
Voltage-Gated T-Type Calcium Channel Modulation by Kinases and Phosphatases: The Old Ones, the New Ones, and the Missing Ones

1
Department of Biotechnology, Indian Institute of Technology Hyderabad (IITH), Kandi 502284, Telangana, India
2
National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London W12 0NN, UK
*
Authors to whom correspondence should be addressed.
Cells 2023, 12(3), 461; https://doi.org/10.3390/cells12030461
Submission received: 29 November 2022
/
Revised: 14 January 2023
/
Accepted: 29 January 2023
/
Published: 31 January 2023
(This article belongs to the Special Issue Exclusive Review Papers in "Cell Signaling")
Abstract
:Calcium (Ca2+) can regulate a wide variety of cellular fates, such as proliferation, apoptosis, and autophagy. More importantly, changes in the intracellular Ca2+ level can modulate signaling pathways that control a broad range of physiological as well as pathological cellular events, including those important to cellular excitability, cell cycle, gene-transcription, contraction, cancer progression, etc. Not only intracellular Ca2+ level but the distribution of Ca2+ in the intracellular compartments is also a highly regulated process. For this Ca2+ homeostasis, numerous Ca2+ chelating, storage, and transport mechanisms are required. There are also specialized proteins that are responsible for buffering and transport of Ca2+. T-type Ca2+ channels (TTCCs) are one of those specialized proteins which play a key role in the signal transduction of many excitable and non-excitable cell types. TTCCs are low-voltage activated channels that belong to the family of voltage-gated Ca2+ channels. Over decades, multiple kinases and phosphatases have been shown to modulate the activity of TTCCs, thus playing an indirect role in maintaining cellular physiology. In this review, we provide information on the kinase and phosphatase modulation of TTCC isoforms Cav3.1, Cav3.2, and Cav3.3, which are mostly described for roles unrelated to cellular excitability. We also describe possible potential modulations that are yet to be explored. For example, both mitogen-activated protein kinase and citron kinase show affinity for different TTCC isoforms; however, the effect of such interaction on TTCC current/kinetics has not been studied yet.
1. Introduction
1.1. Voltage-Gated Calcium Channels
In the cell, voltage-gated Ca2+ channels (VGCCs) play two distinct roles: they can depolarize the membrane potential to promote electrical excitability, and they can also activate transient cytoplasmic Ca2+ signals. Action potentials and subthreshold depolarizing impulses cause Ca2+ influx through VGCCs, which are found in many different cell types [1]. The Ca2+ ions entering the cells can act as secondary messengers, thus accounting for various physiological roles such as ATP synthesis, fertilization, cell cycle, differentiation, proliferation, apoptosis, neurotransmitter/hormone release [2,3,4], activation of gene transcription [5], and muscle contraction [6] thus indicating VGCC roles in non-excitable cells as well. Different cascade effects on cell activities, such as cell proliferation, motility, or even cell death, may result from variations in Ca2+ channel transcript levels [7]. Thus, VGCCs play a critical role in controlling the function of several organs, such as the brain, heart, and muscles. Malfunctions of VGCCs can result in a variety of pathophysiological conditions, from cardiovascular diseases to neurological and psychiatric disorders like epilepsy, pain, and autism [8]. Over the last decade or so, VGCCs have been shown to be involved in the disorders of non-excitable cells as well, such as diabetes and cancer [9,10]. VGCCs are classified into two major categories: low-voltage activated (LVA) TTCCs that are activated below the action potential threshold and mainly regulate the excitability of the cells, and high-voltage activated (HVA) channels (L, N, P/Q, and R-types) that are activated after the initiation of an action potential and are therefore important effectors for transient Ca2+ signaling [11]. Voltage-gated calcium channels are classified into three subfamilies (Cav1, Cav2, Cav3). Each family has an internal sequence identity of >80% and an interfamily sequence identity of 52% (Cav1 & Cav2) and 28% (Cav3 & Cav1/Cav2), as shown in Figure 1 [12,13,14,15]. TTCCs, being LVA, also have an important role in Ca2+ homeostasis through calcium influx near resting membrane potential in several cell types.
1.2. T-Type Calcium Channels
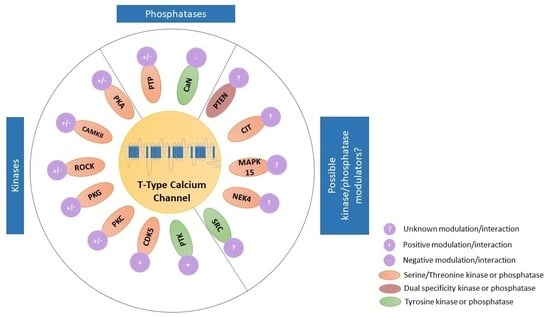
TTCCs are LVA channels that belong to the family of VGCCs. There are three isoforms of TTCCs, Cav3.1, Cav3.2, and Cav3.3, that are encoded by the genes CACNA1G, CACNA1H, and CACNA1I, respectively. TTCCs are formed by α1/Cav3 pore-forming subunit, consisting of four homologous domains, each of those made up of six transmembrane helices (S1 to S6). The four domains are linked together with intracellular loops (I-II, II-III, and III-IV) between the S6 segment of the preceding domain and the S1 segment of the following domain. The Ca2+ selectivity is due to the acidic residues (glutamate or aspartate) in the pore-forming segments (P loop, connecting segments S5 and S6 of each domain [16,17]. It is known that multiple kinases and phosphatases interact with and thereby modulate TTCCs. These specific kinases and phosphatases are shown in Figure 2 [18,19]. The modulation of TTCCs by these kinases and phosphatases is discussed in detail in the later sections of the article.
TTCCs can operate very close to resting membrane potentials, with Cav3.1 having the most rapid inactivation kinetics (Cav3.1 > Cav3.2 > Cav3.3) [20]. TTCCs have wide expression throughout several organs (Table 1), where they maintain some vital functions such as neuronal excitability, cardiac pacemaking, arterial constriction, hormone release, cell proliferation, cell cycle, etc. [21,22,23,24,25]. In addition, they are also involved in pathological conditions not associated with cellular excitability, such as cancer. Some of the pathological roles of TTCCs are mentioned below in Table 2.
1.3. Protein Kinases
Protein kinases (PKs) are molecular switches that exist either in an “on” state with maximum activity or an “off” inactive state. Remarkably, PKs have striking structural similarities in the “on” state while different PKs in the “off” state can adopt distinct conformations [52]. All PKs catalyze the same reaction, the transfer of the γ -phosphate of ATP to the hydroxyl group of serine, threonine, or tyrosine. Eukaryotic PKs (EPKs) constitute one of the largest recognized protein families represented in the human genome [53]. About 1.7% of all human genes, or 518 distinct PKs, are thought to be encoded by the human genome [54]. The catalytic domain of EPK contains 12 preserved subdomains. The invariant aspartate in the subdomain VIB, which is most likely the catalytic base in the phosphotransferase activity, and the invariant lysine in subdomain II, which binds and orients ATP, are indispensable for the catalytic function of EPKs.
In terms of substrate selectivity, PKs may be divided into three major categories: Ser/Thr PKs (PKs that utilize serine/threonine residue as the principal phosphate acceptor site), protein tyrosine kinases (PKs that utilize tyrosine residue as the principal phosphate acceptor site), and dual-specificity PKs (e.g., mitogen-activated protein kinase (MEK) that phosphorylate both threonine and tyrosine residues on target proteins) [55]. PKs are essential for controlling various cellular events such as cell–cell adhesion, cell–substrate adhesion, and hormone signaling that are associated with the dysregulation of kinase-related signaling and may lead to cancer and developmental disorders [56,57].
1.4. Protein Phosphatases
The action of PKs in the cell is balanced by protein phosphatases (PPs), which remove phosphate groups from the amino acid residues to manage the regulation of protein function. In contrast to PKs, eukaryotes utilize a number of unique molecular paradigms for the construction of the catalytic components in the PPs with which they carry out the dephosphorylation of phosphoproteins. PPs have high evolutionary diversity and complexity based on different/unrelated ancestors compared to PKs [58,59,60]. They also have an intrinsic substrate profile, as phosphatases can recognize proteins, lipids, or carbohydrates. PPs have been divided into protein Ser/Thr phosphatases (PSTPs or PSPs) and protein tyrosine phosphatases (PTPs) based on their unique catalytic processes and substrate selectivity [61,62].
2. Kinase Modulators of T-Type Ca2+ Channels
2.1. Modulation of T-Type Ca2+ Channels by Serine/Threonine Kinase Family
2.1.1. Modulation of T-Type Ca2+ Channels by Protein Kinase A
Protein kinase A (PKA) is one of the first PKs to be identified [63], sequenced [64], cloned [65], and had crystal structure determined [66]. It is a Ser/Thr kinase that participates in a number of biological processes through phosphorylating and, thus, regulates the activity of many proteins, which also includes the regulation of TTCCs. In mammalian cells (tsA-201-kidney cell line and Chinese hamster ovary cell line), PKA enhanced Ca2+ current through all three TTCC isoforms in a temperature-dependent manner. PKA effects were observed at physiological temperatures ranging from 30–37 °C but not at room temperature (22–27 °C). This could be due to the impaired kinase translocation at room temperature [67]. Several studies demonstrated PKA-mediated upregulation of current through TTCCs though very few of them demonstrated isoform-specific or direct modulation of TTCCs. In rodent trigeminal ganglion neurons, activation of both neuromedin B receptor and brain-derived neurotrophic factor receptor activated PKA, which stimulated current through Cav3.2 but not Cav3.1 and Cav3.3 TTCCs to induce hyperexcitability and pain sensitivity. Blockade of TTCC reduced these effects [68,69]. In mice, trigeminal ganglion neuronal cells, G-protein coupled receptor (GPCR) 30 (GPR30) estrogen receptor activation also led to the activation of PKA, which further enhanced current through TTCCs [70], indicating that downstream effects of GPCRs are also mediated by TTCCs in neuronal tissues. Serotonin-induced activation of 5-hydroxytryptamine receptor-7 (5HT7) receptors in rat glomerulosa cells enhanced current through TTCCs via a PKA-mediated pathway, which led to aldosterone release [25,71], indicating a role of TTCCs in hormone release in non-excitable cells. Further evidence for the PKA role in hormone release via TTCCs in non-excitable cells comes from human adrenocortical cells where cortisol secretion upon activation of 5HT4 receptors was dependent on the activation of cAMP-dependent PKs that enhanced membrane Ca2+ influx through TTCCs to activate other VGCCs ultimately leading to the increased intracellular Ca2+ and, thus cortisol secretion [72]. A similar observation was made in the primary cultured ovine and human somatotrophs, where growth hormone-releasing hormone (GHRH)-induced T-type current was blocked by a PKA inhibitor. This PKA-mediated influx of Ca2+ served as a key step in growth hormone exocytosis [73]. Luteinizing hormone (LH), a major regulator of testosterone synthesis, stimulates Leydig cells, which involves the augmentation of TTCC [74]. The effect of LH is completely blocked upon application of PKA inhibitors H89 and a synthetic inhibitory peptide (IP-20), indicating a PKA-mediated augmentation of T-type current [75]. In an attempt to identify the structural domains that were essential for PKA modulation of TTCCs, chimeric Cav3.2 channel was over-expressed in Xenopus oocytes, which revealed that PKA increases Cav3.2 channel current by a possible modulation in its II-III cytoplasmic loop [76] which was later identified as ser1107. This information is lacking for other Cav3.1 and Cav3.3. Fibroblast growth factor-23 (FGF-23), when bound to the fibroblast growth factor receptor type-1 (FGFR-1) in adult mice, selectively increased TTCC. A PI3K-dependent PKA signaling cascade was discovered through pharmacological manipulation. Because TTCCs are known to play a role in pain transmission, FGFR1 antagonists can be effective analgesics for FGF-23-induced nociception. [77]. An interesting study on NIH 3T3 cells stably expressing five muscarinic receptor subtypes (m1-m5) demonstrated a differential modulation of T-type current upon acetylcholine (Ach) application. Application of Ach on cells expressing m3 and m5 receptors showed an increase in the T-type current. PKA inhibitors blocked this effect, indicating a PKA-mediated augmentation of TTCC upon m3 and m5 receptor activation. While m1 receptor stimulation activated PKA, it did not increase T-type current. An increase in T-type current was observed upon preincubation of cells with PKC inhibitors implying a crosstalk between PKA and PKC. Hence, fine-tuning the activity of these kinases is essential for the modulation of TTCC.s On the other hand, no significant change in the amplitude of T-type current was observed upon m2 and m4 receptor activation [78]. Recently, a study with mesenchymal stem cells (MSCs) transfected with high mobility group box 1 (HMGB1) demonstrated an enhanced T-type current via a PKA-mediated pathway. Here PKA was not shown to interact directly with TTCC, but it modulated TTCC indirectly by phosphorylating β-catenin, which increased its translocation to the nucleus. Phosphorylated β- catenin increased the transcription of γ- cystathionase, which catalyzes H2S production, in turn augmenting TTCC, specifically Cav3.2. This augmentation of Cav3.2 TTCC by HMGB1 increased the mobility of MSCs, which can help in clinical applications in tissue repair and regeneration [79]. Protein expression of Cav3.1 and Cav3.3 protein was not detected in transfected MSCs and therefore ruled out PKA modulation of Cav3.1 and Cav3.3 in MSCs.
PKA modulation of TTCCs does not always enhance current through TTCCs as described above, but a handful of studies have demonstrated PKA-mediated downregulation of current through TTCCs. Dopamine, a neurotransmitter and hormone, inhibited T-type currents in bass retinal horizontal cells, an effect that is prevented by PKA inhibitors and mimicked by 8-CPT, a cAMP analog. PKA modulation of these currents may influence the response properties of horizontal cells [80,81]. A similar observation was made in newt olfactory cells where adrenalin stimulation inhibited T-type current in PKA dependent manner and enhanced odorant contrast in olfactory perception [82]. A subsequent study in H295R cells (human adrenocortical carcinoma cells) revealed that dopamine binding to dopamine receptors 1 and 2 inhibits Cav3.2 channel currents by Gβγ dimers. PKA functions as a molecular switch for the function of Gβγ dimers where inhibition by Gβγ dimers requires PKA-mediated phosphorylation of Cav3.2 at ser1107 residue in the II-III loop of the channel. Failure of this mechanism might be a contributing factor for aldosterone excess and loss of dopaminergic tone in some forms of human hypertension, thus implicating PKA regulation of Cav3.2 as a causative factor. Notably, PKA phosphorylation site ser1107 is specific to Cav3.2 since swapping the II-III loop of Cav3.2 with Cav3.1 eliminated the PKA modulation of the Cav3.2 channel [83]. This is surprising given the fact that ser1107 is conserved across all three isoforms and, thus, warrants further investigation for identifying the phosphorylation sites in Cav3.1 and Cav3.3. In mouse dorsal root ganglion (DRG) neurons, neuromedin U, a neuropeptide with diverse physiological functions, inhibited current through TTCCs in a dose-dependent manner that required PKA activity. This revealed a novel role for neuromedin U peptide in neuronal hypo-excitability in small DRG neurons in mice [84]. PKA-mediated inhibition of current through TTCCs was also observed upon muscarinic M4 receptor stimulation by alpha-cobratoxin (a neurotoxic protein) in DRG neurons [85]. In these neurons, pre-treatment of cells with a PKA inhibitor or selective muscarinic M4 receptor antagonist, tropicamide, abolished the inhibition of TTCC current observed upon alpha-cobratoxin application, suggesting that the analgesic effects of alpha-cobratoxin are mediated by its effect on TTCC current in DRG neurons. Reduction in TTCC current via the action of PKA was observed in rat retinal ganglion cells, which was mediated by activation of G-protein coupled cannabinoid receptors CB2 but not cannabinoid receptors CB1. Cannabinoid receptors are involved in multiple neuronal functions, and these functions are in part regulated by their effect on downstream targets such as TTCCs. This study also points to the fact that the action of PKA on TTCCs is determined by the specific GPCR type and isoform [86].
Overall, we observed that PKA showed differential regulation of TTCCs, and enhancement or reduction of current, which seems to be tissue and GPCR-specific. Therefore, a generalization cannot be made on PKA regulation of TTCCs. Reduction in the TTCC current was observed when binding the ligand to a GPCR led to the activation of a G-inhibitory alpha subunit, whereas activation of the G-stimulatory alpha subunit led to the enhancement of current through TTCCs [70,84].
2.1.2. Modulation of T-Type Ca2+ Channels by Ca2+/Calmodulin-Dependent Protein Kinase II
Calmodulin is a 16.7 kDa protein highly conserved in eukaryotes. It was discovered as a Ca2+ regulator in the brain. When Ca2+ binds to calmodulin, it induces a structural modification, forming a Ca2+/calmodulin complex [87]. The Ca2+/calmodulin complex activates Ca2+/calmodulin-dependent protein kinase II (CaMKII), which participates in a number of signaling cascades, including those that regulate chloride transport [88], immune cell activation [89] and Ca2+ homeostasis [90]. It plays a significant role in long-term potentiation [91], memory, and cardiac function [92]. CaMKII dysregulation is also associated with a number of cardiac diseases, such as dilated cardiomyopathy and arrhythmia [93,94]. CaMKII works in a feedback loop where its activity is regulated by a number of Ca2+ channels [95], and in turn, it regulates multiple channel proteins [96]. In calf adrenal glomerulosa cells, the elevation of intracellular Ca2+ concentration led to the enhancement of TTCC current, which was abolished upon application of a CaMKII inhibitor, suggesting that CaMKII could enhance current through TTCCs. In addition, CaMKII also induced a leftward shift in the activation threshold of TTCCs, rendering them suited to activation by a low amplitude depolarizing stimulus [97]. In fact, this could be how TTCCs can be involved in maintaining calcium influx at near resting membrane potentials. CaMKII regulation could further enhance current through TTCCs in pathological conditions where cells are slightly depolarized. Subsequent experiments in adrenal glomerulosa cells also reiterated that CaMKII could enhance the unitary current amplitude of TTCCs, which was dependent on intracellular Ca2+ concentration [98]. CaMKII, however, showed differential regulation of Cav3 family members, augmenting only Cav3.2 but not the Cav3.1 isoform in a heterologous overexpression system using HEK293 cells [99]. This augmentation of TTCC current is predicted to be necessary for sustaining stimulated aldosterone production by glomerulosa cells. Mechanistic studies using the heterologous over-expression system HEK293 revealed that CaMKII could phosphorylate serine residue in the Cav3.2 II-III intracellular loop, which is absent in the Cav3.1 subunit, thus providing a basis for the differential regulation of these channels by CaMKII [100]. This mechanism was later confirmed in vivo in rat adrenal glands where angiotensin II receptor stimulation activated CaMKII, which then phosphorylated ser1198 in Cav3.2 channels leading to the augmentation of Cav3.2 current and sustained aldosterone secretion [101]. The involvement of TTCCs in aldosterone release has great significance in cardiovascular disease, where there is a marked activation of the renin-angiotensin-aldosterone axis. TTCC regulation by the CaMKII pathway is involved in several different functions other than hormone secretion. It was observed that spiro[imidazo [1,2-a] pyridine-3,2-indan]-2(3H)-one (ST101), a cognitive enhancer, augments calcium influx through TTCCs in the brain cortex, thereby causing autophosphorylation of CaMKII leading to long-term potentiation as observed in rat cortical slices but not in hippocampal slices. This is also an example where activation of CaMKII is dependent on TTCC activity, contrary to CaMKII augmentation of TTCC currents. This CaMKII activation was necessary for direct phosphorylation of AMPA-type glutamate receptor subunit 1 (GluR1) in AMPA receptors for long-term potentiation (cognitive enhancement), thus suggesting a role of TTCCs in the cognitive enhancement and a potential therapeutic target in diseases such as Alzheimer’s [102]. SAK3, another spiroimidazopyridine derivative that is also an enhancer of TTCC Cav3.1, showed anti-depressant-like activity, which was in part mediated by CaMKII. SAK3 enhancement of Cav3.1 in olfactory bulbectomized mice led to the rescue of reduction of CaMKII phosphorylation levels that were observed in untreated olfactory bulbectomized mice [103]. This reiterates the involvement of TTCCs in cognitive disorders and suggests that modulation of TTCCs may be targeted for therapeutic purposes. Similar to PKA-mediated reduction in TTCC current upon activation of cannabinoid receptors, CaMKII has also been shown to dampen the activity of TTCCs. In retinal muller cells, CaMKII inhibited currents carried by TTCCs upon activation of cannabinoid receptors, both CB1 and CB2, unlike PKA, which reduced TTCC current only upon activation of CB2 receptors, as mentioned in the previous section. This demonstrated that endogenous cannabinoids could regulate TTCCs by acting on multiple receptors and by employing multiple pathways [86]. The mechanism underlying the off-target neurotoxic effect of vincristine (VCR), a chemotherapeutic drug, was recently investigated using a mouse model, which revealed that VCR-induced neuropathic pain was associated with astrocyte activation. This astrocyte activation was accompanied by an increase in the expression of several genes, including CaMKII, Cav3.2, and connexin-43. CaMKII increased Ca2+ influx via the Cav3.2 channel, resulting in connexin-43-dependent inflammation. However, siRNA-mediated knockdown of connexin-43 had no effect on astrocyte activation or the expression of Cav3.2 and CaMKII [104], indicating a rather consequential pathway.
In summary, it is observed that CaMKII maintains Ca2+ homeostasis in both excitable and non-excitable cells via a feedback loop and by acting on several protein substrates, one of which is TTCCs. By regulating TTCCs, CaMKII participates in a variety of cell functions such as cognitive enhancement, endocannabinoid signaling, aldosterone secretion, etc. CaMKII, unlike other kinases, targets only Cav3.2 and not Cav3.1, as demonstrated in a heterologous expression system and an in vivo rat model. CaMKII-mediated modulation of TTCCs may serve as a potential therapeutic target in cardiovascular and cognitive disorders due to its involvement in multiple aforementioned pathways. Whether CaMKII can phosphorylate Cav3.3 remains elusive. Also remaining undetermined is the modulation of TTCCs by other CaMKs.
2.1.3. Modulation of T-Type Ca2+ Channels by Rho/Rho-Kinase
Rho kinases (ROCK) are recognized as downstream effector proteins of the small GTPase Rho [105]. They primarily participate in cytoskeleton rearrangement and cytoskeleton-associated processes [106,107,108,109,110]. The signaling lipid molecule lysophosphatidic acid (LPA) is an endogenous ligand for a class of GPCRs that mediate downstream effects by activating ROCK. Studies on LPA receptors using LPA receptor knockout mice and ROCK inhibitors suggested that Rho kinase is associated with neuropathic pain [111]. Since TTCCs are also involved in neuropathic pain, the association of TTCCs and ROCK was investigated though data is very limited. Application of LPA in cells overexpressing Cav3.1 and Cav3.3 TTCCs inhibited TTCC current via phosphorylation of amino acid residues in the II-III linker region of the channel that was dependent on ROCK activity as TTCC inhibition was abolished upon using ROCK inhibitors. Cav3.2 TTCC showed a more complex regulation by ROCK where both inhibition and augmentation of Cav3.2 current could be observed with depolarizing shift which awaits further investigation. LPA reduced the native current carried by TTCCs in Y29 retinoblastoma and lateral habenular neurons. This modulation was found to be cell-specific, as in DRG, the current carried by native Cav3.2 was found to be upregulated [112].
Further, a study in rat cardiomyocytes showed hypoxia-induced selective upregulation of the Cav3.2 channel and an increased ROCK1 expression. This stabilization of Cav3.2 mRNA and subsequent increase in T-type current is mediated by a hypoxia-induced factor 1α (HIF-1α). Application of C3 transferase, a Rho inhibitor, partially blocked HIF-1α stabilization of Cav3.2 mRNA, whereas HIF-1α mutant did not affect RhoA activity which indicated an upstream regulation of HIF-1α by RhoA. Moreover, C3 transferase and some ROCK inhibitors, Y27632 and fasudil, significantly reduced hypoxia-induced upregulation of Cav3.2 mRNA. These findings suggest that ROCK1 regulates hypoxia-induced upregulation of Cav3.2 current via HIF-1 α, which might be involved in the increased arrhythmias seen in ischemic conditions as well as the pathogenesis of diseases associated with hypoxic Ca2+ overload [113]. Potent regulation of TTCCs by ROCK activation suggests that this modulation could be important for neuronal physiology. However, more investigations using in vivo models are needed before we can exploit this modulation for any therapeutics. In addition, ROCK has two isoforms, ROCK1 and ROCK2. It would be interesting to determine if any specific isoform of ROCK is involved in the modulation of TTCCs by ROCK, which needs isoform-specific blockers of ROCK.
2.1.4. Modulation of T-Type Ca2+ Channels by Protein Kinase G
Protein kinase G (PKG) is a PK that is activated by cGMP and regulates a number of downstream targets. PKG plays a crucial role in the control of blood pressure. PKG also mediates Ca2+ homeostasis [114,115] and modulates platelet function and adhesion [116,117]. It also facilitates the release of renin [118] and causes relaxation of vascular smooth muscle accompanied by a decrease in the TTCC current. PKG-mediated downregulation of TTCC was also observed in rat cerebral arterial smooth muscle upon nitric oxide (NO), a vasodilator, application. NO-mediated suppression of TTCCs via PKG influences Ca2+ dynamics, restraining its entry through TTCCs and limiting its contribution to myogenic tone. Hence NO/PKG/TTCC controls smooth muscle function where the loss of NO-induced suppression of TTCC may serve as an underlying mechanism for cerebral vasospasm [119,120]. Data regarding PKG regulation of TTCCs is very limited. In an early study using NG108-15 cells (a neuroblastoma x glioma hybrid cell line), it was observed that cGMP had no effect on TTCC activity [121]. However, further studies using Newt olfactory cells revealed that cGMP increased Ni2+-sensitive current carried by TTCCs. This enhancing effect of cGMP was mimicked by the application of a cGMP phosphodiesterase inhibitor or the permeant cGMP analog, CPT-cGMP. Whereas KT5823, a PKG inhibitor, abolished the cGMP-mediated increase in TTCC current, indicating a PKG-mediated increase in the TTCC current. It is suggested that cGMP via augmentation of TTCCs may lower the threshold in olfactory perception and also prevent the saturation of odor signals by increasing the maximum spike frequency [122]. As with other Ser/Thr kinases, PKG also showed dual regulation where, in addition to augmentation as described above, it also mediated inhibition of TTCC current via activation of GPCR sst5 in acutely dissociated retinal ganglion cells [123]. Since TTCCs are important determinants of neuronal excitability, this may provide a mechanistic basis for the protection of ganglion cells against Ca2+ overload and, thus, injury. Studies are required for the elucidation of PKG phosphorylation sites in TTCCs and isoform-specific modulation of TTCCs by PKG if any.
2.1.5. Modulation of T-Type Ca2+ Channels by Protein Kinase C
In the heart, activation of various GPCRs is important to maintain cellular excitability and homeostasis. It is thus important to know the downstream effectors of such GPCR activation. The angiotensin pathway is already a target in cardiovascular disorders; therefore, the effect of the angiotensin pathway on TTCC current was investigated. Since Protein kinase C (PKC) is also activated upon activation of angiotensin receptors [124], the crosstalk between receptor activation and TTCC was investigated. PKC is a Ser/Thr kinase that selectively regulates Ca2+ and K+ currents [125]. Just like PKA, PKC is shown to modulate all three isoforms of TTCCs in a highly temperature-dependent manner [67]. TTCC isoforms are also differentially regulated upon PKC activation. In both human and chick cardiomyocytes, angiotensin II enhanced current through TTCCs, an effect that was mimicked by a PKC activator, phorbol 12,13-dibutyrate [124]. Further studies on cardiomyocytes revealed PKC-dependent elevation of Cav3.2 current but not Cav3.1 current in response to lysophosphatidylcholine-induced stimulation of PKC in a concentration-dependent manner [126]. These studies are important in light of the fact that PKC inhibitors are also being researched as therapeutic drugs in diabetes-related hypertension or endothelial insulin resistance [127], that is, disorders where Ca2+ channels are also implicated. Upregulation of current through TTCCs was observed in Leydig cells upon luteinizing hormone induction. Although this effect is mostly mediated by PKA, partial blockage of TTCC current by chelerythrine (PKC inhibitor) indicated a role also for PKC in Leydig cell Ca2+ dynamics [75]. PKC-mediated augmentation of TTCC was found to be involved in endothelin 1-induced calcium entry in neonatal rat ventricular myocytes [128]. Endothelin 1 is a potent endogenous vasoconstrictor and might regulate blood pressure and contribute to hypertrophy through this pathway. Studies on Xenopus oocytes revealed that phorbol 12-myristate 13-acetate (PMA), a PKC activator augmented the current amplitude of all three TTCC isoforms but to a varying degree. This augmentation was inhibited by PKC inhibitors. However, a similar effect of PMA was not seen in other mammalian cell lines indicating a species-specific PKC regulation. An increase in the Cav3.1 current by PMA in xenopus oocytes was not brought about by a change in channel surface density. It could be due to direct PKC phosphorylation of Cav3.1 in the intracellular II-III loop region or by indirect consequence of phosphorylation of other signaling effectors [129,130].
Numerous investigations have also demonstrated PKC-mediated suppression or downregulation of TTCCs and currents through them though this was mostly observed in either heterologous overexpression systems or neuronal tissues. Earliest studies on chicken DRG neurons, rat hippocampal neurons, canine cardiac Purkinje and ventricular cells, rat DRG neurons, and rat pituitary GH3 cells showed an inhibitory effect of PKC activators-diacylglycerol (DAG) analog 1-oleoyl-2-acetyl-sn-glycerol (OAG) and PMA on the Ni2+ sensitive T-type current [131,132,133,134,135,136,137]. In contrast to cardiomyocytes, angiotensin II inhibited TTCC in bovine adrenal glomerulosa cells, and this effect was mimicked by PKC activators (phorbol esters and DAG), causing a reduced cytoplasmic Ca2+ level indicating a PKC-mediated decrease in the intracellular Ca2+ and subsequent release of aldosterone in these cells [138]. Hence, PKC can serve as a potential target for hypertension. In an MN9D dopaminergic cell line, the application of corticotropin-releasing factor (CRF) related peptide, urocortin 1, reversibly inhibited current through TTCCs. Patch clamp studies showed that CRF receptor activation and subsequent downregulation of T-type current were blocked by the application of PKC inhibitor chelerythrine and enhanced by PMA [139]. CRF receptors are predominantly expressed in the dopaminergic neurons, where TTCCs are also one of the important determinants of Ca2+ homeostasis and physiology. There is evidence from a rat Parkinson’s model that TTCCs are involved in the pathology of Parkinson’s disease since the application of TTCC blockers showed reduced motor deficits in the Parkinson’s rats. [140]. Therefore, this PKC-mediated suppression of TTCCs via CRF receptor activation could be of significance in diseases such as Parkinson’s, where dopaminergic neurons are affected. In heterologous over-expression systems, PKC is also shown to inhibit TTCC current mainly through inhibition of Cav3.2 channel current upon activation of neurokinin 1 receptor [141]. In an interesting study using HEK293 cells and DRG neurons, ethanol was shown to reduce the current density of Cav3.2 TTCC isoform and produce a hyperpolarizing shift in the steady-state inactivation of the channels leading to an increase in the window current. Since DRG neurons are known to be enriched in Cav3.2, it is speculated that results from HEK293 cells corroborated well with the native channels. The inhibitory effect of ethanol was blocked by myristoylated PKC peptide inhibitor (MPI), whereas a lower concentration of PMA mimicked this effect, suggesting the PKC-dependent activity of ethanol [142]. In contrast, studies with rat cardiomyocytes showed a PKC-mediated enhancement of the TTCC activity upon ethanol administration. A mechanistic study revealed PKC-mediated hyperphosphorylation of GSK3β, which further activated calcineurin-NFAT-Csx/Nkx2.5 signaling. This may serve as a potential mechanism for ‘holiday heart syndrome’ caused by excessive alcohol consumption [143]. In a recent study, the importance of regulation of TTCCs by a specific isoform of PKC was highlighted, where melatonin (a hormone secreted by the pineal gland and important for circadian rhythm) inhibited Cav3.2 TTCC activity through the melatonin receptor 2 coupled to PKC-eta (PKCη) signaling. This downregulation of Cav3.2 decreased the membrane excitability of trigeminal ganglion neurons and pain hypersensitivity in mice, thus providing a mechanistic basis for the protective effects of melatonin. Melatonin did not alter currents through other TTCC isoforms indicating that Cav3.2 is the main target for PKCη [144]. Various studies have suggested crosstalk between PKC and PKA wherein one might inhibit the effects of the other, as seen in T-type current modulation by acetylcholine via muscarinic receptor-1 activation [78]. However, a synergistic effect of both kinases may also be involved in regulating physiological functions [81].
Taken together, it appears that PKC modulation of TTCCs is best suited to the cells or tissues in which they express and based on their contribution to cell physiology. Inhibition of TTCC currents appears to be a protective mechanism employed mainly by neurons to avoid hyperexcitability-related cell death. Upregulation of TTCCs by PKC in cells such as cardiovascular cells appear to be pathogenic and provides a mechanistic basis for the use of GPCR blockers for cardiovascular disorders.
2.1.6. Modulation of T-Type Ca2+ Channels by Cyclin-Dependent Kinase 5
Cyclin-dependent kinase 5 (Cdk5) is a proline-directed Ser/Thr PK meaning this Ser/Thr kinase activity is directed by an adjacent proline, exhibiting a recognition sequence of -X- Ser/Thr-Pro-X-. Based on its 60% sequence similarity and comparable substrate specificities to Cdc2 (a regulator of cell cycle progression), Cdk5 was first assumed to have a role in cell cycle control [145,146,147]. Cdk5 is a neuron-specific kinase and has been most extensively studied for its roles in neuronal migration, neurite outgrowth, axonal guidance, and synaptic plasticity; however, recently, it has been discovered that Cdk5 also plays a major role in circadian clock regulation under physiological conditions, DNA damage, cell cycle re-entry, mitochondrial dysfunction, and oxidative stress [148,149,150,151].
The modulation of TTCCs by Cdk5 was recently shown, where currents through TTCCs were found to be markedly increased by overexpression of Cdk5 in N1E-115 cells (neuroblastoma cell line), while Cdk5 siRNA knockdown significantly decreased TTCC currents. Similarly, overexpression of Cdk5 upregulated Cav3.1 and Cav3.2 TTCC currents in HEK-293 cells expressing Cav3.1 or Cav3.2, respectively [152]. Mechanistically, a phosphorylation site serine 2234 was identified in the c-terminus of Cav3.1 as being phosphorylated by cdk5, whereas the target sites in Cav3.2 were residues serine 561 and serine 1987 [153]. These results suggest that cdk5 may be used to target neuron-specific cellular effects mediated by TTCCs. This warrants further investigation of cell physiology assays modulated by cdk5 and TTCCs together.
2.2. Modulation of T-Type Ca2+ Channels by Protein Tyrosine Kinase Family
Protein tyrosine kinase (PTK) catalyzes the transfer of phosphate from ATP to tyrosine residues on the protein substrates [154]. PTK modulation of TTCCs has been reported in different cell types, which was deduced using PTK blockers. One of the first reports of TTCC modulation by PTK demonstrated that, in mouse spermatogenic cells, TTCC current is enhanced by PTK inhibitors tyrphostin A47 and A25, whereas tyrosine phosphatase inhibitors phenyl arsine oxide and sodium orthovanadate inhibited TTCC currents. In spermatogenic cells, TTCCs are maintained at a low conductance state by tyrosine phosphorylation, and phosphatase activity enhances the conduction, which may serve as a plausible mechanism for sperm activity during the early stage of mammalian fertilization [155]. In another study on NG108-15 cells, genistein, along with two other PTK inhibitors, lavendustin A and herbimycin, suppressed current through TTCCs, which was independent of G-protein activation [156]. This study, however, lacks information on the direct effects of PTK inhibitors on TTCC currents since PTK inhibitors can inhibit and alter the kinetics of TTCCs directly and independently of PTKs [157,158]. There is much need for data regarding PTK modulation of TTCCs. Perhaps, better inhibitors are required which do not block the TTCCs directly.
3. Modulation of T-Type Ca2+ Channels by Phosphatases
Protein phosphatases are enzymes that remove phosphate groups from proteins, and together with protein kinases, they modulate the activities of the proteins in a cell, thus indirectly contributing to cellular functions. Several phosphatases also regulate the activity of TTCCs, as described below.
3.1. Modulation of T-Type Ca2+ Channels by Tyrosine Phosphatases
Protein tyrosine phosphatases (PTPs) are a broad class of enzymes found in a wide range of cell types [159]. PTPs are encoded by the largest family of phosphatase genes. The cysteine residue serves as a nucleophile and is required for catalysis in these enzymes, which are identified by the active-site signature motif HCX5R [62]. By opposing the actions of PTKs, PTPs play a significant role in signal transduction systems involving tyrosine phosphorylation. Usually, PTP activities are measured in conjunction with PTK activities where antagonistic results are observed. In a similar study involving TTCCs, where PTK inhibitors (Tyrphostin A47 and A25) enhanced Ni2+-sensitive T-type current in mouse spermatogenic cells, the use of PTP activity antagonists (phenylarsine oxide and sodium orthovanadate) resulted in a blockade of voltage-dependent facilitation of the TTCC current [155]. This data indicates that PTPs, when activated, can enhance TTCCs in mouse spermatogenic cells. Unfortunately, not all studies which have investigated the modulation of TTCCs by PTKs have further explored the role of PTPs, thus amounting to limited data available.
3.2. Modulation of T-Type Ca2+ Channels by Calcineurin
Calcineurin (CaN) is a Ser/Thr phosphatase that is activated by increased intracellular Ca2+ concentrations [160,161]. CaN play a crucial part in several signaling cascades and directly connects Ca2+ signaling to protein phosphorylation states [162]. Structurally CaN is a heterodimer expressed as three different isoforms: αCaN, βCaN, and γCaN. While γCaN is shown to be present in the testis and brain, αCaN and βCaN isoforms are reported to be ubiquitously expressed. In terms of three-dimensional structure, these isoforms are generally made up of two subunits: calcineurin A, which contains the catalytic site and an autoinhibitory domain that is displaced following Ca2+ and calmodulin binding, and calcineurin B, which is a Ca2+-binding protein [163,164,165,166]. Some of the substrates of CaN are transcription factors, receptors, channels, proteins linked to mitochondria, and proteins linked to microtubules [163]. The interaction between CaN and Cav3.2 was discovered via anti-tag immunoprecipitation. It was found that Calmodulin and 0.1 mM Ca2+ enhanced the physical interaction between Cav3.2 and calcineurin, but 2 mM EGTA entirely eliminated it, suggesting that the interaction between Cav3.2 and calcineurin is Ca2+/calmodulin dependent. CaN-binding deficient Cav3.2 TTCCs (Cav3.2-9A and Cav3.2-ΔA, made by deleting the region necessary for CaN binding) conducted larger current in Cav3.2 transfected HEK293 cells [167], providing evidence that calcineurin might have a negative modulatory effect of Cav3.2 channel. CaN is involved in the G1-S transition either by initiating a pathway that leads to the accumulation of cyclin D1 [168] or by dephosphorylating the nuclear factor of activated T cells (NFAT), resulting in its translocation to the nucleus [169]. Cav3.2 was also found to be necessary for the G1-S transition (Table 2). It is intriguing to note that both CaN and TTCC have a role in G1-S transition, yet CaN modulates Cav3.2 negatively. This warrants further investigations.
4. Kinase and Phosphatase Regulation of TTCCs That Is Plausible but Yet to Be Investigated
With the development of sophisticated techniques and advancement in science, there is evidence to suggest several possible kinase modulations of TTCCs by the latest identified kinases; however, experimental data lacks support. We provide such evidences below and have classified them based on the specific isoform of TTCC which may be involved. Experimental investigations in support of the below evidence are required. We also briefly provide information on the kinases and their functions.
4.1. Possible Direct Modulation of Cav3.2 by Mitogen-Activated Protein Kinases
Mitogen-activated protein kinases (MAPKs) are Ser/Thr PKs that regulate evolutionarily conserved signal transduction pathways linked to cellular activities such as proliferation, migration, apoptosis, and differentiation [170]. Abnormal MAPK signaling is extensively implicated in the onset and progression of certain forms of cancer [171]. MAPK15 is the latest identified member of the MAPK family [172] that has recently emerged as a key modulator of autophagy [173], reactive oxygen species generation [174], etc. In fibroblasts that expressed oncogenically active Ras and gain-of-function mutant MEK (MAPK/extracellular signal-regulated kinase (ERK) kinase, which is a direct activator of MAPK), T-type currents were significantly suppressed, and treatment with a MEK-specific inhibitor (PD98059) restored TTCC activity [175]. In a similar study, it was observed that Zinc transporter-1, a putative zinc transporter increases Cav3.1 and Cav3.2 activity by activating Ras-ERK signaling. Endothelin-1, a potent stimulator of Ras-ERK signaling, also increased the surface expression of Cav3.1 channels in murine cardiomyocytes (HL-1 cells), confirmed by surface biotinylation. MAPK cascade inhibitor PD98059 abolished the effect suggesting the involvement of MAPKs [176]. In both these studies, however, the direct effect of MAPK on TTCC was not revealed, although affinity capture-mass spectroscopy has revealed a physical interaction between MAPK15 and TTCC Cav3.2 [177]. The modulation of TTCCs by MAPK, if any, would be interesting to understand as both the proteins have overlapping roles in cancer and vital cellular signaling.
4.2. Possible Modulation of Cav3.2 by Never in Mitosis A—Related Kinase
Never in Mitosis A (NIMA)-related kinases or Nrk or NEK are a large group of homologous kinases with related functions and structures. They participate in cellular processes such as cell cycle, cell division, cilia formation, and the DNA damage response, and any disturbance or abnormal expression of NEKs can cause different disorders such as cancer, diabetes, ciliopathies, central nervous system disorders, and inflammation-related diseases [178]. The activity and levels of NEK family proteins vary during different phases of the cell cycle, with their activity being tightly regulated both transcriptionally and post-translationally by multi-stage phosphorylation and proteolysis, and by these mechanisms, NEK activity is largely restricted to a brief window at the G2–M transition. During the S-phase, NEK proteins are inactive, they become activated by phosphorylation during the G2 phase, and the kinase activity becomes highest during the M-phase when they are hyperphosphorylated. The level of NEK proteins is kept in check by the proteolytic destruction during the end of the M-phase [179,180]. Human NEKs contain 11 isoforms from NEK1 to NEK11.
Affinity capture-mass spectroscopy data again revealed Cav3.2 TTCC & NEK4 as an interacting unit [181]. However, the significance of this association has never been explored in live cells, if any. The significance of this interaction lies in the fact that studies show both Cav3.2 and NEK4 to have a role in apoptosis and cell cycle progression [44,182] Whether they both are communicating with each other to execute such cellular functions is not known and needs to be further experimentally explored.
4.3. Possible Citron Kinase Modulation of Cav3.3
Historically, most of the isoform-specific studies on TTCCs have focused on two other isoforms of TTCCs, which are Cav3.1 and Cav3.2. Very few studies have investigated Cav3.3 isoform-specific regulation. The exact reasons for this disparity are not clear. However, we speculate that the less widespread expression of Cav3.3 and the late discovery of its roles in cellular functions may have been contributory factors. Interestingly, affinity capture-mass spectroscopy revealed a physical interaction between Citron kinase (CIT-K) and Cav3.3 [183]. CIT-K is a rho-interacting Ser/Thr kinase that functions during cytokinesis, where it promotes the actomyosin contractile ring’s constriction [184]. Recently, a mutation in CIT-K has been shown to induce primary microcephaly [185], emphasizing its importance for brain function. Since Cav3.3 channels have a predominant neuronal expression and microcephaly shows symptoms overlapping with symptoms during the dysfunction of TTCCs, it would be really interesting to determine if CIT-K modulates brain Cav3.3 channels.
4.4. Possible Modulation of Cav3.2 by SRC Kinase
Src is the prototype member of non-receptor PTKs whose activity is upregulated in multiple human malignancies. The role of Src is not only associated with maintaining cellular physiology but also in synaptic transmission and remote fear memory in the forebrain [186]. Many studies have shown that activation of kinases of this family mediates Ca2+ release, and in turn, their activation is regulated by Ca2+ signal via calmodulin [187], similar to CaMKII.
Studies regarding the regulation of TTCCs by Src kinase are minimal, and only indirect evidence exists. Gasotransmitter hydrogen sulphide (H2S) was shown to stimulate the growth of neurons by the involvement of TTCCs, Cav3.2 in particular, and Src kinase in NG108-15 cells. This neuritogenic effect of H2S was completely blocked by the application of BAPTA/AM(1,2-Bis(2-aminophenoxy) ethane-N, N, N′, N′-tetra acetic acid tetrakis (acetoxymethyl ester), an intracellular Ca2+ chelator, or by selective Cav3.2 inhibitors, such as zinc chloride and L-ascorbate [188]. Src kinase phosphorylation was also observed upon application of H2S, which was prevented by a Ca2+ chelator and Src kinase inhibitor PP2(4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo [3,4-d] pyrimidine) [189]. Overall, this indicated a role for Ca2+, Src kinase, and TTCCs in neuritogenesis; however, the underlying pathway remains elusive. Whether Src phosphorylates TTCCs or whether Ca2+ influx through TTCCs is responsible for the activation of Src needs further investigation.
4.5. Possible Modulation of Cav3.1 by Phosphatase and Tensin Homolog Deleted on Chromosome 10
Phosphatase and Tensin Homolog deleted on Chromosome 10 (PTEN) is a dual phosphatase with both protein and lipid phosphatase activities [190]. PTEN controls the signals that govern metabolic processes, including glycolysis, gluconeogenesis, glycogen production, lipid metabolism, and mitochondrial metabolism [191]. A significant majority of solid tumors show PTEN mutations, and PTEN is also involved in modeling the tumor immune microenvironment and modulating the DNA damage response [192]. Recently, Cav3.1 mRNA regulation in melanoma cells was shown to be associated with PTEN expression, where Cav3.1 mRNA levels were higher in the case of PTEN-compromised vemurafenib-resistant (Vem-R) melanoma cells, and Cav3.1 mRNA levels were lower in Vem-R cells with PTEN overexpression. This data indicated a negative correlation between PTEN and Cav3.1 expression, which may suggest the downregulation of TTCC expression in Vem-R cells by PTEN [193]. Both PTEN and Cav3.1 have been suggested as protooncogenes, and therefore, it will be interesting to determine if one can regulate the expression of the other, which needs further investigation.
5. Conclusions
Ca2+ can regulate cellular fates such as proliferation, apoptosis, and autophagy linked to cell cycle and even pathologies like cancer and neuronal disorders. Several cellular proteins, including receptors, pumps, and Ca2+ sensing proteins, work together to maintain Ca2+ concentrations inside the cell and cellular compartments. TTCCs specialized membrane proteins that play a crucial role in the signal transduction of many excitable and non-excitable cell types. Scientific literature shows that the activity of TTCCs is regulated by several kinases and phosphatases, leading to the enhancement or inhibition of TTCC currents. A general scheme of TTCC modulation by kinases and phosphatases is shown in Figure 3. A closer look reveals that multiple kinases acting on the same region (not the same amino acids) showed different effects on TTCCs [19,194]. These varied observations point towards several facts that (1) TTCCs may have different phosphorylation sites/regions specific for specific kinases. There is a lot of scope for finding such phosphorylation sites. Most of the studies did not reveal the actual binding site of the kinase or phosphatase to the TTCCs. (2) The modulation seems to be dependent on the tissue and cell type, and the fact that cells may have differential expression of TTCC isoforms complicates the issue. Development of isoform-specific blockers, or activators of TTCCs may address the issue but is equally challenging. Knock-down strategies may be employed to determine the isoform-specific role of TTCCs in modulation by kinases and phosphatases. (3) The modulation of TTCCs by kinases or phosphatases may be altered in pathological conditions. It would be pertinent to determine the level of expression of kinases and phosphatases in the cells/tissues under investigation to normalize the effects across varied cell types. In addition, in pathological conditions, so far, much of the focus has been given to cancerous conditions. Therefore, other pathological states, such as diabetes etc., could be explored where TTCCs are also implicated.
In conclusion, there is still an opportunity to explore different regions of TTCCs for their modulation by biological molecules. With the advent of techniques like affinity capture-mass spectroscopy and anti-tag immunoprecipitation, researchers have revealed some new binding interactions as well. However, the functional significance of such interactions is still to be explored. TTCC modulation by kinases and phosphatases could be important in pathology, where the modulation can lead to altered Ca2+ influx and thus contribute to pathology. An example of this kind exists for L-type Ca2+ channels where phosphorylation of L-type Ca2+ channels by CaMKII during heart failure contributes to the pathology, and inhibiting CaMKII is protective [195]. Much remains to be explored for TTCCs during pathological conditions.
Author Contributions
A.S.: Conceptualization, Literature search, Drafting, Reviewing and Editing, Illustration; G.R.: Literature search, Drafting, Reviewing and Editing, Illustration; A.B.: Conceptualization, Drafting, Reviewing and Editing, Illustration and Funding acquisition; J.G.: Conceptualization, Reviewing and Editing, Funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Indian Institute of Technology Hyderabad (IITH) research grant to A.B., Department of Biotechnology, India, Ph.D. fellowship to A.S., Ministry of Education, India, Ph.D. fellowship to G.R. and British Heart Foundation (grant RG/17/13/33173) to J.G.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Satheesh, N.; Büsselberg, D. The Role of Intracellular Calcium for the Development and Treatment of Neuroblastoma. Cancers 2015, 7, 823–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuttler, L.; Glaum, S.R.; Collins, B.A.; Miller, R.J. Calcium Signalling in Single Growth Hormone-Releasing Factor-Responsive Pituitary Cells. Endocrinology 1992, 130, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. Calcium Control of Neurotransmitter Release. Cold Spring Harb. Perspect. Biol. 2012, 4, a011353. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.G.; Groth, R.D.; Ma, H.; Barrett, C.F.; Owen, S.F.; Safa, P.; Tsien, R.W. CaV1 and CaV2 Channels Engage Distinct Modes of Ca2+ Signaling to Control CREB-Dependent Gene Expression. Cell 2012, 149, 1112–1124. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, T.; Beam, K.G.; Adams, B.A.; Niidome, T.; Numa, S. Regions of the Skeletal Muscle Dihydropyridine Receptor Critical for Excitation–Contraction Coupling. Nature 1990, 346, 567–569. [Google Scholar] [CrossRef]
- Phan, N.N.; Wang, C.-Y.; Chen, C.-F.; Sun, Z.; Lai, M.-D.; Lin, Y.-C. Voltage-Gated Calcium Channels: Novel Targets for Cancer Therapy. Oncol. Lett. 2017, 14, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.W. Targeting Voltage-Gated Calcium Channels in Neurological and Psychiatric Diseases. Nat. Rev. Drug Discov. 2016, 15, 19–34. [Google Scholar] [CrossRef]
- Yang, S.-N.; Berggren, P.-O. The Role of Voltage-Gated Calcium Channels in Pancreatic β-Cell Physiology and Pathophysiology. Endocr. Rev. 2006, 27, 621–676. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-Y.; Lai, M.-D.; Phan, N.N.; Sun, Z.; Lin, Y.-C. Meta-Analysis of Public Microarray Datasets Reveals Voltage-Gated Calcium Gene Signatures in Clinical Cancer Patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef]
- Dolphin, A.C. Voltage-Gated Calcium Channels: Their Discovery, Function and Importance as Drug Targets. Brain Neurosci. Adv. 2018, 2, 239821281879480. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Clapham, D.E. Ion Channels—Basic Science and Clinical Disease. N. Engl. J. Med. 1997, 336, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.M.; Nimigean, C.M. Voltage-Gated Potassium Channels: A Structural Examination of Selectivity and Gating. Cold Spring Harb. Perspect. Biol. 2016, 8, a029231. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Wang, J.; Jiang, H.; Shi, L.; Xie, J. Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels: An Emerging Role in Neurodegenerative Diseases. Front. Mol. Neurosci. 2019, 12, 141. [Google Scholar] [CrossRef] [Green Version]
- Lambert, R.C.; Maulet, Y.; Mouton, J.; Beattie, R.; Volsen, S.; De Waard, M.; Feltz, A. T-Type Ca2+ Current Properties Are Not Modified by Ca2+ Channel β Subunit Depletion in Nodosus Ganglion Neurons. J. Neurosci. 1997, 17, 6621–6628. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Striessnig, J.; Snutch, T.P.; Perez-Reyes, E. International Union of Pharmacology. XL. Compendium of Voltage-Gated Ion Channels: Calcium Channels. Pharmacol. Rev. 2003, 55, 579–581. [Google Scholar] [CrossRef] [Green Version]
- Blesneac, I.; Chemin, J.; Bidaud, I.; Huc-Brandt, S.; Vandermoere, F.; Lory, P. Phosphorylation of the Cav3.2 T-Type Calcium Channel Directly Regulates Its Gating Properties. Proc. Natl. Acad. Sci. USA 2015, 112, 13705–13710. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jiang, X.; Snutch, T.P.; Tao, J. Modulation of Low-Voltage-Activated T-Type Ca2+ Channels. Biochim. Biophys. Acta BBA-Biomembr. 2013, 1828, 1550–1559. [Google Scholar] [CrossRef] [Green Version]
- Weiss, N.; Zamponi, G.W. T-Type Calcium Channels: From Molecule to Therapeutic Opportunities. Int. J. Biochem. Cell Biol. 2019, 108, 34–39. [Google Scholar] [CrossRef]
- Billups, S.J.; Carter, B.L. Mibefradil: A New Class of Calcium-Channel Antagonists. Ann. Pharmacother. 1998, 32, 659–671. [Google Scholar] [CrossRef]
- Hayashi, K.; Wakino, S.; Homma, K.; Sugano, N.; Saruta, T. Pathophysiological Significance of T-Type Ca2+ Channels: Role of T-Type Ca2+ Channels in Renal Microcirculation. J. Pharmacol. Sci. 2005, 99, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Timic Stamenic, T.; Todorovic, S.M. Thalamic T-Type Calcium Channels as Targets for Hypnotics and General Anesthetics. Int. J. Mol. Sci. 2022, 23, 2349. [Google Scholar] [CrossRef]
- Iftinca, M. Neuronal T–Type Calcium Channels: What’s New? Iftinca: T–Type Channel Regulation. J. Med. Life 2011, 4, 126–138. [Google Scholar]
- Lenglet, S.; Louiset, E.; Delarue, C.; Vaudry, H.; Contesse, V. Activation of 5-HT7 Receptor in Rat Glomerulosa Cells Is Associated with an Increase in Adenylyl Cyclase Activity and Calcium Influx through T-Type Calcium Channels. Endocrinology 2002, 143, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Talley, E.M.; Cribbs, L.L.; Lee, J.-H.; Daud, A.; Perez-Reyes, E.; Bayliss, D.A. Differential Distribution of Three Members of a Gene Family Encoding Low Voltage-Activated (T-Type) Calcium Channels. J. Neurosci. 1999, 19, 1895–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteil, A.; Chemin, J.; Bourinet, E.; Mennessier, G.; Lory, P.; Nargeot, J. Molecular and Functional Properties of the Human A1G Subunit That Forms T-Type Calcium Channels. J. Biol. Chem. 2000, 275, 6090–6100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, G.; Moosmang, S.; Conrad, H.; Ludwig, A.; Hofmann, F.; Klugbauer, N. Expression of T- and L-Type Calcium Channel MRNA in Murine Sinoatrial Node. FEBS Lett. 2000, 481, 73–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, H.; Bhattacharjee, A.; Hu, F.; Zhang, M.; Goswami, T.; Wang, L.; Wu, S.; Berggren, P.O.; Li, M. Cloning of a T-Type Ca2+ Channel Isoform in Insulin-Secreting Cells. Diabetes 2000, 49, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Vignali, S.; Leiss, V.; Karl, R.; Hofmann, F.; Welling, A. Characterization of Voltage-Dependent Sodium and Calcium Channels in Mouse Pancreatic A- and B-Cells. J. Physiol. 2006, 572, 691–706. [Google Scholar] [CrossRef]
- Levitsky, K.L.; López-Barneo, J. Developmental Change of T-Type Ca2+ Channel Expression and Its Role in Rat Chromaffin Cell Responsiveness to Acute Hypoxia: T-Type Channels and Chromaffin Cell O2 Sensing. J. Physiol. 2009, 587, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.E.; Lunardi, N.; Boscolo, A.; Dong, X.; Erisir, A.; Jevtovic-Todorovic, V.; Todorovic, S.M. Immunohistological Demonstration of CaV3.2 T-Type Voltage-Gated Calcium Channel Expression in Soma of Dorsal Root Ganglion Neurons and Peripheral Axons of Rat and Mouse. Neuroscience 2013, 250, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, L.L.; Lee, J.-H.; Yang, J.; Satin, J.; Zhang, Y.; Daud, A.; Barclay, J.; Williamson, M.P.; Fox, M.; Rees, M.; et al. Cloning and Characterization of A1H From Human Heart, a Member of the T-Type Ca2+ Channel Gene Family. Circ. Res. 1998, 83, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barghouth, M.; Ye, Y.; Karagiannopoulos, A.; Ma, Y.; Cowan, E.; Wu, R.; Eliasson, L.; Renström, E.; Luan, C.; Zhang, E. The T-Type Calcium Channel CaV3.2 Regulates Insulin Secretion in the Pancreatic β-Cell. Cell Calcium 2022, 108, 102669. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Washburn, M.S.; Hans, M.; Urrutia, A.; Brust, P.F.; Prodanovich, P.; Harpold, M.M.; Stauderman, K.A. Structure and Functional Characterization of a Novel Human Low-Voltage Activated Calcium Channel. J. Neurochem. 1999, 72, 791–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrier, A.D.; Wang, H.; Talley, E.M.; Perez-Reyes, E.; Barrett, P.Q. α 1H T-Type Ca2+ Channel Is the Predominant Subtype Expressed in Bovine and Rat Zona Glomerulosa. Am. J. Physiol.-Cell Physiol. 2001, 280, C265–C272. [Google Scholar] [CrossRef]
- Lee, J.-H.; Daud, A.N.; Cribbs, L.L.; Lacerda, A.E.; Pereverzev, A.; Klöckner, U.; Schneider, T.; Perez-Reyes, E. Cloning and Expression of a Novel Member of the Low Voltage-Activated T-Type Calcium Channel Family. J. Neurosci. 1999, 19, 1912–1921. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Bao, W.; Wang, Z.; Nattel, S. Comparison of Ion-Channel Subunit Expression in Canine Cardiac Purkinje Fibers and Ventricular Muscle. Circ. Res. 2002, 91, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Toyota, M.; Ho, C.; Ohe-Toyota, M.; Baylin, S.B.; Issa, J.-P.J. Inactivation of CACNA1G, a T-Type Calcium Channel Gene, by Aberrant Methylation of Its 5′ CpG Island in Human Tumors1. Cancer Res. 1999, 59, 4535–4541. [Google Scholar]
- Ohkubo, T. T-Type Voltage-Activated Calcium Channel Cav3.1, but Not Cav3.2, Is Involved in the Inhibition of Proliferation and Apoptosis in MCF-7 Human Breast Cancer Cells. Int. J. Oncol. 2012, 41, 267–275. [Google Scholar] [CrossRef]
- Chemin, J.; Siquier-Pernet, K.; Nicouleau, M.; Barcia, G.; Ahmad, A.; Medina-Cano, D.; Hanein, S.; Altin, N.; Hubert, L.; Bole-Feysot, C.; et al. De Novo Mutation Screening in Childhood-Onset Cerebellar Atrophy Identifies Gain-of-Function Mutations in the CACNA1G Calcium Channel Gene. Brain 2018, 141, 1998–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Pushparaj, C.; Bahí, N.; Sorolla, A.; Herreros, J.; Pamplona, R.; Vilella, R.; Matias-Guiu, X.; Martí, R.M.; Cantí, C. Functional Expression of Voltage-Gated Calcium Channels in Human Melanoma: Functional Expression of VGCCs in Human Melanoma. Pigment Cell Melanoma Res. 2012, 25, 200–212. [Google Scholar] [CrossRef]
- Li, R.-F.; Man, Q.-W.; Liu, J.-Y.; Zheng, Y.-Y.; Gao, X.; Liu, H.-M. Overexpression of T-Type Calcium Channel Cav3.1 in Oral Squamous Cell Carcinoma: Association with Proliferation and Anti-Apoptotic Activity. J. Mol. Histol. 2021, 52, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Dziegielewska, B.; Casarez, E.V.; Yang, W.Z.; Gray, L.S.; Dziegielewski, J.; Slack-Davis, J.K. T-Type Ca2+ Channel Inhibition Sensitizes Ovarian Cancer to Carboplatin. Mol. Cancer Ther. 2016, 15, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Chen, H.; Zhou, C.; Liu, S.; Guo, M.; Chen, P.; Zhuang, H.; Xie, D.; Wu, S. T-Type Ca2+ Channel Expression in Human Esophageal Carcinomas: A Functional Role in Proliferation. Cell Calcium 2008, 43, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cruickshanks, N.; Yuan, F.; Wang, B.; Pahuski, M.; Wulfkuhle, J.; Gallagher, I.; Koeppel, A.F.; Hatef, S.; Papanicolas, C.; et al. Targetable T-Type Calcium Channels Drive Glioblastoma. Cancer Res. 2017, 77, 3479–3490. [Google Scholar] [CrossRef] [Green Version]
- Fukami, K.; Sekiguchi, F.; Yasukawa, M.; Asano, E.; Kasamatsu, R.; Ueda, M.; Yoshida, S.; Kawabata, A. Functional Upregulation of the H2S/Cav3.2 Channel Pathway Accelerates Secretory Function in Neuroendocrine-Differentiated Human Prostate Cancer Cells. Biochem. Pharmacol. 2015, 97, 300–309. [Google Scholar] [CrossRef]
- Basson, M.D.; Zeng, B.; Downey, C.; Sirivelu, M.P.; Tepe, J.J. Increased Extracellular Pressure Stimulates Tumor Proliferation by a Mechanosensitive Calcium Channel and PKC-β. Mol. Oncol. 2015, 9, 513–526. [Google Scholar] [CrossRef]
- Rzhepetskyy, Y.; Lazniewska, J.; Blesneac, I.; Pamphlett, R.; Weiss, N. CACNA1H Missense Mutations Associated with Amyotrophic Lateral Sclerosis Alter Ca v 3.2 T-Type Calcium Channel Activity and Reticular Thalamic Neuron Firing. Channels 2016, 10, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Daniil, G.; Fernandes-Rosa, F.L.; Chemin, J.; Blesneac, I.; Beltrand, J.; Polak, M.; Jeunemaitre, X.; Boulkroun, S.; Amar, L.; Strom, T.M.; et al. CACNA1H Mutations Are Associated With Different Forms of Primary Aldosteronism. EBioMedicine 2016, 13, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Pera, E.; Kaemmerer, E.; Milevskiy, M.J.G.; Yapa, K.T.D.S.; O’Donnell, J.S.; Brown, M.A.; Simpson, F.; Peters, A.A.; Roberts-Thomson, S.J.; Monteith, G.R. The Voltage Gated Ca2+-Channel Cav3.2 and Therapeutic Responses in Breast Cancer. Cancer Cell Int. 2016, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, M.; Kuriyan, J. The Conformational Plasticity of Protein Kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Kostich, M.; English, J.; Madison, V.; Gheyas, F.; Wang, L.; Qiu, P.; Greene, J.; Laz, T.M. Human Members of the Eukaryotic Protein Kinase Family. Genome Biol. 2002, 3, research0043.1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Hanks, S.K. Genomic Analysis of the Eukaryotic Protein Kinase Superfamily: A Perspective. Genome Biol. 2003, 4, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P. The Role of Protein Phosphorylation in Human Health and Disease.: Delivered on June 30th 2001 at the FEBS Meeting in Lisbon. Eur. J. Biochem. 2001, 268, 5001–5010. [Google Scholar] [CrossRef]
- Levinson, A.D.; Oppermann, H.; Levintow, L.; Varmus, H.E.; Bishop, J.M. Evidence That the Transforming Gene of Avian Sarcoma Virus Encodes a Protein Kinase Associated with a Phosphoprotein. Cell 1978, 15, 561–572. [Google Scholar] [CrossRef]
- Li, X.; Wilmanns, M.; Thornton, J.; Köhn, M. Elucidating Human Phosphatase-Substrate Networks. Sci. Signal. 2013, 6, rs10. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein Tyrosine Phosphatases—From Housekeeping Enzymes to Master Regulators of Signal Transduction. FEBS J. 2013, 280, 346–378. [Google Scholar] [CrossRef] [Green Version]
- Stoker, A.W. Protein Tyrosine Phosphatases and Signalling. J. Endocrinol. 2005, 185, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Brautigan, D.L. Protein Ser/ Thr Phosphatases—The Ugly Ducklings of Cell Signalling: Protein Ser/Thr Phosphatases. FEBS J. 2013, 280, 324–325. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein Tyrosine Phosphatases: From Genes, to Function, to Disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Walsh, D.A.; Perkins, J.P.; Krebs, E.G. An Adenosine 3′,5′-Monophosphate-Dependant Protein Kinase from Rabbit Skeletal Muscle. J. Biol. Chem. 1968, 243, 3763–3765. [Google Scholar] [CrossRef] [PubMed]
- Shoji, S.; Ericsson, L.H.; Walsh, K.A.; Fischer, E.H.; Titani, K. Amino Acid Sequence of the Catalytic Subunit of Bovine Type II Adenosine Cyclic 3′,5′-Phosphate-Dependent Protein Kinase. Biochemistry 1983, 22, 3702–3709. [Google Scholar] [CrossRef] [PubMed]
- Uhler, M.D.; Carmichael, D.F.; Lee, D.C.; Chrivia, J.C.; Krebs, E.G.; McKnight, G.S. Isolation of CDNA Clones Coding for the Catalytic Subunit of Mouse CAMP-Dependent Protein Kinase. Proc. Natl. Acad. Sci. USA 1986, 83, 1300–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.-H.; Taylor, S.S.; Sowadski, J.M. Crystal Structure of the Catalytic Subunit of Cyclic Adenosine Monophosphate-Dependent Protein Kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Chemin, J.; Mezghrani, A.; Bidaud, I.; Dupasquier, S.; Marger, F.; Barrère, C.; Nargeot, J.; Lory, P. Temperature-Dependent Modulation of CaV3 T-Type Calcium Channels by Protein Kinases C and A in Mammalian Cells. J. Biol. Chem. 2007, 282, 32710–32718. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wei, Y.; Pu, Y.; Jiang, D.; Jiang, X.; Zhang, Y.; Tao, J. Brain-Derived Neurotrophic Factor Stimulation of T-Type Ca2+ Channels in Sensory Neurons Contributes to Increased Peripheral Pain Sensitivity. Sci. Signal. 2019, 12, eaaw2300. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, Z.; Jiang, D.; Sun, Y.; Gao, S.; Jiang, X.; Wang, H.; Tao, J. Neuromedin B Receptor Stimulation of Cav3.2 T-Type Ca2+ Channels in Primary Sensory Neurons Mediates Peripheral Pain Hypersensitivity. Theranostics 2021, 11, 9342–9357. [Google Scholar] [CrossRef]
- Yue, J.; Zhang, Y.; Li, X.; Gong, S.; Tao, J.; Jiang, X. Activation of G-Protein-Coupled Receptor 30 Increases T-Type Calcium Currents in Trigeminal Ganglion Neurons via the Cholera Toxin-Sensitive Protein Kinase A Pathway. Pharm. 2014, 69, 804–808. [Google Scholar]
- Lenglet, S.; Louiset, E.; Delarue, C.; Vaudry, H.; Contesse, V. Involvement of T-type calcium channels in the mechanism of action of 5-HT in rat glomerulosa cells: A novel signaling pathway for the 5-HT7 receptor. Endocr. Res. 2002, 28, 651–655. [Google Scholar] [CrossRef]
- Louiset, E.; Duparc, C.; Lenglet, S.; Gomez-Sanchez, C.E.; Lefebvre, H. Role of CAMP/PKA Pathway and T-Type Calcium Channels in the Mechanism of Action of Serotonin in Human Adrenocortical Cells. Mol. Cell. Endocrinol. 2017, 441, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Xu, R.; Clarke, I.J.; Ruan, M.; Loneragan, K.; Roh, S. Diverse Intracellular Signalling Systems Used by Growth Hormone-releasing Hormone in Regulating Voltage-gated Ca2+ or K + Channels in Pituitary Somatotropes. Immunol. Cell Biol. 2000, 78, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.R.; Varanda, W.A. Intracellular Calcium Changes in Mice Leydig Cells Are Dependent on Calcium Entry through T-Type Calcium Channels: T-Type Calcium Channels in Leydig Cells. J. Physiol. 2007, 585, 339–349. [Google Scholar] [CrossRef]
- Costa, R.R.; dos Reis, R.I.; Aguiar, J.F.; Varanda, W.A. Luteinizing Hormone (LH) Acts through PKA and PKC to Modulate T-Type Calcium Currents and Intracellular Calcium Transients in Mice Leydig Cells. Cell Calcium 2011, 49, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-A.; Park, J.-Y.; Kang, H.-W.; Huh, S.-U.; Jeong, S.-W.; Lee, J.-H. Augmentation of Ca v 3.2 T-Type Calcium Channel Activity by CAMP-Dependent Protein Kinase A. J. Pharmacol. Exp. Ther. 2006, 318, 230–237. [Google Scholar] [CrossRef]
- Si, W.; Zhang, Y.; Chen, K.; Hu, D.; Qian, Z.; Gong, S.; Li, H.; Hao, Y.; Tao, J. Fibroblast Growth Factor Type 1 Receptor Stimulation of T-Type Ca2+ Channels in Sensory Neurons Requires the Phosphatidylinositol 3-Kinase and Protein Kinase A Pathways, Independently of Akt. Cell. Signal. 2018, 45, 93–101. [Google Scholar] [CrossRef]
- Pemberton, K.E.; Hill-Eubanks, L.J.; Jones, S.V.P. Modulation of Low-Threshold T-Type Calcium Channels by the Five Muscarinic Receptor Subtypes in NIH 3T3 Cells. Pflüg. Arch.-Eur. J. Physiol. 2000, 440, 452–461. [Google Scholar] [CrossRef]
- Wu, H.; Xie, X.; Sun, M.; Chen, M.; Tao, X.; Fang, X.; Meng, X.; Wei, W.; Yu, M. Modification of Mesenchymal Stem Cells by HMGB1 Promotes the Activity of Cav3.2 T-Type Calcium Channel via PKA/β-Catenin/γ-Cystathionase Pathway. Stem Cell Res. Ther. 2022, 13, 4. [Google Scholar] [CrossRef]
- Pfeiffer-Linn, C.; Lasater, E.M. Dopamine Modulates in a Differential Fashion T- and L-Type Calcium Currents in Bass Retinal Horizontal Cells. J. Gen. Physiol. 1993, 102, 277–294. [Google Scholar] [CrossRef]
- Pfeiffer-Linn, C.L.; Lasater, E.M. Multiple Second-Messenger System Modulation of Voltage-Activated Calcium Currents in Teleost Retinal Horizontal Cells. J. Neurophysiol. 1998, 80, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Kawai, F.; Kurahashi, T.; Kaneko, A. Adrenaline Enhances Odorant Contrast by Modulating Signal Encoding in Olfactory Receptor Cells. Nat. Neurosci. 1999, 2, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; DePuy, S.D.; Yao, J.; McIntire, W.E.; Barrett, P.Q. Protein Kinase A Activity Controls the Regulation of T-Type CaV3.2 Channels by Gβγ Dimers. J. Biol. Chem. 2009, 284, 7465–7473. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, Y.; Jiang, X.; Zhang, Y.; Zhang, L.; Gong, S.; Liu, C.; Zhou, L.; Tao, J. Neuromedin U Inhibits T-Type Ca2+ Channel Currents and Decreases Membrane Excitability in Small Dorsal Root Ganglia Neurons in Mice. Cell Calcium 2011, 49, 12–22. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Jiang, D.; Reid, P.F.; Jiang, X.; Qin, Z.; Tao, J. Alpha-Cobratoxin Inhibits T-Type Calcium Currents through Muscarinic M4 Receptor and Go-Protein Βγ Subunits-Dependent Protein Kinase A Pathway in Dorsal Root Ganglion Neurons. Neuropharmacology 2012, 62, 1062–1072. [Google Scholar] [CrossRef]
- Qian, W.-J.; Yin, N.; Gao, F.; Miao, Y.; Li, Q.; Li, F.; Sun, X.-H.; Yang, X.-L.; Wang, Z. Cannabinoid CB1 and CB2 Receptors Differentially Modulate L- and T-Type Ca2+ Channels in Rat Retinal Ganglion Cells. Neuropharmacology 2017, 124, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Chao, A.C.; Kouyama, K.; Heist, E.K.; Dong, Y.J.; Gardner, P. Calcium- and CaMKII-Dependent Chloride Secretion Induced by the Microsomal Ca(2+)-ATPase Inhibitor 2,5-Di-(Tert-Butyl)-1,4-Hydroquinone in Cystic Fibrosis Pancreatic Epithelial Cells. J. Clin. Investig. 1995, 96, 1794–1801. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.Y.; Zal, T.; Ch’en, I.L.; Gascoigne, N.R.J.; Hedrick, S.M. A Pivotal Role for the Multifunctional Calcium/Calmodulin-Dependent Protein Kinase II in T Cells: From Activation to Unresponsiveness. J. Immunol. 2005, 174, 5583–5592. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Zhao, W.; Li, B.; Desantiago, J.; Picht, E.; Kaetzel, M.A.; Schultz, J.E.J.; Kranias, E.G.; Bers, D.M.; Dedman, J.R. Targeted Inhibition of Sarcoplasmic Reticulum CaMKII Activity Results in Alterations of Ca2+ Homeostasis and Cardiac Contractility. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H599–H606. [Google Scholar] [CrossRef] [Green Version]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII Action in Long-Term Potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, W.; Lee, J.; Chen, X.; Díaz-Alonso, J.; Zhou, J.; Pleasure, S.; Nicoll, R.A. Synaptic Memory Requires CaMKII. eLife 2021, 10, e60360. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.; Greer-Short, A.; Satroplus, T.; Patel, N.; Nassal, D.; Mohler, P.J.; Hund, T.J. CaMKII-Dependent Late Na + Current Increases Electrical Dispersion and Arrhythmia in Ischemia-Reperfusion. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H794–H801. [Google Scholar] [CrossRef] [PubMed]
- Luczak, E.D.; Wu, Y.; Granger, J.M.; Joiner, M.A.; Wilson, N.R.; Gupta, A.; Umapathi, P.; Murphy, K.R.; Reyes Gaido, O.E.; Sabet, A.; et al. Mitochondrial CaMKII Causes Adverse Metabolic Reprogramming and Dilated Cardiomyopathy. Nat. Commun. 2020, 11, 4416. [Google Scholar] [CrossRef] [PubMed]
- Pasek, J.G.; Wang, X.; Colbran, R.J. Differential CaMKII Regulation by Voltage-Gated Calcium Channels in the Striatum. Mol. Cell. Neurosci. 2015, 68, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudmon, A.; Schulman, H.; Kim, J.; Maltez, J.M.; Tsien, R.W.; Pitt, G.S. CaMKII Tethers to L-Type Ca2+ Channels, Establishing a Local and Dedicated Integrator of Ca2+ Signals for Facilitation. J. Cell Biol. 2005, 171, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.K.; Fern, R.J.; Nee, J.J.; Barrett, P.Q. Ca(2+)-Dependent Activation of T-Type Ca2+ Channels by Calmodulin-Dependent Protein Kinase II. Am. J. Physiol.-Ren. Physiol. 1994, 267, F183–F189. [Google Scholar] [CrossRef] [PubMed]
- Barrett, P.Q.; Lu, H.-K.; Colbran, R.; Czernik, A.; Pancrazio, J.J. Stimulation of Unitary T-Type Ca2+ Channel Currents by Calmodulin-Dependent Protein Kinase II. Am. J. Physiol.-Cell Physiol. 2000, 279, C1694–C1703. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, J.T.; Wang, H.; Perez-Reyes, E.; Barrett, P.Q. Stimulation of Recombinant Ca v 3.2, T-type, Ca2+ Channel Currents by CaMKIIγ C. J. Physiol. 2002, 538, 343–355. [Google Scholar] [CrossRef]
- Welsby, P.J.; Wang, H.; Wolfe, J.T.; Colbran, R.J.; Johnson, M.L.; Barrett, P.Q. A Mechanism for the Direct Regulation of T-Type Calcium Channels by Ca2+ /Calmodulin-Dependent Kinase II. J. Neurosci. 2003, 23, 10116–10121. [Google Scholar] [CrossRef] [Green Version]
- Yao, J. Molecular Basis for the Modulation of Native T-Type Ca2+ Channels in Vivo by Ca2+/Calmodulin-Dependent Protein Kinase II. J. Clin. Investig. 2006, 116, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, S.; Shioda, N.; Yamamoto, Y.; Tagashira, H.; Fukunaga, K. The T-Type Voltage-Gated Calcium Channel as a Molecular Target of the Novel Cognitive Enhancer ST101: Enhancement of Long-Term Potentiation and CaMKII Autophosphorylation in Rat Cortical Slices: ST101 Enhances Cortical LTP and CaMKII Activity via T-Type VGCCs. J. Neurochem. 2012, 121, 44–53. [Google Scholar] [CrossRef]
- Xu, J.; Yabuki, Y.; Yu, M.; Fukunaga, K. T-Type Calcium Channel Enhancer SAK3 Produces Anti-Depressant-like Effects by Promoting Adult Hippocampal Neurogenesis in Olfactory Bulbectomized Mice. J. Pharmacol. Sci. 2018, 137, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hu, Y.; Lu, Y.; Yang, Q.; Fu, D.; Chen, F.; Li, Y. CaMKII and CaV3.2 T-Type Calcium Channel Mediate Connexin-43-Dependent Inflammation by Activating Astrocytes in Vincristine-Induced Neuropathic Pain. Cell Biol. Toxicol. 2021, 1–24. [Google Scholar] [CrossRef]
- Leung, T.; Manser, E.; Tan, L.; Lim, L. A Novel Serine/Threonine Kinase Binding the Ras-Related RhoA GTPase Which Translocates the Kinase to Peripheral Membranes. J. Biol. Chem. 1995, 270, 29051–29054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, M.; Ohashi, K.; Sasaki, Y.; Goshima, Y.; Niwa, R.; Uemura, T.; Mizuno, K. Control of Growth Cone Motility and Morphology by LIM Kinase and Slingshot via Phosphorylation and Dephosphorylation of Cofilin. J. Neurosci. 2003, 23, 2527–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, Y.; Oshiro, N.; Kinoshita, N.; Kawano, Y.; Matsuoka, Y.; Bennett, V.; Matsuura, Y.; Kaibuchi, K. Phosphorylation of Adducin by Rho-Kinase Plays a Crucial Role in Cell Motility. J. Cell Biol. 1999, 145, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Kano, Y.; Amano, M.; Onishi, H.; Kaibuchi, K.; Fujiwara, K. Rho-Kinase--Mediated Contraction of Isolated Stress Fibers. J. Cell Biol. 2001, 153, 569–584. [Google Scholar] [CrossRef]
- Leung, T.; Chen, X.Q.; Manser, E.; Lim, L. The P160 RhoA-Binding Kinase ROK Alpha Is a Member of a Kinase Family and Is Involved in the Reorganization of the Cytoskeleton. Mol. Cell. Biol. 1996, 16, 5313–5327. [Google Scholar] [CrossRef] [Green Version]
- Marlow, F.; Topczewski, J.; Sepich, D.; Solnica-Krezel, L. Zebrafish Rho Kinase 2 Acts Downstream of Wnt11 to Mediate Cell Polarity and Effective Convergence and Extension Movements. Curr. Biol. 2002, 12, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Rashid, M.H.; Fujita, R.; Contos, J.J.A.; Chun, J.; Ueda, H. Initiation of Neuropathic Pain Requires Lysophosphatidic Acid Receptor Signaling. Nat. Med. 2004, 10, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Iftinca, M.; Hamid, J.; Chen, L.; Varela, D.; Tadayonnejad, R.; Altier, C.; Turner, R.W.; Zamponi, G.W. Regulation of T-Type Calcium Channels by Rho-Associated Kinase. Nat. Neurosci. 2007, 10, 854–860. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Falcón, D.; Castro, M.J.; Ureña, J.; López-Barneo, J.; Castellano, A. Hypoxic Induction of T-Type Ca2+ Channels in Rat Cardiac Myocytes: Role of HIF-1α and RhoA/ROCK Signalling: HIF-1α and RhoA/ROCK Signalling in Cardiac Myocytes. J. Physiol. 2015, 593, 4729–4745. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, D.; Wang, F.; Shi, S.; Chen, Y.; Yang, B.; Tang, Y.; Huang, C. Exendin-4 Inhibits Structural Remodeling and Improves Ca2+ Homeostasis in Rats with Heart Failure via the GLP-1 Receptor through the ENOS/CGMP/PKG Pathway. Peptides 2017, 90, 69–77. [Google Scholar] [CrossRef]
- Krill, J.L.; Dawson-Scully, K. Characterization of a Novel Stimulus-Induced Glial Calcium Wave in Drosophila Larval Peripheral Segmental Nerves and Its Role in PKG-Modulated Thermoprotection. J. Neurogenet. 2021, 35, 221–235. [Google Scholar] [CrossRef]
- Weinmeister, P.; Lukowski, R.; Linder, S.; Traidl-Hoffmann, C.; Hengst, L.; Hofmann, F.; Feil, R. Cyclic Guanosine Monophosphate-Dependent Protein Kinase I Promotes Adhesion of Primary Vascular Smooth Muscle Cells. Mol. Biol. Cell 2008, 19, 4434–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, L.S.; Elbatarny, H.S.; Crawley, S.W.; Bennett, B.M.; Maurice, D.H. Compartmentation and Compartment-Specific Regulation of PDE5 by Protein Kinase G Allows Selective CGMP-Mediated Regulation of Platelet Functions. Proc. Natl. Acad. Sci. USA 2008, 105, 13650–13655. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Schweda, F.; Sequeira-Lopez, M.L.S.; Hofmann, F.; Sandner, P.; Schlossmann, J. Protein Kinase G Is Involved in Acute but Not in Long-Term Regulation of Renin Secretion. Front. Pharmacol. 2019, 10, 800. [Google Scholar] [CrossRef] [Green Version]
- Harraz, O.F.; Brett, S.E.; Welsh, D.G. Nitric Oxide Suppresses Vascular Voltage-Gated T-Type Ca2+ Channels through CGMP/PKG Signaling. Am. J. Physiol.-Heart Circ. Physiol. 2014, 306, H279–H285. [Google Scholar] [CrossRef] [Green Version]
- Samuel, S.; Zhang, K.; Tang, Y.-D.; Gerdes, A.M.; Carrillo-Sepulveda, M.A. Triiodothyronine Potentiates Vasorelaxation via PKG/VASP Signaling in Vascular Smooth Muscle Cells. Cell. Physiol. Biochem. 2017, 41, 1894–1904. [Google Scholar] [CrossRef] [Green Version]
- Buisson, B.; Laflamme, L.; Bottari, S.P.; de Gasparo, M.; Gallo-Payet, N.; Payet, M.D. A G Protein Is Involved in the Angiotensin AT2 Receptor Inhibition of the T-Type Calcium Current in Non-Differentiated NG108-15 Cells. J. Biol. Chem. 1995, 270, 1670–1674. [Google Scholar] [CrossRef] [Green Version]
- Kawai, F.; Miyachi, E. Modulation by CGMP of the Voltage-Gated Currents in Newt Olfactory Receptor Cells. Neurosci. Res. 2001, 39, 327–337. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Wu, N.; Yin, N.; Sun, X.-H.; Wang, Z. Activation of Somatostatin Receptor 5 Suppresses T-Type Ca2+ Channels through NO/CGMP/PKG Signaling Pathway in Rat Retinal Ganglion Cells. Neurosci. Lett. 2019, 708, 134337. [Google Scholar] [CrossRef]
- Bkaily, G.; Sculptoreanu, A.; Wang, S.; Nader, M.; Hazzouri, K.M.; Jacques, D.; Regoli, D.; D’Orleans-Juste, P.; Avedanian, L. Angiotensin II-Induced Increase of T-Type Ca2+ Current and Decrease of L-Type Ca2+ Current in Heart Cells. Peptides 2005, 26, 1410–1417. [Google Scholar] [CrossRef]
- Doerner, D.; Pitler, T.; Alger, B. Protein Kinase C Activators Block Specific Calcium and Potassium Current Components in Isolated Hippocampal Neurons. J. Neurosci. 1988, 8, 4069–4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Wang, Y.; Kang, L.; Shimaoka, T.; Marni, F.; Ono, K. Intracellular Ca2+- and PKC-Dependent Upregulation of T-Type Ca2+ Channels in LPC-Stimulated Cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 48, 131–139. [Google Scholar] [CrossRef]
- Lu, X.; Bean, J.S.; Kassab, G.S.; Rekhter, M.D. Protein Kinase C Inhibition Ameliorates Functional Endothelial Insulin Resistance and Vascular Smooth Muscle Cell Hypersensitivity to Insulin in Diabetic Hypertensive Rats. Cardiovasc. Diabetol. 2011, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, T.; Ito, H.; Nitta, J.; Tsujino, M.; Adachi, S.; Hiroe, M.; Marumo, F.; Sawanobori, T.; Hiraoka, M. Endothelin-1 Enhances Calcium Entry through T-Type Calcium Channels in Cultured Neonatal Rat Ventricular Myocytes. Circ. Res. 1992, 71, 1242–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-Y.; Kang, H.-W.; Moon, H.-J.; Huh, S.-U.; Jeong, S.-W.; Soldatov, N.M.; Lee, J.-H. Activation of Protein Kinase C Augments T-Type Ca2+ Channel Activity without Changing Channel Surface Density. J. Physiol. 2006, 577, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Jeong, S.-W.; Perez-Reyes, E.; Lee, J.-H. Modulation of Ca v 3.2 T-Type Ca2+ Channels by Protein Kinase C. FEBS Lett. 2003, 547, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Herrington, J.; Lingle, C.J. Kinetic and Pharmacological Properties of Low Voltage-Activated Ca2+ Current in Rat Clonal (GH3) Pituitary Cells. J. Neurophysiol. 1992, 68, 213–232. [Google Scholar] [CrossRef]
- Hockberger, P.; Toselli, M.; Swandulla, D.; Lux, H.D. A Diacylglycerol Analogue Reduces Neuronal Calcium Currents Independently of Protein Kinase C Activation. Nature 1989, 338, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Brown, A.M. Protein Kinase Activator 1-Oleoyl-2-Acetyl-Sn-Glycerol Inhibits Two Types of Calcium Currents in GH3 Cells. Am. J. Physiol.-Cell Physiol. 1988, 254, C206–C210. [Google Scholar] [CrossRef]
- Schroeder, J.; Fischbach, P.; McCleskey, E. T-Type Calcium Channels: Heterogeneous Expression in Rat Sensory Neurons and Selective Modulation by Phorbol Esters. J. Neurosci. 1990, 10, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Toselli, M.; Lux, H.D. Opposing Effects of Acetylcholine on the Two Classes of Voltage-Dependent Calcium Channels in Hippocampal Neurons. In Central Cholinergic Synaptic Transmission; Frotscher, M., Misgeld, U., Eds.; Experientia Supplementum; Birkhäuser Basel: Basel, Switzerland, 1989; Volume 57, pp. 97–103. ISBN 978-3-0348-9922-2. [Google Scholar]
- Tseng, G.N.; Boyden, P.A. Different Effects of Intracellular Ca and Protein Kinase C on Cardiac T and L Ca Currents. Am. J. Physiol.-Heart Circ. Physiol. 1991, 261, H364–H379. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Wang, F.; Zhang, Y.; Wang, J.; Qin, Z.; Jiang, X.; Tao, J. Activation of M3 Muscarinic Receptors Inhibits T-Type Ca2+ Channel Currents via Pertussis Toxin-Sensitive Novel Protein Kinase C Pathway in Small Dorsal Root Ganglion Neurons. Cell. Signal. 2011, 23, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Rossier, M.F.; Aptel, H.B.C.; Python, C.P.; Burnay, M.M.; Vallotton, M.B.; Capponi, A.M. Inhibition of Low Threshold Calcium Channels by Angiotensin II in Adrenal Glomerulosa Cells through Activation of Protein Kinase C. J. Biol. Chem. 1995, 270, 15137–15142. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Park, M.K.; Uhm, D.-Y.; Chung, S. Modulation of T-Type Ca2+ Channels by Corticotropin-Releasing Factor through Protein Kinase C Pathway in MN9D Dopaminergic Cells. Biochem. Biophys. Res. Commun. 2007, 358, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.-H.; Yang, Y.-C.; Pan, M.-K.; Huang, C.-S.; Kuo, C.-C. Modulation of Subthalamic T-Type Ca2+ Channels Remedies Locomotor Deficits in a Rat Model of Parkinson Disease. J. Clin. Investig. 2011, 121, 3289–3305. [Google Scholar] [CrossRef]
- Rangel, A.; Sánchez-Armass, S.; Meza, U. Protein Kinase C-Mediated Inhibition of Recombinant T-Type Ca V 3.2 Channels by Neurokinin 1 Receptors. Mol. Pharmacol. 2010, 77, 202–210. [Google Scholar] [CrossRef]
- Shan, H.Q.; Hammarback, J.A.; Godwin, D.W. Ethanol Inhibition of a T-Type Ca2+ Channel Through Activity of Protein Kinase C. Alcohol. Clin. Exp. Res. 2013, 37, 1333–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Morishima, M.; Li, D.; Takahashi, N.; Saikawa, T.; Nattel, S.; Ono, K. Binge Alcohol Exposure Triggers Atrial Fibrillation Through T-Type Ca2+ Channel Upregulation via Protein Kinase C (PKC)/Glycogen Synthesis Kinase 3β (GSK3β)/Nuclear Factor of Activated T-Cells (NFAT) Signaling—An Experimental Account of Holiday Heart Syndrome. Circ. J. 2020, 84, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ji, H.; Wang, J.; Sun, Y.; Qian, Z.; Jiang, X.; Snutch, T.P.; Sun, Y.; Tao, J. Melatonin-Mediated Inhibition of Cav3.2 T-Type Ca2+ Channels Induces Sensory Neuronal Hypoexcitability through the Novel Protein Kinase C-Eta Isoform. J. Pineal Res. 2018, 64, e12476. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Pant, H.C.; Wada, E.; Battey, J.F. Neuronal Cdc2-like Kinase: A Cdc2-Related Protein Kinase with Predominantly Neuronal Expression. Proc. Natl. Acad. Sci. USA 1992, 89, 10867–10871. [Google Scholar] [CrossRef] [Green Version]
- Lew, J.; Beaudette, K.; Litwin, C.M.; Wang, J.H. Purification and Characterization of a Novel Proline-Directed Protein Kinase from Bovine Brain. J. Biol. Chem. 1992, 267, 13383–13390. [Google Scholar] [CrossRef]
- Meyerson, M.; Enders, G.H.; Wu, C.L.; Su, L.K.; Gorka, C.; Nelson, C.; Harlow, E.; Tsai, L.H. A Family of Human Cdc2-Related Protein Kinases. EMBO J. 1992, 11, 2909–2917. [Google Scholar] [CrossRef] [PubMed]
- Kesavapany, S.; Li, B.-S.; Pant, H.C. Cyclin-Dependent Kinase 5 in Neurofilament Function and Regulation. Neurosignals 2003, 12, 252–264. [Google Scholar] [CrossRef]
- Lai, K.-O.; Ip, N. Cdk5: A Key Player at Neuronal Synapse with Diverse Functions. Mini-Rev. Med. Chem. 2015, 15, 390–395. [Google Scholar] [CrossRef]
- Pao, P.-C.; Tsai, L.-H. Three Decades of Cdk5. J. Biomed. Sci. 2021, 28, 79. [Google Scholar] [CrossRef]
- Su, S.C.; Tsai, L.-H. Cyclin-Dependent Kinases in Brain Development and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 465–491. [Google Scholar] [CrossRef]
- Calderón-Rivera, A.; Sandoval, A.; González-Ramírez, R.; González-Billault, C.; Felix, R. Regulation of Neuronal Cav3.1 Channels by Cyclin-Dependent Kinase 5 (Cdk5). PLoS ONE 2015, 10, e0119134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, K.; Calderón-Rivera, A.; Sandoval, A.; González-Ramírez, R.; Vargas-Parada, A.; Ojeda-Alonso, J.; Granados-Soto, V.; Delgado-Lezama, R.; Felix, R. Cdk5-Dependent Phosphorylation of Ca V 3.2 T-Type Channels: Possible Role in Nerve Ligation-Induced Neuropathic Allodynia and the Compound Action Potential in Primary Afferent C Fibers. J. Neurosci. 2020, 40, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R.; Till, J.H. Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, C.; Lemos, J.R.; Florman, H.M. Voltage-Dependent Modulation of T-Type Calcium Channels by Protein Tyrosine Phosphorylation. EMBO J. 1997, 16, 1593–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, H.; Fukuda, K.; Mima, H.; Shoda, T.; Kato, S.; Mori, K. Tyrosine Kinase Inhibitors Suppress N-Type and T-Type Ca2+ Channel Currents in NG108-15 Cells. Pflugers Arch. Eur. J. Physiol. 1998, 436, 127–132. [Google Scholar] [CrossRef]
- Tao, J.; Zhang, Y.; Li, S.; Sun, W.; Soong, T.W. Tyrosine Kinase-Independent Inhibition by Genistein on Spermatogenic T-Type Calcium Channels Attenuates Mouse Sperm Motility and Acrosome Reaction. Cell Calcium 2009, 45, 133–143. [Google Scholar] [CrossRef]
- Kurejová, M.; Lacinová, L. Effect of Protein Tyrosine Kinase Inhibitors on the Current through the CaV3.1 Channel. Arch. Biochem. Biophys. 2006, 446, 20–27. [Google Scholar] [CrossRef]
- Lau, K.H.W.; Farley, J.R.; Baylink, D.J. Phosphotyrosyl Protein Phosphatases. Biochem. J. 1989, 257, 23–36. [Google Scholar] [CrossRef]
- Klee, C.B.; Ren, H.; Wang, X. Regulation of the Calmodulin-Stimulated Protein Phosphatase, Calcineurin. J. Biol. Chem. 1998, 273, 13367–13370. [Google Scholar] [CrossRef] [Green Version]
- Rusnak, F.; Mertz, P. Calcineurin: Form and Function. Physiol. Rev. 2000, 80, 1483–1521. [Google Scholar] [CrossRef]
- Park, Y.-J.; Yoo, S.-A.; Kim, M.; Kim, W.-U. The Role of Calcium–Calcineurin–NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Creamer, T.P. Calcineurin. Cell Commun. Signal. 2020, 18, 137. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, C.S.; Heitman, J. Calcineurin: Structure, Function, and Inhibition. Cell Biochem. Biophys. 1999, 30, 115–151. [Google Scholar] [CrossRef]
- Jorgensen, K.A.; Koefoed-Nielsen, P.B.; Karamperis, N. Calcineurin Phosphatase Activity and Immunosuppression. A Review on the Role of Calcineurin Phosphatase Activity and the Immunosuppressive Effect of Cyclosporin A and Tacrolimus. Scand. J. Immunol. 2003, 57, 93–98. [Google Scholar] [CrossRef]
- Perrino, B.A.; Wilson, A.J.; Ellison, P.; Clapp, L.H. Substrate Selectivity and Sensitivity to Inhibition by FK506 and Cyclosporin A of Calcineurin Heterodimers Composed of the α or β Catalytic Subunit: Enzymatic Characteristics of Calcineurin Isoforms. Eur. J. Biochem. 2002, 269, 3540–3548. [Google Scholar] [CrossRef]
- Huang, C.-H.; Chen, Y.-C.; Chen, C.-C. Physical Interaction between Calcineurin and Cav3.2 T-type Ca2+ Channel Modulates Their Functions. FEBS Lett. 2013, 587, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Tomono, M.; Toyoshima, K.; Ito, M.; Amano, H.; Kiss, Z. Inhibitors of Calcineurin Block Expression of Cyclins A and E Induced by Fibroblast Growth Factor in Swiss 3T3 Fibroblasts. Arch. Biochem. Biophys. 1998, 353, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional Regulation by Calcium, Calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Ahmadi, A.; Mortezaee, K. Extracellular-signal-regulated Kinase/Mitogen-activated Protein Kinase Signaling as a Target for Cancer Therapy: An Updated Review. Cell Biol. Int. 2019, 43, 1206–1222. [Google Scholar] [CrossRef]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK Pathways in the Regulation of Metabolic Reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [Green Version]
- Abe, M.K.; Saelzler, M.P.; Espinosa, R.; Kahle, K.T.; Hershenson, M.B.; Le Beau, M.M.; Rosner, M.R. ERK8, a New Member of the Mitogen-Activated Protein Kinase Family. J. Biol. Chem. 2002, 277, 16733–16743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colecchia, D.; Dapporto, F.; Tronnolone, S.; Salvini, L.; Chiariello, M. MAPK15 Is Part of the ULK Complex and Controls Its Activity to Regulate Early Phases of the Autophagic Process. J. Biol. Chem. 2018, 293, 15962–15976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Colecchia, D.; Ilardi, G.; Acunzo, M.; Nigita, G.; Sasdelli, F.; Celetti, A.; Strambi, A.; Staibano, S.; Croce, C.M.; et al. MAPK15 Upregulation Promotes Cell Proliferation and Prevents DNA Damage in Male Germ Cell Tumors. Oncotarget 2016, 7, 20981–20998. [Google Scholar] [CrossRef] [PubMed]
- Strobeck, M.W.; Okuda, M.; Yamaguchi, H.; Schwartz, A.; Fukasawa, K. Morphological Transformation Induced by Activation of the Mitogen-Activated Protein Kinase Pathway Requires Suppression of the T-Type Ca2+ Channel. J. Biol. Chem. 1999, 274, 15694–15700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, M.; Beharier, O.; Levy, S.; Kahn, J.; Dror, S.; Blumenthal, D.; Gheber, L.A.; Peretz, A.; Katz, A.; Moran, A.; et al. ZnT-1 Enhances the Activity and Surface Expression of T-Type Calcium Channels through Activation of Ras-ERK Signaling. Am. J. Physiol.-Cell Physiol. 2012, 303, C192–C203. [Google Scholar] [CrossRef] [Green Version]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The Protein Interaction Landscape of the Human CMGC Kinase Group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef] [Green Version]
- Peres de Oliveira, A.; Kazuo Issayama, L.; Betim Pavan, I.C.; Riback Silva, F.; Diniz Melo-Hanchuk, T.; Moreira Simabuco, F.; Kobarg, J. Checking NEKs: Overcoming a Bottleneck in Human Diseases. Molecules 2020, 25, 1778. [Google Scholar] [CrossRef]
- Fry, A.M.; Nigg, E.A. Cell Cycle: The NIMA Kinase Joins Forces with Cdc2. Curr. Biol. 1995, 5, 1122–1125. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, M.J.; Krien, M.J.E.; Hunter, T. Never Say Never. The NIMA-Related Protein Kinases in Mitotic Control. Trends Cell Biol. 2003, 13, 221–228. [Google Scholar] [CrossRef]
- Basei, F.L.; Meirelles, G.V.; Righetto, G.L.; dos Santos Migueleti, D.L.; Smetana, J.H.C.; Kobarg, J. New Interaction Partners for Nek4.1 and Nek4.2 Isoforms: From the DNA Damage Response to RNA Splicing. Proteome Sci. 2015, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Antal, L.; Martin-Caraballo, M. T-Type Calcium Channels in Cancer. Cancers 2019, 11, 134. [Google Scholar] [CrossRef] [Green Version]
- Capalbo, L.; Bassi, Z.I.; Geymonat, M.; Todesca, S.; Copoiu, L.; Enright, A.J.; Callaini, G.; Riparbelli, M.G.; Yu, L.; Choudhary, J.S.; et al. The Midbody Interactome Reveals Unexpected Roles for PP1 Phosphatases in Cytokinesis. Nat. Commun. 2019, 10, 4513. [Google Scholar] [CrossRef] [Green Version]
- Madaule, P.; Eda, M.; Watanabe, N.; Fujisawa, K.; Matsuoka, T.; Bito, H.; Ishizaki, T.; Narumiya, S. Role of Citron Kinase as a Target of the Small GTPase Rho in Cytokinesis. Nature 1998, 394, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Basit, S.; Al-Harbi, K.M.; Alhijji, S.A.M.; Albalawi, A.M.; Alharby, E.; Eldardear, A.; Samman, M.I. CIT, a Gene Involved in Neurogenic Cytokinesis, Is Mutated in Human Primary Microcephaly. Hum. Genet. 2016, 135, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhang, B.; Lu, W.; Peng, L.; Yang, Q.; Cao, W.; Lin, S.; Yu, W.; Li, X.; Ke, Y.; et al. Increased Src Family Kinase Activity Disrupts Excitatory Synaptic Transmission and Impairs Remote Fear Memory in Forebrain Shp2-Deficient Mice. Mol. Neurobiol. 2017, 54, 7235–7250. [Google Scholar] [CrossRef] [PubMed]
- Stateva, S.R.; Salas, V.; Anguita, E.; Benaim, G.; Villalobo, A. Ca2+/Calmodulin and Apo-Calmodulin Both Bind to and Enhance the Tyrosine Kinase Activity of c-Src. PLoS ONE 2015, 10, e0128783. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, K.; Tarui, T.; Yoshida, S.; Sekiguchi, F.; Matsunami, M.; Ohi, A.; Fukami, K.; Ichida, S.; Nishikawa, H.; Kawabata, A. Hydrogen Sulfide Evokes Neurite Outgrowth and Expression of High-Voltage-Activated Ca2+ Currents in NG108-15 Cells: Involvement of T-Type Ca2+ Channels. J. Neurochem. 2009, 108, 676–684. [Google Scholar] [CrossRef]
- Tarui, T.; Fukami, K.; Nagasawa, K.; Yoshida, S.; Sekiguchi, F.; Kawabata, A. Involvement of Src Kinase in T-Type Calcium Channel-Dependent Neuronal Differentiation of NG108-15 Cells by Hydrogen Sulfide: Src-Dependent Neuronal Differentiation by H2S. J. Neurochem. 2010, 114, 512–519. [Google Scholar] [CrossRef]
- Lee, J.-O.; Yang, H.; Georgescu, M.-M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal Structure of the PTEN Tumor Suppressor. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Fusco, N.; Sajjadi, E.; Venetis, K.; Gaudioso, G.; Lopez, G.; Corti, C.; Guerini Rocco, E.; Criscitiello, C.; Malapelle, U.; Invernizzi, M. PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes 2020, 11, 719. [Google Scholar] [CrossRef] [PubMed]
- Barceló, C.; Sisó, P.; Maiques, O.; García-Mulero, S.; Sanz-Pamplona, R.; Navaridas, R.; Megino, C.; Felip, I.; Urdanibia, I.; Eritja, N.; et al. T-Type Calcium Channels as Potential Therapeutic Targets in Vemurafenib-Resistant BRAFV600E Melanoma. J. Investig. Dermatol. 2020, 140, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Huc, S.; Monteil, A.; Bidaud, I.; Barbara, G.; Chemin, J.; Lory, P. Regulation of T-Type Calcium Channels: Signalling Pathways and Functional Implications. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2009, 1793, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alonso, J.L.; Bhargava, A.; O’Hara, T.; Glukhov, A.V.; Schobesberger, S.; Bhogal, N.; Sikkel, M.B.; Mansfield, C.; Korchev, Y.E.; Lyon, A.R.; et al. Microdomain-Specific Modulation of L-Type Calcium Channels Leads to Triggered Ventricular Arrhythmia in Heart Failure. Circ. Res. 2016, 119, 944–955. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Classification of voltage-gated ion channels based on the activation potential. *Cav1.3 channel isoforms are the exception in L-type channels as they can also be activated at low-voltages.
Figure 1.
Classification of voltage-gated ion channels based on the activation potential. *Cav1.3 channel isoforms are the exception in L-type channels as they can also be activated at low-voltages.

Figure 2.
Schematic representation of Cav3 pore-forming subunit showing interaction sites of different kinases and phosphatases. Each of the four domains have transmembrane segments labelled from 1-6. Target residues of PTK, PKG, ROCK, and PKC are not yet identified; however, they are found to be associated with the II-III loop region of the Cav3 channel. PKA phosphorylates ser1107 residue of Cav3.2, while its interaction with other isoforms is not yet known. Ser 1198 of Cav3.2 acts as a modulatory site for CaMKII. Cdk5 interacts with both Cav3.1 (ser 2234 in the C-terminus) and Cav3.2 (ser 561 (I-II loop) & ser 1987 in the C-terminus). Calcineurin recognizes PCISVE (2190–2195) & LTVP (2261–2264) motifs in the C-terminus.
Figure 2.
Schematic representation of Cav3 pore-forming subunit showing interaction sites of different kinases and phosphatases. Each of the four domains have transmembrane segments labelled from 1-6. Target residues of PTK, PKG, ROCK, and PKC are not yet identified; however, they are found to be associated with the II-III loop region of the Cav3 channel. PKA phosphorylates ser1107 residue of Cav3.2, while its interaction with other isoforms is not yet known. Ser 1198 of Cav3.2 acts as a modulatory site for CaMKII. Cdk5 interacts with both Cav3.1 (ser 2234 in the C-terminus) and Cav3.2 (ser 561 (I-II loop) & ser 1987 in the C-terminus). Calcineurin recognizes PCISVE (2190–2195) & LTVP (2261–2264) motifs in the C-terminus.

Figure 3.
General scheme of TTCC modulation by Kinases and phosphatases. Pathway 1 shows the phosphatase activation via increase in intracellular Ca2+ levels, which then modulates the TTCC activity. Pathway 2 shows the kinase activation via GPCR, which in turn modulates TTCC activity. Question mark indicates that direct experimental evidence is awaited.
Figure 3.
General scheme of TTCC modulation by Kinases and phosphatases. Pathway 1 shows the phosphatase activation via increase in intracellular Ca2+ levels, which then modulates the TTCC activity. Pathway 2 shows the kinase activation via GPCR, which in turn modulates TTCC activity. Question mark indicates that direct experimental evidence is awaited.


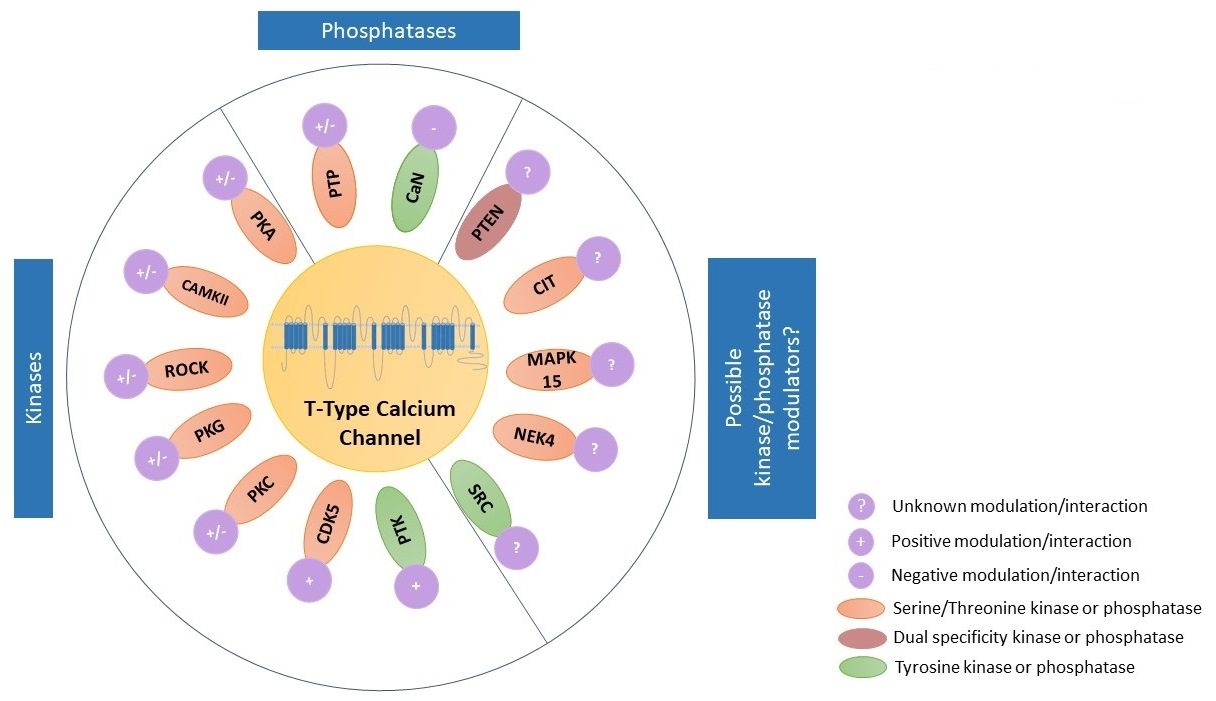{kind=link}
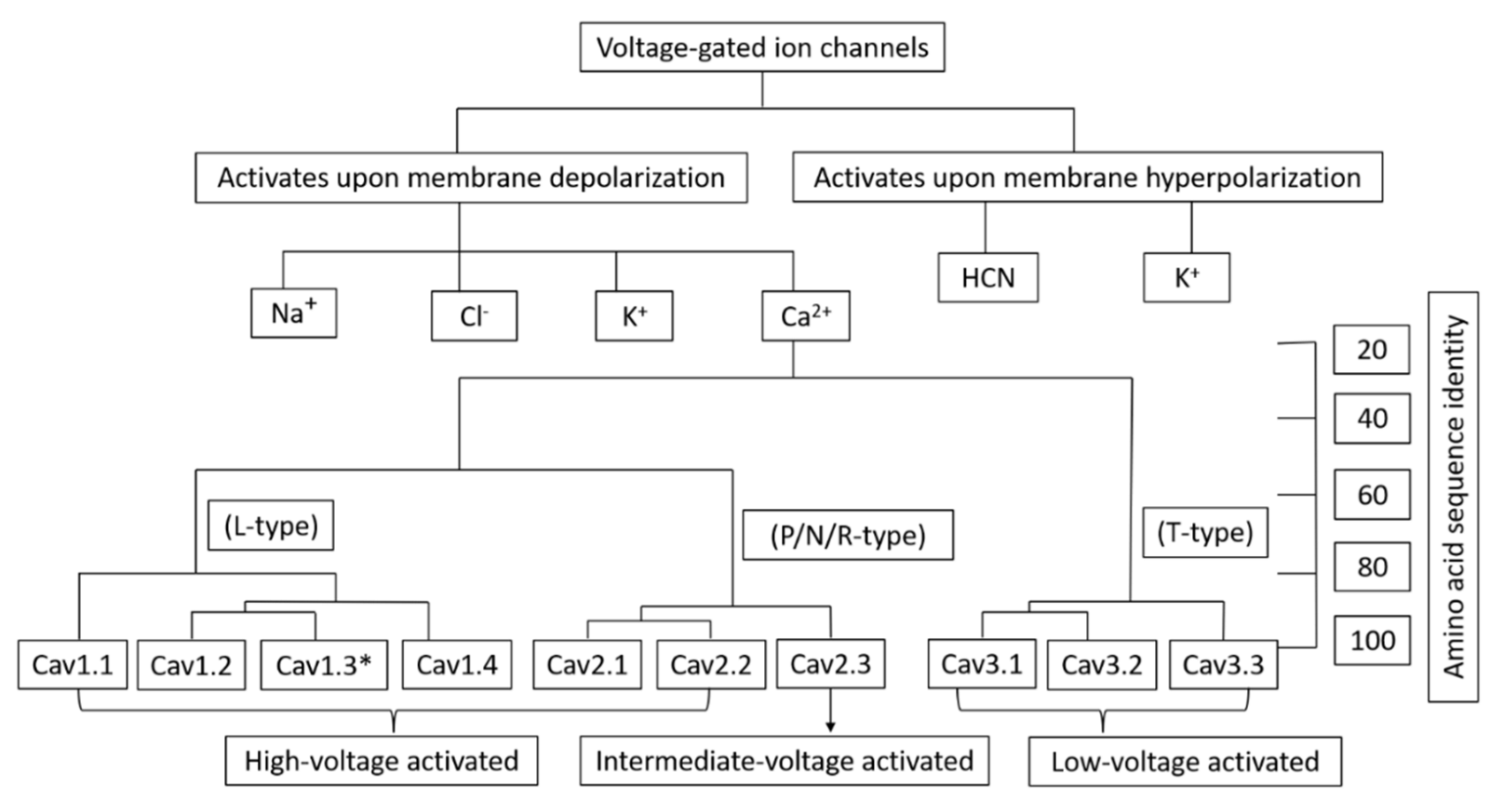{kind=link}
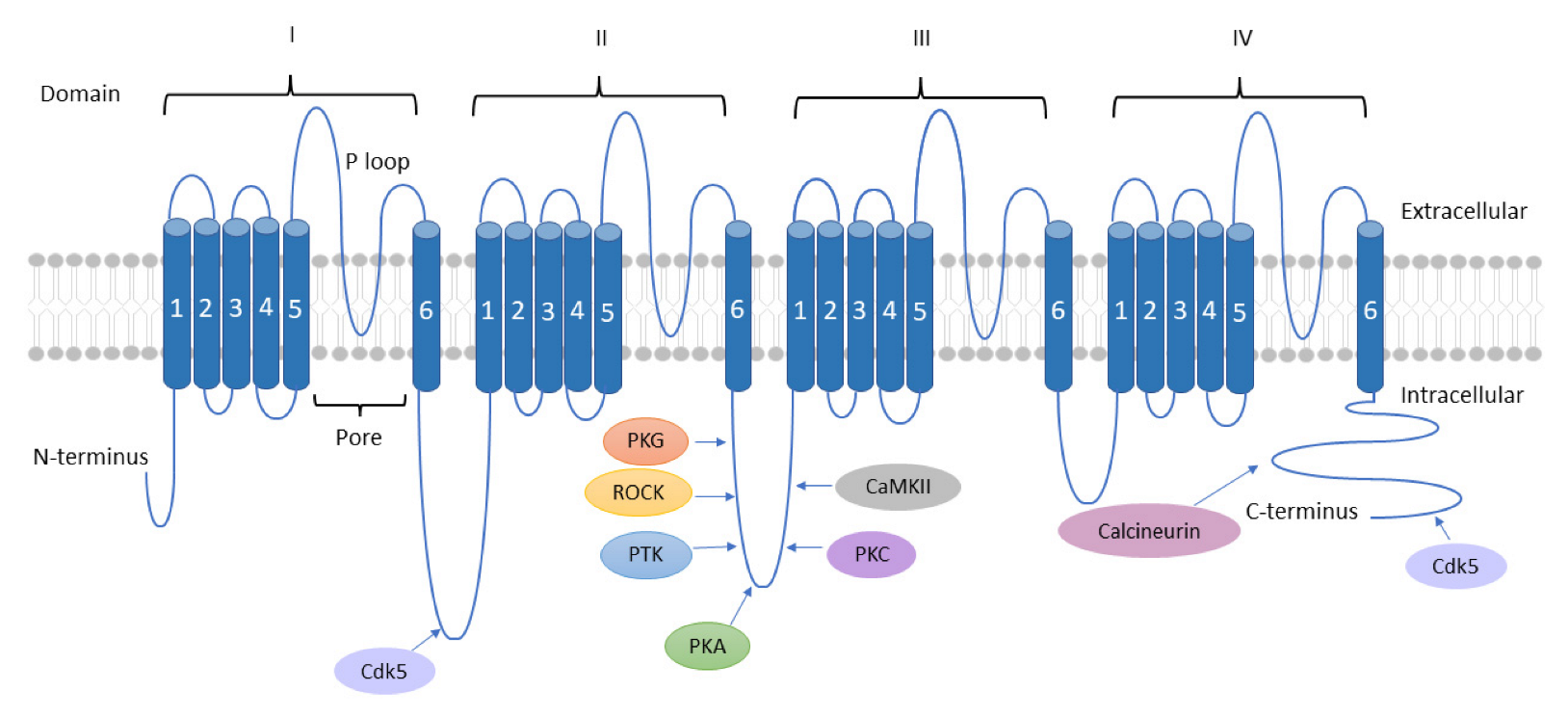{kind=link}
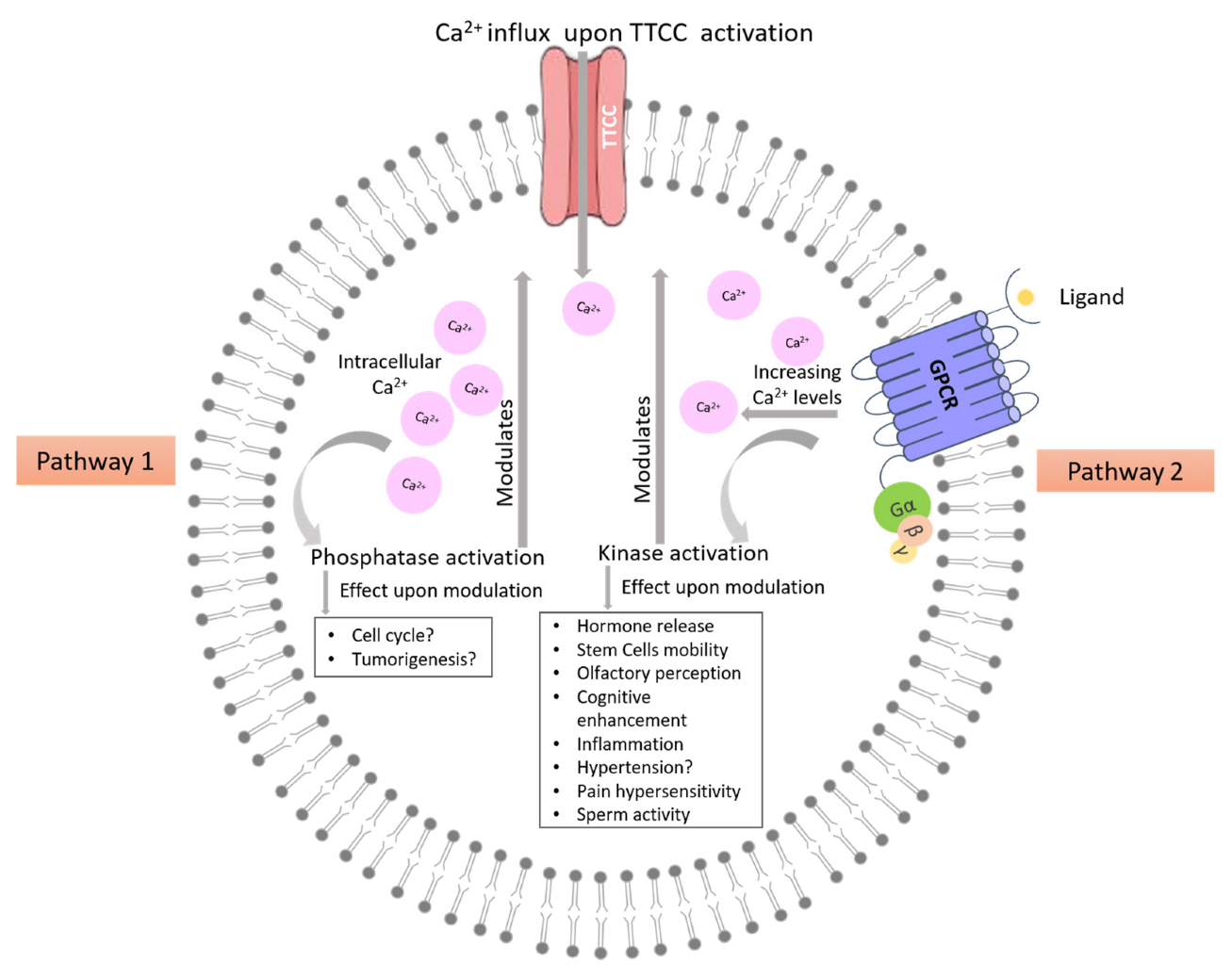{kind=link}
Table 1.
Major location of T-type Ca2+ channels.
| T-Type Ca2+ Channel Isoform | Location | Reference |
|---|---|---|
| Cav3.1 | Brain and Peripheral nervous system | [26] |
| Ovary and Placenta | [27] | |
| Heart | [28] | |
| Pancreatic islets and Beta cells | [29,30] | |
| Adrenal medulla | [31] | |
| Cav3.2 | Brain and Peripheral nervous system | [26,32] |
| Heart | [28,33] | |
| Pancreatic islets and Beta cells | [30,34] | |
| Kidney and liver | [35] | |
| Adrenal cortex | [36] | |
| Cav3.3 | Brain and Peripheral nervous system | [26,37] |
| Pancreatic islets | [30,34] | |
| Cardiac Purkinje fibers | [38] |
Table 2.
Pathological roles of T-type Ca2+ channels.
| T-Type Ca2+ Channel Isoform | Roles | Reference |
|---|---|---|
| Cav3.1 (Encoded by CACNA1G) | CACNA1G as a tumor suppressor gene | [39,40] |
| Cav3.1 gain-of-function mutations are associated with childhood-onset cerebellar atrophy. | [41] | |
| Cav3.1, Cav3.2 & Cav3.3 | Cav3.1, Cav3.2 & Cav3.3 are involved in the cancer cell proliferation across several cancer types, such as oral squamous cell carcinoma, melanoma, ovarian cancer, colorectal cancer, glioblastoma, neuroendocrine tumors, and esophageal cancer cells as shown by the pharmacological blockade. | [10,42,43,44,45,46,47,48] |
| Cav3.2 (Encoded by CACNA1H) | CACNA1H mutations are associated with amyotrophic lateral sclerosis and Primary Aldosteronism | [49,50] |
| Cav3.2 may be a potential differential biomarker for survival and treatment response in specific breast cancer subtypes | [51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharma, A.; Rahman, G.; Gorelik, J.; Bhargava, A. Voltage-Gated T-Type Calcium Channel Modulation by Kinases and Phosphatases: The Old Ones, the New Ones, and the Missing Ones. Cells 2023, 12, 461. https://doi.org/10.3390/cells12030461
AMA Style
Sharma A, Rahman G, Gorelik J, Bhargava A. Voltage-Gated T-Type Calcium Channel Modulation by Kinases and Phosphatases: The Old Ones, the New Ones, and the Missing Ones. Cells. 2023; 12(3):461. https://doi.org/10.3390/cells12030461
Chicago/Turabian StyleSharma, Ankush, Ghazala Rahman, Julia Gorelik, and Anamika Bhargava. 2023. "Voltage-Gated T-Type Calcium Channel Modulation by Kinases and Phosphatases: The Old Ones, the New Ones, and the Missing Ones" Cells 12, no. 3: 461. https://doi.org/10.3390/cells12030461
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.