

The Microenvironment of the Pathogenesis of Cardiac Hypertrophy
by
 , , , , and
, , , , and
Farhad Bazgir
1,† ,
,
Julia Nau
1,†,
Saeideh Nakhaei-Rad
2,
Ehsan Amin
3 ,
,
Matthew J. Wolf
4,
Jeffry J. Saucerman
5,
Kristina Lorenz
6 and
Mohammad Reza Ahmadian
1,* 1
Institute of Biochemistry and Molecular Biology II, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany
2
Stem Cell Biology, and Regenerative Medicine Research Group, Institute of Biotechnology, Ferdowsi University of Mashhad, Mashhad 91779-48974, Iran
3
Institute of Neural and Sensory Physiology, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany
4
Department of Medicine and Robert M. Berne Cardiovascular Research Center, University of Virginia, Charlottesville, VA 22908, USA
5
Department of Biomedical Engineering, University of Virginia, Charlottesville, VA 22908, USA
6
Institute of Pharmacology and Toxicology, University of Würzburg, Leibniz Institute for Analytical Sciences, 97078 Würzburg, Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2023, 12(13), 1780; https://doi.org/10.3390/cells12131780
Submission received: 7 May 2023
/
Revised: 22 June 2023
/
Accepted: 29 June 2023
/
Published: 4 July 2023
(This article belongs to the Special Issue Cardiomyopathy: Pathogenesis, Diagnosis, and Treatment)
Abstract
:Pathological cardiac hypertrophy is a key risk factor for the development of heart failure and predisposes individuals to cardiac arrhythmia and sudden death. While physiological cardiac hypertrophy is adaptive, hypertrophy resulting from conditions comprising hypertension, aortic stenosis, or genetic mutations, such as hypertrophic cardiomyopathy, is maladaptive. Here, we highlight the essential role and reciprocal interactions involving both cardiomyocytes and non-myocardial cells in response to pathological conditions. Prolonged cardiovascular stress causes cardiomyocytes and non-myocardial cells to enter an activated state releasing numerous pro-hypertrophic, pro-fibrotic, and pro-inflammatory mediators such as vasoactive hormones, growth factors, and cytokines, i.e., commencing signaling events that collectively cause cardiac hypertrophy. Fibrotic remodeling is mediated by cardiac fibroblasts as the central players, but also endothelial cells and resident and infiltrating immune cells enhance these processes. Many of these hypertrophic mediators are now being integrated into computational models that provide system-level insights and will help to translate our knowledge into new pharmacological targets. This perspective article summarizes the last decades’ advances in cardiac hypertrophy research and discusses the herein-involved complex myocardial microenvironment and signaling components.
1. General Introduction
Myocardial remodeling associated with cardiac hypertrophy is one of the critical causes in the development of heart failure. The pathogenesis of heart dysfunction is one of the primary causes of morbidity and mortality in elderly people [1].
Cardiac hypertrophy is the most frequently compensatory or adaptive process to numerous physiological or pathological conditions (Table 1) [2]. Hypertrophic enlargement is characterized by an increase in the cell size of cardiomyocytes. The heart can dynamically change its muscle mass to cope with the stimuli of development, physiological conditions of exercise and pregnancy, or pathological disease stimuli (Table 1) [3]. Increased workload as a consequence of volume or pressure overload due to pathological or physiological stimuli increases tension on the cardiac walls of the heart chambers [4,5]. This ultimately triggers stress signals released by different cell types of the microenvironment to compensate for the wall tension increase, resulting in a hypertrophic growth response [4,5]. Individual cardiomyocytes can increase in length and/or width in response to hypertrophic stimuli depending on the intracellular signaling cascades involved [6,7,8].
Hypertrophy, or the enlargement of individual muscle fibers, is the primary mechanism by which skeletal muscle mass increases during postnatal development. A similar process may be induced in adult skeletal muscle in response to contractile activity, satellite cell proliferation and fusion, which increases the number of myonuclei. This event may also play a role in muscle growth during early but not late stages of postnatal development and in some forms of muscle hypertrophy [9]. Likewise, the number of endothelium and mesenchymal cells can also increase from birth through early adulthood, but on the other hand the entire complement of cardiomyocytes is created during pregnancy and remains nearly constant throughout the human lifespan. Early infancy has the highest levels of cardiomyocyte exchange, which steadily declines during life to 1% per year in maturity in processes that increase very modestly in the vicinity of cardiac injury [10,11]. Important to note that cell duplication is not always a result of cardiomyocyte cell cycle activity. In contrast, multinucleation and polyploidization occur during various phases of development including heart development as a result of premature cell cycle exit [12].
Physiological and pathological cardiac hypertrophy are associated with distinct molecular characteristics (Table 1) involving alterations in the expression of fetal genes, and contractile and calcium-handling proteins [13]. A major molecular characteristic of pathological hypertrophy is the re-expression of fetal genes. Pathological settings such as hypertension cause the induction of the stress program that involves increased expression of atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), and alpha-skeletal actin (α-sk actin) [14]. In contrast, an important characteristic of physiological hypertrophy is the absence of molecular stress programs [15]. In addition, expression of cardiac contractile proteins, such as alpha- and beta-myosin heavy chain and calcium-handling proteins, e.g., sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) remain unchanged during physiological cardiac hypertrophy, whereas pathological cardiac hypertrophy is closely associated with alterations in the above-named genes and proteins [14].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Characteristics of physiological and pathological cardiac hypertrophy 1.
| Characteristic | Physiological Cardiac Hypertrophic | Pathological Cardiac Hypertrophic |
|---|---|---|
| Stimuli | exercise, pregnancy | i.a. pressure or volume overload |
| Cardiomyocyte size | increased | increased |
| Concentric or eccentric | eccentric > concentric | concentric or eccentric |
| Adaptivity | yes | initially yes/advanced maladaptive |
| Contractility | preserved or increased | preserved or decreased |
| Cardiac metabolism | ||
| Fatty acid oxidation | increased | decreased |
| Glycolysis | increased | increased |
| Structural and functional | ||
| Replacement | no | yes |
| Interstitial fibrosis | no | yes |
| Cardiomyocyte apoptosis | no | yes |
| Capillary network | sufficient | insufficient |
| Molecular characteristics | ||
| Fetal gene expression | unmodified | upregulated |
| Contractile linked genes Inflammation | normal or increased unmodified | downregulated increased |
| Cardiac function | normal or increased | depressed |
| Reversible | yes | no |
| Heart failure | unlikely | prone |
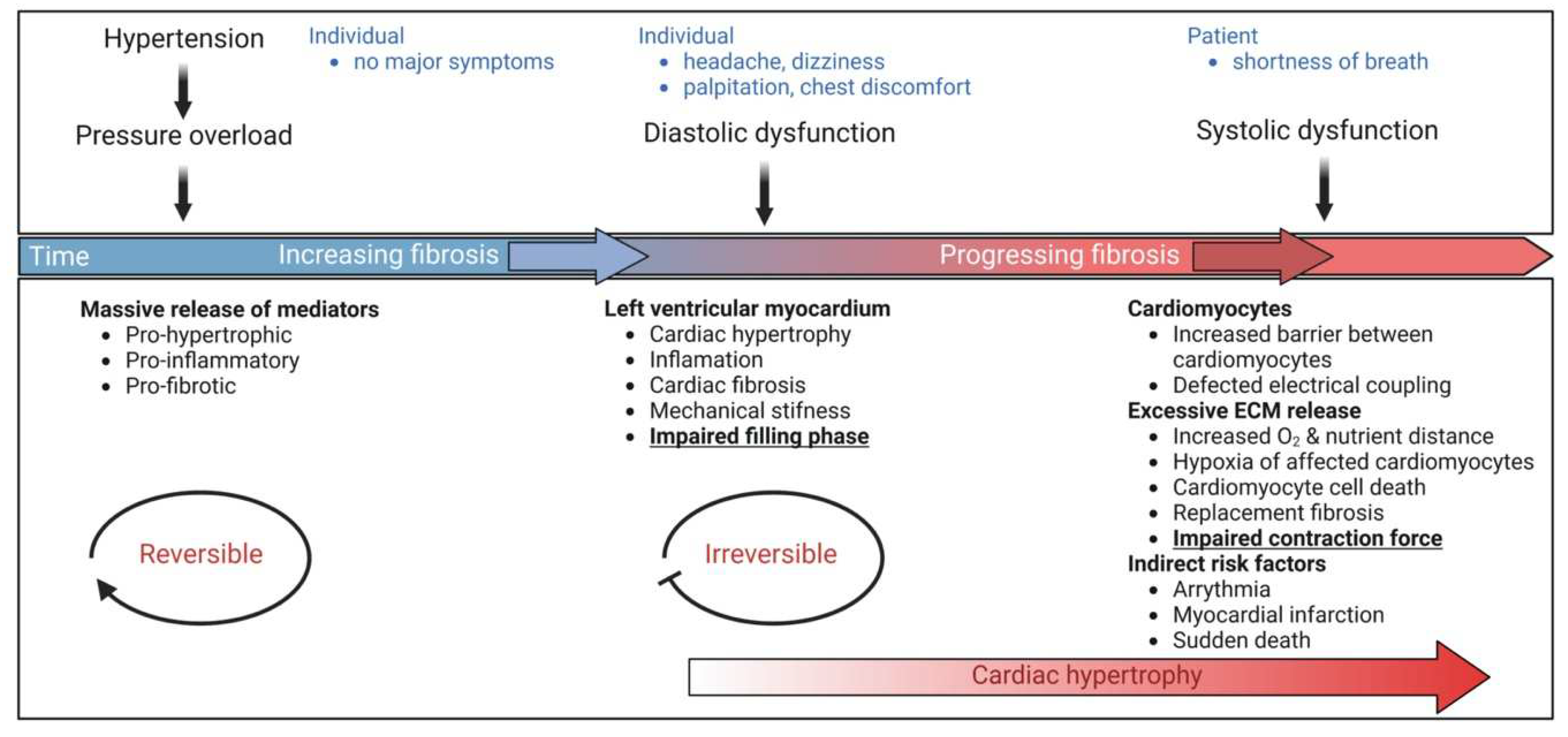
While the events associated with physiological hypertrophy are generally reversible, those associated with pathological cardiac hypertrophy are commonly irreversible and impose a high risk of heart failure (Table 1). A common disease stimulus, such as long-standing hypertension usually causes pressure overload and increases systolic wall stress [16]. In this case, individual cardiomyocytes typically grow in width more than in length, leading to the thickening of the cardiac walls, a condition referred to as concentric hypertrophy [3,17]. Hypertrophic changes have been rationalized employing Laplace’s law, which says wall stress (or tension) is an inverse function of wall thickness (tension = [pressure × radius]/2 × wall thickness). Thus, compensated growth of the cardiac muscle is a physiological response to decrease wall tension and thereby maintain cardiac pump function [4]. Prolonged pathological stress, however, causes maladaptive changes at the cellular and molecular level resulting in pathological cardiac hypertrophy. Untreated pathological cardiac hypertrophy predisposes individuals to heart failure, arrhythmia, and sudden death [6,7].
Triggers of pathological cardiac hypertrophy include extrinsic drives such as pressure overload due to long-standing hypertension or valvular stenosis, as well as volume overload due to mitral regurgitation or aortic insufficiency (Table 1), loss of contractile mass (myocardial infarction), or intrinsic causes such as hereditary defects [2,18]. Although a notable feature of physiological and pathological cardiac hypertrophy is the increase in heart size, pathological cardiac hypertrophy involves the loss of myocytes and fibrotic replacement, leading to cardiac dysfunction, heart failure, and/or sudden death [19,20]. Despite views that the length of stress has a significant impact on the distinction between pathological and physiological cardiac hypertrophy, the nature of stress and the intracellular signaling cascades involved are thought to be more important in the development of maladaptive cardiac dysfunction than the chronic duration of exposure [21].
The role of the cardiac microenvironment and intercell communication in the cardiac niche is becoming increasinglyevident for future studies and therapeutic interventions, as also recently highlighted by Tazhor et al. [22], challenging the traditional view of the heart as a cardiomyocyte-centric and targeted regenerative and therapeutic target in cardiovascular disease. Future therapeutic and mechanistic investigations should take into account the fact that immune cells, fibroblasts, and endothelial cells collectively outnumber cardiomyocytes by a significant margin as the resident cells in the heart, making this viewpoint increasingly important as a crucial element in the study of the intercellular communications and the treatment of heart disease. Therefore, the aim of this review article was to focus on these processes related to the onset, progression, and pathogenesis of hypertrophic cardiomyopathy and to complement previous work by incorporating molecular axes and details of intercellular communication in the cardiac microenvironment that have not yet been illuminated.
2. An Interplay of Different Cells in Hypertrophic Remodeling
The heart consists of various cell types, including myocytes, endothelial cells, fibroblasts, vascular smooth muscle cells, sympathetic neurons, and immune cells, which collectively account for a synchronized cardiac function [23,24]. However, it has been shown that owing to their enormous size, cardiomyocytes in particular account for the majority of heart mass, increase in size and reprogram transcription in the process of cardiac hypertrophy [2,25]. Communications between cardiomyocytes and non-myocytes lead to the secretion of bioactive mediators, which operate in an autocrine and paracrine manner (Table 2). This is followed by microenvironmental stimulation of different cell types and the activation of various signaling pathways within the cells (Figure 1 and Figure 2) [26,27]. Altogether these complex processes result in cardiomyocyte hypertrophy, fibroblast hyperplasia, interstitial tissue composition changes, and remodeling of the ventricular chambers [28].
2.1. Fibroblast Remodeling
Pressure overload triggers resident cardiac fibroblasts originating from the epicardium and endocardium to undergo rapid expansion and activation, rather than previously reported hematopoietic precursor-derived fibroblasts or endothelial-to-mesenchymal transition (EndMT) as a contributing source (Figure 1 and Figure 3) [29,30]. Despite this, the exact origins of cardiac fibroblasts as well as the delineation of their characteristics and plasticity remain a field of current investigation and controversy [31]. Like cardiomyocytes, fibroblasts respond to external stress stimuli, but in a slightly different manner. Mechanical stress promotes fibroblast differentiation to a myofibroblast-like phenotype (Figure 1 and Figure 3) [32,33], which has been shown to develop from tissue-derived fibroblasts rather than endothelial or smooth muscle cells [30]. Myofibroblasts overproduce and release extracellular matrix (ECM) components and pro-hypertrophic mediators, including Transforming growth factor-beta (TGF-ß) (Table 2), and are engaged in a wide range of pathological conditions, particularly fibrosis and tissue remodeling [34]. Enhanced release of ECM by myofibroblasts contributing to mechanical stiffness accompanied by increasing fibrosis evolves into severe consequences causing cardiac diastolic dysfunction [35]. Moreover, progressing fibrosis can affect systolic function by building a barrier between the resident cardiomyocytes, thereby provoking defective electrical coupling within the myocardium [36]. Additionally, an increased level of ECM, such as collagen, can disrupt the oxygen diffusion capacity leading to hypoxia in the affected myocytes a process that may further enhance pathological remodeling [37]. In conclusion, cardiac fibroblasts react to pressure overload-induced injury with activation, accumulation, and excessive ECM deposition (Figure 1 and Figure 3). The resulting conditions including mechanical stiffness, myocyte uncoupling, and ischemia comprise key contributors to heart failure [29]. These lines of evidence also emphasize the identification of mechanical stress in cardiac hypertrophy as an independent risk factor for arrhythmias, myocardial infarction, and sudden death [19].
2.2. Endothelial Cell Activation
In response to pressure overload, cardiac endothelial cells, similar to cardiac fibroblasts, are capable of changing their phenotype (Figure 1). It has been reported that endothelial cells can undergo an EndMT, differentiate into myofibroblast-like cells, and thereby contribute to cardiac fibrosis [38]. Others outlined that EndMT recruits circulating hematopoietic progenitors to the heart thereby generating significant numbers of cardiac fibroblasts (reviewed in [39]) but also their origin from tissue-resident fibroblasts is being discussed [29,30]. Altogether, left ventricular myocardial tissue of end-stage cardiac failure patients revealed dramatically increased expression levels of EndMT-related genes [40], indicating the need for further investigation to clarify the exact contribution of EndMT.
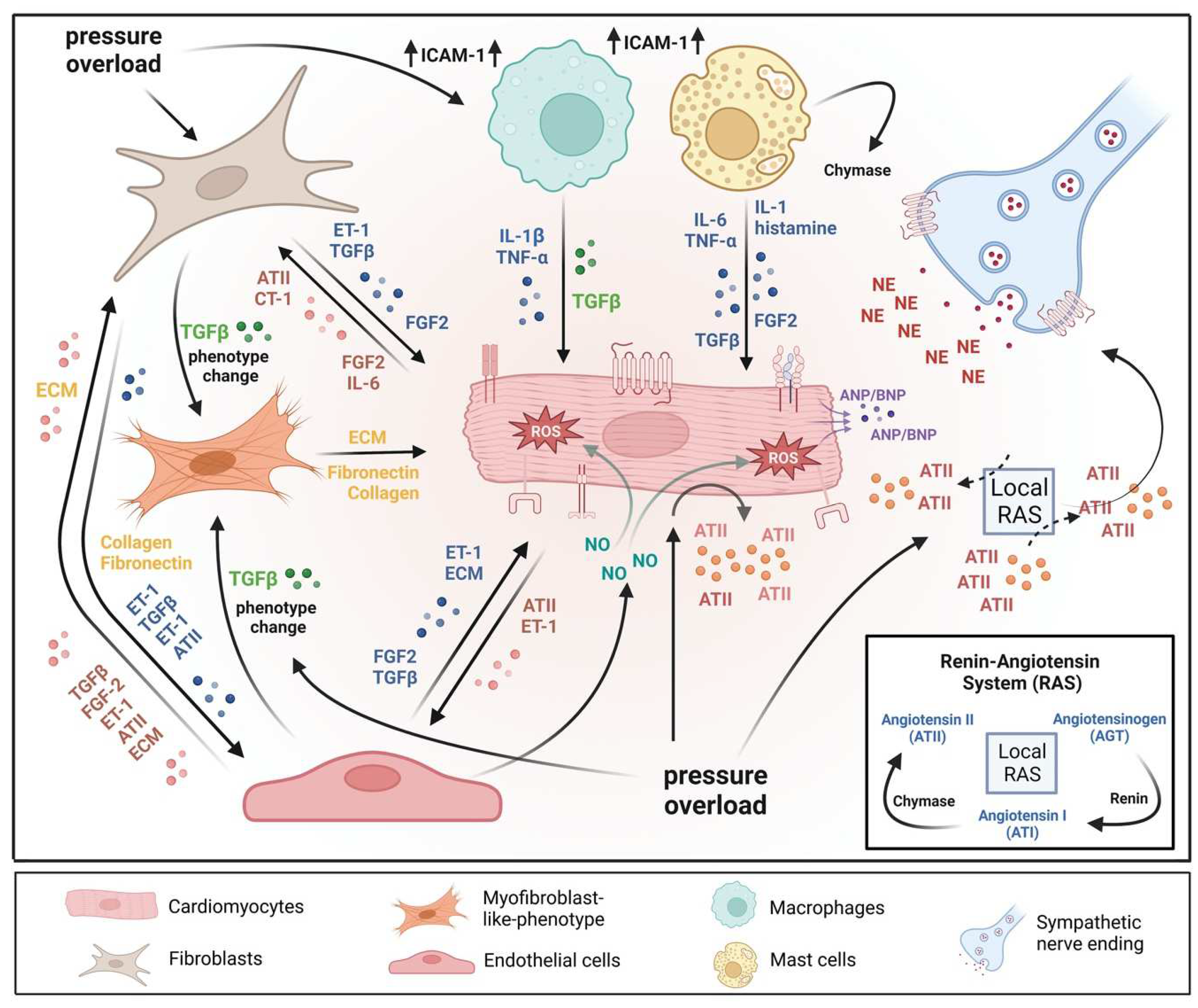
![Cells 12 01780 g001]()
Figure 1.
A microenvironmental model of pressure overload-induced cardiac hypertrophy. The model also illustrates multiple cell types’ substantial roles and reciprocal interactions in the myocardium. In response to pressure overload cardiomyocyte and non-myocardial cells are transformed into an “activated state”, releasing numerous pro-hypertrophic, pro-fibrotic, and pro-inflammatory mediators. In addition, vasoactive hormones, various growth factors, cytokines, and the local renin-angiotensin system (RAS) act in an autocrine and/or paracrine mode. Collectively, the above-mentioned mechanisms orchestrate effects that contribute to pathological remodeling processes leading to cardiac hypertrophy, fibrosis, and inflammation. AT II: angiotensin II; CT-1: cardiotrophin-1; ECM: extracellular matrix; ET-1: endothelin-1; FGF-2: fibroblast growth factor-2; ICAM-1: intercellular adhesion molecule-1; IL-1: interleukin-1; IL-6: interleukin-6; NE: norepinephrine; TGF-ß: transforming growth factor-ß; TNFα: tumor necrosis factor-α.
Figure 1.
A microenvironmental model of pressure overload-induced cardiac hypertrophy. The model also illustrates multiple cell types’ substantial roles and reciprocal interactions in the myocardium. In response to pressure overload cardiomyocyte and non-myocardial cells are transformed into an “activated state”, releasing numerous pro-hypertrophic, pro-fibrotic, and pro-inflammatory mediators. In addition, vasoactive hormones, various growth factors, cytokines, and the local renin-angiotensin system (RAS) act in an autocrine and/or paracrine mode. Collectively, the above-mentioned mechanisms orchestrate effects that contribute to pathological remodeling processes leading to cardiac hypertrophy, fibrosis, and inflammation. AT II: angiotensin II; CT-1: cardiotrophin-1; ECM: extracellular matrix; ET-1: endothelin-1; FGF-2: fibroblast growth factor-2; ICAM-1: intercellular adhesion molecule-1; IL-1: interleukin-1; IL-6: interleukin-6; NE: norepinephrine; TGF-ß: transforming growth factor-ß; TNFα: tumor necrosis factor-α.

Major factors secreted by cardiac endothelial cells (Table 2) comprise nitric oxide (NO), endothelin 1 (ET-1), prostaglandin I2 (PI2), and angiotensin II (AT-II), which directly influence cardiac metabolism, growth, contractile performance, and rhythmicity of the adult heart [41]. In response to various stimuli, activated endothelial cells express adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM), which attract and further promote the infiltration of immune cells into the myocardium (Figure 1). One major mediator produced and secreted by endothelial cells is NO (Table 2; Figure 1). Among the numerous functional influences of NO are cardiac-related functions, including key regulators of vasodilation, reduction of permeability and thrombogenesis, and inhibition of inflammation [42]. Another active mediator secreted by endothelial cells is CNP (Table 2). Together, NO and C-type natriuretic peptide (CNP) contribute to the suppression of cardiac hypertrophy by up-regulating cyclic GMP (cGMP)-dependent protein kinase 1 (PKGI) signaling [43] by inhibiting calcineurin (Figure 2).
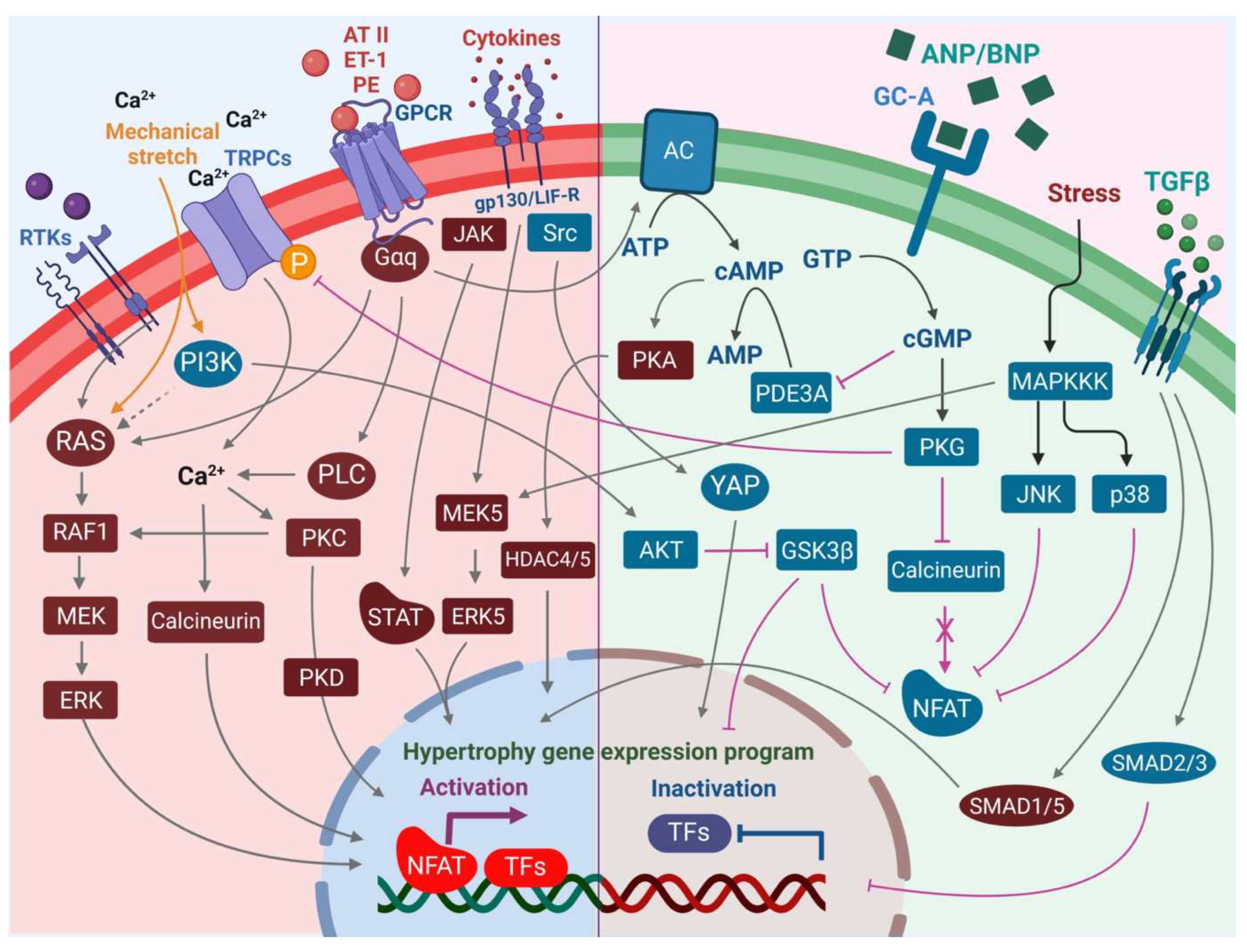
![Cells 12 01780 g002]()
Figure 2.
An overview of the pro-hypertrophic (left panel) and anti-hypertrophic (right panel) signaling pathways regulating the hypertrophic response in the cardiomyocyte. Increased intracellular Ca2+ levels mediated by TRPCs and Ca2+ import promote pro-hypertrophic transcriptional signaling events via calcineurin-NFAT and activation of PKC. PLC may contribute to these axes in the activation of alpha-adrenergic receptor signaling. Although canonical MAPK signaling via RTKs including FGFR-1 promotes pro-hypertrophic signaling, the PI3K-AKT axis plays an opposing role in hypertrophic signaling via inhibition of GSK3β and activation of YAP transcriptional activity. Increased secretion of cytokines promotes transcriptional activation of the pro-hypertrophic gene program in the nucleus not only via JAK-STAT but also the MEK5-ERK5 axis. On the other hand, increased pressure overload in cardiac tissue promotes secretion of ANP and BNP by cardiomyocytes, leading to vasodilation and an anti-hypertrophic response in cells via an increase in intracellular cGMP levels, which leads to activation of PKG, in turn mediating reduced hypertrophic growth. Activation of JNK and p38 stress signaling events in the cardiomyocytes, although leading to cardiomyopathy and heart failure, results in inhibition of NFAT through phosphorylation events that prevent its nuclear localization and pro-hypertrophic transcriptional regulation activities, thereby blocking the calcineurin axis. Increased secretion of TGF-β during increased pressure stress can lead to mixed responses, with canonical TGF-β-SMAD2/SMAD3 signaling leading to anti-hypertrophic responses, whereas activation of non-canonical SMAD1/SMAD5 leads to pro-hypertrophic responses.
Figure 2.
An overview of the pro-hypertrophic (left panel) and anti-hypertrophic (right panel) signaling pathways regulating the hypertrophic response in the cardiomyocyte. Increased intracellular Ca2+ levels mediated by TRPCs and Ca2+ import promote pro-hypertrophic transcriptional signaling events via calcineurin-NFAT and activation of PKC. PLC may contribute to these axes in the activation of alpha-adrenergic receptor signaling. Although canonical MAPK signaling via RTKs including FGFR-1 promotes pro-hypertrophic signaling, the PI3K-AKT axis plays an opposing role in hypertrophic signaling via inhibition of GSK3β and activation of YAP transcriptional activity. Increased secretion of cytokines promotes transcriptional activation of the pro-hypertrophic gene program in the nucleus not only via JAK-STAT but also the MEK5-ERK5 axis. On the other hand, increased pressure overload in cardiac tissue promotes secretion of ANP and BNP by cardiomyocytes, leading to vasodilation and an anti-hypertrophic response in cells via an increase in intracellular cGMP levels, which leads to activation of PKG, in turn mediating reduced hypertrophic growth. Activation of JNK and p38 stress signaling events in the cardiomyocytes, although leading to cardiomyopathy and heart failure, results in inhibition of NFAT through phosphorylation events that prevent its nuclear localization and pro-hypertrophic transcriptional regulation activities, thereby blocking the calcineurin axis. Increased secretion of TGF-β during increased pressure stress can lead to mixed responses, with canonical TGF-β-SMAD2/SMAD3 signaling leading to anti-hypertrophic responses, whereas activation of non-canonical SMAD1/SMAD5 leads to pro-hypertrophic responses.

Another endothelium-derived factor next to NO and CNP is ET-1 (Table 2) [44], which contributes to cardiac hypertrophy and fibrosis as a major growth factor. Aside from endothelial cells, ET-1 is, amongst others, also expressed in non-endothelial cells, such as fibroblasts and cardiomyocytes (Table 2; Figure 1). Functioning in an autocrine and paracrine manner, ET-1 seems to have important effects during the development of cardiac hypertrophy [45]. ET-1 exhibits a positive inotropic effect as well as triggers cardiac hypertrophy responses [46]. Moreover, cardiac endothelial cells carry enzymes with protease activities, like the angiotensin-converting enzyme (ACE) and chymase (Table 2; Figure 1), which may contribute to changes in local levels of AT-II [47]. Besides fibroblasts, endothelial cells may also contribute to cardiac fibrosis (Figure 3). For example, it is known that endothelial cells and pericytes as the capillary lining cells wrapped around them, control cardiac fibroblast numbers [38]. Whether this contribution is similarly relevant as the proliferation and activation of resident fibroblasts upon exposure to pressure overload await further investigation [29].
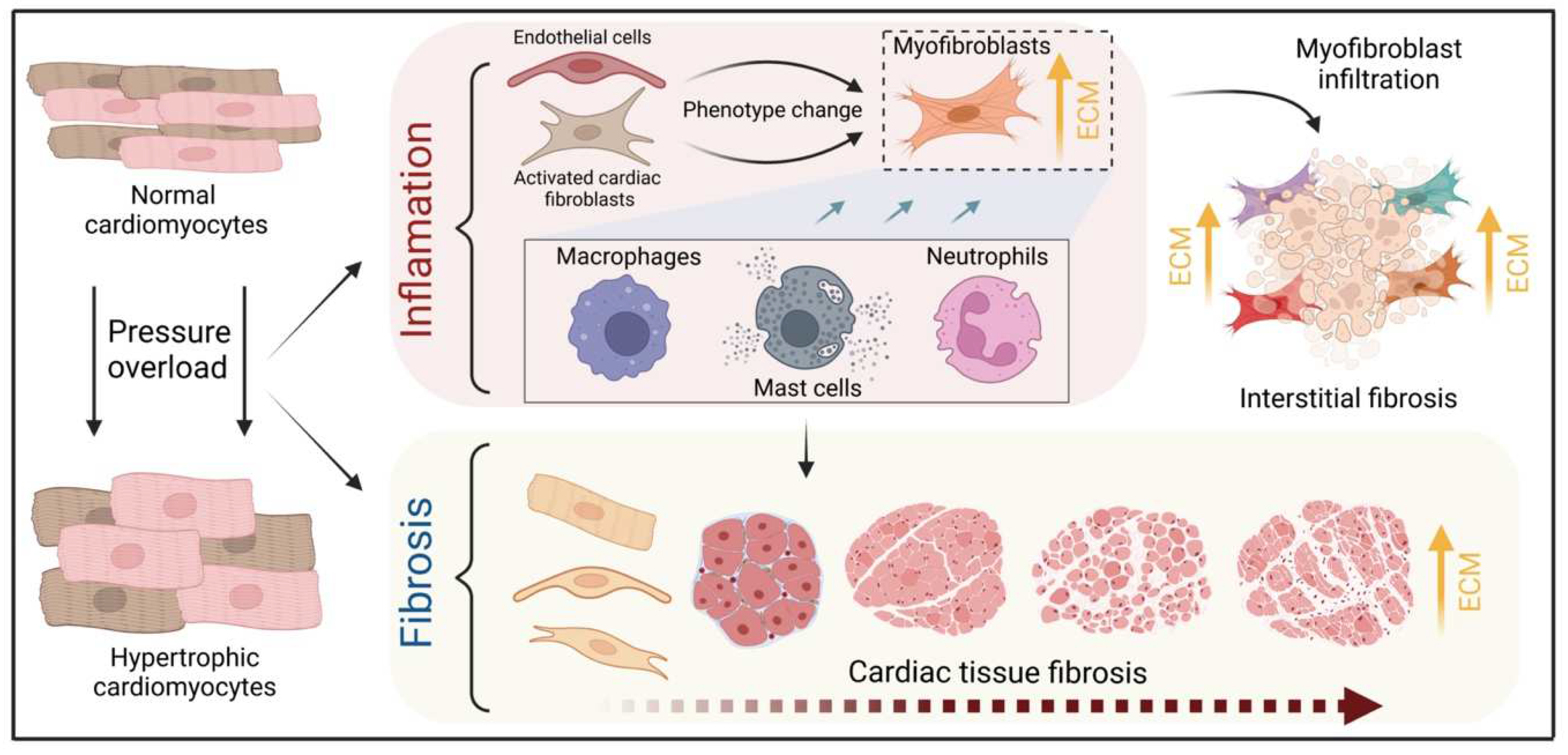
![Cells 12 01780 g003]()
Figure 3.
Schematic illustration of the process of fibrotic scar formation at the cellular level. The myocardium develops cardiomyocyte hypertrophy under pressure overload conditions, triggering concomitant inflammatory processes and fibrotic scar formation. The evidence discussed in the text suggests a central role for resident fibroblasts, nonetheless cardiac endothelial cells may also contribute to myofibroblast-like cells and drive cardiac fibrosis. Resident and infiltrating immune cells, including mast cells, macrophages, and neutrophils, enhance this phenotype change by releasing TGFß while mediating tissue inflammation via cytokines such as TNFα, IL-6, and IL-1. These mechanisms increase the number of myofibroblasts and the accumulation of collagen, which accelerates fibrotic scar formation in the microenvironment of cardiac hypertrophy.
Figure 3.
Schematic illustration of the process of fibrotic scar formation at the cellular level. The myocardium develops cardiomyocyte hypertrophy under pressure overload conditions, triggering concomitant inflammatory processes and fibrotic scar formation. The evidence discussed in the text suggests a central role for resident fibroblasts, nonetheless cardiac endothelial cells may also contribute to myofibroblast-like cells and drive cardiac fibrosis. Resident and infiltrating immune cells, including mast cells, macrophages, and neutrophils, enhance this phenotype change by releasing TGFß while mediating tissue inflammation via cytokines such as TNFα, IL-6, and IL-1. These mechanisms increase the number of myofibroblasts and the accumulation of collagen, which accelerates fibrotic scar formation in the microenvironment of cardiac hypertrophy.

3. The Role of Immune Cells in Cardiac Hypertrophy
The pathogenesis of pressure overload and heart failure has been suggested to be in close context with the activation of inflammatory cells and the release of inflammatory mediators (Figure 1) [48]. Early inflammation in hypertrophic cardiomyopathy (HCM) may be brought on by cardiomyocyte disorganization, sarcomere damage, mitochondrial oxidative stress, and microvascular dysfunction [49]. Numerous investigations have shown that HCM patients have leucocyte infiltration in the myocardium and elevated levels of inflammatory cytokines, which may be crucial to the condition of HCM and its development to the dilated-phase end stage [50,51,52]. Additionally, genetic deletion of IL-6 has been shown to mitigate TAC-induced LV dysfunction and hypertrophy, demonstrating a crucial role for IL-6 in the pathophysiology of LV hypertrophy in response to pressure overload [53]. The nodal point for integrating hypertrophic and inflammatory signals in the myocardium is CaMKII, whose activity is elevated in MI hearts and which promotes cardiac hypertrophy and inflammation, processes that are persistently stimulated by cardiac injury [54].
3.1. Cardiac Mast Cells
Identification of the presence of mast cells in the heart tissue of animals and humans [55,56], as well as the discovery of mast cells as the source of an array of mediators (Table 2) [57], clearly emphasize the crucial participation of innate immune cells, especially cardiac mast cells, in cardiac hypertrophy and remodeling (reviewed in [48,58]).
Table 2.
Mediators influencing the microenvironment in cardiac hypertrophy.
| Vasoactive Peptides | Secretion from/Location | References |
|---|---|---|
| AT-II | cardiomyocyte | [59,60] |
| AT-II | endothelial cell | [41] |
| ET-1 | cardiomyocyte | [45,61] |
| ET-1 | fibroblast | [45,61,62,63] |
| ET-1 | endothelial cell | [41,45,61,64] |
| Catecholamines | ||
| NE | sympathetic nerve ending | [65,66] |
| Growth factors | ||
| FGF (aFGF, bFGF) | cardiomyocyte | [67,68] |
| FGF (aFGF, bFGF) | non-myocyte | [69] |
| FGF-2 (bFGF) | fibroblast | [67,68,70] |
| FGF-2 (bFGF) | endothelial cell | [71,72,73] |
| FGF-2 (bFGF) | mast cell | [57,74,75] |
| High-FGF-2 (Hi-bFGF) | fibroblast | [70] |
| TGF-ß | cardiomyocyte | [76,77,78] |
| TGF-ß | fibroblast | [45,62,70,76,77,78] |
| TGF-ß | endothelial cell | [76,77,78] |
| TGF-ß | mast cell | [57,75,79,80] |
| TGF-ß | myofibroblast | [81,82,83] |
| Cytokines | ||
| IL-6, CT-1, LIF | cardiomyocyte | [84,85,86,87,88] |
| IL-6, CT-1, LIF | fibroblast | [85,86,87,88] |
| IL-6 | mast cell | [57,75] |
| IL-1 | mast cell | [57,75] |
| TNFα | mast cell | [57,75,89,90,91,92] |
| Various other components | ||
| VCAM-1, ICAM-1 | endothelial cell | [42] |
| ECM components | cardiomyocyte | [93] |
| ECM components | fibroblast | [93] |
| ECM components | endothelial cell | [93] |
| ECM components | myofibroblast | [81,94] |
| Histamine | mast cell | [55] |
| Chemotactic factors | mast cell | [57,75] |
| Anti-hypertrophic peptides | ||
| ANP, BNP | cardiomyocyte | [8,43,95] |
| NO | endothelial cell | [41,42] |
| CNP | endothelial cell | [43] |
| Enzymatic activities | ||
| Local RAS | cardiac tissue | [96,97,98] |
| AGT, renin, ACE, AT1, AT2 | cardiac tissue | [99,100] |
| AGT | cardiomyocyte | [101] |
| AGT | fibroblast | [101] |
| Renin | mast cell | [65,102] |
| ACE | endothelial cell | [103] |
| Chymase (alternative ACE) | endothelial cell | [103] |
| Chymase (alternative ACE) | mast cell | [57,75,104] |
Activated cardiac mast cells were identified in spontaneously hypertensive rats as a major source of growth factors (Figure 1), such as TGF-ß and bFGF, in areas of myocardial fibrosis [105]. This is consistent with findings that the release of TGF-ß provokes an increase in collagen production alongside the differentiation of fibroblasts to myofibroblasts (Figure 1) [106], and indicates that cardiac mast cells also contribute to the key steps of cardiac tissue fibrosis [107]. Another major mediator that is released upon mast cell degranulation in the heart is histamine (Table 2) [55]. Histamine is a neurohormonal mediator that binds to histamine H1, H2, and H3 receptors, thereby inducing various cellular functions [108,109] as well as cardiac hypertrophy (Figure 1) [110]. Notably, cardiomyocytes express the histamine H2 receptor, which is coupled to the beta receptor and Gs proteins [111,112,113,114,115]. Consistently, histamine triggers positive inotropic effects [116,117]. In contrast, blocking the histamine H2 receptors decreases cardiac output [116]. The application of famotidine, a histamine H2 receptor antagonist, in chronic heart failure (CHF) patients, was found to decrease left ventricular remodeling [118].
Another characteristic of mast cells involves their strategic location often at a perivascular site, thereby exerting regulatory functions on endothelial cells. Mast cells synthesize several endothelial cell activators comprising, amongst others, the platelet-activating factor (PAF), IL-1ß, IL-4, and tumor necrosis factors alpha (TNFα) [119,120,121]. Several studies have indicated mast cell degranulation as a major source of TNFα (Table 2; Figure 1) [89,90,91,92]. Even though many cardiac cells have been described to generate TNFα, cardiac mast cells appear to constitutively express TNFα [89,91] and activate TNFα/nuclear factor kappa B (NF-κB)/IL-6 cascades [105]. Activation of the TNFα/NF-κB axis leads to the activation of p38-MAPK (Figure 2), collectively causing hypertrophy and dysfunction of the heart [122,123]. Moreover, mast cells release other cytokines including IL-1 and IL-6 (Table 2; Figure 1) [57,75]. IL-6 cytokine family binds the common co-receptor glycoprotein 130 (gp130) and thereby potentially takes an active role in cardiac hypertrophy induction via the JAK/STAT pathway (Figure 2) [124,125].
Although several studies suggest that cardiac mast cells are a source of renin, released upon mast cell degranulation (Table 2) [65,102], the major source of renin in the myocardial microenvironment is complex [126,127]. Mast cells release the proteolytic enzyme chymase, which catalyzes, independently of ACE, the conversion of AT-I to AT-II (Figure 1) [104]. Thus, mast cell renin and chymase may serve as an alternative way to upregulate AT-II levels in the myocardial microenvironment, and it has been demonstrated in rat models that mast cell inhibition using mast cell stabilizer cromolyn sodium reduces pathological left ventricular remodeling [128].
3.2. Monocytes & Macrophages
Healthy and injured cardiac tissues possess heterogeneous populations of macrophages, in both humans and mice (Figure 1) [129]. Most macrophages within the heart are established embryonically from the yolk sac and fetal liver progenitors, similar to tissue macrophages of the liver or brain. Local proliferation in contrast to monocyte recruitment serves to maintain resident macrophage subsets [130,131]. In the absence of disease, self-renewal serves to maintain local tissue macrophage populations [132]. Despite this, in response to pressure overload or ischemic injuries, the majority of macrophages are derived from the recruitment and differentiation of blood monocytes [133].
Cardiac macrophages are key effector cells mediating tissue remodeling and fibrosis (Figure 3) [134]. The initial and significant event for vascular lesion formation results from inflammatory cytokine- and growth factor-producing migrating macrophages (Figure 1) [135]. The accumulation of macrophages has been found in the perivascular space, where they co-localize with fibroblasts collectively producing collagen during cardiac hypertrophy (Figure 3) [136,137]. Consistent with this, other studies have found that pressure overload initiates endothelial cells of the intramyocardial arteries to exhibit intercellular adhesion molecule (ICAM)-1 and that accumulation of macrophages occurs adjacent to the ICAM-1 expressing arteries in the perivascular space (Figure 1) [138]. Additionally, vascular cells and monocytes synthesize and express monocyte chemoattractant protein (MCP)-1, a potent monocyte chemoattractant [139], primarily regulating the recruitment of macrophages to the vessels [140]. For example, the continuous infusion of the AT-II or norepinephrine in hypertensive rats demonstrated that MCP-1 induction was associated with adventitial macrophage accumulation in the aortic wall [139].
Collectively, this suggests that resident and recruited macrophages actively take part in the early responses to stress preceding hypertrophic remodeling.
3.3. Neutrophils
Under normal reparative conditions, neutrophil granulocytes are recruited to areas of acute inflammation, where they perform functions such as the clearance of dead cells and matrix debris (Figure 3) [141,142]. As key components of the inflammatory response, neutrophils also act on the recruitment, activation, and programming of antigen-presenting cells (APCs). Specifically, they attract monocytes and dendritic cells (DCs) by generating chemotactic signals, thereby influencing the differentiation of macrophages into a predominantly pro- or anti-inflammatory state [143,144,145]. Because neutrophil granulocytes are one of the most important cellular components of the body for the destruction of microorganisms, there is also the possibility that these cells damage host cells and tissues [146]. Accordingly, they may have deleterious effects on cardiac tissue when recruited to sites where pressure overload is present (Figure 3).
Several studies have reported that in response to hypertrophy triggered by pressure overload, the first leukocytes to appear in the myocardium within 3 days of injury are neutrophils (Figure 3) [147,148]. Activation of endothelial cells and subsequent expression of adhesion molecules allow the transmigration of neutrophils (Figure 3) [149,150]. In addition, inflammatory mediators such as TNFα, IL-1ß, and mast cell-derived histamine enhance this process [151,152,153]. Additionally, macrophage and neutrophil infiltration appeared in the first 3 days after injury next to ICAM-1 containing coronary arteries in the left and right ventricle, using a mouse model with inter-renal aortic banding. Moreover, these alterations of macrophage and neutrophil content occurred ahead in perivascular fibrosis (10 days), and cardiomyocyte hypertrophy (28 day) [154].
Neutrophils have been described to produce cytokines such as TNFα that drive macrophage and dendritic cell differentiation [143,145,155]. Additionally, neutrophilic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase gets activated in response to pressure overload injury [156], resulting in the degranulation of neutrophils and thereby release of pro-fibrotic proteases (Figure 3) as well as reactive oxygen species (ROS) [157].
3.4. Lymphocytes
A growing body of research indicates that systemic inflammation may play a significant pathophysiologic role in the etiology of cardiac disease development, including HCM, and may have an impact on the severity of the phenotypic and clinical outcomes, including heart failure. A high neutrophil-to-lymphocyte ratio (NLR), a marker of oxidative stress damage, has been linked to an increased 5-year risk of sudden cardiac death associated with HCM [158,159], which supported further the prognostic significance of inflammation. In addition, the lymphocyte-to-monocyte ratio (LMR) and survival in patients with hypertrophic cardiomyopathy have been correlated, with a lower LMR being associated with a lower mortality rate [160].
However, in angiotensin II-induced HF models, the absence of B cells led to less hypertrophy and collagen deposition, the preservation of left ventricular function, and, in conjunction with these changes, a decrease in the expression of proinflammatory cytokines and apoptosis in the myocardium [161]. Different studies have also reported that activation of NK T cells improved cardiac remodeling events and failure in mice by increasing the expression of cardioprotective cytokines, including IL-10 [162,163]. Additionally, the activation of invariant natural killer T (iNKT) cells may act as a preventative measure against HF brought on by pressure overload as their disruption was shown to worsen cardiac hypertrophy [164]. Importantly, Ayach et al. emphasized the crucial role of c-KIT signaling in preventing ventricular dilation and hypertrophy as well as the maintenance of cardiac function after infarction by mediating the mobilization of NK and angiogenic cells derived from bone marrow, which helps with improved remodeling after MI [165].
On the other hand, a case study of a primary cardiac lymphoma (PCL) patient with T cell-lymphoma was shown to be in association with hypertrophic cardiomyopathy [166], besides a different patient with acute lymphoblastic leukemia (ALL) being reported in another case study to have developed HCM after cell therapy interventions using T cells [167]. Apart from these reports, in conclusion, the majority of the evidence points to lymphocytes having a favorable influence on HCM remodeling events.
3.5. Sympathetic Neurons
Sympathetic neurons that innervate the heart and release norepinephrine (NE) also express the endothelin receptor A (ET-A) [168,169]. ET-resulted in a tremendous NE release in cocultured cardiomyocytes and sympathetic neurons with exaggerated hypertrophy of cardiomyocytes compared to monocultured cardiomyocytes. In contrast, mice lacking the ET-A receptor exclusively in sympathetic neurons showed less adverse structural remodeling, and cardiac dysfunction when exposed to pathological pressure overload [170].
Substantial amounts of renin released in the cardiac microenvironment upon cardiac mast cell degranulation [65] result in both AT-II formation within striking distance of AT1 receptor-expressing cardiac sympathetic nerve terminals and enhanced NE release (Figure 1) and arrhythmias (Figure 4) [171,172]. The fact that these events can be prevented by mast cell-stabilizing agents confirms the central role of cardiac mast cell-derived renin in AT1 receptor signaling [65]. Locally produced AT-II thus activates the AT1 receptor at sympathetic nerve endings, resulting in increased NE release (Figure 1) [65].
4. Mediators of Cardiac Remodeling
Mechanical stretch and neurohumoral mechanisms identify the most proximal stimuli for initiating hypertrophic signaling pathways (Figure 1) [3]. Due to hemodynamic overload, cardiomyocytes undergo mechanical stress and thereby release autocrine and paracrine signaling factors, such as growth factors, hormones, cytokines, and chemokines (Table 2) [14]. Furthermore, mechanical stress is sensed by both cardiac fibroblasts resulting in the production and release of signaling mediators (Figure 1) [173], and cardiac endothelial cells, which communicate with cardiomyocytes by secretion of autocrine and paracrine mediators [43,174]. Cardiomyocytes sense these ligands through a multitude of G-protein-coupled receptors (GPCRs), growth factor receptors, and cytokine receptors (Figure 1 and Figure 2) [3]. Orchestrated mechanisms of the induction, maintenance, and progression of cardiac hypertrophy, particularly left ventricular hypertrophy, underlie a series of events that follows the activation of cardiomyocytes upon pressure overload/mechanical stress.
4.1. Activation of the Local Renin-Angiotensin System (RAS)
In addition to the classical circulating renin-angiotensin system (RAS) [96], the heart has a local RAS that mediates autocrine, paracrine, and intracrine effects (Figure 1) [97,98]. Components of the RAS, including angiotensinogen (AGT), renin, ACE, AT-I, and AT-II, are expressed in the heart [99,100], and component expression is upregulated in cardiomyocytes in vitro in response to stretch [175,176]. Several studies have indicated that hemodynamic overload activates the local RAS and highlighted the crucial role of the AT1 receptor in strain-induced cardiac hypertrophy [177,178,179,180]. Thus, mechanical stress can be considered the major upstream trigger that activates the local RAS and leads to increased AT-II levels throughout the microenvironment.
4.2. Reactive Oxygen Species (ROS)
Reactive oxygen species (ROS) such as superoxide anion (O−2), hydroxyl (OH), and hydrogen peroxide (H2O2), and reactive nitrogen species including nitric oxide (NO) and peroxynitrite (ONOO−) classify reactive species involved in redox signaling. The latter results from the reaction of (O−2) with NO [181]. Data suggest that both direct and indirect mechanisms resulting from redox signaling within and between endothelial cells and cardiomyocytes are responsible for functional communication between these cells [23]. Moreover, redox signaling not only influences many physiological processes in the heart but also plays an important role in pathological cardiac remodeling [182,183].
In cardiac cells, several sources of ROS have been described, such as mitochondria [184], xanthine oxidase (XO) [185], uncoupled NO synthases (NOS) [186], and NADPH oxidases (NOXs) [187]. The interactions of NOX proteins with NOS-derived NO have been highlighted to be particularly important for redox signaling in the development of heart failure (Figure 1) [187,188,189].
An increase in the cardiac generation of ROS and therefore an increase in oxidative stress has been implicated in pressure-overload-induced left ventricular cardiac hypertrophy (LVH) and heart failure (Figure 1) [13,190]. Additionally, the development of cellular hypertrophy and remodeling has been found to implicate increased ROS production, and activation of the mitogen-activated protein kinase (MAPK) superfamily, where redox-sensitive protein kinases, are known to be partly responsible. Moreover, cardiomyocyte apoptosis and necrosis may be due to increased oxidative stress (Figure 4), which is described to be associated with the transition from compensated pressure-overload-induced hypertrophy to heart failure. Furthermore, alterations in the redox-sensitive activity of several key proteins including sarcolemma ion channels and exchangers and sarcoplasmic reticulum calcium release channels, which collectively account for excitation-contraction coupling, contribute to myocardial contractile dysfunction (Figure 4). Beyond that, the consequent generation of peroxynitrite (ONOO−) as a result of increased inactivation of NO has been attributed to indirect effects of ROS, leading to coronary vascular endothelial dysfunction and peroxynitrite-induced inhibition of myocardial respiration [191].
Several mediators including AT-II, ET-1, alpha-adrenergic agonists, TNFα, and mechanical forces trigger NOX2 activation. Via induction of four cytosolic regulatory subunits (p47phox, p67phox, p40phox, and RAC1), these mediators initiate O2− production [192], indicating that pressure overload subsequently increases O2− levels (Figure 1). Excessive O2− levels interact extremely rapidly with NO, resulting in peroxynitrite formation, thereby disrupting physiological NO signaling [189,193]. Hence, pressure overload shifts the balance towards increased ROS (Figure 1), a condition that suppresses the physiological functions of NO. Consistently, O−2 has long been recognized to be implicated in severe cardiovascular diseases. Moreover, reports indicate that NOS may generate O2− instead of NO, a condition referred to as uncoupled NOS. The switch to O2− generation appears as a consequence of tetrahydrobiopterin (BH4) depletion (usually through oxidation to BH2) or as NOS enzymes undergo post-translational modification [194]. Consistent, increased levels of O2− and ONOO− may be implicated in an amplifying mechanism that aggravates NOS uncoupling through oxidation of BH4 [195].
Hence as outlined above, reactive oxygen species should be considered as a group of key mediators driving pathological remodeling in the microenvironment of cardiac hypertrophy (Figure 1), especially regarding pressure overload.
4.3. Endogenous Storage Pools of AT-II in Secretory Granules
AT-II secretion into the culture medium upon mechanical stress of isolated cardiomyocytes has been observed and provides some evidence supporting the concept of increased local concentrations of AT-II [175]. Potential autocrine and paracrine regulatory mechanisms of AT-II may activate the AT1 receptor on cardiomyocytes and surrounding cells (Table 2) [196,197]. This in turn has been proposed to induce the release of autocrine and paracrine mediators, including vasoactive peptides, growth factors, cytokines, and ECM components, such as collagen (Figure 1) [45,62,70,198]. Potentiated or sustained AT1 receptor activation is likely associated with cardiomyocyte hypertrophy, fibroblast hyperplasia, and fibrosis (Figure 4) [59,199,200]. Alternative mechanisms have been proposed to contribute to the activation of the AT1 receptor upon binding of AT-II [201], including membrane stretch and mechanoactivation that can in turn promote distinct conformational rearrangements in the receptor, leading to alternative signaling outcomes [202,203]. Several proteins have been implicated as sensors of mechanical stretches, such as muscle LIM proteins, integrins, and their associated signaling pathways [204,205]. Network models have been developed to predict how these mechano-sensitive proteins work together to coordinate cardiomyocyte hypertrophy [206,207]. Mechanisms that integrate these events and propagate the stress signal to the AT1 receptor after activation by mechanical stress remain areas of active investigation. Interestingly, despite the absence of AT-II/AT1 signaling, cardiac hypertrophy, systolic dysfunction, and fibrosis occurred in response to pressure overload (Figure 4) [200].
4.4. The Two Faces of the TGF-ß Signaling
AT-II-activated fibroblasts release TGF-ß and ET-1 in a paracrine manner into cardiomyocytes, leading to hypertrophy (Table 2) [45]. Similar to mechanical stress, autocrine TGF-ß signaling promotes fibroblast proliferation and ECM production (Figure 1), especially collagen and fibronectin, whereas degradation of these components is reduced [208]. Several studies report that the canonical TGF-ß/SMAD2/3 signaling pathways (Figure 2) induce the expression of genes related to collagen, fibronectin, and other ECM proteins [209,210,211,212], which concomitantly contribute to cardiac fibrosis (Figure 1) [76]. Experiments using pressure-overload rats demonstrated that a TGF-ß neutralizing antibody inhibited fibroblast activation and proliferation, and diastolic dysfunction [76]. These data suggest TGF-ß as a central target and the inhibition of TGF-ß signaling as beneficial. In line with this, cardiac fibrosis was attenuated in SMAD3 deficient mice subjected to cardiac pressure overload, but interestingly cardiac hypertrophy and cardiac dysfunction were aggravated [213]. Also, another rat model revealed that worsened cardiac remodeling and increased mortality correlate with a reduction of ECM using a TGF-ß neutralizing antibody after myocardial infarction [214]. TGF-ß-activated kinase 1 (TAK1) binds directly to type II (TBRII) TGF-ß receptors. Identification of this interaction links TAK1 to the TGF-ß signaling cascade, implicating an additional way of hypertrophy induction in cardiomyocytes by TGF-ß signaling [215]. Thus, aside from contributing to cardiac fibrosis, the non-canonical TGF-ß/TAK 1 signaling pathway has also been reported to promote cardiac hypertrophy (Figure 2) [216]. Altogether, TGF-ß is released from cardiomyocytes, fibroblasts, and endothelial cells in the healthy heart (Table 2) [77,78] and in the context of injury and repair also from myofibroblasts and infiltrating immune cells [82,83]. Thus, TGF-ß seems to be involved in adaptive or maladaptive processes most likely depending on the context, and may locally trigger interactions between different cell types such as cardiomyocytes and fibroblasts (Figure 1) and thereby impact cardiac hypertrophy, fibrosis, and the development of heart failure (Figure 4).
4.5. Endothelin-1 Effects
Endothelin-1 (ET-1) is an endothelium-derived vasoconstrictor of 21 amino acids. Later, two additional homologs (ET-2 and ET-3) were identified. ET-1 is released from vascular endothelium and other cells including cardiomyocytes (Figure 1) after cleavage from a large precursor peptide [217]. ET-1 is the predominant endothelin in the heart and is identified as a potent hypertrophic stimulus in neonatal cardiomyocytes [218]. ET-1 is a ligand for two GPCRs: ET-A and ET-B where 90% of the endothelin receptors on cardiomyocytes belong to the ET-A subtype (Figure 2) [219]. In rat hearts, the ET-A is predominant and identified to be coupled to both the Gq and Gi subfamily of G-proteins (Figure 2) [220,221]. In addition, a characteristic pattern of gene expression is induced by ET-1 in ventricular neonatal rat cardiomyocytes (NRC) including immediate early genes (c-FOS, c-JUN, EGR-1), early genes (ANF, β-MHC, α-sk actin), and later on, ventricular MLC-2 and α-cd actin [222]. The Gq-RAS-RAF-ERK pathway may be involved in these transcriptional changes (Figure 2). Furthermore, ET-1 activates the Ras-MEKK1-SEK-JNK pathway contributing to the hypertrophy-associated gene expression program field [223].
ET-1 causes cell damage in cardiomyocytes in vivo, and experiments with long-term treatment with the ET-A receptor blocker BQ-123 showed improved survival of rats with heart failure [224].
The release of ANP and BNP from cardiomyocytes can also be triggered by AT-II and ET-1, though cardiomyocyte stretch is the main regulatory mechanism for ANP and BNP production [225].
4.6. FGF-2 Effects in Scar Formation
In general, considering the epigenetic state and very low proliferative potential of adult cardiomyocytes, consensus exists that there is only a small ability to regenerate injured myocardium through the proliferation of cardiomyocytes [226,227]. Instead, scar formation occurs through infiltrating highly proliferative cardiac fibroblasts (Figure 1 and Figure 3) [228]. A key player is FGF-2 (bFGF), which is expressed by numerous cell types in the adult myocardium. FGF-2 is released upon cardiac injury from its “storage site” (Table 2) thereby potentially activating cell surface receptors, such as FGFR (Figure 2) [229]. Moreover, AT-II, ET-1, and FGF-2 itself are known to promote FGF-2 gene expression (Table 2) [67,230]. Accordingly, FGF-2 increases both fibroblast and myofibroblast proliferation [231], therefore contributing to both enhanced scar formation and stiffness during cardiac injury (Figure 3). Noteworthy, FGF-2 exists as an isoform with a high molecular weight (Hi-FGF-2) and low molecular weight (Lo-FGF-2), thus it is important to determine the potential effects of both in the context of cardiac hypertrophy and tissue remodeling. In the past, several in vitro studies revealed evidence for an important role of FGF-2 in cardiac hypertrophy (Figure 1). Consistent with reports Lo-FGF-2 alters the gene profile of contractile proteins from “adult” to “fetal” programs when added to cultured neonatal cardiomyocytes, a distinct characteristic that is attributed to pressure overload-induced cardiac hypertrophy in vivo. Although data seems contradictory as others reported that cardiomyocyte hypertrophy is stimulated only by Hi-FGF-2, both in vivo and in vitro [198,232]. Hi-FGF-2 accumulates preferentially in response to stress stimuli (Figure 1), including AT-II [233] and oxidative stress [234]. This is further supported by others who found that Hi-FGF-2 is preferentially accumulated and released by cardiac fibroblasts which induce paracrine cardiomyocyte hypertrophy (Table 2) [70]. Once released, Hi-FGF-2 may directly interact and activate the tyrosine kinase receptor FGFR-1 (Figure 2) [235], and downstream MAPK signaling [70,236]. Lo-FGF-2 exhibits cardioprotective effects, especially against post-ischemic cardiac dysfunction [237]. One mechanism for the effects of Lo-FGF-2 is its potent angiogenic activity that may increase resistance to ischemic injury and cardioprotection [67,238,239]. In conclusion, these data imply that Hi-FGF-2 is a contributor to cardiac hypertrophy, fibrosis, and heart failure (Figure 4), while Lo-FGF-2 seems to exert opposite functions as a component of adaptive responses in the injured myocardium [240].
FGF-2 null mice had a marked reduction of the hypertrophic response in cardiomyocytes in response to pressure overload [241]; however, questions remain whether the entire blockade of FGF-2 signaling is therapeutically beneficial. Considering data highlighting the role of FGF-2 in the progression of many cancer types [242,243,244,245,246], blocking of FGF-2 may have beneficial effects as shown in reports on the elimination of tumor angiogenesis [247]. But, in the context of ischemic heart disease, inhibition of FGF-2 signaling may be detrimental, since an angiogenic effect by Lo-FGF-2 upregulation may be desirable [67,238,239]. Although data suggests functions for Hi-FGF-2 and Lo-FGF-2 in the myocardium, further investigations are certainly needed to understand (a) the precise outcomes of targeting one or the other isoform, (b) the effects on exact organs/cells, and (c) to define the precise function of the isoforms in the context of cardiomyocyte hypertrophy and fibrosis. Additionally, unwanted effects of Hi-FGF-2 and Lo-FGF-2 need to be considered. Moreover, in addition to FGF-2, TGF-ß, AT-II, catecholamines, and other molecules as well orchestrate the response to hemodynamic stress (Figure 1), which suggests that targeting just one mediator may not be sufficient.
ERK as the most prominent downstream effector of FGF-2 signaling plays a predominant role in the development of both physiological and pathological cardiac hypertrophy (Figure 2). While cytosolic functions of ERK upon activation through pressure overload and mediators are believed to promote the development of physiological hypertrophic conditions, nuclear transcriptional activations mediated by ERK promote a pathological hypertrophic response in CMs (Figure 2) [248,249]. Hypertrophic stimuli such as AT-II, ET-1, cytokines, catecholamines, and biomechanical stress may also contribute to detrimental ROS formation in cardiomyocytes, and additional autophosphorylation of ERK1/2 has been reported to trigger pathological ERK1/2-mediated cardiac hypertrophy (Figure 2) [250,251]. These changes can then activate several hypertrophic signaling mediators regulated by ERK1/2 [249,252].
Hyperactivation of ERK1/2 activity is most frequently linked to HCMs caused by genetic abnormalities [253,254]. While genetic variant-induced hyperactivation of ERK is closely linked to pathogenic remodeling, normalization of ERK activation by simvastatin treatment restores contractility and protects against fibrosis in animal models [255,256].
One study reported different cardiac hypertrophic responses using both mice that completely lacked ERK1/2 protein in the heart and mice that expressed an activated MEK1 in the heart. Inhibiting MEK-ERK1/2 in mice lacking ERK1/2 in the heart causes eccentric cardiac growth with elongated cardiomyocytes, whereas activation of MEK1-ERK1/2 signaling by the overexpression of an active MEK1 mutant appears to be responsible for the concentric type of hypertrophy with thicker cells [257]. Thus, increased pre- versus afterload have been described to result in typical hypertrophic responses, and ERK1/2 seem to exhibit a central role, partially regulating the underlying molecular mechanisms. Induction of ERK1/2 translocation to the nucleus in adult rat myocytes, corresponded to reduced myocyte lengths and increased width, under both baseline and chronic pacing conditions [258], pointing to the critical role played by ERK signaling in balancing concentric and eccentric hypertrophic growth (Figure 2).
4.7. Cytokines and Inflammasome in Cardiac Remodeling
Cytokines of the interleukin-6 (IL-6) family are key molecules for the local regulation of hypertrophic responses in cardiomyocytes (Figure 1). Pressure overload acts as a strong trigger for the upregulation of genes related to leukemia inhibitory factor (LIF) and cardiotrophin-1 (CT-1) in the adult human myocardium [259,260]. Cardiomyocytes and cardiac fibroblasts produce leukemia LIF and CT-1 (Table 2) [261]. The release of Hi-FGF-2 from cardiac fibroblasts (Table 2) has been suggested to act in an autocrine way and trigger the release of pro-hypertrophic CT-1 [70,262]. Moreover, cardiomyocytes also express autocrine-acting CT-1, and CT-1 induces hypertrophy of cardiomyocytes in vitro [263]. Increased production and release of LIF, CT-1, and IL-6 in cardiac fibroblasts in response to AT-II can contribute to cardiomyocyte hypertrophy by paracrine activation of the gp130-linked downstream signaling (Figure 2) [264]. Interestingly, IL-6 contributes to the induction of massive collagen release by cardiac fibroblasts in response to AT-II and norepinephrine stimulation [265,266], consistent with a pro-hypertrophic response. Alternatively, LIF stimulates several beneficial effects including reduction of collagen production and matrix metalloproteinase activity in cardiac fibroblasts, resulting in an inhibition of differentiation of cardiac fibroblast to myofibroblast [267]. Likewise, the role of CT-1 seems unclear as consistent with reports describing CT-1 as having a potent hypertrophic effect on cultured cardiomyocytes [268] in addition to cardioprotective effects such as promoting cardiomyocyte survival [269]. In conclusion, during the process of developing cardiac hypertrophy, cytokine release is increased in response to a variety of stress stimuli, including pressure overload, injury, and mediators like AT-II. However, since IL-6 has a negative inotropic effect, its function is still unclear, suggesting the possibility of detrimental impacts by IL-6 driving hypertrophy toward heart failure.
According to data binding of all IL-6-type cytokines to their common receptor subunit gp130 potently activates STAT3 and to a lesser extent STAT1 (Figure 2) [270]. Transgenic mice with cardiac-specific STAT3 over-expression found that STAT3 holds a key role in hypertrophic and protective signaling, respectively. STAT3 induced the expression of cardiac protective factors and guarded against decreases in the expression rates of cardiac contractile genes in the case of doxorubicin-induced cardiomyopathy [271]. In line with this, another study that used pressure overload on ventricular-restricted gp130 receptor knockout mice found a rapid onset of dilated cardiomyopathy and induction of cardiomyocyte apoptosis. In comparison, a normal cardiac structure and function were found under basal conditions, and compensated hypertrophy was found in control mice under pressure overload [272]. These observations suggest a key role of the gp130/STAT pathway in cardiomyocytes for transmitting adaptive and protective functions in response to pressure overload and injury. However, a study on transgenic mice that expressed a dominant negative mutant of gp130 (to decrease activation of this pathway) reported concomitant to a suppressed STAT3 activation a significantly smaller hypertrophic response when subjected to pressure overload [273], suggesting a pro-hypertrophic function for STAT3. Whether the effects of the gp130 signaling pathway are beneficial or detrimental remains unclear. Since pressure overload triggers hypertrophic responses in cardiomyocytes via GPCRs in turn activating PKC and PKD (Figure 2) [274], potential crosstalk of signaling pathways could be involved. Likewise, neonatal rat cardiomyocytes showed that stretch induces a transient activation in a sequential time order on PKC and other downstream targets as the successive components of the MAPK signaling cascade (Figure 2) [275].
In contrast, YAP1, a downstream effector of Hippo signaling regulating proliferation, survival, and organogenesis in mammalian cells, that can also be activated through SRC-mediated gp130 activation in cardiomyocytes [276], is involved in cardio-protective mechanisms against pressure overload stimulation of cardiac hypertrophy (Figure 2). Under chronic pressure overload conditions, activation of YAP transcriptional activity reduces the development of cardiac hypertrophy. Additionally, apoptosis and fibrosis effects on cardiomyocytes that can be prerequisites for myocardial infarction are reduced [277]. The transcriptional activity of YAP mediates compensatory cardiac hypertrophy under pressure overload conditions [278] to stop the progression of wall stress into myocardial infarction, while CMs are driven toward heart failure by the detrimental effects of YAP signaling loss-of-function [279].
Concomitant hypertrophic responses via activation of PKC and MAP kinases can also be triggered by AT-II (Figure 2). Cardiomyocytes under mechanical stress secrete AT-II [280]. Here, active PKC, with its numerous nuclear and cytosolic substrates, specifies the extensive crosstalk of signaling pathways in response to pressure overload. The alpha-isoform of PKC directly activates RAF1 [281], providing evidence for a complex link between the signaling pathway downstream of growth factor receptors in the context of cardiac hypertrophy. Others have reported that GPCR signaling can be linked directly to RAS GTPase (Figure 2) [282], and GTP-bound RAS interacts with many downstream effectors which in turn transmit the signal for activating multiple signaling pathways [283,284], potentially promoting hypertrophic responses in cardiomyocytes. Additionally, others reported that the C-terminus of the AT1 receptor associates with JAK2 upon binding of the ligand, resulting in JAK2/STAT3 pathway activation [285,286], indicating another example of the crosstalk of signaling pathways in response to hypertrophy-associated stress signals. These lines of evidence indicate that the discrepancy of data regarding the gp130 signaling pathway may be due to the extensive crosstalk between intracellular signaling pathways (Figure 2). Taken together, there are contradictory reports regarding the individual effects of IL-6, LIF, CT-1 and their signaling via the gp130 pathway in cardiac hypertrophy, thus further investigation is necessary for elucidating the exact mechanisms.
4.8. Calcineurin/NFAT in Cardiac Hypertrophy
Calcineurin as a Ca2+-dependent serine/threonine protein-phosphatase has been found to exhibit central pro-hypertrophic functions in the myocardium (Figure 2) [287,288]. Calcineurin contains two subunits: the 57–61-kDa catalytic subunit (CnA) and the 19-kDa regulatory subunit (CnB). Activation of this dimeric protein occurs through direct binding of the Ca2+-saturated adaptor protein calmodulin [289]. The mammalian heart only expresses CnAα, CnAβ, and CnB1, although there are three genes including CnAα, β γ encoding for CnA, and two genes (CnB1 and B2) encode for CnB. Calcineurin becomes activated in response to increased Ca2+ levels, which enables binding to transcription factors of the nuclear factor of activated T cells (NFAT) family (Figure 2) [289].
Pro-hypertrophic gene expression is activated upon binding, and through dephosphorylation of conserved serine residues at the N-terminus of NFAT by calcineurin, resulting in NFAT translocating into the nucleus (Figure 2). Here, NFAT regulates the expression of cardiac genes via association with GATA4 and myocyte enhancer factor 2 (MEF2), which are also transcription factors [290,291]. Noteworthy, several studies indicate that NFAT transcription factors act as primary calcineurin effectors in the heart, as they have been identified as necessary and sufficient mediators promoting cardiac hypertrophy [287,290,292]. Moreover, cardiomyocytes contain structural proteins located in the repetitive Z-disc that have been found to regulate calcineurin in addition to the activation via increased Ca2+ [293,294].
GPCR stimulation with hypertrophic agonists, including AT-II and PE on cultured neonatal rat cardiomyocytes indicated an increase in calcineurin enzymatic activity, which was induced by increased calcineurin Aß (CnAβ) mRNA and protein, compared to CnAα or CnAγ [295]. By that, human hypertrophied and failing hearts (Figure 4) also exhibit increased calcineurin activity [296], as well as in ventricular muscle with exposure to AT-II, ET-1, and Urotensin II in human failing heart [297]. Significantly, hypertrophied hearts in rodents subjected to aortic banding displayed upregulated calcineurin activity [298,299] and profound cardiac hypertrophy with rapid progression to dilated cardiomyopathy, extensive fibrosis, congestive heart failure, and sudden death (Figure 4) were observed in active calcineurin expressing transgenic mice [292].
Upregulated NFAT activity has been observed upon both physiological stimuli (exercise training, growth hormone-IGF1 infusion) and pathological stimuli (pressure overload, myocardial infarction) (Table 1) [15]. In contrast, the hypertrophic response to pressure overload and GPCR agonists was impaired in a model of transgenic mice exhibiting a targeted inactivation of calcineurin Aβ [300] and in transgenic mice expressing a dominant negative form of calcineurin A [298]. Furthermore, cardiac hypertrophy was prevented in a model using pharmacological inhibition of calcineurin A activity on transgenic mice with constitutively active calcineurin A [289,292].
These lines of evidence taken together indicate that calcineurin/NFAT plays a major role in the conversion of pathogenic stimuli into pathological cardiac remodeling, suggesting it is a key target in the setup of clinical prevention of cardiac hypertrophic (Figure 4). But data seems contradictory, as a study reported accentuated hypertrophy, impaired histopathology as well as risk for early death when applying calcineurin inhibitors [301]. Thus, further investigation is necessary to clarify if calcineurin/NFAT could be considered as a key target.
4.9. ANP/BNP in Cardiac Hypertrophy
Development of pathological cardiac hypertrophy is frequently linked to increased mRNA expression of atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP), according to studies in both human and animal models [302,303], as well as an increase in the plasma levels of ANP and BNP with the severity of heart failure. Under critical conditions, more BNP than ANP is secreted, largely in the ventricles and atria, respectively. However, as heart failure worsens, ANP is also secreted in the ventricles; for this reason, the ventricles are crucial locations for both BNP and ANP [304]. Both ANP and BNP, as well as their more stable cleavage products, NT-proANP and NT-proBNP, respectively, are efficient biomarkers in the clinical diagnosis and management of heart failure (Figure 4) [305,306].
Besides the physiological effects of ANP and BNP such as vasodilation, regulation of sodium reabsorption and water balance as well as inhibition of the renin-angiotensin-aldosterone (RAA) system, collectively directed towards responding to cardiac pressure and volume dynamics and suppression of heart failure [307,308], ANP/BNP causes the cGMP-dependent PKG to be activated (Figure 2), which in turn prompts the Ca2+/calmodulin-dependent endothelial nitric oxide (NO) synthase to aid in the production of more NO, which relaxes the vascular smooth muscle cells and lowers systemic blood pressure [307,309,310]. ANP/BNP and NO can also counteract NE effects on the size expansion of cardiomyocytes, presumably through the cGMP-PKG-mediated cardioprotective axis resulting in the reduction of NE-stimulated Ca2+ influx [309,311].
Moreover, while ANP and BNP expression is being regulated by pro-hypertrophic transcriptional activation of NFAT, on the other hand, ANP and BNP can counteract as negative regulators of hypertrophy by PKG-mediated inhibition of calcineurin to curb nuclear translocation of NFAT (Figure 2) [312,313,314].
5. Mathematical Modeling of Cardiac Remodeling
Many mediators and pathways implicated in cardiac hypertrophy hinder the field’s ability to integrate individual findings into a common framework. Mathematical modeling has also been useful for the elucidation of intracellular and intercellular signaling that controls cardiac functionality.
5.1. Computational Models of Cardiac Hypertrophy
Several computational models have been developed to address this, providing systems-level insight into how cardiac hypertrophy is regulated. In the first model of hypertrophic signaling, Cooling et al. examined the factors that control the kinetics of IP3 [315]. They found that ET-1 induced a much more sustained IP3 signal than AT-II, which was best explained by differences in receptor kinetics. To obtain a more global view of hypertrophic signaling, Ryall et al. used a logic-based modeling framework [316] to simulate 193 reactions integrated across 14 pathways [317]. Comprehensive knockout simulations supported the conclusion that RAS GTPase is the hub of a bow-tie control structure, which integrates signals from many receptors and stimulates hypertrophy through partially redundant MAPK pathways. This was validated in new experiments comparing the effects of inhibition of RAS GTPase, MEK, p38, and JNK [317].
While neonatal cardiomyocytes have been extremely useful in the study of cardiac hypertrophy, it is well-known that they show limited maturity compared to adult cells [318]. But quantitatively, to what extent are in vitro data predictive of in vivo cardiac hypertrophy? To address this question comprehensively, Frank et al. used the model of neonatal cardiomyocyte hypertrophy [317] to attempt to predict the in vivo hypertrophy for 52 cardiac-specific transgenic mice [319]. Strikingly, they found that the model correctly predicted 78% of cardiac outputs, including four double-transgenic mouse models. Differences between model predictions and in vivo experiments may indicate differences between in vitro and in vivo mechanisms or specific transgenic mice whose hypertrophic phenotype depends on specific contexts (e.g., hormones, genetic background).
Indeed, examination of context-dependent regulation can elucidate new aspects of signaling networks. Khalilimeybodi et al. developed a computational method called CLASSED to systematically revise the previous model of Ryall et al. [316] using context-dependent experimental data from 550 experimental data from 230 literature articles. Examining areas of a model-experiment disagreement using CLASSED, they identified the reactions that should be removed or added from the network. They also found new crosstalks between Gβγ and CaMKII or calcineurin, which were validated in neonatal cardiomyocytes [316]. Most recently, models of cardiomyocyte signaling are being incorporated into models of multiscale integration of mechanics and signaling in pressure overload, hormones [320], or pregnancy [321]. Recently, researchers have attempted to mature iPSC-CMs by prolonged culture duration, metabolic substrates, and mechanical and electrical stimulation to model HCM and measure cellular morphology, contractility, electrophysiological property, calcium handling, and metabolism [322,323]. Therefore, network model reparameterization for iPSC-CM will be advantageous from the perspective of translational applications. Cardiac hypertrophy is associated with increased ventricular arrhythmia [324]. Interestingly, several nodes in the signaling network of hypertrophy (such as CaMKII, PKA, and calcineurin) modulate the ion channels [325,326]. Therefore, the involvement of these node states in multiscale electromechanical models may predict the association of hypertrophy and arrhythmia.
5.2. Computational Modeling of Fibrosis
As illustrated in Figure 2, the complexity of intracellular networks often prohibits the identification of the signaling mechanisms that control cellular responses to biochemical or mechanical stimuli upon hypertrophy. To address this challenge, Zeigler et al. developed a logic-based differential equation model of the cardiac fibroblast signaling network, which was successfully validated against 80% of 41 papers from the literature not used in model development [327]. This model predicted that stretch-mediated myofibroblast activation was mediated not by any single path from integrins to α-SMA expression, but by an autocrine TGF-β autocrine loop. They validated this new prediction in new experiments by using a TGF-β receptor inhibitor to block cardiac myofibroblast activation in mechanically-restrained collagen gels [327]. This model was later extended to predict the in vivo fibroblast dynamics after myocardial infarction, predicting how IL-1 can paradoxically enhance collagen production through the above autocrine TGF-β loop but suppress it through activation of NFkB and BAMBI [328]. To make patient-specific predictions, Rogers et al. connected the logic-based model to transcriptional responses from valvular interstitial cells treated with patient serum samples [329]. They found that endothelin-1, IL-6, and TGF-β were most important for explaining patient-specific fibroblast activation.
To predict therapeutic approaches, the fibroblast network model was integrated with DrugBank to predict FDA-approved drugs that could be repurposed against cardiac fibrosis [330]. Interestingly, the combination drug Entresto (valsartan/sacubitril) was predicted to be particularly effective due to combined suppression of ERK through valsartan and enhancement of PKG through sacubitril [330]. This prediction was validated by independent studies showing that Entresto decreases fibrosis due to pressure overload in rats [331,332] and heart failure in humans [333]. Watts et al. extended this model with estrogen signaling, predicting that the effects of some drugs may be sex specific [334], building on previous experimental studies of how estrogen affects cardiac fibroblast signaling and activation [335]. Others have combined network modeling with an experimental drug screen through machine learning to identify pathways by which drugs regulate new fibroblast phenotypes such as stress fiber organization [336].
While fibroblasts play a central role, they regulate fibrosis through communication with many other cell types. Jin et al. developed a mathematical model that simulated the communication between monocytes, macrophages, and fibroblasts that lead to fibrosis after myocardial infarction [337]. Their model was validated against dynamics of inflammation and collagen content after myocardial infarction and then applied to predict how the strength or timing of perturbations to TGF-β or MMP9 can modulate the kinetics of post-MI fibrosis. Using a similar paradigm, Chowkwale et al. developed a model of the post-MI communication between neutrophils, monocytes, macrophages, cardiomyocytes, and fibroblasts [338]. They validated the model against 61 of 84 experiments not used to build the model. Using this model, they identified key dynamic features that control inflammation, fibrosis, and a new concept of inflammation-fibrosis coupling. Specifically, they predicted that inflammation is amplified by positive feedback between neutrophils and IL-1β, macrophage phagocytosis of cardiomyocytes is critical for inflammation to drive fibrosis, and that fibroblast proliferation acts as an ultrasensitive switch to amplify collagen deposition [338]. Intriguingly, this dual-amplification control system identified for inflammation-fibrosis coupling [338] appears analogous to that in excitation-contraction coupling [339]. Intercellular models illustrate complex dynamic relationships that should be experimentally validated for potential therapeutic strategies.
6. Concluding Remarks and Future Directions
The microenvironment involved in the development of cardiac hypertrophy involves cardiomyocytes and non-myocardial cells, and the accompanying release of numerous pro-hypertrophic, pro-fibrotic, and pro-inflammatory mediators facilitating reciprocal interactions.
Cardiac fibroblasts are the main players in the development of fibrosis, nevertheless, endothelial cells that can undergo EndMT toward a myofibroblast-like phenotype are closely involved as well. Resident and infiltrating immune cells (mast cells, macrophages, neutrophils) enhance these processes while simultaneously contributing to tissue inflammation. Thus, considering all these mechanisms in the hypertrophic microenvironment, tailoring an efficient treatment regimen appears extremely complex. Sophisticated strategies and most likely multidirectional approaches are needed and should be well-approachable using computational modeling systems that allow the integration of all signaling components.
Since it is not feasible to discuss every cellular and molecular process involved in the development of different types of cardiac hypertrophy, we aimed to outline the main drivers of the hypertrophic microenvironment and the respective signaling pathways being affected. A necessary future approach will be the identification of the precise involvement of different cell types, cellular mediators released by them, and the respective activation of second messengers. This will allow us to evaluate the known and thus far unrecognized molecular signaling axes during disease development. Moreover, such data collection within a computational model will help to guide effective and selective targeting strategies in cardiac hypertrophy. Given the high prevalence of heart disease in the Western world, an important future effort should be to translate the knowledge gained into new pharmacological targets that help to delay or even stop the remodeling process and the severe consequences that patients experience after diagnosis of diastolic or systolic dysfunction.
Author Contributions
Conceptualization, F.B., J.N. and M.R.A.; methodology, F.B. and J.N.; software and figures, F.B.; resources, F.B. and J.N.; writing—original draft preparation, F.B., J.N.; writing—review and editing, all authors; funding acquisition, M.R.A., M.J.W., J.J.S. and K.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the International Research Training Group 1902 Intra- and Interorgan Communication of the Cardiovascular System (Grant number: IRTG 1902-P6) funded by the German Research Foundation; the German Federal Ministry of Education and Research (BMBF): German Network of RASopathy Research (GeNeRARe, grant numbers: 01GM1902C) and Cardioprotective targeting of ERK (ERK-CASTing; grant number: 16GW0262K); the European Network on Noonan Syndrome and Related Disorders (NSEuroNet, grant number: 01GM1602B); the German Research Foundation (grant numbers: SFB1525/453989101, AH 92/8-3 and TRR 296/1 2020); the National Institutes of Health (grant number: NIH R01HL162925).
Acknowledgments
We thank Joachim Schmitt of the Institute of Pharmacology, Faculty of Medicine, Heinrich Heine University, Düsseldorf, for the critical review of the manuscript. Figures were Created with “BioRender.com”.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
AC, adenylate cyclase; ACE, angiotensin converting enzyme; α-cd actin, alpha-cardiac actin; aFGF, acidic fibroblast growth factor; AGT, angiotensinogen; AKT/PKB, protein kinase B; α-MHC, α-myosin heavy chain; AMP, adenosine monophosphate; ANP, atrial natriuretic peptide; APCs, antigen-presenting cells; α-sk actin, alpha skeletal muscle actin; α-sm actin, alpha smooth muscle actin; AT-I, angiotensin I; AT-II, angiotensin II; AT1R, angiotensin II type 1 receptor; ATP, adenosine triphosphate; bFGF, basic fibroblast growth factor; FGF-2, fibroblast growth factor 2; BH4, tetrahydrobiopterin; ß-MHC, ß-myosin heavy chain; BNP, brain natriuretic peptide; Ca2+, Calcium ion; CaMK, calmodulin-dependent kinase; cAMP, cyclic adenosine monophosphate; CHF, chronic heart failure; CMs, cardiomyocytes; CnAβ, calcineurin Aß; CNP, C-type natriuretic peptide; CR, cytokine receptor; CT-1, cardiotrophin-1; DCs, dendritic cells; ECM, extracellular-matrix; EndMT, endothelial-to-mesenchymal transition; ERK, extracellular signal regulated kinase; ET-1, endothelin-1; FGFR, FGF receptor; GPCR, G-protein-coupled receptor; gp130, glycoprotein 130; GSK3ß, glycogen synthase kinase-3ß; GTP, guanosine-5′-triphosphate; HCM, hypertrophic cardiomyopathy; HDAC4/5, histone deacetylase-4 and -5; Hi-bFGF, high molecular weight FGF-2; H2O2,hydroxyl and hydrogen peroxide; ICAM-1, intercellular adhesion molecule-1; IGF1, Insulin like growth factor-1; IL-1, Interleukin-1; IL-1b, Interleukin-1b; IP3, inositol-1,4,5-trisphosphate; iPSC, induced pluripotent stem cells; Janus kinase; JNK, c-Jun N-terminal kinase; LIF, leukemia inhibitory factor; Lo-FGF-2, low molecular weight FGF-2; MAPK, mitogen-activated protein kinase; MAPKKK, mitogen-activated kinase kinase kinase; MCP-1, monocyte chemoattractant protein-1; MEF2, myocyte enhancer factor 2; MEK, mitogen activated ERK activating kinase; MLC, myosin light chain; NADPH, nicotinamide adenine dinucleotide phosphate; NFAT, nuclear factor of activated T cells; NE, norepinephrine; NF-κB, nuclear factor kappa B; NO, nitric oxide; NOS, uncoupled NO synthases; NOX, nicotinamide adenine dinucleotide phosphate oxidase; NRCs, neonatal rat cardiomyocytes; O2−, superoxide anion; ONOO−, peroxynitrite; PDE3A, phosphodiesterase-3a; PAF, platelet-activating factor; PDGF, platelet-derived growth factor; PE, phenylephrine; PI2, prostaglandin I2; PKA, protein kinase-A; PKC, protein kinase-C; PKD, protein kinase-D; PI3K, phosphoinositide 3-kinase; PKGI, cyclic GMP (cGMP)-dependent protein kinase 1; PLC, phospholipase-C; RAAS, renin-angiotensin-aldosterone system; RAC1, RAS-related C3 botulinum toxin substrate-1; RAF, rapidly accelerated fibrosarcoma; RAS, renin-angiotensin system; RAS GTPase, rat sarcoma guanosine triphosphatase; ROS, reactive oxygen species; SERCA2, Calcium pump of sarcoplasmic reticulum; SMAD, Suppressor of Mothers Against Decapentaplegic; SRC, sarcoma; STAT, signal transducer and activator of transcription; TGF-ß, transforming growth factor beta; TFs, transcription factors; TNFα, tumor necrosis factor-α; VCAM-1, vascular cell adhesion molecule-1; LVH, ventricular cardiac hypertrophy; XO, xanthine oxidase; YAP, yes-associated protein.
References
- Zhu, L.; Li, C.; Liu, Q.; Xu, W.; Zhou, X. Molecular biomarkers in cardiac hypertrophy. J. Cell. Mol. Med. 2019, 23, 1671–1677. [Google Scholar] [CrossRef]
- Schaub, M.C.; Hefti, M.A.; Harder, B.A.; Eppenberger, H.M. Various hypertrophic stimuli induce distinct phenotypes in cardiomyocytes. J. Mol. Med. 1997, 75, 901–920. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovchinnikova, E.; Hoes, M.; Ustyantsev, K.; Bomer, N.; de Jong, T.V.; van der Mei, H.; Berezikov, E.; van der Meer, P. Modeling Human Cardiac Hypertrophy in Stem Cell-Derived Cardiomyocytes. Stem Cell Rep. 2018, 10, 794–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenji, K.; Drazner, M.H.; Rothermel, B.A.; Hill, J.A. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H8–H16. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.W.; Larson, M.G.; Benjamin, E.J.; Levy, D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 1998, 32, 1454–1459. [Google Scholar] [CrossRef] [Green Version]
- Nakhaei-Rad, S.; Bazgir, F.; Dahlmann, J.; Busley, A.V.; Buchholzer, M.; Haghighi, F.; Schänzer, A.; Hahn, A.; Kötter, S.; Schanze, D. Alteration of myocardial structure and function in RAF1-associated Noonan syndrome: Insights from cardiac disease modeling based on patient-derived iPSCs. bioRxiv 2022. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C. Dynamics of cell generation and turnover in the human heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.-D.; Guerquin-Kern, J.-L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O. Clearing Up the Mist: Cardiomyocyte Renewal in Human Hearts; Oxford University Press: Oxford, UK, 2019. [Google Scholar]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004, 94, 110–118. [Google Scholar] [CrossRef]
- Huston, J.H.; Shah, S.J. Understanding the pathobiology of pulmonary hypertension due to left heart disease. Circ. Res. 2022, 130, 1382–1403. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., II. The fuzzy logic of physiological cardiac hypertrophy. Hypertension 2007, 49, 962–970. [Google Scholar] [CrossRef] [Green Version]
- Shenasa, M.; Shenasa, H. Hypertension, left ventricular hypertrophy, and sudden cardiac death. Int. J. Cardiol. 2017, 237, 60–63. [Google Scholar] [CrossRef]
- Levy, D.; Garrison, R.J.; Savage, D.D.; Kannel, W.B.; Castelli, W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 1990, 322, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Prasad, S.V.N.; Mao, L.; Noma, T.; Yan, Z.; Kim, H.-S.; Smithies, O.; Rockman, H.A. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J. Clin. Investig. 2006, 116, 1547–1560. [Google Scholar] [CrossRef] [Green Version]
- Tzahor, E.; Dimmeler, S. A coalition to heal—The impact of the cardiac microenvironment. Science 2022, 377, eabm4443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Wang, D.; Zhou, X.; Chen, L.; Feng, K.; Xu, X.; Huang, T.; Li, Z.; Cai, Y. Predicting heart cell types by using transcriptome profiles and a machine learning method. Life 2022, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.K.; Bjerke, M.A.; Leinwand, L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell 2016, 27, 2149–2160. [Google Scholar] [CrossRef]
- Takeda, N.; Manabe, I. Cellular Interplay between Cardiomyocytes and Nonmyocytes in Cardiac Remodeling. Int. J. Inflamm. 2011, 2011, 535241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hefti, M.A.; Harder, B.A.; Eppenberger, H.M.; Schaub, M.C. Signaling pathways in cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2873–2892. [Google Scholar] [CrossRef]
- Nikolov, A.; Popovski, N. Extracellular matrix in heart disease: Focus on circulating collagen type I and III derived peptides as biomarkers of myocardial fibrosis and their potential in the prognosis of heart failure: A concise review. Metabolites 2022, 12, 297. [Google Scholar] [CrossRef]
- Moore-Morris, T.; Guimaraes-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; J Lin, S.-C.; Aronow, B.J.; Tallquist, M.D. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [Green Version]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Sun, C.; Tian, X.; Jia, Y.; Yang, M.; Li, Y.; Fernig, D.G. Functions of exogenous FGF signals in regulation of fibroblast to myofibroblast differentiation and extracellular matrix protein expression. Open Biol. 2022, 12, 210356. [Google Scholar] [CrossRef] [PubMed]
- Ragazzini, S.; Scocozza, F.; Bernava, G.; Auricchio, F.; Colombo, G.I.; Barbuto, M.; Conti, M.; Pesce, M.; Garoffolo, G. Mechanosensor YAP cooperates with TGF-β1 signaling to promote myofibroblast activation and matrix stiffening in a 3D model of human cardiac fibrosis. Acta Biomater. 2022, 152, 300–312. [Google Scholar] [CrossRef]
- Chaturvedi, R.R.; Herron, T.; Simmons, R.; Shore, D.; Kumar, P.; Sethia, B.; Chua, F.; Vassiliadis, E.; Kentish, J.C. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation 2010, 121, 979–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spach, M.S.; Boineau, J.P. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: A major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. 1997, 20, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Sharov, V.G.; Lesch, M.; Goldstein, S. Progression of heart failure: A role for interstitial fibrosis. Mol. Cell. Biochem. 1995, 147, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Cheng, W.; Li, X.; Liu, D.; Cui, C.; Wang, X. Endothelial-to-mesenchymal transition: Role in cardiac fibrosis. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 3–11. [Google Scholar] [CrossRef]
- Xu, X.; Tan, X.; Tampe, B.; Nyamsuren, G.; Liu, X.; Maier, L.S.; Sossalla, S.; Kalluri, R.; Zeisberg, M.; Hasenfuss, G.; et al. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc. Res. 2015, 105, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Brutsaert, D.L. Cardiac endothelial-myocardial signaling: Its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 2003, 83, 59–115. [Google Scholar] [CrossRef]
- Esper, R.J.; Nordaby, R.A.; Vilarino, J.O.; Paragano, A.; Cacharron, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Cardiology: A big-hearted molecule. Nature 2015, 519, 416–417. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial dysfunction and diabetic cardiomyopathy. Front. Endocrinol. 2022, 13, 851941. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.O.; Long, C.S.; Kalinyak, J.E.; Li, H.T.; Karliner, J.S. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc. Res. 1998, 40, 352–363. [Google Scholar] [CrossRef]
- Drawnel, F.M.; Archer, C.R.; Roderick, H.L. The role of the paracrine/autocrine mediator endothelin-1 in regulation of cardiac contractility and growth. Br. J. Pharm. 2013, 168, 296–317. [Google Scholar] [CrossRef] [Green Version]
- Froogh, G.; Kandhi, S.; Duvvi, R.; Le, Y.; Weng, Z.; Alruwaili, N.; Ashe, J.O.; Sun, D.; Huang, A. The contribution of chymase-dependent formation of ANG II to cardiac dysfunction in metabolic syndrome of young rats: Roles of fructose and EETs. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H985–H993. [Google Scholar] [CrossRef]
- Liu, X.; Shi, G.P.; Guo, J. Innate Immune Cells in Pressure Overload-Induced Cardiac Hypertrophy and Remodeling. Front. Cell Dev. Biol. 2021, 9, 659666. [Google Scholar] [CrossRef]
- Monda, E.; Palmiero, G.; Rubino, M.; Verrillo, F.; Amodio, F.; Di Fraia, F.; Pacileo, R.; Fimiani, F.; Esposito, A.; Cirillo, A.; et al. Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. Int. J. Mol. Sci. 2020, 21, 6462. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Ellims, A.H.; Beale, A.L.; Taylor, A.J.; Murphy, A.; Dart, A.M. Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy. Am. J. Transl. Res. 2017, 9, 5063–5073. [Google Scholar] [PubMed]
- Kuusisto, J.; Kärjä, V.; Sipola, P.; Kholová, I.; Peuhkurinen, K.; Jääskeläinen, P.; Naukkarinen, A.; Ylä-Herttuala, S.; Punnonen, K.; Laakso, M. Low-grade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart 2012, 98, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Högye, M.; Mándi, Y.; Csanády, M.; Sepp, R.; Buzás, K. Comparison of circulating levels of interleukin-6 and tumor necrosis factor-alpha in hypertrophic cardiomyopathy and in idiopathic dilated cardiomyopathy. Am. J. Cardiol. 2004, 94, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cheng, G.; Jin, R.; Afzal, M.R.; Samanta, A.; Xuan, Y.-T.; Girgis, M.; Elias, H.K.; Zhu, Y.; Davani, A. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ. Res. 2016, 118, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.V.; Anderson, M.E. Is CaMKII a link between inflammation and hypertrophy in heart? J. Mol. Med. 2011, 89, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, A.M. Mast-cell degranulation in human hearts. N. Engl. J. Med. 1986, 315, 969–970. [Google Scholar]
- Ingason, A.B.; Mechmet, F.; Atacho, D.A.M.; Steingrímsson, E.; Petersen, P.H. Distribution of mast cells within the mouse heart and its dependency on Mitf. Mol. Immunol. 2019, 105, 9–15. [Google Scholar] [CrossRef]
- Mekori, Y.A.; Metcalfe, D.D. Mast cells in innate immunity. Immunol. Rev. 2000, 173, 131–140. [Google Scholar] [CrossRef]
- Balakumar, P.; Singh, A.P.; Ganti, S.S.; Krishan, P.; Ramasamy, S.; Singh, M. Resident cardiac mast cells: Are they the major culprit in the pathogenesis of cardiac hypertrophy? Basic Clin. Pharmacol. Toxicol. 2008, 102, 5–9. [Google Scholar] [CrossRef]
- Sadoshima, J.; Izumo, S. Molecular characterization of angiotensin II—Induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 1993, 73, 413–423. [Google Scholar] [CrossRef]
- Ramos-Kuri, M.; Meka, S.H.; Salamanca-Buentello, F.; Hajjar, R.J.; Lipskaia, L.; Chemaly, E.R. Molecules linked to Ras signaling as therapeutic targets in cardiac pathologies. Biol. Res. 2021, 54, 23. [Google Scholar] [CrossRef]
- Heiden, S.; Vignon-Zellweger, N.; Masuda, S.; Yagi, K.; Nakayama, K.; Yanagisawa, M.; Emoto, N. Vascular endothelium derived endothelin-1 is required for normal heart function after chronic pressure overload in mice. PLoS ONE 2014, 9, e88730. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Itoh, H.; Nakagawa, O.; Ogawa, Y.; Miyamoto, Y.; Kuwahara, K.; Ogawa, E.; Igaki, T.; Yamashita, J.; Masuda, I.; et al. Significance of ventricular myocytes and nonmyocytes interaction during cardiocyte hypertrophy: Evidence for endothelin-1 as a paracrine hypertrophic factor from cardiac nonmyocytes. Circulation 1997, 96, 3737–3744. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Duangrat, R.; Parichatikanond, W.; Likitnukul, S.; Mangmool, S. Endothelin-1 induces cell proliferation and myofibroblast differentiation through the ETAR/Gαq/ERK signaling pathway in human cardiac fibroblasts. Int. J. Mol. Sci. 2023, 24, 4475. [Google Scholar] [CrossRef] [PubMed]
- Mackins, C.J.; Kano, S.; Seyedi, N.; Schafer, U.; Reid, A.C.; Machida, T.; Silver, R.B.; Levi, R. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J. Clin. Investig. 2006, 116, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Seravalle, G.; Grassi, G. Sympathetic nervous system and hypertension: New evidences. Auton. Neurosci. 2022, 238, 102954. [Google Scholar] [CrossRef] [PubMed]
- Detillieux, K.A.; Sheikh, F.; Kardami, E.; Cattini, P.A. Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc. Res. 2003, 57, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardami, E.; Koleini, N. The Role of FGF2 isoforms in Cell Survival in the Heart. In Biochemistry of Apoptosis and Autophagy; Springer: Cham, Switzerland, 2022; pp. 269–283. [Google Scholar]
- Kardami, E.; Fandrich, R.R. Basic fibroblast growth factor in atria and ventricles of the vertebrate heart. J. Cell Biol. 1989, 109, 1865–1875. [Google Scholar] [CrossRef]
- Pellieux, C.; Foletti, A.; Peduto, G.; Aubert, J.F.; Nussberger, J.; Beermann, F.; Brunner, H.R.; Pedrazzini, T. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J. Clin. Investig. 2001, 108, 1843–1851. [Google Scholar] [CrossRef]
- Cheng, G.C.; Briggs, W.H.; Gerson, D.S.; Libby, P.; Grodzinsky, A.J.; Gray, M.L.; Lee, R.T. Mechanical strain tightly controls fibroblast growth factor-2 release from cultured human vascular smooth muscle cells. Circ. Res. 1997, 80, 28–36. [Google Scholar] [CrossRef]
- Ku, P.T.; D’Amore, P.A. Regulation of basic fibroblast growth factor (bFGF) gene and protein expression following its release from sublethally injured endothelial cells. J. Cell. Biochem. 1995, 58, 328–343. [Google Scholar] [CrossRef]
- Dolivo, D.M. Anti-fibrotic effects of pharmacologic FGF-2: A review of recent literature. J. Mol. Med. 2022, 100, 847–860. [Google Scholar] [CrossRef]
- Qu, Z.; Liebler, J.M.; Powers, M.R.; Galey, T.; Ahmadi, P.; Huang, X.N.; Ansel, J.C.; Butterfield, J.H.; Planck, S.R.; Rosenbaum, J.T. Mast cells are a major source of basic fibroblast growth factor in chronic inflammation and cutaneous hemangioma. Am. J. Pathol. 1995, 147, 564–573. [Google Scholar] [PubMed]
- Prussin, C.; Metcalfe, D.D. 5. IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol. 2006, 117, S450–S456. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Euler, G. Good and bad sides of TGFbeta-signaling in myocardial infarction. Front. Physiol. 2015, 6, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, D.W.; Lopez, A.R.; Thomas, P.S.; Peck, C.; Gold, W.M. Dog mastocytoma cells produce transforming growth factor beta 1. J. Clin. Investig. 1992, 90, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, K.A.; Wang, Y.; Shiota, N.; Saarinen, J.; Hyytiainen, M.; Kokkonen, J.O.; Keski-Oja, J.; Kovanen, P.T. Activation of paracrine TGF-beta1 signaling upon stimulation and degranulation of rat serosal mast cells: A novel function for chymase. FASEB J. 2001, 15, 1377–1388. [Google Scholar] [CrossRef] [Green Version]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, G.E.; Chambers, R.C.; Papakrivopoulou, J.; Dawson, S.J.; Jacobsen, M.C.; Bishop, J.E.; Laurent, G.J. Activation of fibroblast procollagen alpha 1(I) transcription by mechanical strain is transforming growth factor-beta-dependent and involves increased binding of CCAAT-binding factor (CBF/NF-Y) at the proximal promoter. J. Biol. Chem. 2002, 277, 6153–6161. [Google Scholar] [CrossRef] [Green Version]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Wollert, K.C.; Taga, T.; Saito, M.; Narazaki, M.; Kishimoto, T.; Glembotski, C.C.; Vernallis, A.B.; Heath, J.K.; Pennica, D.; Wood, W.I.; et al. Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series VIA gp130/leukemia inhibitory factor receptor-dependent pathways. J. Biol. Chem. 1996, 271, 9535–9545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, K.L.; Winer, J.; Phillips, D.M.; Quach, J.; Williams, P.M.; Mather, J.P. Phenylephrine, endothelin, prostaglandin F2alpha’ and leukemia inhibitory factor induce different cardiac hypertrophy phenotypes in vitro. Endocrine 1998, 9, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ancey, C.; Corbi, P.; Froger, J.; Delwail, A.; Wijdenes, J.; Gascan, H.; Potreau, D.; Lecron, J.C. Secretion of IL-6, IL-11 and LIF by human cardiomyocytes in primary culture. Cytokine 2002, 18, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Ancey, C.; Menet, E.; Corbi, P.; Fredj, S.; Garcia, M.; Rucker-Martin, C.; Bescond, J.; Morel, F.; Wijdenes, J.; Lecron, J.C.; et al. Human cardiomyocyte hypertrophy induced in vitro by gp130 stimulation. Cardiovasc. Res. 2003, 59, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Saito, Y.; Harada, M.; Ishikawa, M.; Ogawa, E.; Miyamoto, Y.; Hamanaka, I.; Kamitani, S.; Kajiyama, N.; Takahashi, N.; et al. Involvement of cardiotrophin-1 in cardiac myocyte-nonmyocyte interactions during hypertrophy of rat cardiac myocytes in vitro. Circulation 1999, 100, 1116–1124. [Google Scholar] [CrossRef] [Green Version]
- Gilles, S.; Zahler, S.; Welsch, U.; Sommerhoff, C.P.; Becker, B.F. Release of TNF-alpha during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovasc. Res. 2003, 60, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.R.; Galli, S.J. Mast cells as a source of both preformed and immunologically inducible TNF-alpha/cachectin. Nature 1990, 346, 274–276. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Kaartinen, M.; Penttila, A.; Kovanen, P.T. Mast cells in rupture-prone areas of human coronary atheromas produce and store TNF-alpha. Circulation 1996, 94, 2787–2792. [Google Scholar] [CrossRef]
- Bowers, S.L.; Banerjee, I.; Baudino, T.A. The extracellular matrix: At the center of it all. J. Mol. Cell. Cardiol. 2010, 48, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Kanekar, S.; Hirozanne, T.; Terracio, L.; Borg, T.K. Cardiac fibroblasts form and function. Cardiovasc. Pathol. 1998, 7, 127–133. [Google Scholar] [CrossRef]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, D.J. Circulating and tissue angiotensin systems. J. Clin. Investig. 1987, 79, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Dinh, D.T.; Frauman, A.G.; Johnston, C.I.; Fabiani, M.E. Angiotensin receptors: Distribution, signalling and function. Clin. Sci. 2001, 100, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Lindpaintner, K.; Ganten, D. The cardiac renin-angiotensin system. An appraisal of present experimental and clinical evidence. Circ. Res. 1991, 68, 905–921. [Google Scholar] [CrossRef]
- Baker, K.M.; Booz, G.W.; Dostal, D.E. Cardiac actions of angiotensin II: Role of an intracardiac renin-angiotensin system. Annu. Rev. Physiol. 1992, 54, 227–241. [Google Scholar] [CrossRef]
- Lee, M.A.; Bohm, M.; Paul, M.; Ganten, D. Tissue renin-angiotensin systems. Their role in cardiovascular disease. Circulation 1993, 87, IV7–IV13. [Google Scholar] [PubMed]
- Campbell, D.J.; Habener, J.F. Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat. J. Clin. Investig. 1986, 78, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.B.; Reid, A.C.; Mackins, C.J.; Askwith, T.; Schaefer, U.; Herzlinger, D.; Levi, R. Mast cells: A unique source of renin. Proc. Natl. Acad. Sci. USA 2004, 101, 13607–13612. [Google Scholar] [CrossRef]
- Urata, H.; Hoffmann, S.; Ganten, D. Tissue angiotensin II system in the human heart. Eur. Heart J. 1994, 15, 68–78. [Google Scholar] [CrossRef]
- McEuen, A.R.; Sharma, B.; Walls, A.F. Regulation of the activity of human chymase during storage and release from mast cells: The contributions of inorganic cations, pH, heparin and histamine. Biochim. Biophys. Acta 1995, 1267, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, N.; Rysa, J.; Kovanen, P.T.; Ruskoaho, H.; Kokkonen, J.O.; Lindstedt, K.A. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J. Hypertens. 2003, 21, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.V.; Fagard, R.H.; Lijnen, P.J. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension 2002, 39, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, K.T. Fibrosis and hypertensive heart disease. Curr. Opin. Cardiol. 2000, 15, 264–272. [Google Scholar] [CrossRef]
- Leurs, R.; Bakker, R.A.; Timmerman, H.; de Esch, I.J. The histamine H3 receptor: From gene cloning to H3 receptor drugs. Nat. Rev. Drug. Discov. 2005, 4, 107–120. [Google Scholar] [CrossRef]
- Hough, L.B. Genomics meets histamine receptors: New subtypes, new receptors. Mol. Pharm. 2001, 59, 415–419. [Google Scholar] [CrossRef]
- Levick, S.P. Histamine receptors in heart failure. Heart Fail. Rev. 2022, 27, 1355–1372. [Google Scholar] [CrossRef]
- Matsuda, N.; Jesmin, S.; Takahashi, Y.; Hatta, E.; Kobayashi, M.; Matsuyama, K.; Kawakami, N.; Sakuma, I.; Gando, S.; Fukui, H.; et al. Histamine H1 and H2 receptor gene and protein levels are differentially expressed in the hearts of rodents and humans. J. Pharm. Exp. Ther. 2004, 309, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; Ganellin, C.R.; Timmerman, H.; Schwartz, J.C.; Shankley, N.P.; Young, J.M.; Schunack, W.; Levi, R.; Haas, H.L. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharm. Rev. 1997, 49, 253–278. [Google Scholar]
- Eckel, L.; Gristwood, R.W.; Nawrath, H.; Owen, D.A.; Satter, P. Inotropic and electrophysiological effects of histamine on human ventricular heart muscle. J. Physiol. 1982, 330, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.Y.; Schoemaker, R.G.; Bax, W.A.; Bos, E.; Saxena, P.R. Effects of histamine on porcine isolated myocardium: Differentiation from effects on human tissue. J. Cardiovasc. Pharm. 1993, 22, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y. Cardiac histamine receptors: Their pharmacological consequences and signal transduction pathways. Methods Find. Exp. Clin. Pharm. 1999, 21, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kirch, W.; Halabi, A.; Hinrichsen, H. Hemodynamic effects of quinidine and famotidine in patients with congestive heart failure. Clin. Pharm. Ther. 1992, 51, 325–333. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Cai, W.-K.; Yin, S.-J.; Wang, P.; Li, Z.-R.; Yang, Q.; Zhou, T.; Meng, R.; Yang, M.; Guo, Y. Histamine H2 receptor antagonist exposure was related to decreased all-cause mortality in critical ill patients with heart failure: A cohort study. Eur. J. Prev. Cardiol. 2022, 29, 1854–1865. [Google Scholar] [CrossRef]
- Kim, J.; Ogai, A.; Nakatani, S.; Hashimura, K.; Kanzaki, H.; Komamura, K.; Asakura, M.; Asanuma, H.; Kitamura, S.; Tomoike, H.; et al. Impact of blockade of histamine H2 receptors on chronic heart failure revealed by retrospective and prospective randomized studies. J. Am. Coll. Cardiol. 2006, 48, 1378–1384. [Google Scholar] [CrossRef] [Green Version]
- Galli, S.J. New concepts about the mast cell. N. Engl. J. Med. 1993, 328, 257–265. [Google Scholar] [CrossRef]
- Bradding, P.; Feather, I.H.; Howarth, P.H.; Mueller, R.; Roberts, J.A.; Britten, K.; Bews, J.P.; Hunt, T.C.; Okayama, Y.; Heusser, C.H.; et al. Interleukin 4 is localized to and released by human mast cells. J. Exp. Med. 1992, 176, 1381–1386. [Google Scholar] [CrossRef] [Green Version]
- Ohkawara, Y.; Yamauchi, K.; Tanno, Y.; Tamura, G.; Ohtani, H.; Nagura, H.; Ohkuda, K.; Takishima, T. Human lung mast cells and pulmonary macrophages produce tumor necrosis factor-alpha in sensitized lung tissue after IgE receptor triggering. Am. J. Respir. Cell. Mol. Biol. 1992, 7, 385–392. [Google Scholar] [CrossRef]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Sugden, P.H.; Clerk, A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ. Res. 1998, 83, 345–352. [Google Scholar] [CrossRef]
- Yamauchi-Takihara, K.; Hirota, H.; Kunisada, K.; Matsui, H.; Fujio, Y.; Taga, T.; Kishimoto, T. Roles of gp130 signaling pathways in cardiac myocytes: Recent advances and implications for cardiovascular disease. J. Card. Fail. 1996, 2, S63–S68. [Google Scholar] [CrossRef]
- Plenz, G.; Song, Z.F.; Tjan, T.D.; Koenig, C.; Baba, H.A.; Erren, M.; Flesch, M.; Wichter, T.; Scheld, H.H.; Deng, M.C. Activation of the cardiac interleukin-6 system in advanced heart failure. Eur. J. Heart Fail. 2001, 3, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Dostal, D.E.; Baker, K.M. The cardiac renin-angiotensin system: Conceptual, or a regulator of cardiac function? Circ. Res. 1999, 85, 643–650. [Google Scholar] [CrossRef]
- Krop, M.; Danser, A.H. Circulating versus tissue renin-angiotensin system: On the origin of (pro)renin. Curr. Hypertens. Rep. 2008, 10, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; da Silva, J.; Alencar, A.; Zapata-Sudo, G.; Lin, M.S.; Sun, X.; Ahmad, S.; Ferrario, C.M.; Groban, L. Mast cell inhibition attenuates cardiac remodeling and diastolic dysfunction in middle-aged, ovariectomized Fischer344× Brown Norway rats. J. Cardiovasc. Pharmacol. 2016, 68, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Azzawi, M.; Kan, S.W.; Hillier, V.; Yonan, N.; Hutchinson, I.V.; Hasleton, P.S. The distribution of cardiac macrophages in myocardial ischaemia and cardiomyopathy. Histopathology 2005, 46, 314–319. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Hinglais, N.; Heudes, D.; Nicoletti, A.; Mandet, C.; Laurent, M.; Bariety, J.; Michel, J.B. Colocalization of myocardial fibrosis and inflammatory cells in rats. Lab. Investig. 1994, 70, 286–294. [Google Scholar] [PubMed]
- Nicoletti, A.; Heudes, D.; Mandet, C.; Hinglais, N.; Bariety, J.; Michel, J.B. Inflammatory cells and myocardial fibrosis: Spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc. Res. 1996, 32, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Niiyama, H.; Tahara, N.; Kusaba, K.; Takemiya, K.; Jalalidin, A.; Koga, M.; Nagata, T.; et al. Roles of intercellular adhesion molecule-1 in hypertensive cardiac remodeling. Hypertension 2003, 41, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Capers, Q.t.; Alexander, R.W.; Lou, P.; De Leon, H.; Wilcox, J.N.; Ishizaka, N.; Howard, A.B.; Taylor, W.R. Monocyte chemoattractant protein-1 expression in aortic tissues of hypertensive rats. Hypertension 1997, 30, 1397–1402. [Google Scholar] [CrossRef]
- Reape, T.J.; Groot, P.H. Chemokines and atherosclerosis. Atherosclerosis 1999, 147, 213–225. [Google Scholar] [CrossRef]
- Bratton, D.L.; Henson, P.M. Neutrophil clearance: When the party is over, clean-up begins. Trends Immunol. 2011, 32, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef]
- Bennouna, S.; Bliss, S.K.; Curiel, T.J.; Denkers, E.Y. Cross-talk in the innate immune system: Neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J. Immunol. 2003, 171, 6052–6058. [Google Scholar] [CrossRef] [PubMed]
- Chertov, O.; Ueda, H.; Xu, L.L.; Tani, K.; Murphy, W.J.; Wang, J.M.; Howard, O.M.; Sayers, T.J.; Oppenheim, J.J. Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J. Exp. Med. 1997, 186, 739–747. [Google Scholar] [CrossRef]
- Tsuda, Y.; Takahashi, H.; Kobayashi, M.; Hanafusa, T.; Herndon, D.N.; Suzuki, F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 2004, 21, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Bui, T.A.; Jickling, G.C.; Winship, I.R. Neutrophil dynamics and inflammaging in acute ischemic stroke: A transcriptomic review. Front. Aging Neurosci. 2022, 14, 1041333. [Google Scholar] [CrossRef] [PubMed]
- Weisheit, C.; Zhang, Y.; Faron, A.; Kopke, O.; Weisheit, G.; Steinstrasser, A.; Frede, S.; Meyer, R.; Boehm, O.; Hoeft, A.; et al. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure-overload. PLoS ONE 2014, 9, e112710. [Google Scholar] [CrossRef] [PubMed]
- Weisheit, C.K.; Kleiner, J.L.; Rodrigo, M.B.; Niepmann, S.T.; Zimmer, S.; Duerr, G.D.; Coburn, M.; Kurts, C.; Frede, S.; Eichhorn, L. CX3CR1 is a prerequisite for the development of cardiac hypertrophy and left ventricular dysfunction in mice upon transverse aortic constriction. PLoS ONE 2021, 16, e0243788. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.D. Neutrophil transendothelial migration: Updates and new perspectives. Blood 2019, 133, 2149–2158. [Google Scholar] [CrossRef]
- Woodfin, A.; Voisin, M.B.; Imhof, B.A.; Dejana, E.; Engelhardt, B.; Nourshargh, S. Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2, JAM-A, and PECAM-1. Blood 2009, 113, 6246–6257. [Google Scholar] [CrossRef] [Green Version]
- Sahni, A.; Sahni, S.K.; Francis, C.W. Endothelial cell activation by IL-1beta in the presence of fibrinogen requires alphavbeta3. Arter. Thromb. Vasc. Biol. 2005, 25, 2222–2227. [Google Scholar] [CrossRef] [Green Version]
- Mackay, F.; Loetscher, H.; Stueber, D.; Gehr, G.; Lesslauer, W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993, 177, 1277–1286. [Google Scholar] [CrossRef] [Green Version]
- Asako, H.; Kurose, I.; Wolf, R.; DeFrees, S.; Zheng, Z.L.; Phillips, M.L.; Paulson, J.C.; Granger, D.N. Role of H1 receptors and P-selectin in histamine-induced leukocyte rolling and adhesion in postcapillary venules. J. Clin. Investig. 1994, 93, 1508–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashiyama, H.; Sugai, M.; Inoue, H.; Mizuyachi, K.; Kushida, H.; Asano, S.; Kinoshita, M. Histopathological study of time course changes in inter-renal aortic banding-induced left ventricular hypertrophy of mice. Int. J. Exp. Pathol. 2007, 88, 31–38. [Google Scholar] [CrossRef] [PubMed]
- van Gisbergen, K.P.; Sanchez-Hernandez, M.; Geijtenbeek, T.B.; van Kooyk, Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J. Exp. Med. 2005, 201, 1281–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Ciz, M.; Denev, P.; Kratchanova, M.; Vasicek, O.; Ambrozova, G.; Lojek, A. Flavonoids inhibit the respiratory burst of neutrophils in mammals. Oxid. Med. Cell. Longev. 2012, 2012, 181295. [Google Scholar] [CrossRef] [Green Version]
- Ozyilmaz, S.; Akgul, O.; Uyarel, H.; Pusuroglu, H.; Gul, M.; Satilmisoglu, M.H.; Bolat, I.; Ozyilmaz, I.; Uçar, H.; Yildirim, A. The importance of the neutrophil-to-lymphocyte ratio in patients with hypertrophic cardiomyopathy. Rev. Port. Cardiol. 2017, 36, 239–246. [Google Scholar] [CrossRef]
- Fries, R.C.; Kadotani, S.; Stack, J.P.; Kruckman, L.; Wallace, G. Prognostic Value of Neutrophil-to-Lymphocyte Ratio in Cats With Hypertrophic Cardiomyopathy. Front. Vet. Sci. 2022, 9, 813524. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Z.; Li, Y.; Chen, X.; He, S. Relation between lymphocyte to monocyte ratio and survival in patients with hypertrophic cardiomyopathy: A retrospective cohort study. PeerJ 2022, 10, e13212. [Google Scholar] [CrossRef]
- Cordero-Reyes, A.M.; Youker, K.A.; Trevino, A.R.; Celis, R.; Hamilton, D.J.; Flores-Arredondo, J.H.; Orrego, C.M.; Bhimaraj, A.; Estep, J.D.; Torre-Amione, G. Full expression of cardiomyopathy is partly dependent on B-cells: A pathway that involves cytokine activation, immunoglobulin deposition, and activation of apoptosis. J. Am. Heart Assoc. 2016, 5, e002484. [Google Scholar] [CrossRef] [Green Version]
- Sobirin, M.A.; Kinugawa, S.; Takahashi, M.; Fukushima, A.; Homma, T.; Ono, T.; Hirabayashi, K.; Suga, T.; Azalia, P.; Takada, S. Activation of natural killer T cells ameliorates postinfarct cardiac remodeling and failure in mice. Circ. Res. 2012, 111, 1037–1047. [Google Scholar] [CrossRef]
- Wang, H.-X.; Li, W.-J.; Hou, C.-L.; Lai, S.; Zhang, Y.-L.; Tian, C.; Yang, H.; Du, J.; Li, H.-H. CD1d-dependent natural killer T cells attenuate angiotensin II-induced cardiac remodelling via IL-10 signalling in mice. Cardiovasc. Res. 2019, 115, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kinugawa, S.; Takada, S.; Kakutani, N.; Furihata, T.; Sobirin, M.A.; Fukushima, A.; Obata, Y.; Saito, A.; Ishimori, N. The disruption of invariant natural killer T cells exacerbates cardiac hypertrophy and failure caused by pressure overload in mice. Exp. Physiol. 2020, 105, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Ayach, B.B.; Yoshimitsu, M.; Dawood, F.; Sun, M.; Arab, S.; Chen, M.; Higuchi, K.; Siatskas, C.; Lee, P.; Lim, H. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc. Natl. Acad. Sci. USA 2006, 103, 2304–2309. [Google Scholar] [CrossRef]
- Santhosh, S.; Bahl, A.; Saikia, U.N.; Lad, D.; Mittal, B.R.; Malhotra, P.; Varma, S. FDG PET/CT in the staging and follow-up of primary cardiac ‘T’cell lymphoma presenting as hypertrophic cardiomyopathy. J. Nucl. Cardiol. 2016, 23, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Suzuki, N.; Mizue, N.; Hatakeyama, N.; Takamuro, M.; Tsutsumi, H. Relapse of T-cell all after stem cell transplant presenting as hypertrophic cardiomyopathy: The value of non-invasive diagnostic imaging in detecting cardiac leukemia. Pediatr. Blood Cancer 2006, 46, 108–111. [Google Scholar] [CrossRef]
- Isaka, M.; Kudo, A.; Imamura, M.; Kawakami, H.; Yasuda, K. Endothelin receptors, localized in sympathetic nerve terminals of the heart, modulate norepinephrine release and reperfusion arrhythmias. Basic Res. Cardiol. 2007, 102, 154–162. [Google Scholar] [CrossRef]
- Lehmann, L.H.; Stanmore, D.A.; Backs, J. The role of endothelin-1 in the sympathetic nervous system in the heart. Life Sci. 2014, 118, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.H.; Rostosky, J.S.; Buss, S.J.; Kreusser, M.M.; Krebs, J.; Mier, W.; Enseleit, F.; Spiger, K.; Hardt, S.E.; Wieland, T.; et al. Essential role of sympathetic endothelin A receptors for adverse cardiac remodeling. Proc. Natl. Acad. Sci. USA 2014, 111, 13499–13504. [Google Scholar] [CrossRef]
- Reid, A.C.; Mackins, C.J.; Seyedi, N.; Levi, R.; Silver, R.B. Coupling of angiotensin II AT1 receptors to neuronal NHE activity and carrier-mediated norepinephrine release in myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1448–H1454. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, M.; Lubian-Gutierrez, M.; Cascales-Poyatos, H.M.; Perez-Reviriego, A.A.; Castellano-Martinez, A. Role of the Renin–Angiotensin–Aldosterone System in Dystrophin-Deficient Cardiomyopathy. Int. J. Mol. Sci. 2020, 22, 356. [Google Scholar] [CrossRef]
- MacKenna, D.; Summerour, S.R.; Villarreal, F.J. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc. Res. 2000, 46, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadoshima, J.; Xu, Y.; Slayter, H.S.; Izumo, S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 1993, 75, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Sadoshima, J.; Brosius III, F.C.; Izumo, S. Mechanical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes in vitro. Circ. Res. 1999, 85, 137–146. [Google Scholar] [CrossRef]
- Cohn, J.N.; Tognoni, G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N. Engl. J. Med. 2001, 345, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Poole-Wilson, P.A.; Segal, R.; Martinez, F.A.; Dickstein, K.; Camm, A.J.; Konstam, M.A.; Riegger, G.; Klinger, G.H.; Neaton, J. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: Randomised trial—The Losartan Heart Failure Survival Study ELITE II. Lancet 2000, 355, 1582–1587. [Google Scholar] [CrossRef]
- Watanabe, H.; Martini, A.G.; Brown, E.A.; Liang, X.; Medrano, S.; Goto, S.; Narita, I.; Arend, L.J.; Sequeira-Lopez, M.L.S.; Gomez, R.A. Inhibition of the renin-angiotensin system causes concentric hypertrophy of renal arterioles in mice and humans. JCI Insight 2021, 6, e154337. [Google Scholar] [CrossRef]
- Fan, Z.; Wu, G.; Yue, M.; Ye, J.; Chen, Y.; Xu, B.; Shu, Z.; Zhu, J.; Lu, N.; Tan, X. Hypertension and hypertensive left ventricular hypertrophy are associated with ACE2 genetic polymorphism. Life Sci. 2019, 225, 39–45. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Hafstad, A.D.; Nabeebaccus, A.A.; Shah, A.M. Novel aspects of ROS signalling in heart failure. Basic Res. Cardiol. 2013, 108, 359. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res. 2009, 81, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Carnicer, R.; Crabtree, M.J.; Sivakumaran, V.; Casadei, B.; Kass, D.A. Nitric oxide synthases in heart failure. Antioxid. Redox Signal. 2013, 18, 1078–1099. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tocchetti, C.G.; Krieg, T.; Moens, A.L. Oxidative and nitrosative stress in the maintenance of myocardial function. Free. Radic. Biol. Med. 2012, 53, 1531–1540. [Google Scholar] [CrossRef]
- Nediani, C.; Raimondi, L.; Borchi, E.; Cerbai, E. Nitric oxide/reactive oxygen species generation and nitroso/redox imbalance in heart failure: From molecular mechanisms to therapeutic implications. Antioxid. Redox Signal. 2011, 14, 289–331. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Shah, A.M.; MacCarthy, P.A. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol. Ther. 2000, 86, 49–86. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free. Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Hare, J.M. Nitroso-redox balance in the cardiovascular system. N. Engl. J. Med. 2004, 351, 2112–2114. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [Green Version]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadoshima, J.; Izumo, S. The heterotrimeric G q protein-coupled angiotensin II receptor activates p21 ras via the tyrosine kinase-Shc-Grb2-Sos pathway in cardiac myocytes. EMBO J. 1996, 15, 775–787. [Google Scholar] [CrossRef]
- Kala, P.; Gawrys, O.; Miklovič, M.; Vaňourková, Z.; Škaroupková, P.; Jíchová, Š.; Sadowski, J.; Kompanowska-Jezierska, E.; Walkowska, A.; Veselka, J. Endothelin type A receptor blockade attenuates aorto-caval fistula-induced heart failure in rats with angiotensin II-dependent hypertension. J. Hypertens. 2023, 41, 99–114. [Google Scholar] [CrossRef]
- Jiang, Z.S.; Jeyaraman, M.; Wen, G.B.; Fandrich, R.R.; Dixon, I.M.; Cattini, P.A.; Kardami, E. High- but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. J. Mol. Cell. Cardiol. 2007, 42, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Komuro, I.; Zou, Y.; Kudoh, S.; Kijima, K.; Matsubara, H.; Sugaya, T.; Murakami, K.; Yazaki, Y. Acute pressure overload could induce hypertrophic responses in the heart of angiotensin II type 1a knockout mice. Circ. Res. 1998, 82, 779–785. [Google Scholar] [CrossRef]
- Harada, K.; Komuro, I.; Shiojima, I.; Hayashi, D.; Kudoh, S.; Mizuno, T.; Kijima, K.; Matsubara, H.; Sugaya, T.; Murakami, K.; et al. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 1998, 97, 1952–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, A.D.; Turu, G.; Hunyady, L.; Balla, A. Novel mechanisms of G-protein-coupled receptors functions: AT1 angiotensin receptor acts as a signaling hub and focal point of receptor cross-talk. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hanada, K.; Gareri, C.; Rockman, H.A. Mechanoactivation of the angiotensin II type 1 receptor induces β-arrestin-biased signaling through Gαi coupling. J. Cell. Biochem. 2018, 119, 3586–3597. [Google Scholar] [CrossRef] [Green Version]
- Hunyady, L.; Turu, G. The role of the AT1 angiotensin receptor in cardiac hypertrophy: Angiotensin II receptor or stretch sensor? Trends Endocrinol. Metab. 2004, 15, 405–408. [Google Scholar] [CrossRef]
- Brancaccio, M.; Fratta, L.; Notte, A.; Hirsch, E.; Poulet, R.; Guazzone, S.; De Acetis, M.; Vecchione, C.; Marino, G.; Altruda, F.; et al. Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat. Med. 2003, 9, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Knoll, R.; Hoshijima, M.; Hoffman, H.M.; Person, V.; Lorenzen-Schmidt, I.; Bang, M.L.; Hayashi, T.; Shiga, N.; Yasukawa, H.; Schaper, W.; et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 2002, 111, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saucerman, J.J.; Tan, P.M.; Buchholz, K.S.; McCulloch, A.D.; Omens, J.H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019, 16, 361–378. [Google Scholar] [CrossRef]
- Tan, P.M.; Buchholz, K.S.; Omens, J.H.; McCulloch, A.D.; Saucerman, J.J. Predictive model identifies key network regulators of cardiomyocyte mechano-signaling. PLoS Comput. Biol. 2017, 13, e1005854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Border, W.A.; Noble, N.A. Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar] [CrossRef]
- Yang, Y.C.; Piek, E.; Zavadil, J.; Liang, D.; Xie, D.; Heyer, J.; Pavlidis, P.; Kucherlapati, R.; Roberts, A.B.; Bottinger, E.P. Hierarchical model of gene regulation by transforming growth factor beta. Proc. Natl. Acad. Sci. USA 2003, 100, 10269–10274. [Google Scholar] [CrossRef]
- Verrecchia, F.; Chu, M.L.; Mauviel, A. Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 2001, 276, 17058–17062. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Ren, G.; Kweon, H.J.; Dobaczewski, M.; Reddy, A.; Taffet, G.; Wang, X.F.; Frangogiannis, N.G. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 2007, 116, 2127–2138. [Google Scholar] [CrossRef] [Green Version]
- Ryer, E.J.; Hom, R.P.; Sakakibara, K.; Nakayama, K.I.; Nakayama, K.; Faries, P.L.; Liu, B.; Kent, K.C. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2006, 26, 780–786. [Google Scholar] [CrossRef]
- Divakaran, V.; Adrogue, J.; Ishiyama, M.; Entman, M.L.; Haudek, S.; Sivasubramanian, N.; Mann, D.L. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ. Heart Fail. 2009, 2, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Frantz, S.; Hu, K.; Adamek, A.; Wolf, J.; Sallam, A.; Maier, S.K.; Lonning, S.; Ling, H.; Ertl, G.; Bauersachs, J. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res. Cardiol. 2008, 103, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.J.; Jonker, L.; Arthur, H.M. A direct interaction between TGFbeta activated kinase 1 and the TGFbeta type II receptor: Implications for TGFbeta signalling and cardiac hypertrophy. Cardiovasc. Res. 2006, 69, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gaussin, V.; Taffet, G.E.; Belaguli, N.S.; Yamada, M.; Schwartz, R.J.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000, 6, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.A.; Moukarbel, G.V.; Gupta, R.; Frank, S.M.; Anderson, A.M.; Liu, L.C.; Khouri, S.J. Endothelin 1 is associated with heart failure hospitalization and long-term mortality in patients with heart failure with preserved ejection fraction and pulmonary hypertension. Cardiology 2019, 143, 124–133. [Google Scholar] [CrossRef]
- Jankowich, M.; Choudhary, G. Endothelin-1 levels and cardiovascular events. Trends Cardiovasc. Med. 2020, 30, 1–8. [Google Scholar] [CrossRef]
- Kedzierski, R.M.; Yanagisawa, M. Endothelin system: The double-edged sword in health and disease. Annu. Rev. Pharm. Toxicol. 2001, 41, 851–876. [Google Scholar] [CrossRef]
- Hilal-Dandan, R.; Merck, D.T.; Lujan, J.P.; Brunton, L.L. Coupling of the type A endothelin receptor to multiple responses in adult rat cardiac myocytes. Mol. Pharm. 1994, 45, 1183–1190. [Google Scholar]
- Sano, F.K.; Akasaka, H.; Shihoya, W.; Nureki, O. Cryo-EM structure of the endothelin-1-ETB-Gi complex. Elife 2023, 12, e85821. [Google Scholar] [CrossRef]
- Sugden, P.H.; Bogoyevitch, M.A. Endothelin-1-dependent signaling pathways in the myocardium. Trends Cardiovasc. Med. 1996, 6, 87–94. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Andersson, M.B.; Gillespie-Brown, J.; Clerk, A.; Glennon, P.E.; Fuller, S.J.; Sugden, P.H. Adrenergic receptor stimulation of the mitogen-activated protein kinase cascade and cardiac hypertrophy. Biochem. J. 1996, 314 Pt 1, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, S.; Miyauchi, T.; Kobayashi, M.; Yamaguchi, I.; Goto, K.; Sugishita, Y. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature 1996, 384, 353–355. [Google Scholar] [CrossRef]
- Nishikimi, T.; Nakagawa, Y. B-Type Natriuretic Peptide (BNP) Revisited—Is BNP Still a Biomarker for Heart Failure in the Angiotensin Receptor/Neprilysin Inhibitor Era? Biology 2022, 11, 1034. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zheng, L.; Gao, P.; Yang, H.; Yang, W.-J.; Guo, F.; Liang, R.; Feng, M.; Wang, Z.; Zhang, Z. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. Cell Stem Cell 2022, 29, 545–558.e13. [Google Scholar] [CrossRef] [PubMed]
- Auchampach, J.; Han, L.; Huang, G.N.; Kühn, B.; Lough, J.W.; O’Meara, C.C.; Payumo, A.Y.; Rosenthal, N.A.; Sucov, H.M.; Yutzey, K.E. Measuring cardiomyocyte cell-cycle activity and proliferation in the age of heart regeneration. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H579–H596. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, H.; Hanna, A.; Humeres, C.; Frangogiannis, N.G. Properties and functions of fibroblasts and myofibroblasts in myocardial infarction. Cells 2022, 11, 1386. [Google Scholar] [CrossRef]
- Rao, Z.; Shen, D.; Chen, J.; Jin, L.; Wu, X.; Chen, M.; Li, L.; Chu, M.; Lin, J. Basic fibroblast growth factor attenuates injury in myocardial infarction by enhancing hypoxia-inducible factor-1 alpha accumulation. Front. Pharmacol. 2020, 11, 1193. [Google Scholar] [CrossRef]
- Jimenez, S.K.; Sheikh, F.; Jin, Y.; Detillieux, K.A.; Dhaliwal, J.; Kardami, E.; Cattini, P.A. Transcriptional regulation of FGF-2 gene expression in cardiac myocytes. Cardiovasc. Res. 2004, 62, 548–557. [Google Scholar] [CrossRef]
- Fortier, S.M.; Penke, L.R.; King, D.; Pham, T.X.; Ligresti, G.; Peters-Golden, M. Myofibroblast dedifferentiation proceeds via distinct transcriptomic and phenotypic transitions. JCI Insight 2021, 6, e144799. [Google Scholar] [CrossRef]
- Santiago, J.J.; Ma, X.; McNaughton, L.J.; Nickel, B.E.; Bestvater, B.P.; Yu, L.; Fandrich, R.R.; Netticadan, T.; Kardami, E. Preferential accumulation and export of high molecular weight FGF-2 by rat cardiac non-myocytes. Cardiovasc. Res. 2011, 89, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Myers, J.; Fang, X.; Stachowiak, E.K.; Maher, P.A.; Martins, G.G.; Popescu, G.; Berezney, R.; Stachowiak, M.K. Integrative nuclear FGFR1 signaling (INFS) pathway mediates activation of the tyrosine hydroxylase gene by angiotensin II, depolarization and protein kinase C. J. Neurochem. 2002, 81, 506–524. [Google Scholar] [CrossRef]
- Tong, G.; Liang, Y.; Xue, M.; Chen, X.; Wang, J.; An, N.; Wang, N.; Chen, Y.; Wang, Y.; Jin, L.; et al. The protective role of bFGF in myocardial infarction and hypoxia cardiomyocytes by reducing oxidative stress via Nrf2. Biochem. Biophys. Res. Commun. 2020, 527, 15–21. [Google Scholar] [CrossRef]
- Gualandris, A.; Urbinati, C.; Rusnati, M.; Ziche, M.; Presta, M. Interaction of high-molecular-weight basic fibroblast growth factor with endothelium: Biological activity and intracellular fate of human recombinant M(r) 24,000 bFGF. J. Cell. Physiol. 1994, 161, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Glennon, P.E.; Andersson, M.B.; Lazou, A.; Marshall, C.J.; Sugden, P.H. Acidic fibroblast growth factor or endothelin-1 stimulate the MAP kinase cascade in cardiac myocytes. Biochem. Soc. Trans. 1993, 21, 358S. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Bodmer, J.; Pietras, D.; Azhar, M.; Doetschman, T.; Schultz Jel, J. Biological functions of the low and high molecular weight protein isoforms of fibroblast growth factor-2 in cardiovascular development and disease. Dev. Dyn. 2009, 238, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.S.; Padua, R.R.; Ju, H.; Doble, B.W.; Jin, Y.; Hao, J.; Cattini, P.A.; Dixon, I.M.; Kardami, E. Acute protection of ischemic heart by FGF-2: Involvement of FGF-2 receptors and protein kinase C. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1071–H1080. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.S.; Srisakuldee, W.; Soulet, F.; Bouche, G.; Kardami, E. Non-angiogenic FGF-2 protects the ischemic heart from injury, in the presence or absence of reperfusion. Cardiovasc. Res. 2004, 62, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Santiago, J.-J.; McNaughton, L.J.; Koleini, N.; Ma, X.; Bestvater, B.; Nickel, B.E.; Fandrich, R.R.; Wigle, J.T.; Freed, D.H.; Arora, R.C. High molecular weight fibroblast growth factor-2 in the human heart is a potential target for prevention of cardiac remodeling. PLoS ONE 2014, 9, e97281. [Google Scholar] [CrossRef]
- Schultz, J.E.; Witt, S.A.; Nieman, M.L.; Reiser, P.J.; Engle, S.J.; Zhou, M.; Pawlowski, S.A.; Lorenz, J.N.; Kimball, T.R.; Doetschman, T. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J. Clin. Investig. 1999, 104, 709–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, R.S.; Yamaguchi, F.; Saya, H.; Bruner, J.M.; Yahanda, A.M.; Donehower, L.A.; Berger, M. Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. J. Neurooncol. 1994, 18, 207–216. [Google Scholar] [CrossRef]
- Reed, M.J.; Purohit, A.; Duncan, L.J.; Singh, A.; Roberts, C.J.; Williams, G.J.; Potter, B.V. The role of cytokines and sulphatase inhibitors in regulating oestrogen synthesis in breast tumours. J. Steroid Biochem. Mol. Biol. 1995, 53, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Halaban, R. Growth factors and melanomas. Semin. Oncol. 1996, 23, 673–681. [Google Scholar]
- Kumar-Singh, S.; Weyler, J.; Martin, M.J.; Vermeulen, P.B.; Van Marck, E. Angiogenic cytokines in mesothelioma: A study of VEGF, FGF-1 and -2, and TGF beta expression. J. Pathol. 1999, 189, 72–78. [Google Scholar] [CrossRef]
- Dow, J.K.; deVere White, R.W. Fibroblast growth factor 2: Its structure and property, paracrine function, tumor angiogenesis, and prostate-related mitogenic and oncogenic functions. Urology 2000, 55, 800–806. [Google Scholar] [CrossRef]
- Auguste, P.; Gursel, D.B.; Lemiere, S.; Reimers, D.; Cuevas, P.; Carceller, F.; Di Santo, J.P.; Bikfalvi, A. Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angiogenesis-dependent and -independent mechanisms. Cancer Res. 2001, 61, 1717–1726. [Google Scholar] [PubMed]
- Lorenz, K.; Schmitt, J.P.; Schmitteckert, E.M.; Lohse, M.J. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 2009, 15, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tomasovic, A.; Brand, T.; Schanbacher, C.; Kramer, S.; Hummert, M.W.; Godoy, P.; Schmidt-Heck, W.; Nordbeck, P.; Ludwig, J.; Homann, S.; et al. Interference with ERK-dimerization at the nucleocytosolic interface targets pathological ERK1/2 signaling without cardiotoxic side-effects. Nat. Commun. 2020, 11, 1733. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, A.; Woodman, O.L.; Cao, A.H.; Drummond, G.R.; Marshall, T.; Kaye, D.M.; Ritchie, R.H. Antioxidant actions contribute to the antihypertrophic effects of atrial natriuretic peptide in neonatal rat cardiomyocytes. Cardiovasc. Res. 2006, 72, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, C.; Deiss, K.; Herrmann, S.; Vidal, M.; Oezkur, M.; Gorski, A.; Weidemann, F.; Lohse, M.J.; Lorenz, K. Interference with ERK(Thr188) phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 7440–7445. [Google Scholar] [CrossRef]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [Green Version]
- Alcalai, R.; Seidman, J.G.; Seidman, C.E. Genetic basis of hypertrophic cardiomyopathy: From bench to the clinics. J. Cardiovasc. Electrophysiol. 2008, 19, 104–110. [Google Scholar] [CrossRef]
- Patel, R.; Nagueh, S.F.; Tsybouleva, N.; Abdellatif, M.; Lutucuta, S.; Kopelen, H.A.; Quinones, M.A.; Zoghbi, W.A.; Entman, M.L.; Roberts, R.; et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2001, 104, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.; Liu, X.; Sparrow, A.; Patel, S.; Zhang, Y.H.; Casadei, B.; Watkins, H.; Redwood, C. Hypertrophic cardiomyopathy mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J. Biol. Chem. 2018, 293, 10487–10499. [Google Scholar] [CrossRef] [Green Version]
- Kehat, I.; Davis, J.; Tiburcy, M.; Accornero, F.; Saba-El-Leil, M.K.; Maillet, M.; York, A.J.; Lorenz, J.N.; Zimmermann, W.H.; Meloche, S.; et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 2011, 108, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Davis, L.C.; Correll, R.N.; Makarewich, C.A.; Schwanekamp, J.A.; Moussavi-Harami, F.; Wang, D.; York, A.J.; Wu, H.; Houser, S.R.; et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016, 165, 1147–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemberton, C.J.; Raudsepp, S.D.; Yandle, T.G.; Cameron, V.A.; Richards, A.M. Plasma cardiotrophin-1 is elevated in human hypertension and stimulated by ventricular stretch. Cardiovasc. Res. 2005, 68, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Fukuda, K.; Kodama, H.; Sano, M.; Takahashi, T.; Makino, S.; Kato, T.; Manabe, T.; Hori, S.; Ogawa, S. Involvement of gp130-mediated signaling in pressure overload-induced activation of the JAK/STAT pathway in rodent heart. Heart Vessel. 1998, 13, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ye, D.; Wang, Z.; Pan, H.; Lu, X.; Wang, M.; Xu, Y.; Yu, J.; Zhang, J.; Zhao, M.; et al. The Role of Interleukin-6 Family Members in Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 9, 818890. [Google Scholar] [CrossRef]
- Freed, D.H.; Moon, M.C.; Borowiec, A.M.; Jones, S.C.; Zahradka, P.; Dixon, I.M. Cardiotrophin-1: Expression in experimental myocardial infarction and potential role in post-MI wound healing. Mol. Cell. Biochem. 2003, 254, 247–256. [Google Scholar] [CrossRef]
- Guseh, J.S.; Rosenzweig, A. Size matters: Finding growth pathways that protect the heart. Cell Res. 2017, 27, 1187–1188. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Fukuda, K.; Kodama, H.; Pan, J.; Saito, M.; Matsuzaki, J.; Takahashi, T.; Makino, S.; Kato, T.; Ogawa, S. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J. Biol. Chem. 2000, 275, 29717–29723. [Google Scholar] [CrossRef] [Green Version]
- Briest, W.; Rassler, B.; Deten, A.; Leicht, M.; Morwinski, R.; Neichel, D.; Wallukat, G.; Ziegelhoffer, T.; Zimmer, H.G. Norepinephrine-induced interleukin-6 increase in rat hearts: Differential signal transduction in myocytes and non-myocytes. Pflug. Arch. 2003, 446, 437–446. [Google Scholar] [CrossRef]
- Sarkar, S.; Vellaichamy, E.; Young, D.; Sen, S. Influence of cytokines and growth factors in ANG II-mediated collagen upregulation by fibroblasts in rats: Role of myocytes. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H107–H117. [Google Scholar] [CrossRef]
- Wang, F.; Trial, J.; Diwan, A.; Gao, F.; Birdsall, H.; Entman, M.; Hornsby, P.; Sivasubramaniam, N.; Mann, D. Regulation of cardiac fibroblast cellular function by leukemia inhibitory factor. J. Mol. Cell. Cardiol. 2002, 34, 1309–1316. [Google Scholar] [CrossRef]
- Ping, Y.; Wang, X.; Dai, Y.; Wang, D.; Liu, W.; Yu, P.; Tao, Z. A quantitative detection of Cardiotrophin-1 in chronic heart failure by chemiluminescence immunoassay. J. Clin. Lab. Anal. 2021, 35, e23570. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Knowlton, K.; Chen, J.; Hoshijima, M.; Brown, J.H.; Chien, K.R. Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via a mitogen-activated protein kinase-dependent pathway. Divergence from downstream CT-1 signals for myocardial cell hypertrophy. J. Biol. Chem. 1997, 272, 5783–5791. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S.; Jenkins, B.J.; Garbers, C.; Moll, J.M.; Scheller, J. Targeting IL-6 trans-signalling: Past, present and future prospects. Nat. Rev. Immunol. 2023, 1–16. [Google Scholar] [CrossRef]
- Kunisada, K.; Negoro, S.; Tone, E.; Funamoto, M.; Osugi, T.; Yamada, S.; Okabe, M.; Kishimoto, T.; Yamauchi-Takihara, K. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc. Natl. Acad. Sci. USA 2000, 97, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Hirota, H.; Chen, J.; Betz, U.A.; Rajewsky, K.; Gu, Y.; Ross, J., Jr.; Muller, W.; Chien, K.R. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 1999, 97, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uozumi, H.; Hiroi, Y.; Zou, Y.; Takimoto, E.; Toko, H.; Niu, P.; Shimoyama, M.; Yazaki, Y.; Nagai, R.; Komuro, I. gp130 plays a critical role in pressure overload-induced cardiac hypertrophy. J. Biol. Chem. 2001, 276, 23115–23119. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., II; Force, T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Investig. 2005, 115, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Komuro, I.; Kudoh, S.; Zou, Y.; Shiojima, I.; Mizuno, T.; Takano, H.; Hiroi, Y.; Ueki, K.; Tobe, K.; et al. Mechanical stress activates protein kinase cascade of phosphorylation in neonatal rat cardiac myocytes. J. Clin. Investig. 1995, 96, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Feng, J.; Song, S.; Li, H.; Yang, H.; Zhou, B.; Li, Y.; Yue, Z.; Lian, H.; Liu, L.; et al. gp130 Controls Cardiomyocyte Proliferation and Heart Regeneration. Circulation 2020, 142, 967–982. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.; Del Re, D.P.; Zhai, P.; Ikeda, S.; Shirakabe, A.; Mizushima, W.; Miyamoto, S.; Brown, J.H.; Sadoshima, J. Yes-associated protein (YAP) mediates adaptive cardiac hypertrophy in response to pressure overload. J. Biol. Chem. 2019, 294, 3603–3617. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Del Re, D.P.; Nakano, N.; Sciarretta, S.; Zhai, P.; Park, J.; Sayed, D.; Shirakabe, A.; Matsushima, S.; Park, Y.; et al. miR-206 Mediates YAP-Induced Cardiac Hypertrophy and Survival. Circ. Res. 2015, 117, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Kashihara, T.; Mukai, R.; Oka, S.I.; Zhai, P.; Nakada, Y.; Yang, Z.; Mizushima, W.; Nakahara, T.; Warren, J.S.; Abdellatif, M.; et al. YAP mediates compensatory cardiac hypertrophy through aerobic glycolysis in response to pressure overload. J. Clin. Investig. 2022, 132, e150595. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Dhalla, N.S. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells 2022, 11, 3336. [Google Scholar] [CrossRef]
- Luo, M.; Chen, P.P.; Yang, L.; Wang, P.; Lu, Y.L.; Shi, F.G.; Gao, Y.; Xu, S.F.; Gong, Q.H.; Xu, R.X.; et al. Sodium ferulate inhibits myocardial hypertrophy induced by abdominal coarctation in rats: Involvement of cardiac PKC and MAPK signaling pathways. Biomed. Pharmacother. 2019, 112, 108735. [Google Scholar] [CrossRef]
- Pudewell, S.; Wittich, C.; Kazemein Jasemi, N.S.; Bazgir, F.; Ahmadian, M.R. Accessory proteins of the RAS-MAPK pathway: Moving from the side line to the front line. Commun. Biol. 2021, 4, 696. [Google Scholar] [CrossRef]
- Nauth, T.; Bazgir, F.; Voß, H.; Brandenstein, L.I.; Mosaddeghzadeh, N.; Rickassel, V.; Deden, S.; Gorzelanny, C.; Schlüter, H.; Ahmadian, M.R.; et al. Cutaneous manifestations in Costello syndrome: HRAS p.Gly12Ser affects RIN1-mediated integrin trafficking in immortalized epidermal keratinocytes. Hum. Mol. Genet. 2023, 32, 304–318. [Google Scholar] [CrossRef]
- Nakhaei-Rad, S.; Haghighi, F.; Bazgir, F.; Dahlmann, J.; Busley, A.V.; Buchholzer, M.; Kleemann, K.; Schänzer, A.; Borchardt, A.; Hahn, A.; et al. Molecular and cellular evidence for the impact of a hypertrophic cardiomyopathy-associated RAF1 variant on the structure and function of contractile machinery in bioartificial cardiac tissues. Commun. Biol. 2023, 6, 657. [Google Scholar] [CrossRef] [PubMed]
- Seta, K.; Nanamori, M.; Modrall, J.G.; Neubig, R.R.; Sadoshima, J. AT1 receptor mutant lacking heterotrimeric G protein coupling activates the Src-Ras-ERK pathway without nuclear translocation of ERKs. J. Biol. Chem. 2002, 277, 9268–9277. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.S.; Sayeski, P.P.; Bernstein, K.E. Jak2 acts as both a STAT1 kinase and as a molecular bridge linking STAT1 to the angiotensin II AT1 receptor. J. Biol. Chem. 2000, 275, 15586–15593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunde, I.G.; Aronsen, J.M.; Melleby, A.O.; Strand, M.E.; Skogestad, J.; Bendiksen, B.A.; Ahmed, M.S.; Sjaastad, I.; Attramadal, H.; Carlson, C.R.; et al. Cardiomyocyte-specific overexpression of syndecan-4 in mice results in activation of calcineurin-NFAT signalling and exacerbated cardiac hypertrophy. Mol. Biol. Rep. 2022, 49, 11795–11809. [Google Scholar] [CrossRef]
- Luo, Y.; Jiang, N.; May, H.I.; Luo, X.; Ferdous, A.; Schiattarella, G.G.; Chen, G.; Li, Q.; Li, C.; Rothermel, B.A.; et al. Cooperative Binding of ETS2 and NFAT Links Erk1/2 and Calcineurin Signaling in the Pathogenesis of Cardiac Hypertrophy. Circulation 2021, 144, 34–51. [Google Scholar] [CrossRef]
- Chaklader, M.; Rothermel, B.A. Calcineurin in the heart: New horizons for an old friend. Cell. Signal. 2021, 87, 110134. [Google Scholar] [CrossRef]
- Wilkins, B.J.; De Windt, L.J.; Bueno, O.F.; Braz, J.C.; Glascock, B.J.; Kimball, T.F.; Molkentin, J.D. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol. Cell. Biol. 2002, 22, 7603–7613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Nie, J.; Wang, D.W.; Ni, L. Mechanism of histone deacetylases in cardiac hypertrophy and its therapeutic inhibitors. Front. Cardiovasc. Med. 2022, 9, 931475. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Heineke, J.; Ruetten, H.; Willenbockel, C.; Gross, S.C.; Naguib, M.; Schaefer, A.; Kempf, T.; Hilfiker-Kleiner, D.; Caroni, P.; Kraft, T.; et al. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc. Natl. Acad. Sci. USA 2005, 102, 1655–1660. [Google Scholar] [CrossRef]
- Riaz, M.; Park, J.; Sewanan, L.R.; Ren, Y.; Schwan, J.; Das, S.K.; Pomianowski, P.T.; Huang, Y.; Ellis, M.W.; Luo, J.; et al. Muscle LIM Protein Force-Sensing Mediates Sarcomeric Biomechanical Signaling in Human Familial Hypertrophic Cardiomyopathy. Circulation 2022, 145, 1238–1253. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xia, C.; Yang, Y.; Warusawitharana, H.K.; Liu, X.; Tu, Y. The Prevention Role of Theaflavin-3,3′-digallate in Angiotensin II Induced Pathological Cardiac Hypertrophy via CaN-NFAT Signal Pathway. Nutrients 2022, 14, 1391. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Choukroun, G.; Kang, Z.B.; Ranu, H.; Matsui, T.; Rosenzweig, A.; Molkentin, J.D.; Alessandrini, A.; Woodgett, J.; Hajjar, R.; et al. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol. 2000, 151, 117–130. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Russell, F.D.; Molenaar, P. Activation of calcineurin in human failing heart ventricle by endothelin-1, angiotensin II and urotensin II. Br. J. Pharm. 2005, 145, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Hiroi, Y.; Uozumi, H.; Takimoto, E.; Toko, H.; Zhu, W.; Kudoh, S.; Mizukami, M.; Shimoyama, M.; Shibasaki, F.; et al. Calcineurin plays a critical role in the development of pressure overload-induced cardiac hypertrophy. Circulation 2001, 104, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Fukuzawa, J.; Osaki, J.; Sakuragi, H.; Yao, N.; Haneda, T.; Fujino, T.; Wakamiya, N.; Kikuchi, K.; Hasebe, N. Roles of calcineurin and calcium/calmodulin-dependent protein kinase II in pressure overload-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2003, 35, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Bueno, O.F.; Wilkins, B.J.; Tymitz, K.M.; Glascock, B.J.; Kimball, T.F.; Lorenz, J.N.; Molkentin, J.D. Impaired cardiac hypertrophic response in Calcineurin Abeta -deficient mice. Proc. Natl. Acad. Sci. USA 2002, 99, 4586–4591. [Google Scholar] [CrossRef]
- Fatkin, D.; McConnell, B.K.; Mudd, J.O.; Semsarian, C.; Moskowitz, I.G.; Schoen, F.J.; Giewat, M.; Seidman, C.E.; Seidman, J.G. An abnormal Ca2+ response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J. Clin. Investig. 2000, 106, 1351–1359. [Google Scholar] [CrossRef] [Green Version]
- Iemitsu, M.; Miyauchi, T.; Maeda, S.; Sakai, S.; Kobayashi, T.; Fujii, N.; Miyazaki, H.; Matsuda, M.; Yamaguchi, I. Physiological and pathological cardiac hypertrophy induce different molecular phenotypes in the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R2029–R2036. [Google Scholar] [CrossRef] [Green Version]
- Sangaralingham, S.J.; Kuhn, M.; Cannone, V.; Chen, H.H.; Burnett, J.C. Natriuretic peptide pathways in heart failure: Further therapeutic possibilities. Cardiovasc. Res. 2023, 118, 3416–3433. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Yasue, H.; Ogawa, H. Pathophysiological significance and clinical application of ANP and BNP in patients with heart failure. Can. J. Physiol. Pharm. 2001, 79, 730–735. [Google Scholar] [CrossRef]
- Dunn, M.E.; Manfredi, T.G.; Agostinucci, K.; Engle, S.K.; Powe, J.; King, N.M.; Rodriguez, L.A.; Gropp, K.E.; Gallacher, M.; Vetter, F.J.; et al. Serum Natriuretic Peptides as Differential Biomarkers Allowing for the Distinction between Physiologic and Pathologic Left Ventricular Hypertrophy. Toxicol. Pathol. 2017, 45, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Engle, S.K.; Watson, D.E. Natriuretic Peptides as Cardiovascular Safety Biomarkers in Rats: Comparison With Blood Pressure, Heart Rate, and Heart Weight. Toxicol. Sci. 2016, 149, 458–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.C.; Guo, J.; Zhang, A. The renal and cardiovascular effects of natriuretic peptides. Adv. Physiol. Educ. 2017, 41, 179–185. [Google Scholar] [CrossRef] [Green Version]
- de Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef]
- Gorbe, A.; Giricz, Z.; Szunyog, A.; Csont, T.; Burley, D.S.; Baxter, G.F.; Ferdinandy, P. Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res. Cardiol. 2010, 105, 643–650. [Google Scholar] [CrossRef]
- Elesgaray, R.; Caniffi, C.; Ierace, D.R.; Jaime, M.F.; Fellet, A.; Arranz, C.; Costa, M.A. Signaling cascade that mediates endothelial nitric oxide synthase activation induced by atrial natriuretic peptide. Regul. Pept. 2008, 151, 130–134. [Google Scholar] [CrossRef]
- Calderone, A.; Thaik, C.M.; Takahashi, N.; Chang, D.L.; Colucci, W.S. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J. Clin. Investig. 1998, 101, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, E.; Champion, H.C.; Li, M.; Belardi, D.; Ren, S.; Rodriguez, E.R.; Bedja, D.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 2005, 11, 214–222. [Google Scholar] [CrossRef]
- Fiedler, B.; Lohmann, S.M.; Smolenski, A.; Linnemuller, S.; Pieske, B.; Schroder, F.; Molkentin, J.D.; Drexler, H.; Wollert, K.C. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 11363–11368. [Google Scholar] [CrossRef]
- Li, X.; Lan, Y.; Wang, Y.; Nie, M.; Lu, Y.; Zhao, E. Telmisartan suppresses cardiac hypertrophy by inhibiting cardiomyocyte apoptosis via the NFAT/ANP/BNP signaling pathway. Mol. Med. Rep. 2017, 15, 2574–2582. [Google Scholar] [CrossRef] [Green Version]
- Cooling, M.; Hunter, P.; Crampin, E.J. Modeling hypertrophic IP3 transients in the cardiac myocyte. Biophys. J. 2007, 93, 3421–3433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraeutler, M.J.; Soltis, A.R.; Saucerman, J.J. Modeling cardiac beta-adrenergic signaling with normalized-Hill differential equations: Comparison with a biochemical model. BMC Syst. Biol. 2010, 4, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryall, K.A.; Holland, D.O.; Delaney, K.A.; Kraeutler, M.J.; Parker, A.J.; Saucerman, J.J. Network reconstruction and systems analysis of cardiac myocyte hypertrophy signaling. J. Biol. Chem. 2012, 287, 42259–42268. [Google Scholar] [CrossRef] [Green Version]
- Molkentin, J.D.; Robbins, J. With great power comes great responsibility: Using mouse genetics to study cardiac hypertrophy and failure. J. Mol. Cell. Cardiol. 2009, 46, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.U.; Sutcliffe, M.D.; Saucerman, J.J. Network-based predictions of in vivo cardiac hypertrophy. J. Mol. Cell. Cardiol. 2018, 121, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Estrada, A.C.; Yoshida, K.; Saucerman, J.J.; Holmes, J.W. A multiscale model of cardiac concentric hypertrophy incorporating both mechanical and hormonal drivers of growth. Biomech. Model. Mechanobiol. 2021, 20, 293–307. [Google Scholar] [CrossRef]
- Yoshida, K.; Saucerman, J.J.; Holmes, J.W. Multiscale model of heart growth during pregnancy: Integrating mechanical and hormonal signaling. Biomech. Model. Mechanobiol. 2022, 21, 1267–1283. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Ulfenborg, B.; Andersson, C.X.; Heydarkhan-Hagvall, S.; Jeppsson, A.; Sartipy, P.; Synnergren, J. Cardiac hypertrophy in a dish: A human stem cell based model. Biol. Open 2020, 9, bio052381. [Google Scholar] [CrossRef]
- Li, J.; Feng, X.; Wei, X. Modeling hypertrophic cardiomyopathy with human cardiomyocytes derived from induced pluripotent stem cells. Stem Cell Res. Ther. 2022, 13, 232. [Google Scholar] [CrossRef] [PubMed]
- Bockstall, K.E.; Link, M.S. A primer on arrhythmias in patients with hypertrophic cardiomyopathy. Curr. Cardiol. Rep. 2012, 14, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Saucerman, J.J. Phospholemman is a negative feed-forward regulator of Ca2+ in beta-adrenergic signaling, accelerating beta-adrenergic inotropy. J. Mol. Cell. Cardiol. 2012, 52, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Morotti, S.; Edwards, A.G.; McCulloch, A.D.; Bers, D.M.; Grandi, E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J. Physiol. 2014, 592, 1181–1197. [Google Scholar] [CrossRef]
- Zeigler, A.C.; Richardson, W.J.; Holmes, J.W.; Saucerman, J.J. A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J. Mol. Cell. Cardiol. 2016, 94, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Zeigler, A.C.; Nelson, A.R.; Chandrabhatla, A.S.; Brazhkina, O.; Holmes, J.W.; Saucerman, J.J. Computational model predicts paracrine and intracellular drivers of fibroblast phenotype after myocardial infarction. Matrix Biol. J. Int. Soc. Matrix Biol. 2020, 91–92, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.D.; Aguado, B.A.; Watts, K.M.; Anseth, K.S.; Richardson, W.J. Network modeling predicts personalized gene expression and drug responses in valve myofibroblasts cultured with patient sera. Proc. Natl. Acad. Sci. USA 2022, 119, e2117323119. [Google Scholar] [CrossRef]
- Zeigler, A.C.; Chandrabhatla, A.S.; Christiansen, S.L.; Nelson, A.R.; Holmes, J.W.; Saucerman, J.J. Network model-based screen for FDA-approved drugs affecting cardiac fibrosis. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 377–388. [Google Scholar] [CrossRef]
- Lu, H.I.; Tong, M.S.; Chen, K.H.; Lee, F.Y.; Chiang, J.Y.; Chung, S.Y.; Sung, P.H.; Yip, H.K. Entresto therapy effectively protects heart and lung against transverse aortic constriction induced cardiopulmonary syndrome injury in rat. Am. J. Transl. Res. 2018, 10, 2290–2305. [Google Scholar]
- Burke, R.M.; Lighthouse, J.K.; Mickelsen, D.M.; Small, E.M. Sacubitril/Valsartan Decreases Cardiac Fibrosis in Left Ventricle Pressure Overload by Restoring PKG Signaling in Cardiac Fibroblasts. Circ. Heart Fail. 2019, 12, e005565. [Google Scholar] [CrossRef]
- Cunningham, J.W.; Claggett, B.L.; O’Meara, E.; Prescott, M.F.; Pfeffer, M.A.; Shah, S.J.; Redfield, M.M.; Zannad, F.; Chiang, L.M.; Rizkala, A.R.; et al. Effect of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients With HFpEF. J. Am. Coll. Cardiol. 2020, 76, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.M.; Nichols, W.; Richardson, W.J. Computational Screen for Sex-Specific Drug Effects in a Cardiac Fibroblast Network Model. bioRxiv 2023. [Google Scholar] [CrossRef]
- Watts, K.; Richardson, W.J. Effects of Sex and 17 β-Estradiol on Cardiac Fibroblast Morphology and Signaling Activities In Vitro. Cells 2021, 10, 2564. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Christiansen, S.L.; Naegle, K.M.; Saucerman, J.J. Logic-based mechanistic machine learning on high-content images reveals how drugs differentially regulate cardiac fibroblasts. bioRxiv 2023. [Google Scholar] [CrossRef]
- Jin, Y.F.; Han, H.C.; Berger, J.; Dai, Q.; Lindsey, M.L. Combining experimental and mathematical modeling to reveal mechanisms of macrophage-dependent left ventricular remodeling. BMC Syst. Biol. 2011, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Chowkwale, M.; Lindsey, M.L.; Saucerman, J.J. Intercellular model predicts mechanisms of inflammation-fibrosis coupling after myocardial infarction. J. Physiol. 2022, 601, 2635–2654. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
Figure 4.
Schematic representation of the effects of changes in the microenvironment on cardiac function. Hypertension, a common cardiovascular disease, causes pressure overload followed by a massive release of pro-hypertrophic, pro-fibrotic, and pro-inflammatory mediators. At this stage, when individuals do not experience symptoms, hypertension, and its accompanying microenvironmental complications may be reversible with strategies such as lifestyle modification, however without any intervention, this could evolve into cardiac hypertrophy and fibrotic remodeling. Increasing fibrosis leads to mechanical stiffness and impaired filling phase, both prominent features of diastolic dysfunction. Common symptoms include headache, dizziness, palpitations, and chest discomfort. Notably, this phase is not reversible and requires pharmacological management. Late diagnosis or inadequate treatment leads to progressive fibrosis and detrimental changes at the molecular level, such as a barrier between cardiomyocytes at the cellular level, impaired electrical coupling, and hypoxia of affected cardiomyocytes, collectively resulting in cardiomyocytes’ cell death. The subsequent decreased contractile force characterizes systolic dysfunction while having severe consequences as individuals suffer from shortness of breath. Biomarker identification in a diagnostic screening approach could help detect early onset diastolic dysfunction in affected individuals, setting the platform for early management and preventive course of action to avoid the subsequent detrimental outcomes of the developing condition.
Figure 4.
Schematic representation of the effects of changes in the microenvironment on cardiac function. Hypertension, a common cardiovascular disease, causes pressure overload followed by a massive release of pro-hypertrophic, pro-fibrotic, and pro-inflammatory mediators. At this stage, when individuals do not experience symptoms, hypertension, and its accompanying microenvironmental complications may be reversible with strategies such as lifestyle modification, however without any intervention, this could evolve into cardiac hypertrophy and fibrotic remodeling. Increasing fibrosis leads to mechanical stiffness and impaired filling phase, both prominent features of diastolic dysfunction. Common symptoms include headache, dizziness, palpitations, and chest discomfort. Notably, this phase is not reversible and requires pharmacological management. Late diagnosis or inadequate treatment leads to progressive fibrosis and detrimental changes at the molecular level, such as a barrier between cardiomyocytes at the cellular level, impaired electrical coupling, and hypoxia of affected cardiomyocytes, collectively resulting in cardiomyocytes’ cell death. The subsequent decreased contractile force characterizes systolic dysfunction while having severe consequences as individuals suffer from shortness of breath. Biomarker identification in a diagnostic screening approach could help detect early onset diastolic dysfunction in affected individuals, setting the platform for early management and preventive course of action to avoid the subsequent detrimental outcomes of the developing condition.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bazgir, F.; Nau, J.; Nakhaei-Rad, S.; Amin, E.; Wolf, M.J.; Saucerman, J.J.; Lorenz, K.; Ahmadian, M.R. The Microenvironment of the Pathogenesis of Cardiac Hypertrophy. Cells 2023, 12, 1780. https://doi.org/10.3390/cells12131780
AMA Style
Bazgir F, Nau J, Nakhaei-Rad S, Amin E, Wolf MJ, Saucerman JJ, Lorenz K, Ahmadian MR. The Microenvironment of the Pathogenesis of Cardiac Hypertrophy. Cells. 2023; 12(13):1780. https://doi.org/10.3390/cells12131780
Chicago/Turabian StyleBazgir, Farhad, Julia Nau, Saeideh Nakhaei-Rad, Ehsan Amin, Matthew J. Wolf, Jeffry J. Saucerman, Kristina Lorenz, and Mohammad Reza Ahmadian. 2023. "The Microenvironment of the Pathogenesis of Cardiac Hypertrophy" Cells 12, no. 13: 1780. https://doi.org/10.3390/cells12131780
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.