
Nonsynonymous Mutations in Intellectual Disability and Autism Spectrum Disorder Gene PTCHD1 Disrupt N-Glycosylation and Reduce Protein Stability

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Primary Neurons
2.3. Construct Creation
2.4. Lentivirus Production and Transduction
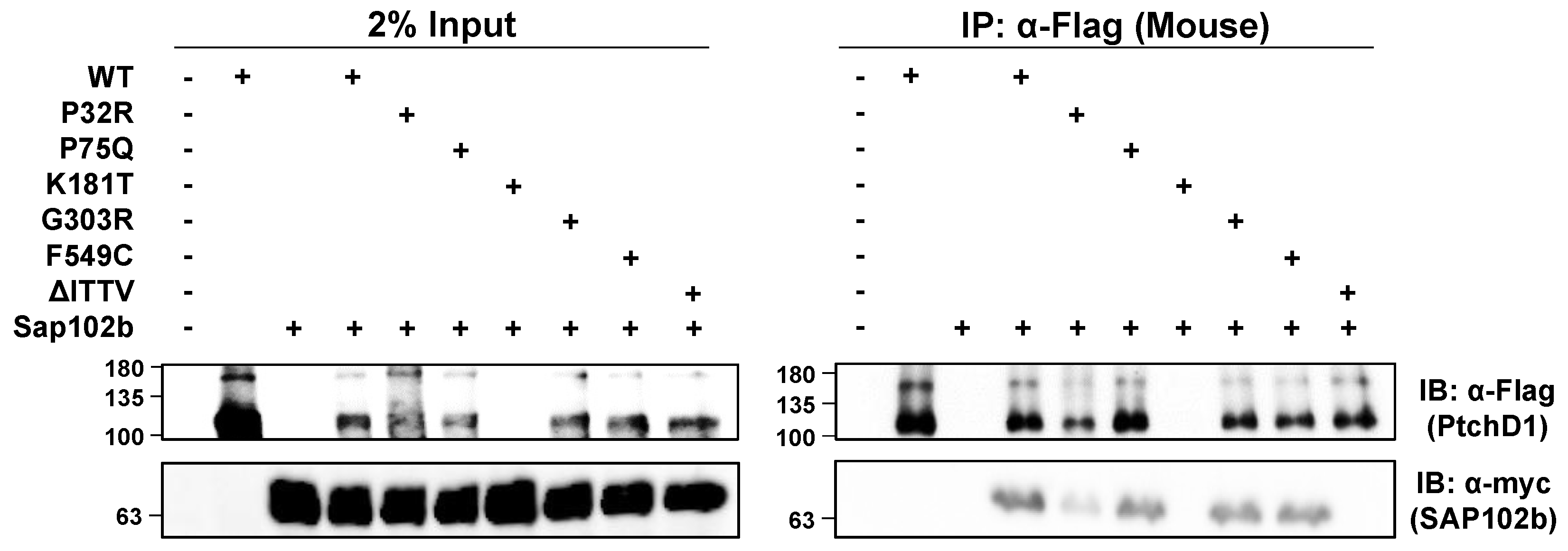
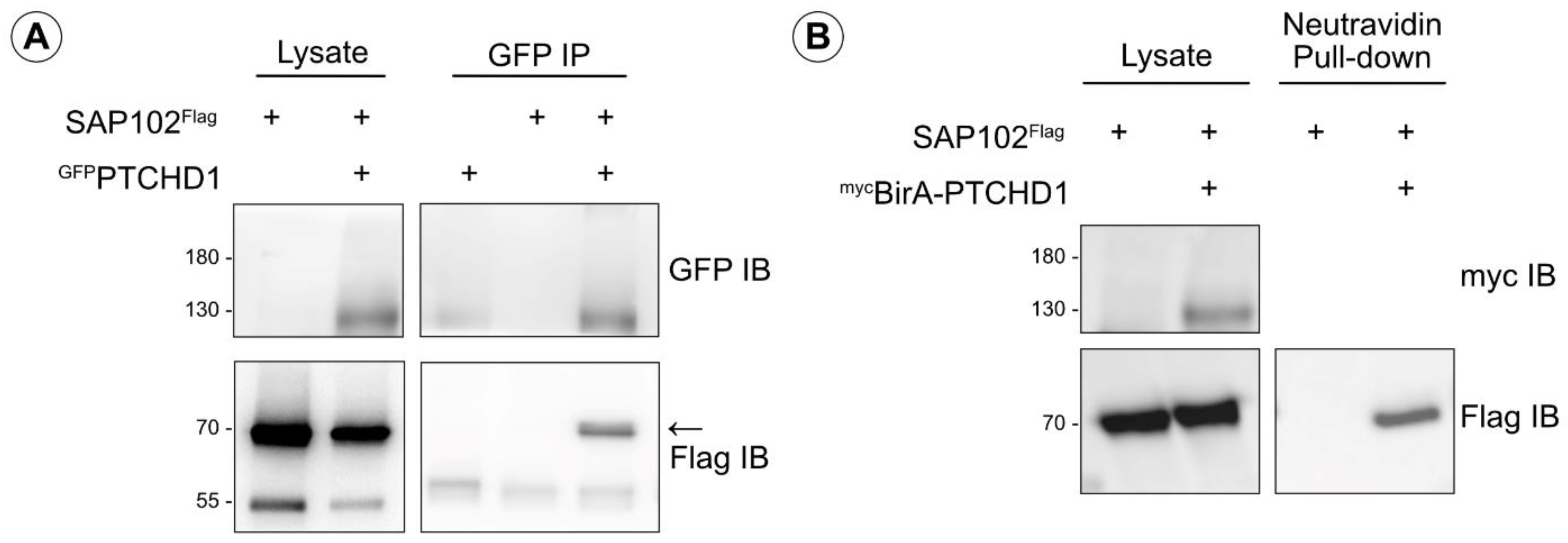
2.5. Western Blotting and Co-Immunoprecipitations
2.6. Glycosylation Assay
2.7. Protein Stability Assay
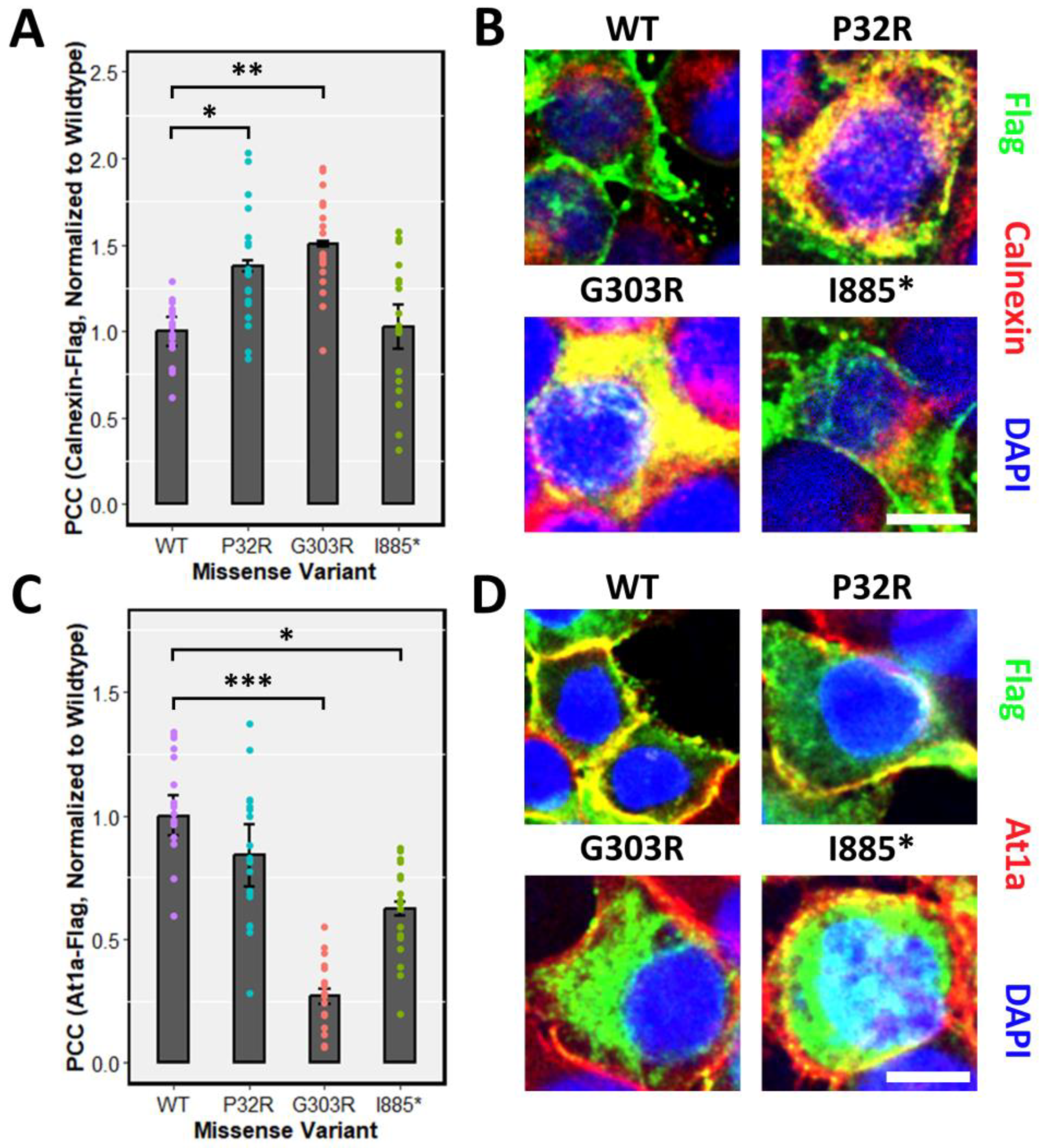
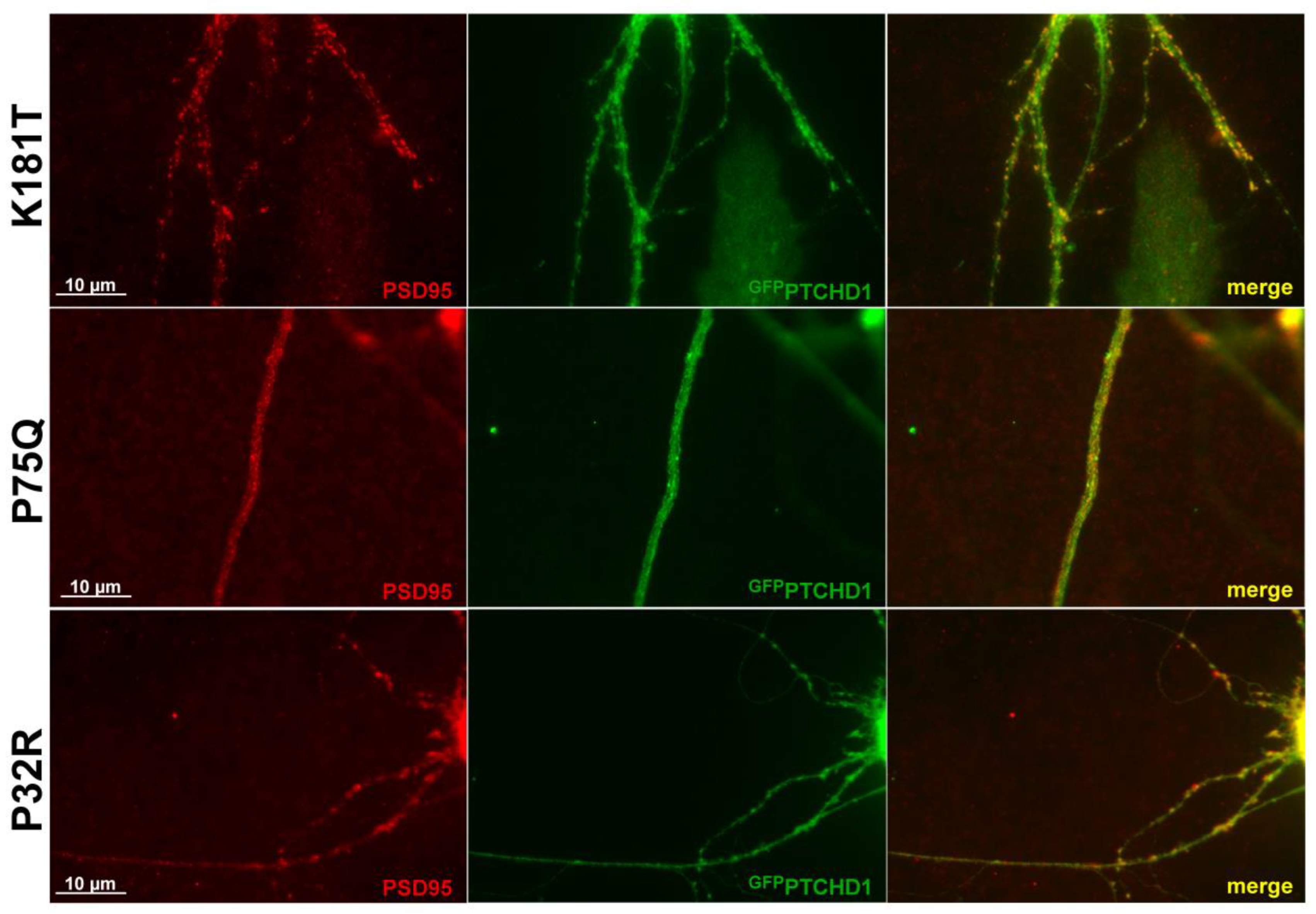
2.8. Co-Localization Image Acquisition and Analysis
3. Results
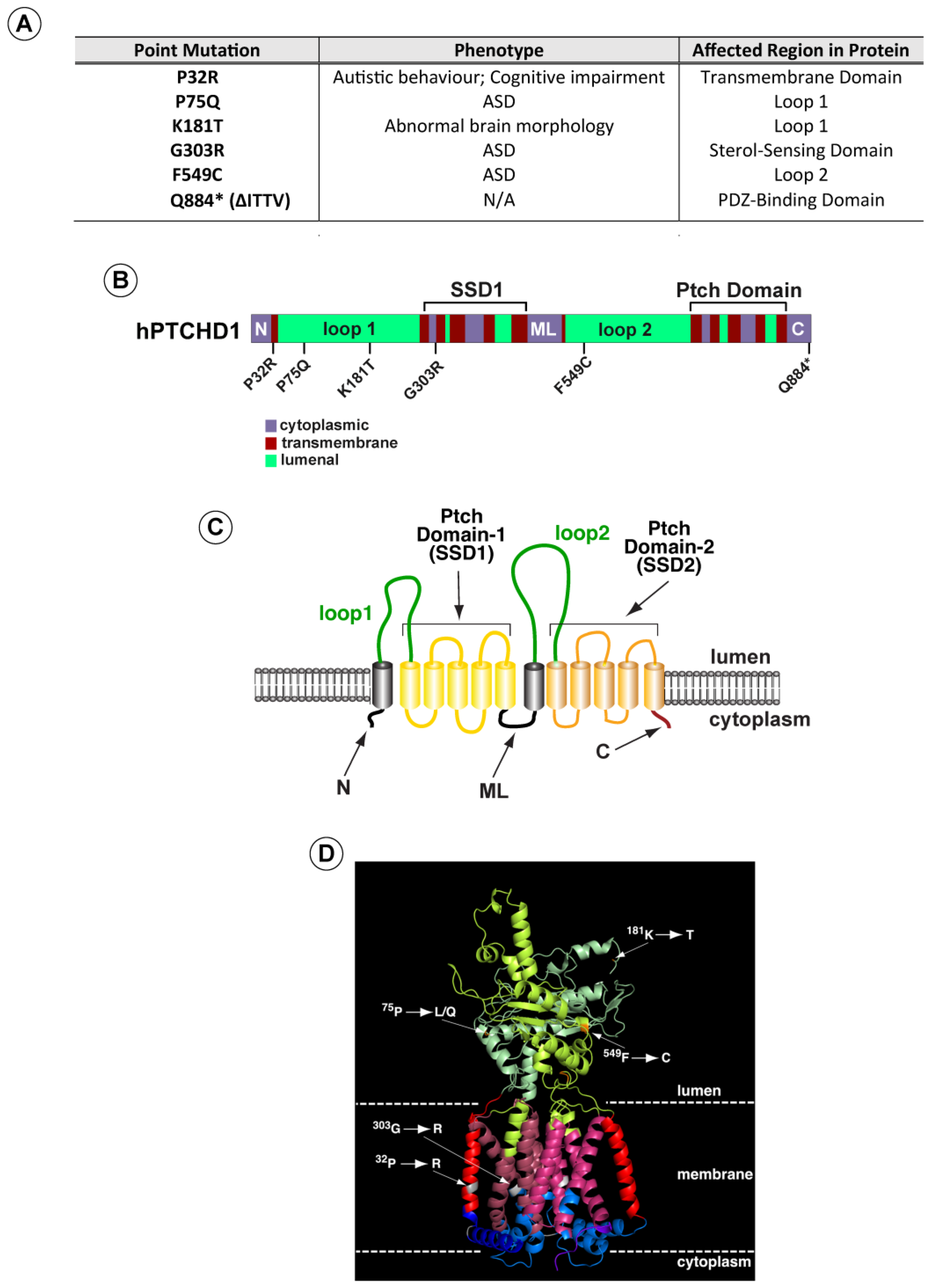
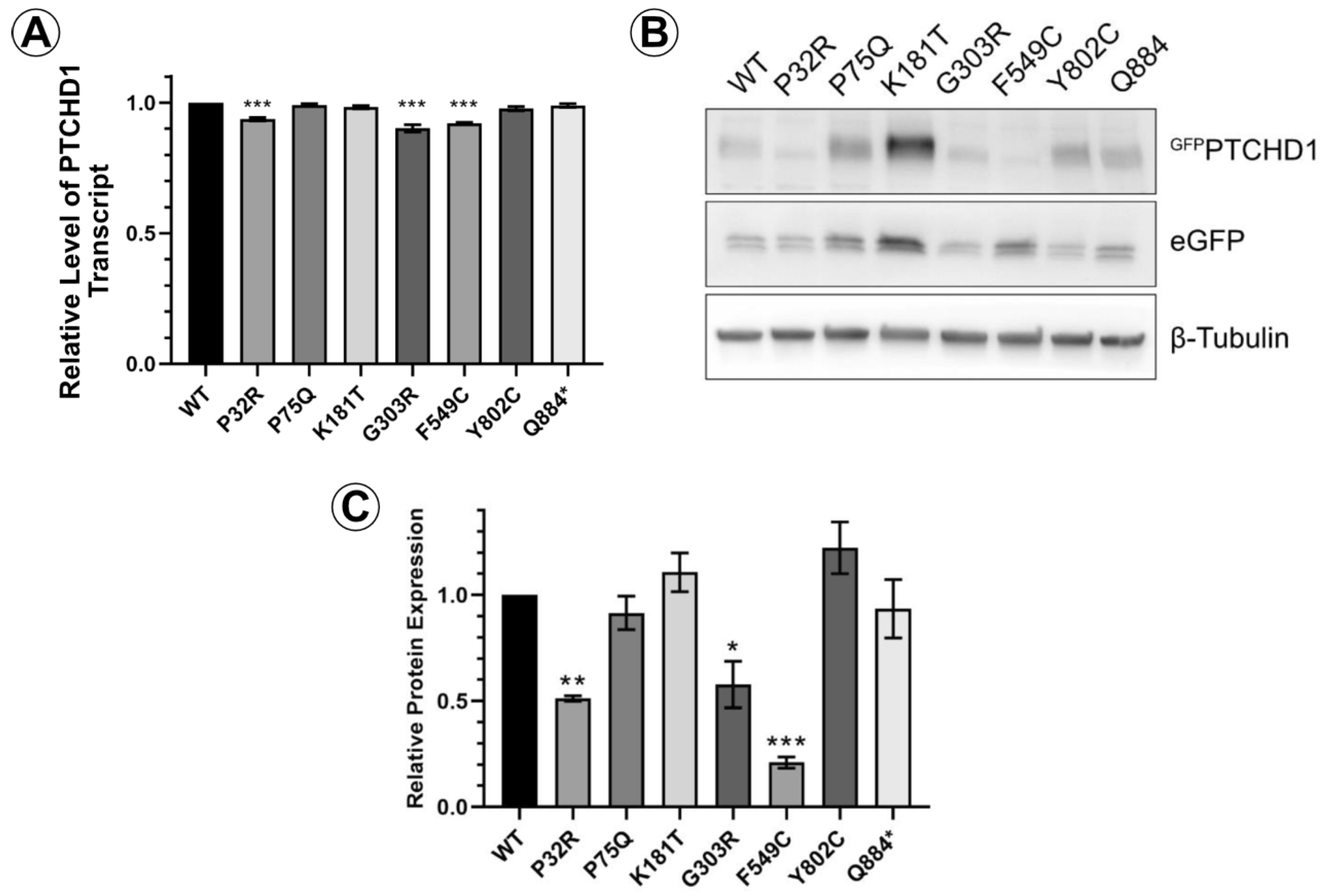
3.1. Characterization of PTCHD1 Mutants
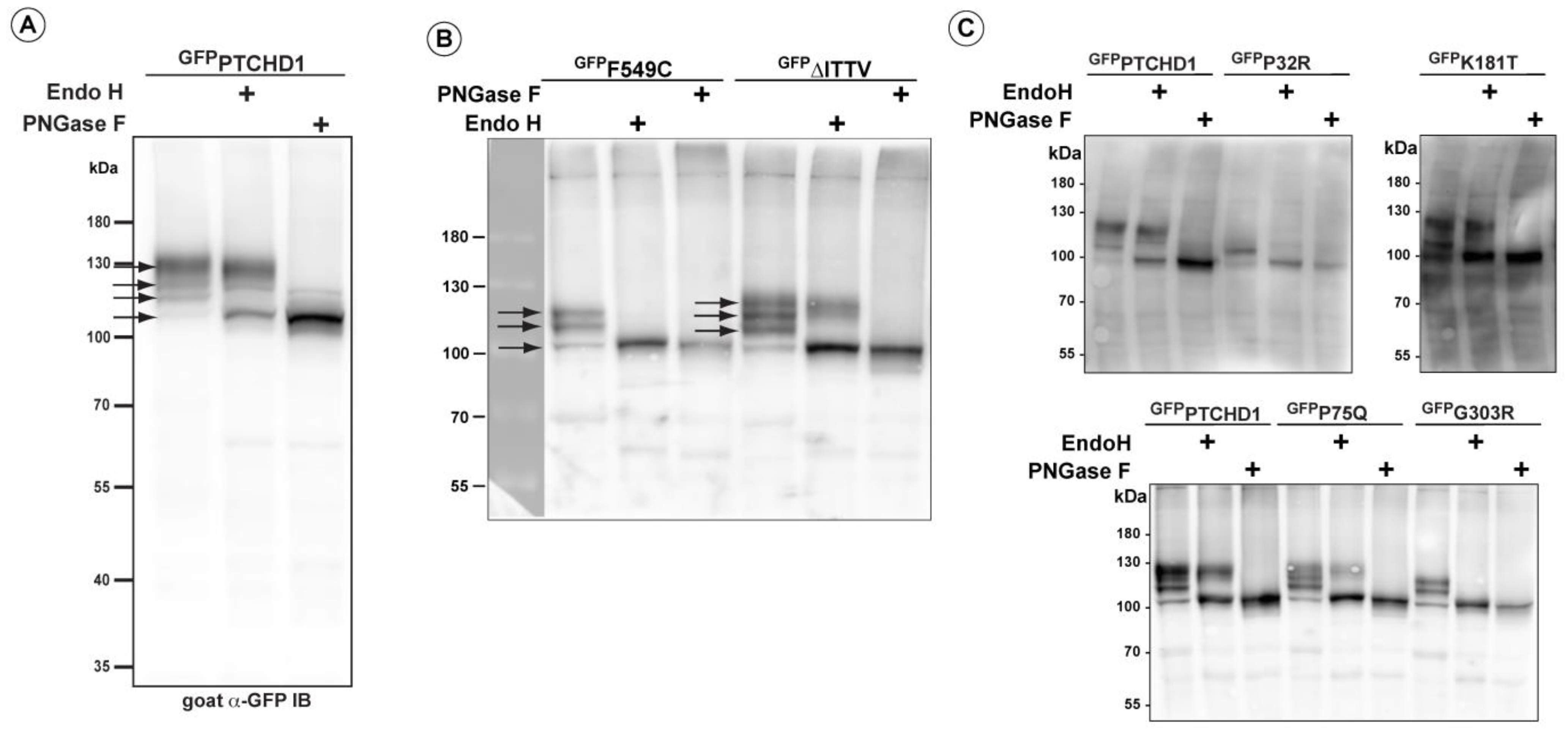
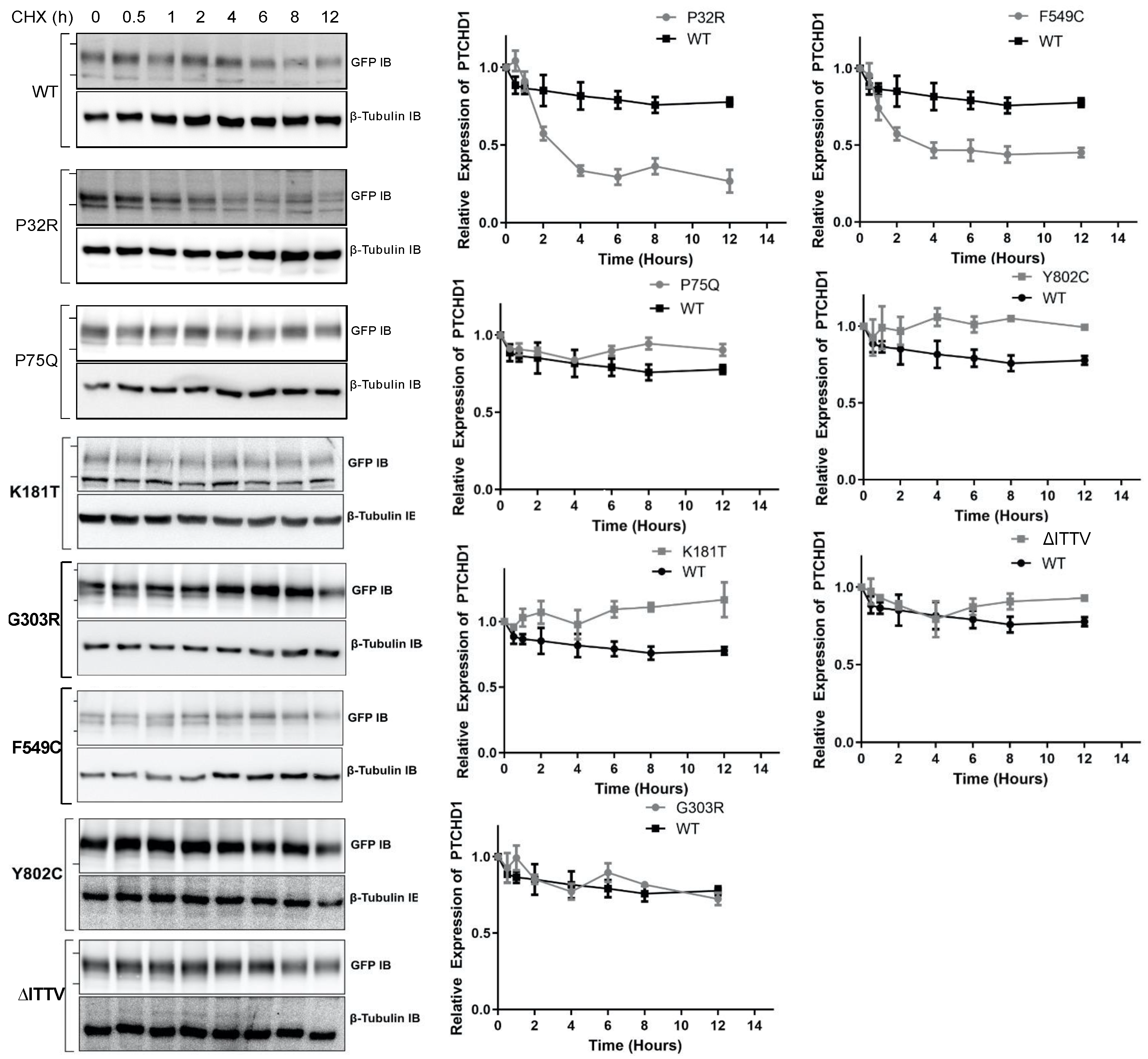
3.2. Distinct Processing of PTCHD1 Mutants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Genomic (hg19); cDNA (NM_173495.2) Coordinates | Source, Subject ID | Inheritance | Minor Allele Frequency (gnomAD)/# Hemizygotes | Prediction: Condel; CADD PHRED-like Scaled C-Score | Reported Phenotype |
|---|---|---|---|---|---|---|
| Pro32Arg | ChrX:23353087; c.95C>G | DECIPHER: 284363 | Mat | 0/0 | Deleterious; 26.8 | Autistic behavior; cognitive impairment |
| Pro75Gln | ChrX:23353216; c.224C>A | MSSNG: AU3794302 | Mat | 0/0 | Deleterious; 27.6 | Autism spectrum disorder |
| Lys181Thr | ChrX:23397898; c.542A>C | Clinvar | nr | 0/0 | Deleterious; 21.9 | Abnormality of brain morphology |
| Gly303Arg | ChrX:23398263; c.907G>A | ClinVar ID 417957 | nr | 0/0 | Deleterious; 27.7 | Autism |
| Phe549Cys | ChrX:23411281; c.1646T>G | Ptchd1-base.com | nr | 0/0 | Deleterious; 28 | Autism spectrum disorder |
| ∆ITTV | ChrX:23412285; c.2650C>T | No subject | na | 0/0 | 38 | na |
| Point Mutation | Sequence |
|---|---|
| (A) | |
| P32R Forward | 5′-CACCCTGTCTTCTTCGCCTCGGCGCGGGTGCTCATCTCCATCCTGCTG-3′ |
| P32R Reverse | 5′-CAGCAGGATGGAGATGAGCACCCGCGCCGAGGCGAAGAAGACAGGGTG-3′ |
| P75Q Forward | 5′-GTTAACAGCCTCTTCCAGGTCAACCGCTCCAAGCACCGTCTCTACTCG-3′ |
| P75Q Reverse | 5′-CGAGTAGAGACGGTGCTTGGAGCGGTTGACCTGGAAGAGGCTGTTAAC-3′ |
| K181T Forward | 5′-ACATACCCAATCACTCACTTAACGGACGGGAGGGCTGTGTACAATGGG-3′ |
| K181T Reverse | 5′-CCCATTGTACACAGCCCTCCCGTCCGTTAAGTGAGTGATTGGGTATGT-3′ |
| G303R Forward | 5′-AAACCCTGGCTAGGCCTGCTCCGATTGGTGACCATAAGCCTGGCC-3′ |
| G303R Reverse | 5′-GGCCAGGCTTATGGTCACCAATCGGAGCAGGCCTAGCCAGGGTTT-3′ |
| F549C Forward | 5′-ACTACTGCCCAGCAAAAGTACTGCAGCAACTACAGTCCTGTGATT-3′ |
| F549C Reverse | 5′-AATCACAGGACTGTAGTTGCTGCAGTACTTTTGCTGGGCAGTAGT-3′ |
| ∆ITTV Forward | 5′-ATATCGATAGTACCCGTGTGGTTGACTAAATTACAACAGTGTGA-3′ |
| ∆ITTV Reverse | 5′-TCACACTGTTGTAATTTAGTCAACCACACGGGTACTATCGATAT-3′ |
| (B) | |
| P32R Forward | 5′-AGAGTGCTCATCTCCATCCTGCTC-3′ |
| P32R Reverse | 5′-CGCCGAAGCAAAGAAGACCG-3′ |
| P75Q Forward | 5′-CAAGTCAACCGCTCCAAGCACC-3′ |
| P75Q Reverse | 5′-GAAGAGGCTGTTGACTAGGTTGCG-3′ |
| K181T Forward | 5′-CAGATGGAAGGGCTGTGTATAATGGG-3′ |
| K181T Reverse | 5′-TTAAGTGAGTGATCGGATATGTGATAGCAAAATTGG-3′ |
| G303R Forward | 5′-CGGTTGGTGACCATAAGCCTAGC-3′ |
| G303R Reverse | 5′-AAGTAGGCCTAACCAGGGTTTGC-3′ |
| F549C Forward | 5′-GTAACAACTACAGTCCTGTTATTGGGTTTTAC-3′ |
| F549C Reverse | 5′-AGTACTTTTGGTGGGCAGTAGTG-3′ |
| ∆ITTV Forward | 5′-TGAATAGGAGTTTAAACCCGCTGATCAGCCTC-3′ |
| ∆ITTV Reverse | 5′-TTGGTCAACCACTCGAGTACTATCAATATCTACC-3′ |
| Ptchd1 Forward (HindIII) | 5′-CGTACGAAGCTTATGCTGCGGCAGGTTCTG-3′ |
| Ptchd1 Reverse (AgeI) | 5′-GTACGACCGGTTCACACTGTGGTTATTTGGTCAACCAC-3′ |
| Antibody | Source | Dilution | |
|---|---|---|---|
| 1° Antibody | Mouse α-Flag-Tag | Applied Biological Materials Inc. (Richmond, BC, Canada) Cat # G191 | 1:1000 WB IB |
| Mouse α-Myc-Tag (9B11) | Cell Signaling Technologies (Danvers, MA, USA) #2276S | 1:1000 WB IB; 1:400 IF | |
| Rabbit α-GFP-Tag | Invitrogen (Waltham, MA, USA) Cat #A11122 | 1:100 WB IB; 1:400 IF | |
| Goat α-GFP-Tag | Applied Biological Materials Inc. (Richmond, BC, Canada) Cat # G096 | 1:1000 WB IB; 1:400 IF | |
| Goat α-Biotin | Cedarlane. (Burlington, ON, Canada) CLAS10-666 | 1:1000 WB IB | |
| Rabbit α-PSD95 | Abcam (Waltham, MA, USA) ab18258 | 1:400 IF | |
| Mouse α-β-Tubulin | Applied Biological Materials Inc. (Richmond, BC, Canada) Cat # G098 | 1:5000 WB IB | |
| Mouse α-GM130 | BD Transduction Laboratories (Mississauga, ON, Canada) Cat # 51-9001978 | 1:400 IF | |
| HRP-Linked 2° Antibody | Donkey α-goat IgG | Santa Cruz Biotechnologies Inc.(Dallas, TX, USA) sc-2020 | 1:5000 |
| Horse α-mouse IgG | Cell Signaling Technologies (Danvers, MA, USA) #7076 | 1:5000 | |
| Goat α-rabbit IgG | Cell Signaling Technologies (Danvers, MA, USA) #7074 | 1:5000 | |
| Immunofluorescence 2° Antibody | Donkey α-rabbit IgG Alexa Fluor 488 | Invitrogen (Waltham, MA, USA) A21206 | 1:400 IF |
| Donkey α-mouse IgG Alexa Fluor 568 | Invitrogen (Waltham, MA, USA) A10037 | 1:400 IF | |
| Donkey α-rabbit IgG Alexa Fluor 568 | Invitrogen (Waltham, MA, USA) A10042 | 1:400 | |
| Donkey α-goat FITC | Jackson ImmunoResearch (West Grove, PA, USA) 705-095-003 | 1:400 | |
| Phalloidin Alexa Fluor 350 | Invitrogen (Waltham, MA, USA) A22281 | 1:20 |
| Study Gene | Putative Protein Interactors | Housekeeping Genes | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Index | Name | Description | Ptchd1 | Snapin | Dlg3 | Dlg4 | Gapdh | Actb | Sdha | Rpl37 |
| 70 | HBCHO3 | Afferent nuclei of cranial nerves VI–XII | 0.86 | 1.4 | 0.346 | 2.24 | 2.34 | 23.2 | 4.68 | 28.3 |
| 81 | HBSER5 | Serotonergic neurons, hindbrain | 0.515 | 0.908 | 0.452 | 2.76 | 1.92 | 10.2 | 3.3 | 41 |
| 167 | HBGLU8 | Excitatory neurons, hindbrain | 0.363 | 0.546 | 1.45 | 2.36 | 7.09 | 17.5 | 8.82 | 16.3 |
| 187 | ENT6 | Cholinergic enteric neurons | 0.313 | 1.08 | 0.0679 | 1.5 | 8.09 | 9.63 | 1.1 | 20.8 |
| 79 | HBSER2 | Serotonergic neurons, hindbrain | 0.266 | 0.836 | 0.384 | 1.86 | 1.72 | 10.9 | 3.39 | 27.9 |
| 82 | HBSER4 | Serotonergic neurons, hindbrain | 0.25 | 0.668 | 0.415 | 2.15 | 1.23 | 7.84 | 2.37 | 30 |
| 165 | HBGLU6 | Excitatory neurons, hindbrain | 0.246 | 0.521 | 0.652 | 1.1 | 3.36 | 8.55 | 5.66 | 11 |
| 183 | ENT2 | Nitrergic enteric neurons | 0.224 | 1.61 | 0.149 | 1.04 | 11.9 | 20.1 | 1.57 | 24.1 |
| 189 | ENT8 | Cholinergic enteric neurons, VGLUT2 | 0.216 | 1.67 | 0.0307 | 1.9 | 14.5 | 16.8 | 1.53 | 38.7 |
| 182 | ENT1 | Nitrergic enteric neurons | 0.214 | 1.29 | 0.213 | 1.21 | 13.3 | 14.6 | 2.29 | 21.4 |
| 68 | DECHO1 | Cholinergic neurons, septal nucleus, Meissnert and diagonal band | 0.202 | 1.57 | 0.498 | 1.75 | 4.4 | 10.3 | 3.16 | 33.1 |
| 166 | HBGLU7 | Excitatory neurons, hindbrain | 0.2 | 0.445 | 0.707 | 1.14 | 2.59 | 8.24 | 3.94 | 8.76 |
| 197 | SYCHO1 | Cholinergic neurons, sympathetic | 0.2 | 3.07 | 0.266 | 1.07 | 14.1 | 5.53 | 3.13 | 23.8 |
| 80 | HBSER3 | Serotonergic neurons, hindbrain | 0.195 | 0.839 | 0.206 | 2.24 | 1.5 | 5.65 | 1.74 | 30.8 |
| 162 | HBCHO2 | Cholinergic neurons, hindbrain | 0.182 | 0.546 | 0.317 | 1.64 | 3.36 | 13 | 5.41 | 14.8 |
| 159 | HBINH6 | Inhibitory neurons, hindbrain | 0.177 | 0.485 | 0.434 | 1.03 | 1.87 | 4.63 | 3.03 | 10.6 |
| 190 | ENT9 | Cholinergic enteric neurons | 0.177 | 1.27 | 0.0238 | 2.05 | 9.82 | 26 | 1.51 | 30.5 |
| 120 | HBGLU2 | Excitatory neurons, hindbrain | 0.165 | 0.421 | 0.244 | 0.864 | 1.3 | 5.82 | 2.21 | 13.7 |
| 115 | SCGLU8 | Excitatory neurons, spinal cord | 0.133 | 0.2 | 0.0667 | 0.533 | 0.598 | 2.27 | 0.801 | 4.6 |
| 150 | HBINH5 | Inhibitory neurons, hindbrain | 0.133 | 0.335 | 0.326 | 1.02 | 1.86 | 5.67 | 3.35 | 9.73 |
| 201 | PSPEP5 | Peptidergic (PEP1.2), DRG | 0.133 | 1.97 | 0.0331 | 0.234 | 1.53 | 2.6 | 1.43 | 6.47 |

References
- Quesnel-Vallières, M.; Weatheritt, R.J.; Cordes, S.P.; Blencowe, B.J. Autism spectrum disorder: Insights into convergent mechanisms from transcriptomics. Nat. Rev. Genet. 2019, 20, 51–63. [Google Scholar] [CrossRef]
- Woodbury-Smith, M.; Scherer, S.W. Progress in the genetics of autism spectrum disorder. Dev. Med. Child. Neurol. 2018, 60, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Filges, I.; Röthlisberger, B.; Blattner, A.; Boesch, N.; Demougin, P.; Wenzel, F.; Huber, A.R.; Heinimann, K.; Weber, P.; Miny, P. Deletion in Xp22.11: PTCHD1 is a candidate gene for X-linked intellectual disability with or without autism. Clin. Genet. 2011, 79, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Noor, A.; Whibley, A.; Marshall, C.R.; Gianakopoulos, P.J.; Piton, A.; Carson, A.R.; Orlic-Milacic, M.; Lionel, A.C.; Sato, D.; Pinto, D.; et al. Disruption at the PTCHD1 locus on Xp22.11 in autism spectrum disorder and intellectual disability. Sci. Transl. Med. 2010, 2, 49ra68. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef]
- Whibley, A.C.; Plagnol, V.; Tarpey, P.S.; Abidi, F.; Fullston, T.; Choma, M.K.; Boucher, C.A.; Shepherd, L.; Willatt, L.; Parkin, G.; et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am. J. Hum. Genet. 2010, 87, 173–188. [Google Scholar] [CrossRef]
- Nikaido, H. RND transporters in the living world. Res. Microbiol. 2018, 169, 363–371. [Google Scholar] [CrossRef]
- Cologna, S.M.; Rosenhouse-Dantsker, A. Insights into the Molecular Mechanisms of Cholesterol Binding to the NPC1 and NPC2 Proteins. Adv. Exp. Med. Biol. 2019, 1135, 139–160. [Google Scholar]
- Millard, E.E.; Gale, S.E.; Dudley, N.; Zhang, J.; Schaffer, J.E.; Ory, D.S. The sterol-sensing domain of the Niemann-Pick C1 (NPC1) protein regulates trafficking of low density lipoprotein cholesterol. J. Biol. Chem. 2005, 280, 28581–28590. [Google Scholar] [CrossRef]
- Pfeffer, S.R. NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 2019, 294, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Fleet, A.J.; Hamel, P.A. The protein-specific activities of the transmembrane modules of Ptch1 and Ptch2 are determined by their adjacent protein domains. J. Biol. Chem. 2018, 293, 16583–16595. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, P.E.; Labouesse, M. The sterol-sensing domain: Multiple families, a unique role? Trends Genet. 2002, 18, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Martín, V.; Carrillo, G.; Torroja, C.; Guerrero, I. The sterol-sensing domain of Patched protein seems to control Smoothened activity through Patched vesicular trafficking. Curr. Biol. 2001, 11, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Tora, D.; Gomez, A.M.; Michaud, J.-F.; Yam, P.T.; Charron, F.; Scheiffele, P. Cellular Functions of the Autism Risk Factor PTCHD1 in Mice. J. Neurosci. 2017, 37, 11993–12005. [Google Scholar] [CrossRef]
- Ung, D.C.; Iacono, G.; Méziane, H.; Blanchard, E.; Papon, M.-A.; Selten, M.; van Rhijn, J.-R.; Montjean, R.; Rucci, J.; Martin, S.; et al. Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Mol. Psychiatry 2018, 23, 1356–1367. [Google Scholar] [CrossRef]
- Li, X.; Saha, P.; Li, J.; Blobel, G.; Pfeffer, S.R. Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc. Natl. Acad. Sci. USA 2016, 113, 10079–10084. [Google Scholar] [CrossRef]
- Gong, X.; Qian, H.; Cao, P.; Zhao, X.; Zhou, Q.; Lei, J.; Yan, N. Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 2018, 361, 6402. [Google Scholar] [CrossRef]
- Qi, X.; Schmiege, P.; Coutavas, E.; Li, X. Two Patched molecules engage distinct sites on Hedgehog yielding a signaling-competent complex. Science 2018, 362, eaas8843. [Google Scholar] [CrossRef]
- Zhang, Y.; Bulkley, D.P.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018, 175, 1352–1364.e14. [Google Scholar] [CrossRef] [PubMed]
- Khlghatyan, J.; Evstratova, A.; Bozoyan, L.; Chamberland, S.; Chatterjee, D.; Marakhovskaia, A.; Soares Silva, T.; Toth, K.; Mongrain, V.; Beaulieu, J.M. Fxr1 regulates sleep and synaptic homeostasis. EMBO J. 2020, 39, e103864. [Google Scholar] [CrossRef] [PubMed]
- Fleet, A.; Lee JP, Y.; Tamachi, A.; Javeed, I.; Hamel, P.A. Activities of the Cytoplasmic Domains of Patched-1 Modulate but Are Not Essential for the Regulation of Canonical Hedgehog Signaling. J. Biol. Chem. 2016, 291, 17557–17568. [Google Scholar] [CrossRef] [PubMed]
- González-Pérez, A.; López-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Qi, X.; Schmiege, P.; Coutavas, E.; Wang, J.; Li, X. Structures of human Patched and its complex with native palmitoylated sonic hedgehog. Nature 2018, 560, 128–132. [Google Scholar] [CrossRef]
- Qian, H.; Cao, P.; Hu, M.; Gao, S.; Yan, N.; Gong, X. Inhibition of tetrameric Patched1 by Sonic Hedgehog through an asymmetric paradigm. Nat. Commun. 2019, 10, 2320. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Wu, X.; Yan, R.; Cao, P.; Qian, H.; Yan, N. Structural advances in sterol-sensing domain-containing proteins. Trends Biochem. Sci. 2022, 47, 289–300. [Google Scholar] [CrossRef]
- Millat, G.; Marçais, C.; Tomasetto, C.; Chikh, K.; Fensom, A.H.; Harzer, K.; Wenger, D.A.; Ohno, K.; Vanier, M.T. Niemann-Pick C1 disease: Correlations between NPC1 mutations, levels of NPC1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop. Am. J. Hum. Genet. 2001, 68, 1373–1385. [Google Scholar] [CrossRef]
- Chidambaram, A.; Goldstein, A.M.; Gailani, M.R.; Gerrard, B.; Bale, S.J.; DiGiovanna, J.J.; Bale, A.E.; Dean, M. Mutations in the human homologue of the Drosophila patched gene in Caucasian and African-American nevoid basal cell carcinoma syndrome patients. Cancer Res. 1996, 56, 4599–4601. [Google Scholar] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Strutt, H.; Thomas, C.; Nakano, Y.; Stark, D.; Neave, B.; Taylor, A.M.; Ingham, P.W. Mutations in the sterol-sensing domain of Patched suggest a role for vesicular trafficking in Smoothened regulation. Curr. Biol. 2001, 11, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Thomas, E.V.; Sanz-Clemente, A.; Roche, K.W. NMDA receptor-dependent regulation of dendritic spine morphology by SAP102 splice variants. J. Neurosci. 2011, 31, 89–96. [Google Scholar] [CrossRef]
- Müller, B.M.; Kistner, U.; Kindler, S.; Chung, W.J.; Kuhlendahl, S.; Fenster, S.D.; Lau, L.F.; Veh, R.W.; Huganir, R.L.; Gundelfinger, E.D.; et al. SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron 1996, 17, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Gray, J.A.; Sanz-Clemente, A.; Wei, Z.; Thomas, E.V.; Nicoll, R.A.; Roche, K.W. SAP102 mediates synaptic clearance of NMDA receptors. Cell Rep. 2012, 2, 1120–1128. [Google Scholar] [CrossRef]
- Liu, M.; Shi, R.; Hwang, H.; Han, K.S.; Wong, M.H.; Ren, X.; Lewis, L.D.; Brown, E.N.; Xu, W. SAP102 regulates synaptic AMPAR function through a CNIH-2-dependent mechanism. J. Neurophysiol. 2018, 120, 1578–1586. [Google Scholar] [CrossRef]
- Sans, N.; Wang, P.Y.; Du, Q.; Petralia, R.S.; Wang, Y.-X.; Nakka, S.; Blumer, J.B.; Macara, I.G.; Wenthold, R.J. mPins modulates PSD-95 and SAP102 trafficking and influences NMDA receptor surface expression. Nat. Cell Biol. 2005, 7, 1179–1190. [Google Scholar] [CrossRef]
- Washbourne, P.; Bennett, J.E.; McAllister, A.K. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat. Neurosci. 2002, 5, 751–759. [Google Scholar] [CrossRef]
- Tarpey, P.; Parnau, J.; Blow, M.; Woffendin, H.; Bignell, G.; Cox, C.; Cox, J.; Davies, H.; Edkins, S.; Holden, S.; et al. Mutations in the DLG3 gene cause nonsyndromic X-linked mental retardation. Am. J. Hum. Genet. 2004, 75, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Zanni, G.; van Esch, H.; Bensalem, A.; Saillour, Y.; Poirier, K.; Castelnau, L.; Ropers, H.H.; de Brouwer, A.P.M.; Laumonnier, F.; Fryns, J.-P.; et al. A novel mutation in the DLG3 gene encoding the synapse-associated protein 102 (SAP102) causes non-syndromic mental retardation. Neurogenetics 2010, 11, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.K.; Sirén, A.; Avela, K.; Somer, M.; Peippo, M.; Ahvenainen, M.; Doagu, F.; Arvio, M.; Kääriäinen, H.; Van Esch, H.; et al. X-exome sequencing in Finnish families with intellectual disability—Four novel mutations and two novel syndromic phenotypes. Orphanet J. Rare Dis. 2014, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Kantojärvi, K.; Kotala, I.; Rehnström, K.; Ylisaukko-Oja, T.; Vanhala, R.; von Wendt, T.N.; von Wendt, L.; Järvelä, I. Fine mapping of Xq11.1-q21.33 and mutation screening of RPS6KA6, ZNF711, ACSL4, DLG3, and IL1RAPL2 for autism spectrum disorders (ASD). Autism Res. 2011, 4, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Perroy, J.; Loll, F.; Perrais, D.; Fagni, L.; Bourgeron, T.; Montcouquiol, M.; Sans, N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 2012, 17, 71–84. [Google Scholar] [CrossRef]
- Hlushchenko, I.; Khanal, P.; Abouelezz, A.; Paavilainen, V.O.; Hotulainen, P. ASD-Associated De Novo Mutations in Five Actin Regulators Show Both Shared and Distinct Defects in Dendritic Spines and Inhibitory Synapses in Cultured Hippocampal Neurons. Front. Cell Neurosci. 2018, 12, 217. [Google Scholar] [CrossRef]
- Wang, L.; Pang, K.; Han, K.; Adamski, C.J.; Wang, W.; He, L.; Lai, J.K.; Bondar, V.V.; Duman, J.G.; Richman, R.; et al. An autism-linked missense mutation in SHANK3 reveals the modularity of Shank3 function. Mol. Psychiatry 2020, 25, 2534–2555. [Google Scholar] [CrossRef]
- Höglinger, D.; Burgoyne, T.; Sanchez-Heras, E.; Hartwig, P.; Colaco, A.; Newton, J.; Futter, C.E.; Spiegel, S.; Platt, F.M.; Eden, E.R. NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun. 2019, 10, 4276. [Google Scholar] [CrossRef]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef]
- Liu, R.; Lu, P.; Chu JW, K.; Sharom, F.J. New Insights into the Drug Binding, Transport and Lipid Flippase Activities of the P-Glycoprotein Multidrug Transporter. J. Biol. Chem. 2009, 284, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Caioli, S.; Saba, L.; Vindigni, G.; Biocca, S.; Canu, N.; Zona, C. Membrane cholesterol depletion in cortical neurons highlights altered NMDA receptor functionality in a mouse model of amyotrophic lateral sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Brachet, A.; Norwood, S.; Brouwers, J.F.; Palomer, E.; Helms, J.B.; Dotti, C.G.; Esteban, J.A. LTP-triggered cholesterol redistribution activates Cdc42 and drives AMPA receptor synaptic delivery. J. Cell Biol. 2015, 208, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Korinek, M.; Vyklicky, V.; Borovska, J.; Lichnerova, K.; Kaniakova, M.; Krausova, B.; Krusek, J.; Balik, A.; Smejkalova, T.; Horak, M.; et al. Cholesterol modulates open probability and desensitization of NMDA receptors. J. Physiol. 2015, 593, 2279–2293. [Google Scholar] [CrossRef] [PubMed]
- Korinek, M.; Gonzalez-Gonzalez, I.M.; Smejkalova, T.; Hajdukovic, D.; Skrenkova, K.; Krusek, J.; Horak, M.; Vyklicky, L. Cholesterol modulates presynaptic and postsynaptic properties of excitatory synaptic transmission. Sci. Rep. 2020, 10, 12651. [Google Scholar] [CrossRef]
- Sibarov, D.A.; Poguzhelskaya, E.E.; Antonov, S.M. Downregulation of calcium-dependent NMDA receptor desensitization by sodium-calcium exchangers: A role of membrane cholesterol. BMC Neurosci. 2018, 19, 73. [Google Scholar] [CrossRef]
- Fanning, A.S.; Anderson, J.M. PDZ domains: Fundamental building blocks in the organization of protein complexes at the plasma membrane. J. Clin. Investig. 1999, 103, 767–772. [Google Scholar] [CrossRef]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, C.T.Y.; Pastore, S.F.; Vincent, J.B.; Frankland, P.W.; Hamel, P.A. Nonsynonymous Mutations in Intellectual Disability and Autism Spectrum Disorder Gene PTCHD1 Disrupt N-Glycosylation and Reduce Protein Stability. Cells 2024, 13, 199. https://doi.org/10.3390/cells13020199
Xie CTY, Pastore SF, Vincent JB, Frankland PW, Hamel PA. Nonsynonymous Mutations in Intellectual Disability and Autism Spectrum Disorder Gene PTCHD1 Disrupt N-Glycosylation and Reduce Protein Stability. Cells. 2024; 13(2):199. https://doi.org/10.3390/cells13020199
Chicago/Turabian StyleXie, Connie T. Y., Stephen F. Pastore, John B. Vincent, Paul W. Frankland, and Paul A. Hamel. 2024. "Nonsynonymous Mutations in Intellectual Disability and Autism Spectrum Disorder Gene PTCHD1 Disrupt N-Glycosylation and Reduce Protein Stability" Cells 13, no. 2: 199. https://doi.org/10.3390/cells13020199