
Medium-Chain Fatty Acids Rescue Motor Function and Neuromuscular Junction Degeneration in a Drosophila Model of Amyotrophic Lateral Sclerosis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fly Stocks and Husbandry
2.2. NA and 4-MOA
2.3. Behavioural Phenotypes
2.4. Immunocytochemistry and Confocal Microscopy
2.5. Electrophysiology
2.6. Statistical Analyses
2.7. Metabolomics
2.7.1. Solvents and Reagents
2.7.2. Metabolite Extraction
2.7.3. Hydrophilic Liquid Interaction Chromatography (HILIC) Analysis of Aqueous Phase
2.7.4. Reversed Phase Analysis of Non-Aqueous Phase
2.7.5. Data Processing and Statistical Analysis
3. Results
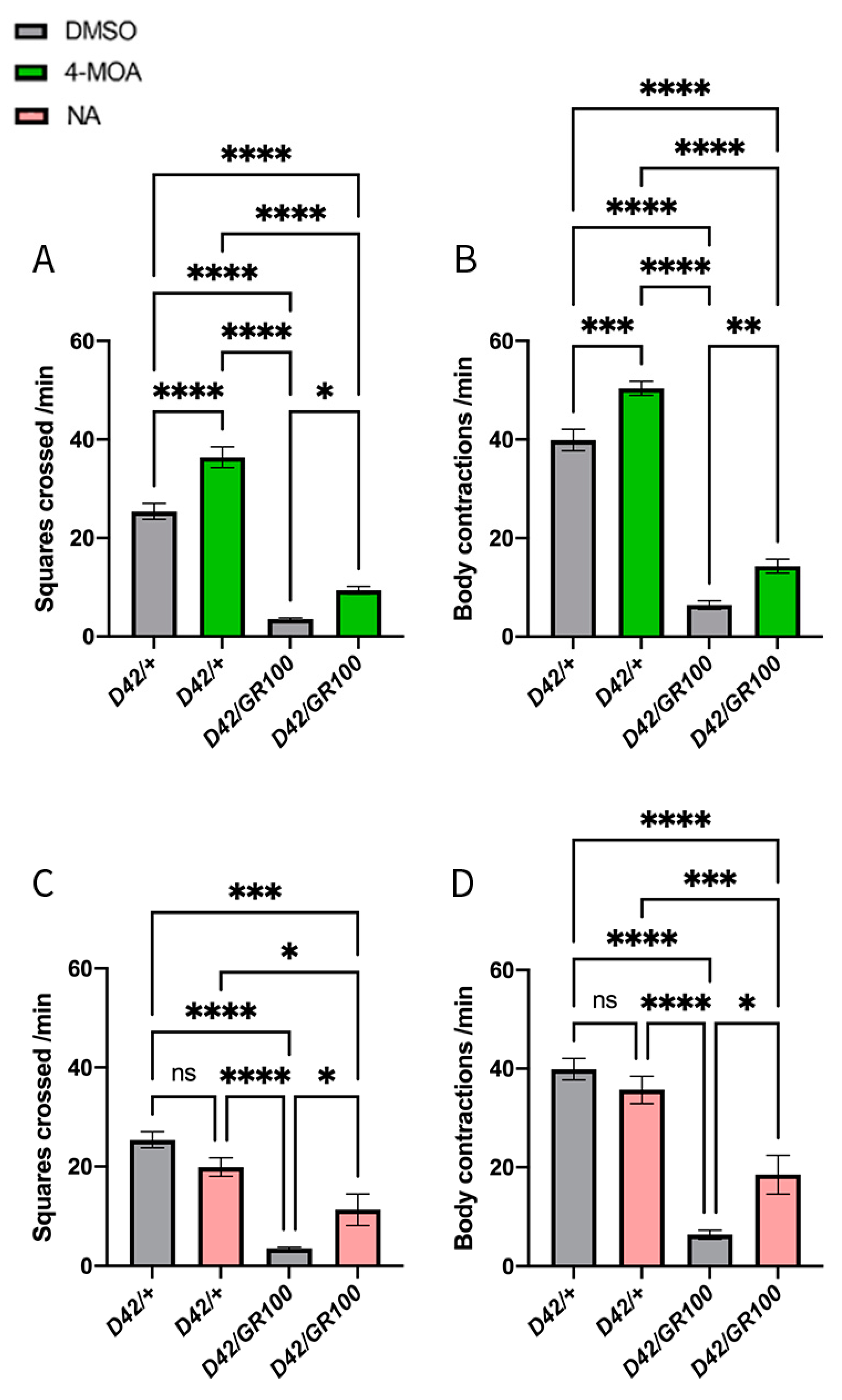
3.1. NA and 4-MOA Partially Reverse the Impaired Motor Phenotype in C9-Model Larvae
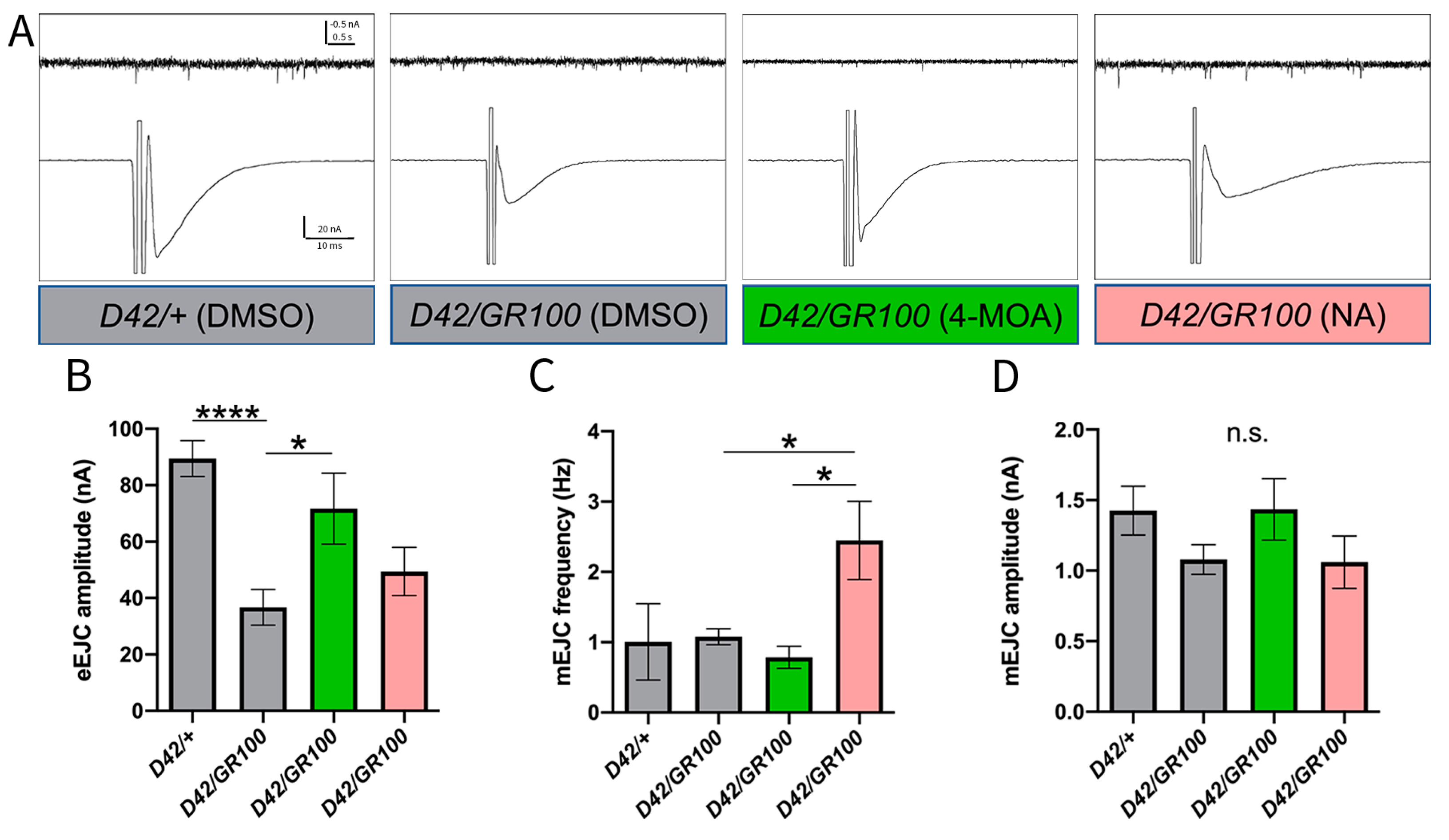
3.2. 4-MOA Rescues Presynaptic Neurotransmitter Release in C9-Model Larvae
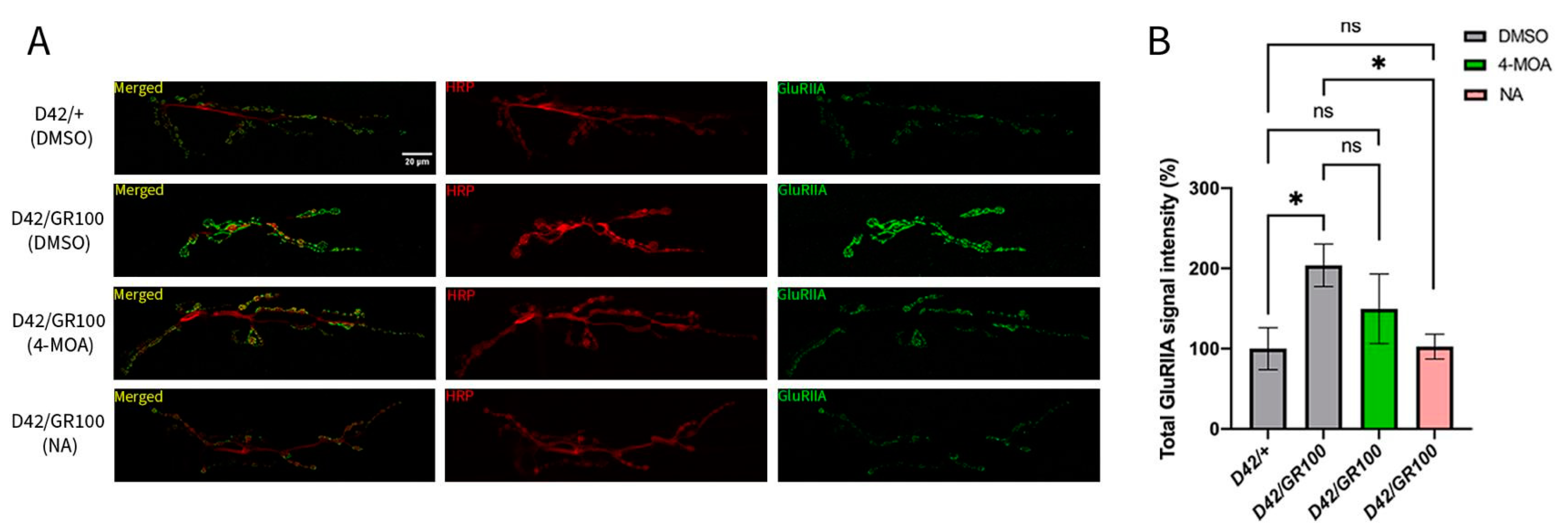
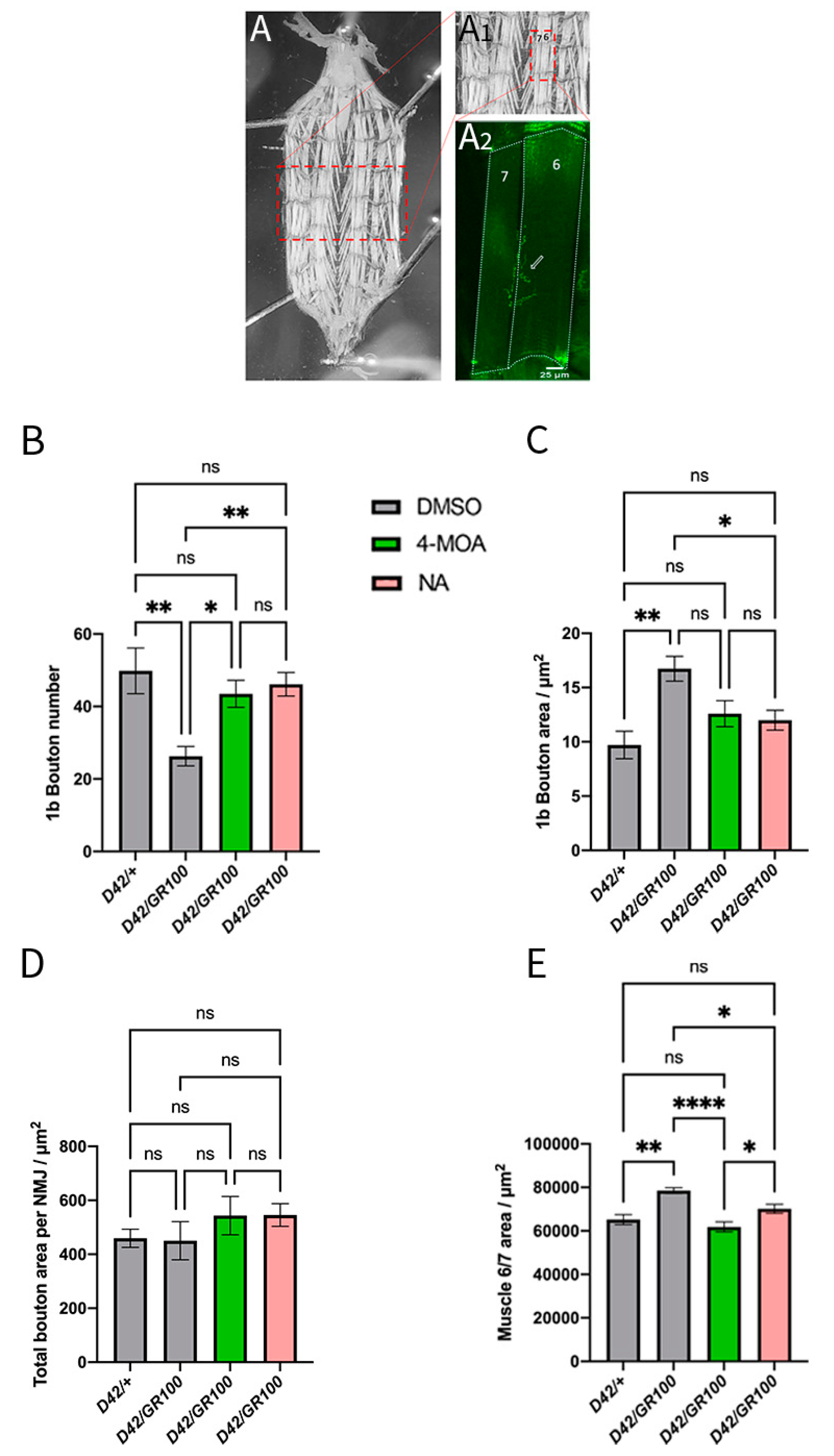
3.3. 4-MOA and NA Modify Neuromuscular Junction Morphology in C9-Model Larvae
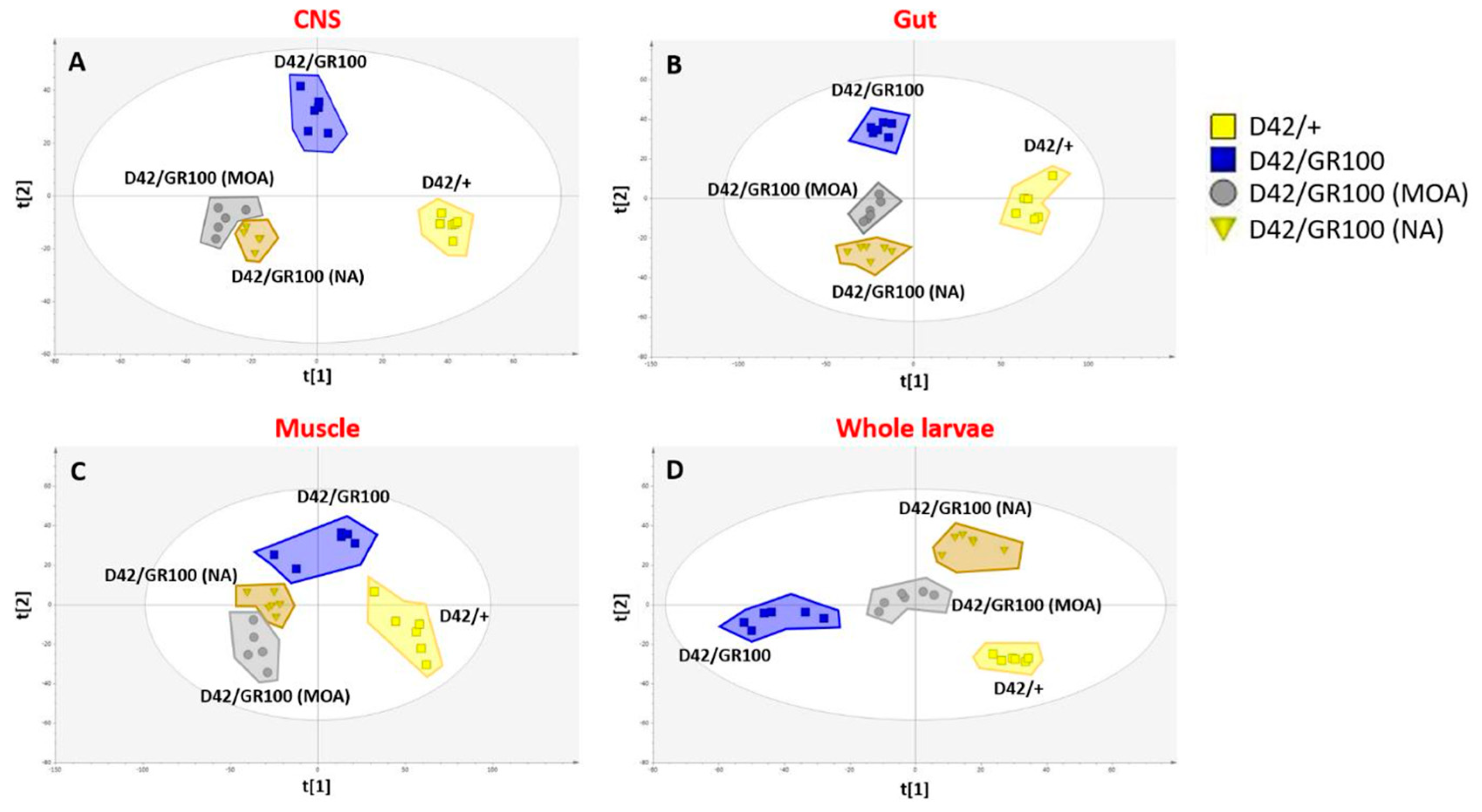
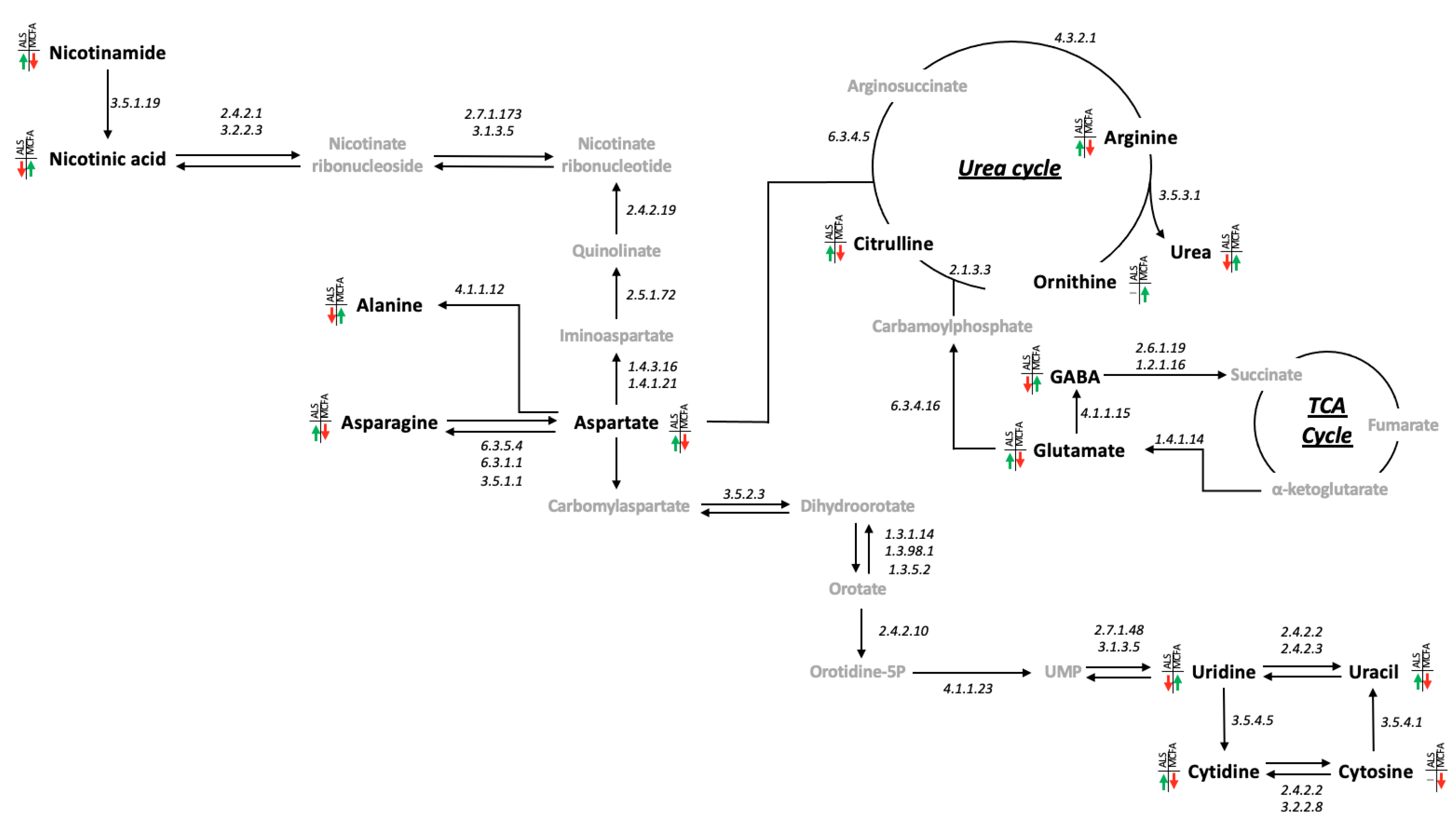
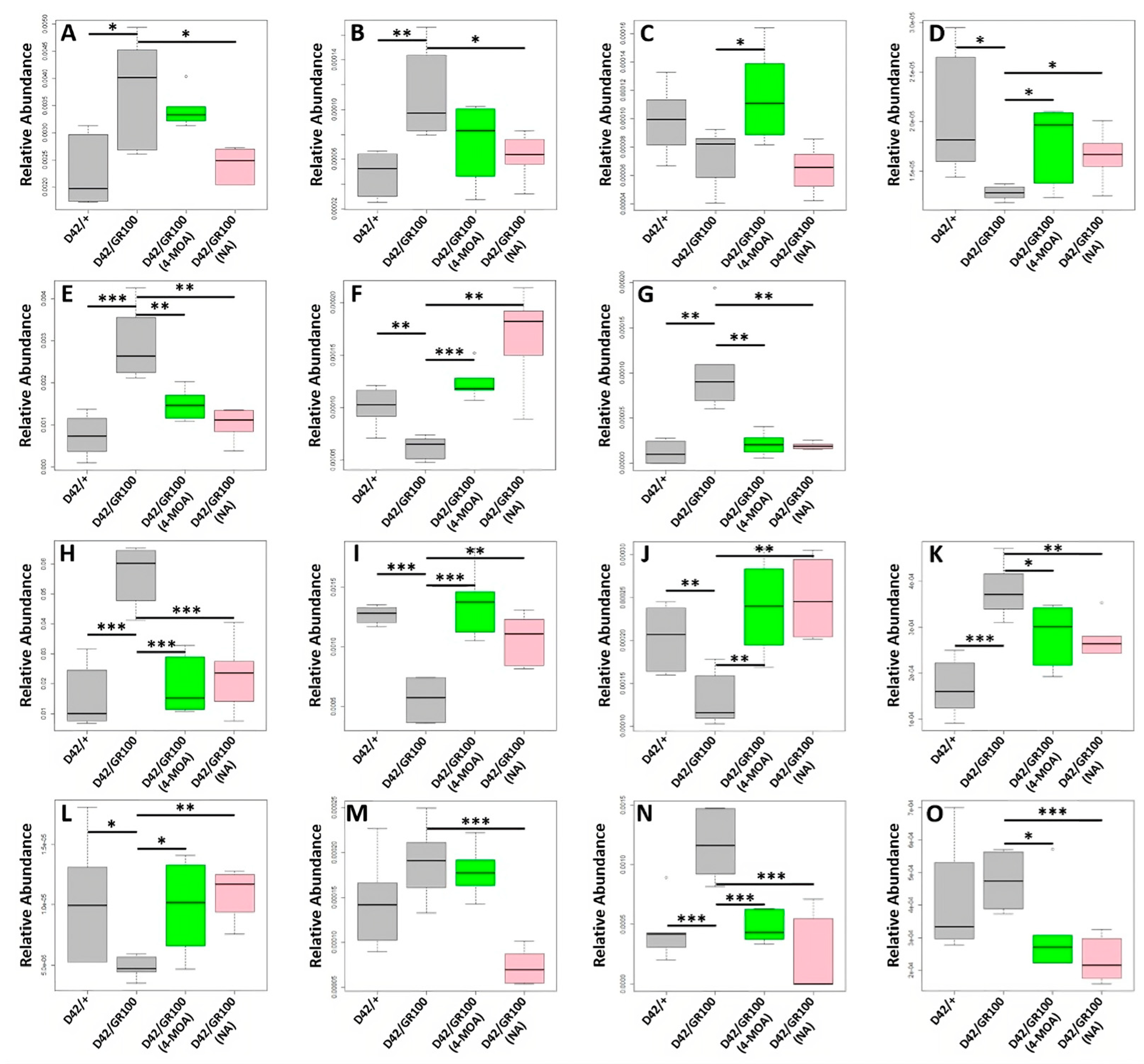
3.4. 4-MOA and NA Alter Metabolism in C9-Model Larvae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barbieri, E. Epidemiology of ALS: Incidence, Prevalence, and Suspected Clusters. TargetALS.org 2022. Available online: https://www.targetals.org/2022/11/22/epidemiology-of-als-incidence-prevalence-and-clusters/ (accessed on 25 July 2023).
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17085. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Marin, B.; Bianchi, E.; Pupillo, E.; Lunetta, C.; Tremolizzo, L.; Logroscino, G.; Chiò, A.; Preux, P.M.; Beghi, E. Non-self-sufficiency as a primary outcome measure in ALS trials. Amyotroph. Lateral Scler. Front. Degener. 2015, 17, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Lunetta, C.; Zuccarino, R.; Vita, G.L.; Sframeli, M.; Lizio, A.; La Foresta, S.; Faraone, C.; Sansone, V.A.; Vita, G.; et al. The 6-min walk test as a new outcome measure in Amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 15580. [Google Scholar] [CrossRef] [PubMed]
- Inam, S.; Vucic, S.; Brodaty, N.E.; Zoing, M.C.; Kiernan, M.C. The 10-metre gait speed as a functional biomarker in amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 2010, 11, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Carreras, I.; Yuruker, S.; Aytan, N.; Hossain, L.; Choi, J.K.; Jenkins, B.G.; Kowall, N.W.; Dedeoglu, A. Moderate exercise delays the motor performance decline in a transgenic model of ALS. Brain Res. 2010, 1313, 192–201. [Google Scholar] [CrossRef]
- Drory, V.E.; Goltsman, E.; Reznik, J.G.; Mosek, A.; Korczyn, A.D. The value of muscle exercise in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 191, 133–137. [Google Scholar] [CrossRef]
- Morioka, H.; Murata, K.; Sugisawa, T.; Shibukawa, M.; Ebina, J.; Sawada, M.; Hanashiro, S.; Nagasawa, J.; Yanagihashi, M.; Hirayama, T.; et al. Effects of Long-term Hybrid Assistive Limb Use on Gait in Patients with Amyotrophic Lateral Sclerosis. Intern. Med. 2022, 61, 1479–1484. [Google Scholar] [CrossRef]
- Fogarty, M.J. Amyotrophic lateral sclerosis as a synaptopathy. Neural Regen. Res. 2019, 14, 189–192. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Arias, N. Synaptopathy Mechanisms in ALS Caused by C9orf72 Repeat Expansion. Front. Cell. Neurosci. 2021, 15, 660693. [Google Scholar] [CrossRef] [PubMed]
- Campanari, M.L.; García-Ayllón, M.S.; Ciura, S.; Sáez-Valero, J.; Kabashi, E. Neuromuscular Junction Impairment in Amyotrophic Lateral Sclerosis: Reassessing the Role of Acetylcholinesterase. Front. Mol. Neurosci. 2016, 9, 160. [Google Scholar] [CrossRef]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Bruneteau, G.; Bauché, S.; Gonzalez de Aguilar, J.L.; Brochier, G.; Mandjee, N.; Tanguy, M.L.; Hussain, G.; Behin, A.; Khiami, F.; Sariali, E.; et al. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2015, 2, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Iyer, S.R.; Shah, S.B.; Lovering, R.M. The Neuromuscular Junction: Roles in Aging and Neuromuscular Disease. Int. J. Mol. Sci. 2021, 22, 8058. [Google Scholar] [CrossRef]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Grammatikopoulou, M.G.; Goulis, D.G.; Gkiouras, K.; Theodoridis, X.; Gkouskou, K.K.; Evangeliou, A.; Dardiotis, E.; Bogdanos, D.P. To Keto or Not to Keto? A Systematic Review of Randomized Controlled Trials Assessing the Effects of Ketogenic Therapy on Alzheimer Disease. Adv. Nutr. 2020, 11, 1583–1602. [Google Scholar] [CrossRef]
- Grammatikopoulou, M.G.; Tousinas, G.; Balodimou, C.; Anastasilakis, D.A.; Gkiouras, K.; Dardiotis, E.; Evangeliou, A.E.; Bogdanos, D.P.; Goulis, D.G. Ketogenic therapy for Parkinson’s disease: A systematic review and synthesis without meta-analysis of animal and human trials. Maturitas 2022, 163, 46–61. [Google Scholar] [CrossRef]
- Bedlack, R.; Barkhaus, P.E.; Barnes, B.; Beauchamp, M.; Bertorini, T.; Bromberg, M.B.; Carter, G.T.; Chaudry, V.; Cudkowicz, M.; Jackson, C.; et al. ALSUntangled #63: Ketogenic diets. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 159–163. [Google Scholar] [CrossRef]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef]
- Chang, P.; Orabi, B.; Deranieh, R.M.; Dham, M.; Hoeller, O.; Shimshoni, J.A.; Yagen, B.; Bialer, M.; Greenberg, M.L.; Walker, M.C.; et al. The antiepileptic drug valproic acid and other medium-chain fatty acids acutely reduce phosphoinositide levels independently of inositol in Dictyostelium. Dis. Models Mech. 2012, 5, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Terbach, N.; Plant, N.; Chen, P.E.; Walker, M.C.; Williams, R.S. Seizure control by ketogenic diet-associated medium chain fatty acids. Neuropharmacology 2013, 69, 105–114. [Google Scholar] [CrossRef]
- Chang, P.; Augustin, K.; Boddum, K.; Williams, S.; Sun, M.; Terschak, J.A.; Hardege, J.D.; Chen, P.E.; Walker, M.C.; Williams, R.S. Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain 2016, 139, 431–443. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Umoh, M.E.; Fournier, C.; Li, Y.; Polak, M.; Shaw, L.; Landers, J.E.; Hu, W.; Gearing, M.; Glass, J.D. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 2016, 87, 1024–1030. [Google Scholar] [CrossRef]
- van der Ende, E.L.; Jackson, J.L.; White, A.; Seelaar, H.; van Blitterswijk, M.; Van Swieten, J.C. Unravelling the clinical spectrum and the role of repeat length in. J. Neurol. Neurosurg. Psychiatry 2021, 92, 502–509. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e717. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.; Han, Y.; Das, A.; Dickman, D. Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Hum. Mol. Genet. 2017, 26, 4153–4167. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef]
- Augustin, H.; McGourty, K.; Steinert, J.R.; Cochemé, H.M.; Adcott, J.; Cabecinha, M.; Vincent, A.; Halff, E.F.; Kittler, J.T.; Boucrot, E.; et al. Myostatin-like proteins regulate synaptic function and neuronal morphology. Development 2017, 144, 2445–2455. [Google Scholar] [CrossRef]
- Robinson, S.W.; Bourgognon, J.M.; Spiers, J.G.; Breda, C.; Campesan, S.; Butcher, A.; Mallucci, G.R.; Dinsdale, D.; Morone, N.; Mistry, R.; et al. Nitric oxide-mediated posttranslational modifications control neurotransmitter release by modulating complexin farnesylation and enhancing its clamping ability. PLoS Biol. 2018, 16, e2003611. [Google Scholar] [CrossRef]
- Stone, A.; Cujic, O.; Rowlett, A.; Aderhold, S.; Savage, E.; Graham, B.; Steinert, J.R. Triose-phosphate isomerase deficiency is associated with a dysregulation of synaptic vesicle recycling in Drosophila melanogaster. Front. Synaptic Neurosci. 2023, 15, 1124061. [Google Scholar] [CrossRef]
- Stewart, B.A.; Atwood, H.L.; Renger, J.J.; Wang, J.; Wu, C.F. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. Physiol. A 1994, 175, 179–191. [Google Scholar] [CrossRef]
- Ebshiana, A.A.; Snowden, S.G.; Thambisetty, M.; Parsons, R.; Hye, A.; Legido-Quigley, C. Metabolomic method: UPLC-q-ToF polar and non-polar metabolites in the healthy rat cerebellum using an in-vial dual extraction. PLoS ONE 2015, 10, e0122883. [Google Scholar] [CrossRef]
- Fernandes, H.J.R.; Kent, J.P.; Bruntraeger, M.; Bassett, A.R.; Koulman, A.; Metzakopian, E.; Snowden, S.G. Mitochondrial and Endoplasmic Reticulum Stress Trigger Triglyceride Accumulation in Models of Parkinson’s Disease Independent of Mutations in MAPT. Metabolites 2023, 13, 112. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef]
- Sanyal, S. Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr. Patterns 2009, 9, 371–380. [Google Scholar] [CrossRef]
- Melom, J.E.; Akbergenova, Y.; Gavornik, J.P.; Littleton, J.T. Spontaneous and evoked release are independently regulated at individual active zones. J. Neurosci. 2013, 33, 17253–17263. [Google Scholar] [CrossRef] [PubMed]
- Peron, S.; Zordan, M.A.; Magnabosco, A.; Reggiani, C.; Megighian, A. From action potential to contraction: Neural control and excitation-contraction coupling in larval muscles of Drosophila. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2009, 154, 173–183. [Google Scholar] [CrossRef] [PubMed]
- DiAntonio, A.; Petersen, S.A.; Heckmann, M.; Goodman, C.S. Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J. Neurosci. 1999, 19, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.A.; DiAntonio, A. Synaptic development: Insights from Drosophila. Curr. Opin. Neurobiol. 2006, 17, 35–42. [Google Scholar] [CrossRef]
- Ruiz-Cañada, C.; Budnik, V. Introduction on the use of the Drosophila embryonic/larval neuromuscular junction as a model system to study synapse development and function, and a brief summary of pathfinding and target recognition. Int. Rev. Neurobiol. 2006, 75, 1–31. [Google Scholar] [CrossRef]
- Sigrist, S.J.; Thiel, P.R.; Reiff, D.F.; Schuster, C.M. The postsynaptic glutamate receptor subunit DGluR-IIA mediates long-term plasticity in Drosophila. J. Neurosci. 2002, 22, 7362–7372. [Google Scholar] [CrossRef]
- Chang, P.; Zuckermann, A.M.; Williams, S.; Close, A.J.; Cano-Jaimez, M.; McEvoy, J.P.; Spencer, J.; Walker, M.C.; Williams, R.S. Seizure control by derivatives of medium chain fatty acids associated with the ketogenic diet show novel branching-point structure for enhanced potency. J. Pharmacol. Exp. Ther. 2015, 352, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.W.; Jung, J.Y.; Lee, I.K.; Kang, S.Y.; Yun, B.S. Nonanoic Acid, an Antifungal Compound from Hibiscus syriacus Ggoma. Mycobiology 2012, 40, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, N.; Xiong, J.; Wei, H.; Jiang, S.; Peng, J. Caprylic acid and nonanoic acid upregulate endogenous host defense peptides to enhance intestinal epithelial immunological barrier function via histone deacetylase inhibition. Int. Immunopharmacol. 2018, 65, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.; Heldreth, B.; Bergfeld, W.F.; Belsito, D.V.; Klaassen, C.D.; Hill, R.; Liebler, D.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; et al. Final report of the Cosmetic Ingredient Review Expert Panel on the safety assessment of pelargonic acid (nonanoic acid) and nonanoate esters. Int. J. Toxicol. 2011, 30, 228S–269S. [Google Scholar] [CrossRef]
- Kamata, Y.; Shiraga, H.; Tai, A.; Kawamoto, Y.; Gohda, E. Induction of neurite outgrowth in PC12 cells by the medium-chain fatty acid octanoic acid. Neuroscience 2007, 146, 1073–1081. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Fomin, V.; Richard, P.; Hoque, M.; Li, C.; Gu, Z.; Fissore-O’Leary, M.; Tian, B.; Prives, C.; Manley, J.L. The C9ORF72 Gene, Implicated in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia, Encodes a Protein That Functions in Control of Endothelin and Glutamate Signaling. Mol. Cell. Biol. 2018, 38, e00155-18. [Google Scholar] [CrossRef]
- Pflanz, N.C.; Daszkowski, A.W.; James, K.A.; Mihic, S.J. Ketone body modulation of ligand-gated ion channels. Neuropharmacology 2019, 148, 21–30. [Google Scholar] [CrossRef]
- Stanback, A.E. The Effects of a Ketone Body on Synaptic Transmission. Theses Diss.-Biol. 2019, 57. [Google Scholar] [CrossRef]
- Burton, B.K. Urea cycle disorders. Clin. Liver Dis. 2000, 4, 815–830. [Google Scholar] [CrossRef]
- Felipo, V.; Butterworth, R.F. Neurobiology of ammonia. Prog. Neurobiol. 2002, 67, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Kerbert, A.J.C.; Habtesion, A.; Hall, A.; Abramov, A.Y.; Jalan, R. Hyperammonaemia induces mitochondrial dysfunction and neuronal cell death. JHEP Rep. 2022, 4, 100510. [Google Scholar] [CrossRef] [PubMed]
- Parekh, B. A(a)LS: Ammonia-induced amyotrophic lateral sclerosis. F1000Research 2015, 4, 119. [Google Scholar] [CrossRef] [PubMed]
- Patten, B.M.; Kurlander, H.M.; Evans, B. Free amino acid concentrations in spinal tissue from patients dying of motor neuron disease. Acta Neurol. Scand. 1982, 66, 594–599. [Google Scholar] [CrossRef]
- Bame, M.; Grier, R.E.; Needleman, R.; Brusilow, W.S. Amino acids as biomarkers in the SOD1(G93A) mouse model of ALS. Biochim. Biophys. Acta 2014, 1842, 79–87. [Google Scholar] [CrossRef]
- Peña-Quintana, L.; Llarena, M.; Reyes-Suárez, D.; Aldámiz-Echevarria, L. Profile of sodium phenylbutyrate granules for the treatment of urea-cycle disorders: Patient perspectives. Patient Prefer. Adherence 2017, 11, 1489–1496. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Brown, R.H.; et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef]
- Kaur, B.; Bhat, A.; Chakraborty, R.; Adlakha, K.; Sengupta, S.; Roy, S.; Chakraborty, K. Proteomic profile of 4-PBA treated human neuronal cells during ER stress. Mol. Omics 2018, 14, 53–63. [Google Scholar] [CrossRef]
- Godfrey, E.W.; Schwarte, R.C. The role of nitric oxide signaling in the formation of the neuromuscular junction. J. Neurocytol. 2003, 32, 591–602. [Google Scholar] [CrossRef]
- Knott, A.B.; Bossy-Wetzel, E. Nitric oxide in health and disease of the nervous system. Antioxid. Redox Signal. 2009, 11, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.A.; Steinert, J.R. Nitric Oxide-Mediated Posttranslational Modifications: Impacts at the Synapse. Oxid. Med. Cell. Longev. 2016, 2016, 5681036. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Brown, G.C. Nitric oxide causes glutamate release from brain synaptosomes. J. Neurochem. 1998, 70, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Brown, G.C. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J. Neurosci. 2001, 21, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.H.; Tsui, K.C.; Chau, S.C.; Chong, P.S.; Lui, S.W.Y.; Aquili, L.; Wong, K.H.; Lim, L.W. Functional Roles of Neuronal Nitric Oxide Synthase in Neurodegenerative Diseases and Mood Disorders. Curr. Alzheimer Res. 2021, 18, 831–840. [Google Scholar] [CrossRef]
- Drechsel, D.A.; Estévez, A.G.; Barbeito, L.; Beckman, J.S. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotox. Res. 2012, 22, 251–264. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Shinobu, L.A.; Schulz, J.B.; Matthews, R.T.; Thomas, C.E.; Kowall, N.W.; Gurney, M.E.; Beal, M.F. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann. Neurol. 1997, 42, 326–334. [Google Scholar] [CrossRef]
- De Paola, M.; Sestito, S.E.; Mariani, A.; Memo, C.; Fanelli, R.; Freschi, M.; Bendotti, C.; Calabrese, V.; Peri, F. Synthetic and natural small molecule TLR4 antagonists inhibit motoneuron death in cultures from ALS mouse model. Pharmacol. Res. 2016, 103, 180–187. [Google Scholar] [CrossRef]
- Martinelli, G.P.; Friedrich, V.L.; Holstein, G.R. L-citrulline immunostaining identifies nitric oxide production sites within neurons. Neuroscience 2002, 114, 111–122. [Google Scholar] [CrossRef]
- Gyawali, A.; Gautam, S.; Hyeon, S.J.; Ryu, H.; Kang, Y.S. L-Citrulline Level and Transporter Activity Are Altered in Experimental Models of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2021, 58, 647–657. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dunn, E.; Steinert, J.R.; Stone, A.; Sahota, V.; Williams, R.S.B.; Snowden, S.; Augustin, H. Medium-Chain Fatty Acids Rescue Motor Function and Neuromuscular Junction Degeneration in a Drosophila Model of Amyotrophic Lateral Sclerosis. Cells 2023, 12, 2163. https://doi.org/10.3390/cells12172163
Dunn E, Steinert JR, Stone A, Sahota V, Williams RSB, Snowden S, Augustin H. Medium-Chain Fatty Acids Rescue Motor Function and Neuromuscular Junction Degeneration in a Drosophila Model of Amyotrophic Lateral Sclerosis. Cells. 2023; 12(17):2163. https://doi.org/10.3390/cells12172163
Chicago/Turabian StyleDunn, Ella, Joern R. Steinert, Aelfwin Stone, Virender Sahota, Robin S. B. Williams, Stuart Snowden, and Hrvoje Augustin. 2023. "Medium-Chain Fatty Acids Rescue Motor Function and Neuromuscular Junction Degeneration in a Drosophila Model of Amyotrophic Lateral Sclerosis" Cells 12, no. 17: 2163. https://doi.org/10.3390/cells12172163