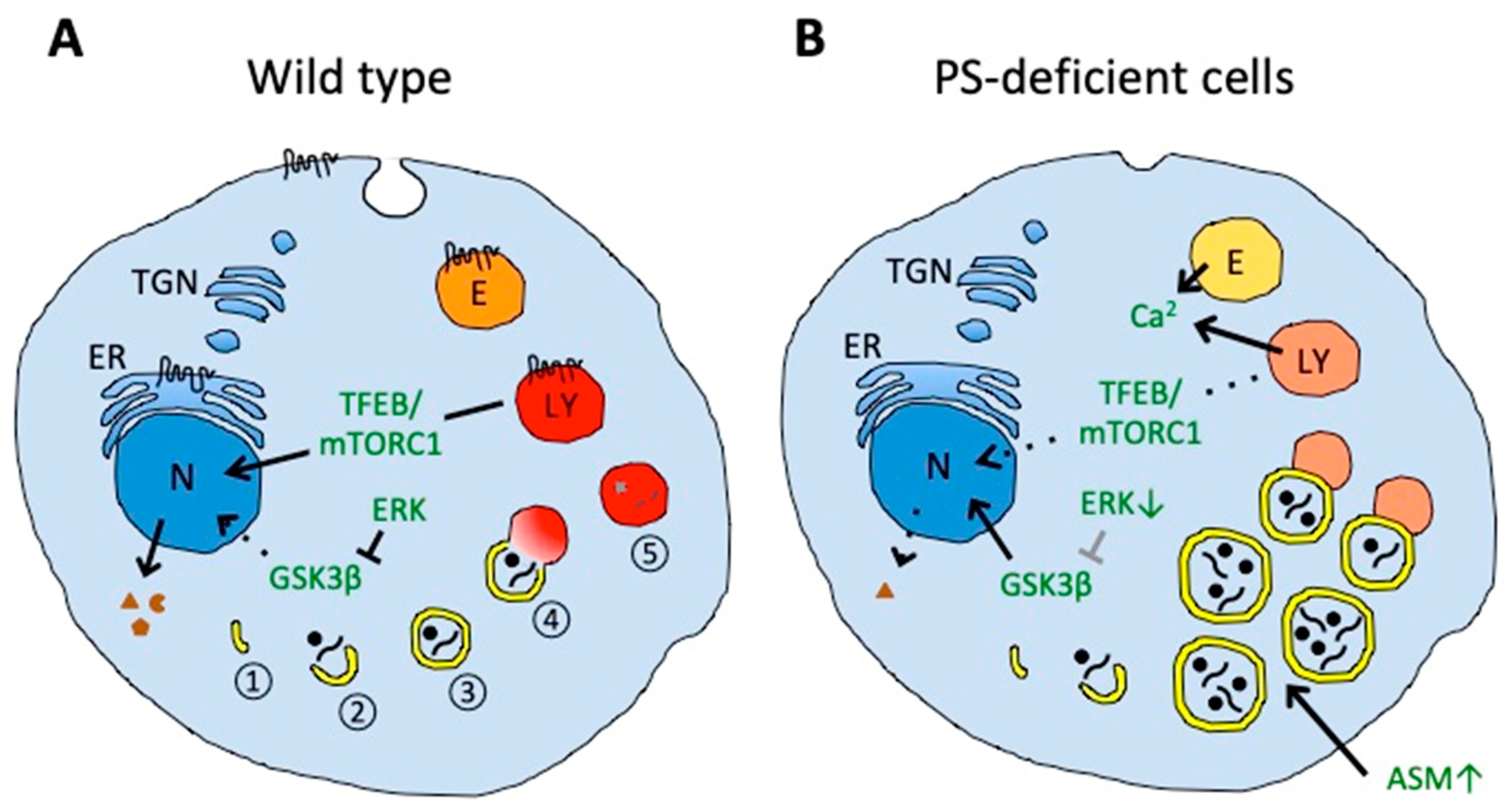
Figure 2.
Impaired autophagy and lysosomal degradation in PS-deficient cells. (A) In wild-type cells, autophagy includes nucleation (1) and elongation of phagophores (2), autophagosome formation by phagophore maturation (3), autolysosome formation by the fusion of autophagosomes and lysosomes (4), and final degradation of the contents (5). (B) In PS-deficient cells, enlarged autophagic vacuoles accumulate and contain undigested engulfed material. The accumulation of autophagic vacuoles could result from the disturbed fusion of autophagosomes with lysosomes, probably caused by impaired lysosomal acidification (illustrated in a pale red color). Aberrant acidification could also affect calcium homeostasis in endolysosomal vesicles that could contribute to impaired vesicle fusion. PS deficiency can also impair amino acid sensing by mTORC1 on lysosomes and decrease activation and nuclear translocation of transcription factor EB (TFEB), thereby decreasing expression of proteins mediating biogenesis of lysosomal and autophagic vesicles. Decreased translocation of glycogen synthase kinase 3β (GSK3β) from the cytosol to the nucleus could also decrease the activation of TFEB. Increased acid sphingomyelinase (ASM) in PS-deficient cells can induce the accumulation of autophagic vacuoles. Decreased endocytosis in PS-deficient cells could also affect membrane protein and lipid homeostasis. Presenilin can be localized in the endoplasmic reticulum (ER), plasma membrane, endosomes (E), and lysosomes (LY). N, nucleus; TGN, trans-Golgi network.
Figure 2.
Impaired autophagy and lysosomal degradation in PS-deficient cells. (A) In wild-type cells, autophagy includes nucleation (1) and elongation of phagophores (2), autophagosome formation by phagophore maturation (3), autolysosome formation by the fusion of autophagosomes and lysosomes (4), and final degradation of the contents (5). (B) In PS-deficient cells, enlarged autophagic vacuoles accumulate and contain undigested engulfed material. The accumulation of autophagic vacuoles could result from the disturbed fusion of autophagosomes with lysosomes, probably caused by impaired lysosomal acidification (illustrated in a pale red color). Aberrant acidification could also affect calcium homeostasis in endolysosomal vesicles that could contribute to impaired vesicle fusion. PS deficiency can also impair amino acid sensing by mTORC1 on lysosomes and decrease activation and nuclear translocation of transcription factor EB (TFEB), thereby decreasing expression of proteins mediating biogenesis of lysosomal and autophagic vesicles. Decreased translocation of glycogen synthase kinase 3β (GSK3β) from the cytosol to the nucleus could also decrease the activation of TFEB. Increased acid sphingomyelinase (ASM) in PS-deficient cells can induce the accumulation of autophagic vacuoles. Decreased endocytosis in PS-deficient cells could also affect membrane protein and lipid homeostasis. Presenilin can be localized in the endoplasmic reticulum (ER), plasma membrane, endosomes (E), and lysosomes (LY). N, nucleus; TGN, trans-Golgi network.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Oikawa, N.; Walter, J. Presenilins and γ-Secretase in Membrane Proteostasis. Cells 2019, 8, 209. https://doi.org/10.3390/cells8030209
AMA Style
Oikawa N, Walter J. Presenilins and γ-Secretase in Membrane Proteostasis. Cells. 2019; 8(3):209. https://doi.org/10.3390/cells8030209
Chicago/Turabian StyleOikawa, Naoto, and Jochen Walter. 2019. "Presenilins and γ-Secretase in Membrane Proteostasis" Cells 8, no. 3: 209. https://doi.org/10.3390/cells8030209
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.