
Who Knew? Dopamine Transporter Activity Is Critical in Innate and Adaptive Immune Responses
 , , , ,
, , , , 
Abstract
:1. Introduction
2. Methods
2.1. Study Design
2.2. Animals
2.3. Measurement of Tissue and Serum Monoamines and Their Metabolites Via HPLC
2.4. LPS Administration
2.5. Blood Collection and PBMC Isolation
2.6. Peritoneal Macrophage Harvest
2.7. Spleen Dissociation
2.8. Flow Cytometry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Supplier | Catalog Number | Purpose | Concentration |
|---|---|---|---|---|
| Ficoll-Paque Plus | GE | 45-001-750 | PBMC isolation | N/A |
| LPS | Sigma | Immune stimulation | 2 µg/g body weight | |
| PBS | In house | N/A | PBMC isolation, FC | 1× |
| K2EDTA Vacutainer | BD | 366643 | Blood collection | N/A |
| FACS tubes | Fisher | FC, mouse PBMC isolation | N/A | |
| Fix/Perm Kit | eBioscience | 88-8824-00 | FC | Stock |
| Leucosep Tube | Grenier BioOne | 227,290P | PBMC isolation | N/A |
| Syringe | Exel | 26016 | IP injection, cardiac puncture blood draw | N/A |
| Isoflurane | Patterson | 07-893-8441 | Anesthesia | 1–5% |
| Phagocytosis beads | Sigma | L3280 | Phagocytosis | 0.5 mL |
| Legendplex | Biolegend | 740150 | Cytokine Analysis | N/A |
| Specificity | Clone/Species | Conjugate | Vendor | Catalog Number | Purpose | Dilution |
|---|---|---|---|---|---|---|
| CD11b | M170/Rat | PerCP-Cy5.5 | Biolegend | 101,228 | FC | 1:100 |
| CD45 | 30-F11/Mouse | FITC | Biolegend | 334,824 | FC | 1:200 |
| CD19 | 6D5/Rat | BV605 | Biolegend | FC | 1:100 | |
| CD27 | LG.3A10/Mouse | APC | Biolegend | 124,212 | FC | 1:100 |
| CD3 | 17A2/Mouse | PacBlue | Biolegend | 100,214 | FC | 1:50 |
| CD4 | GK1.5/Mouse | AF700 | Biolegend | 100,429 | FC | 1:50 |
| CD8a | QA17A07/Mouse | SV538 | Biolegend | 155,020 | FC | 1:50 |
| Ly6C | HK1.4/Rat | BV785 | Biolegend | 128,041 | FC | 1:200 |
| Ly6G | 1A8/Rat | PE | Biolegend | 127,607 | FC | 1:100 |
| CD11b | M1-70/Rat | FITC | Biolegend | 101,206 | FC | 1:100 |
| F4/80 | BM8/Rat | AF700 | Biolegend | 123,129 | FC | 1:100 |
| MHC-II | M5-114.15.2/Rat | APC-Cy7 | Biolegend | 107,602 | FC | 1:100 |
| Zombie Red | N/A | N/A | Biolegend | 423,110 | FC | 1:500–1000 |
2.9. Phagocytosis
2.10. Fluorescence Minus One (FMO) Analysis
2.11. Serum Cytokine Analysis
2.12. Statistical Analysis
| Equipment | Supplier | Part Number | Purpose |
|---|---|---|---|
| Centrifuge | Sorvall | ST8 | PBMC isolation |
| Cytometer | BD | Canto II | FC |
| Spectral Analyzer | Sony | SP6800 | FC |
| Spectral Analyzer | Cytek | Aurora 5 L | FC |
| Flow cytometer | Beckman | Cytoflex LX | FC, 13-plex ELISA |
| Microcentrifuge | Fisher | 59A | FC |
3. Results
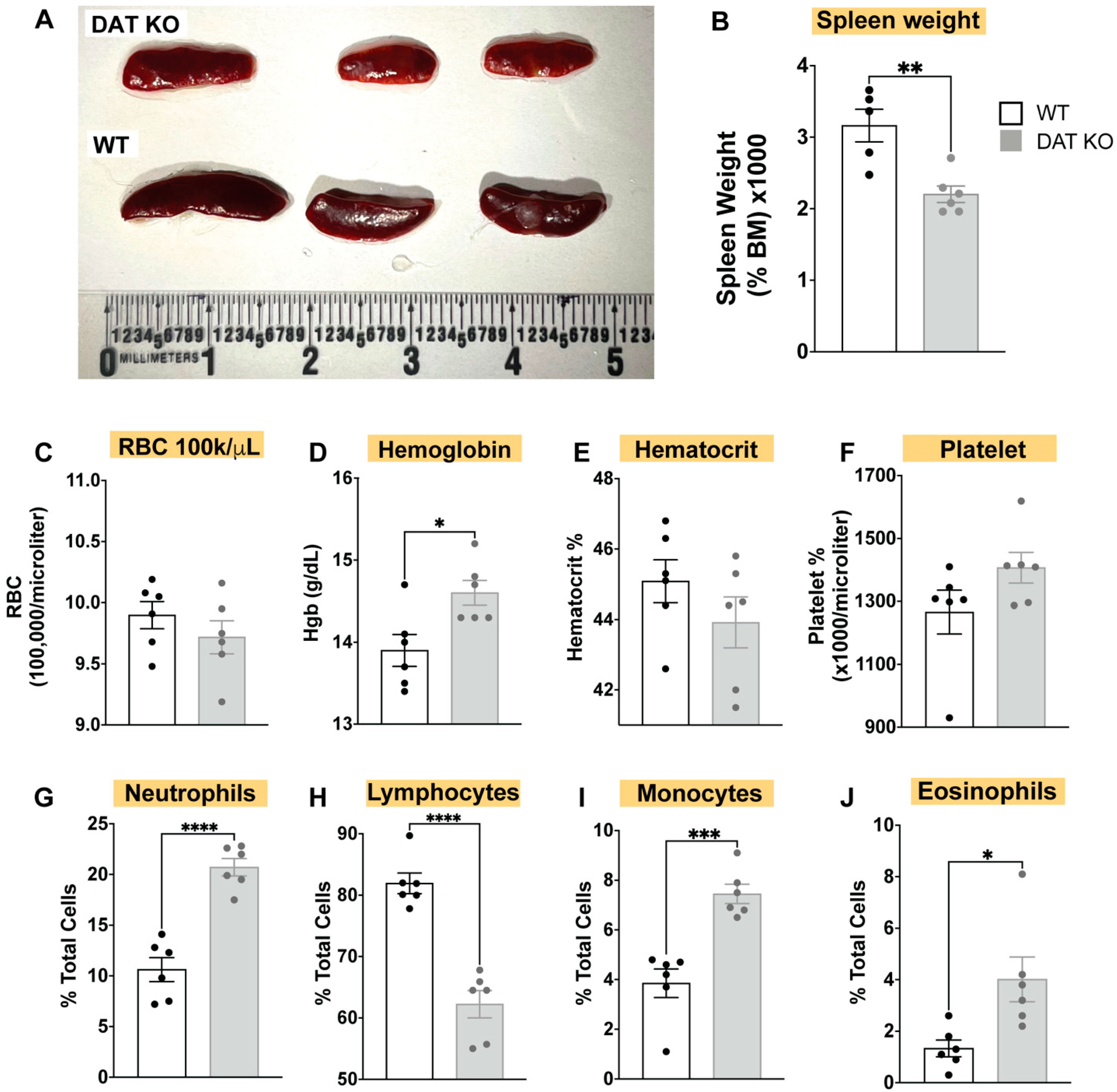
3.1. DAT Deletion Alters the Composition of the Circulating Immune System, Induces Spleen Hypoplasia, and Alters Complete Blood Count (CBC)
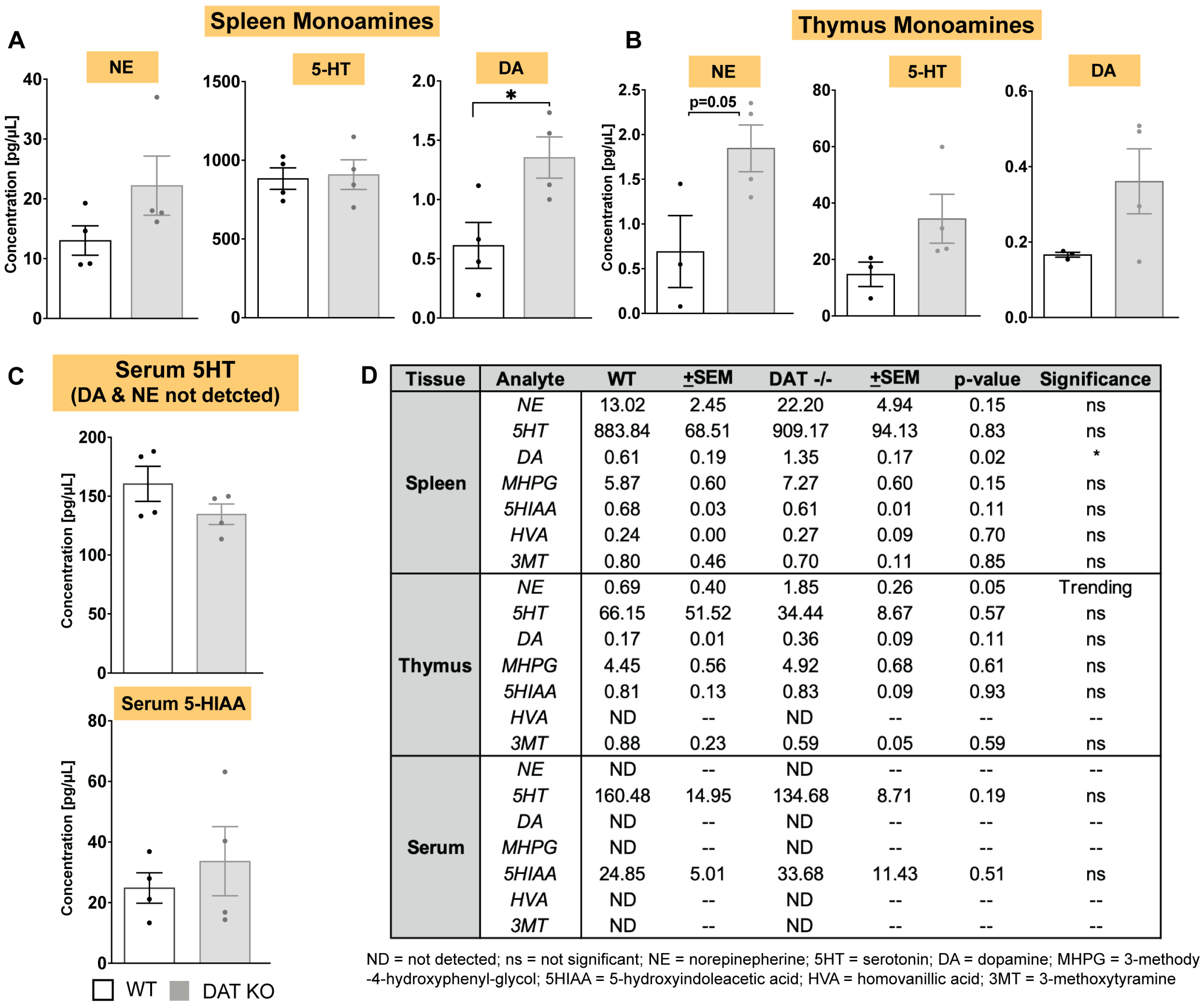
3.2. In DAT−/− Mice, Norepinephrine and Dopamine Levels Are Increased in Spleen and Thymus, but Not in Circulating Serum
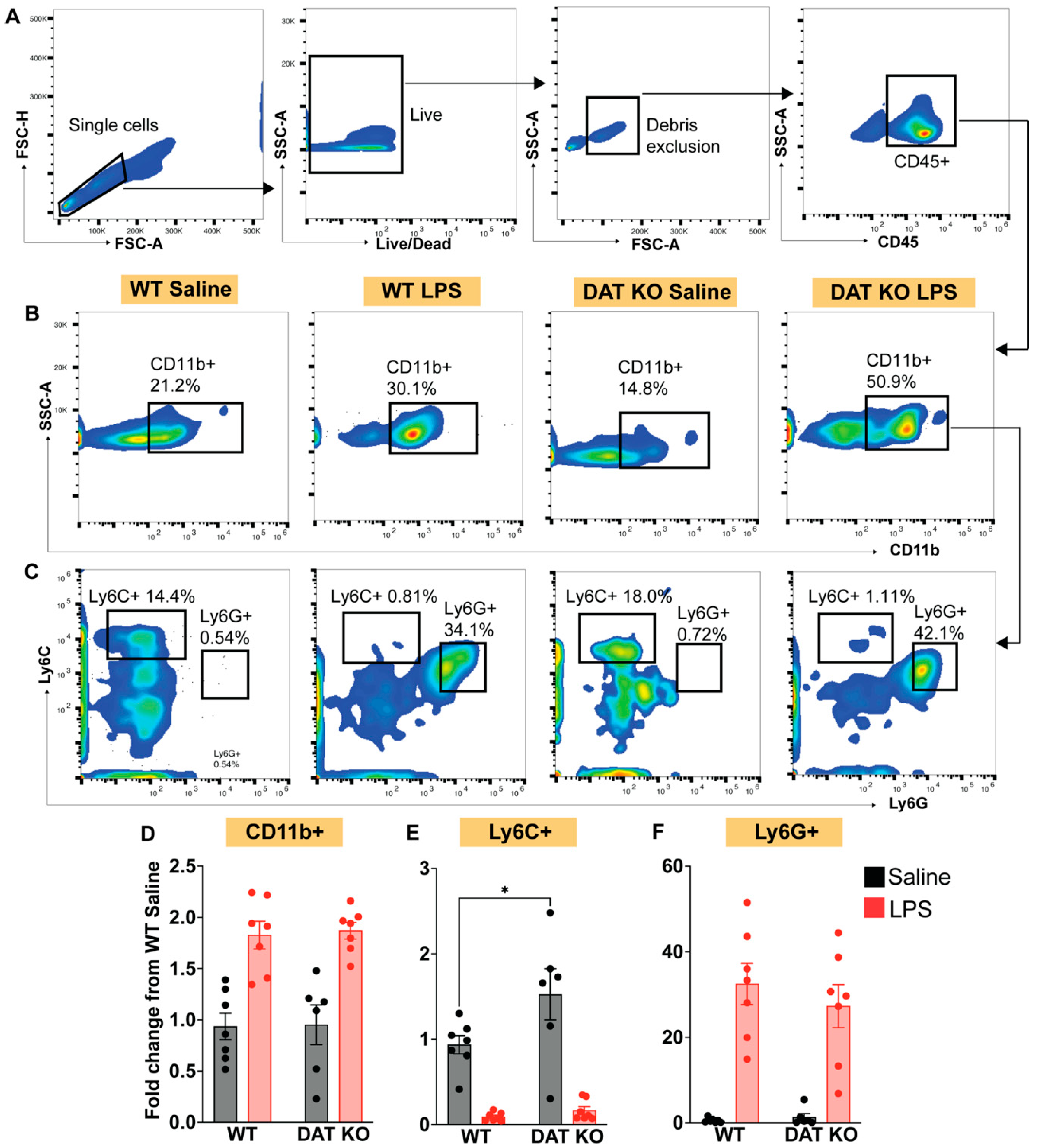
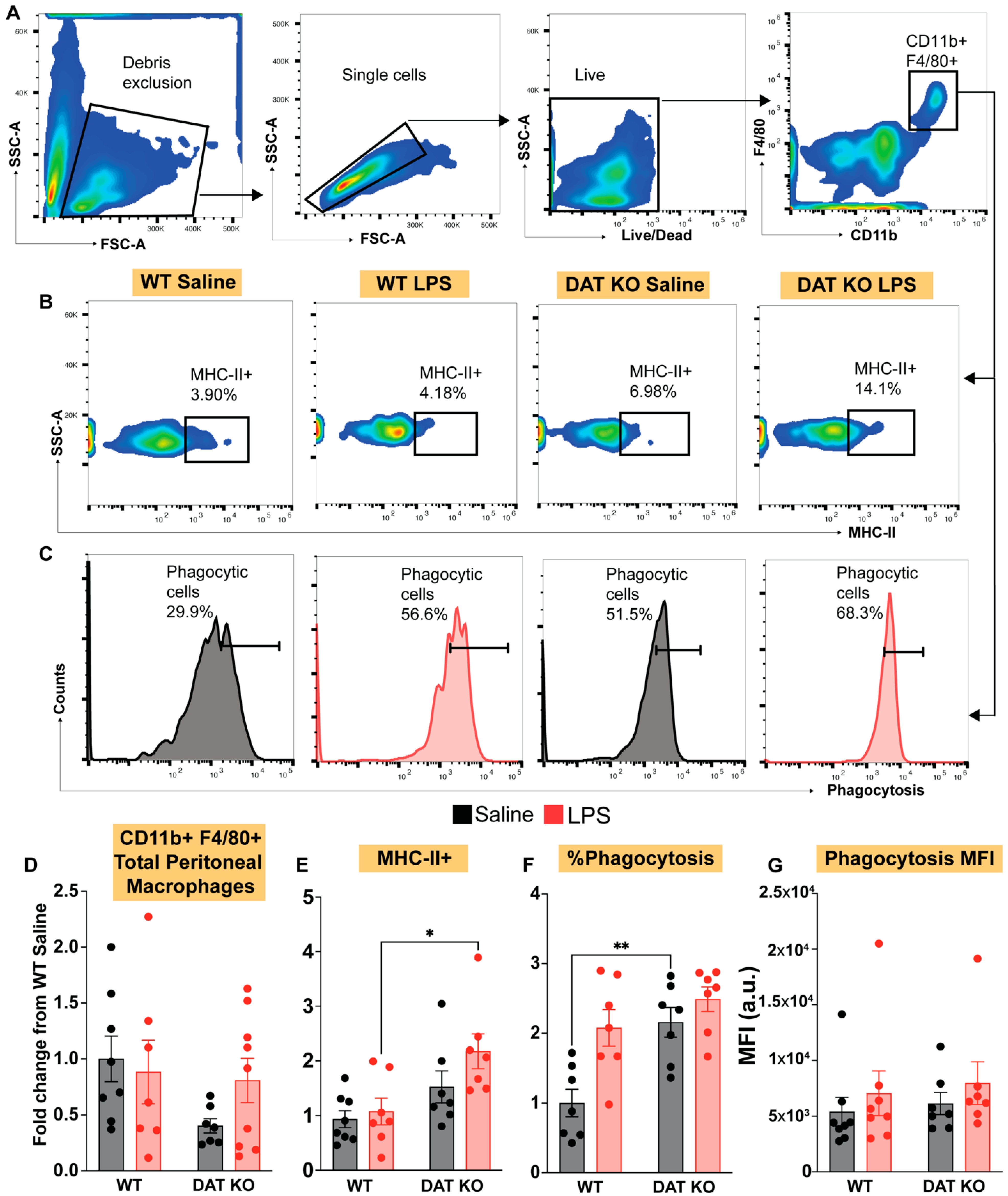
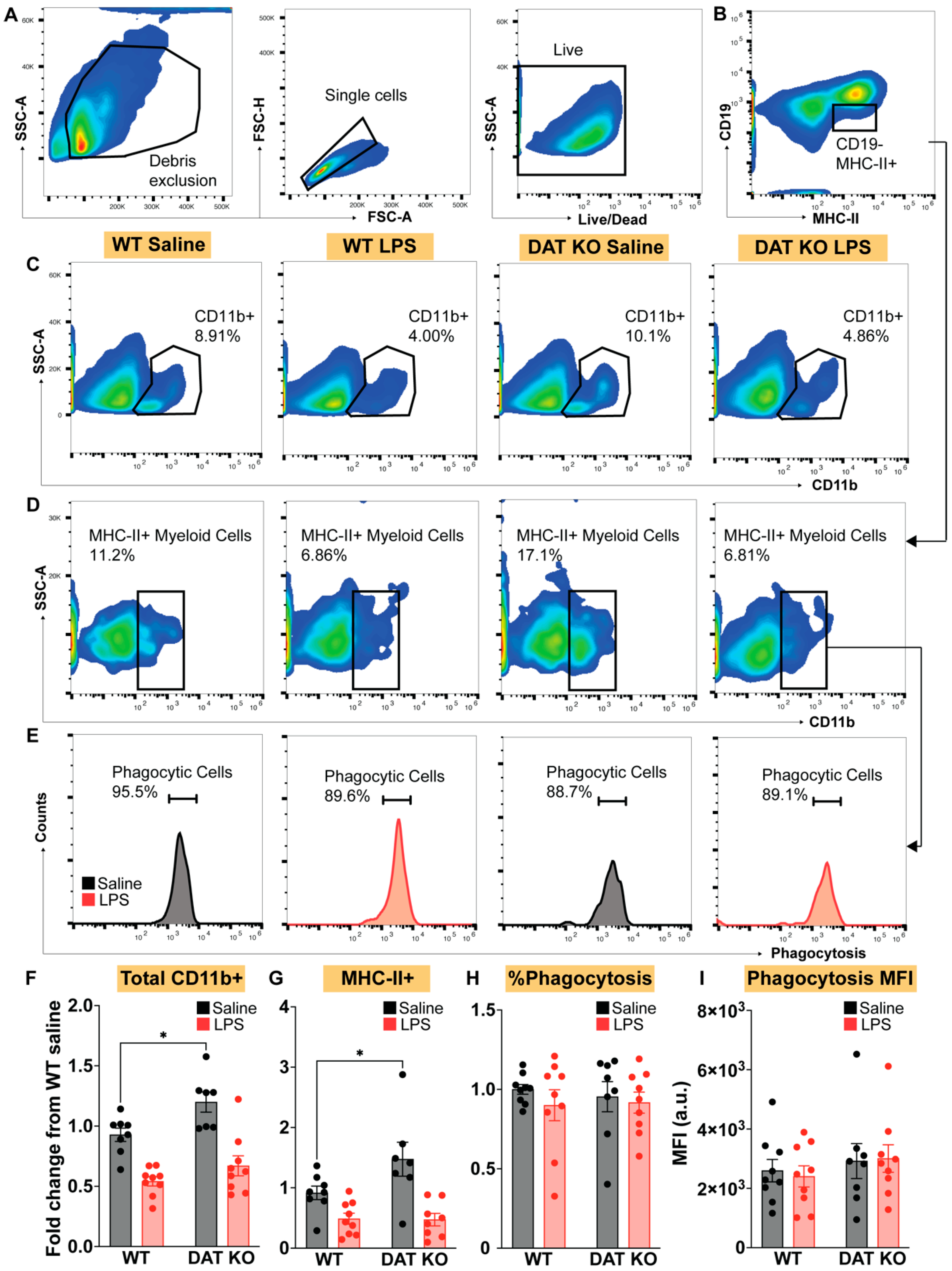
3.3. Loss of DAT Skews the Myeloid Population to a Pro-Inflammatory Phenotype
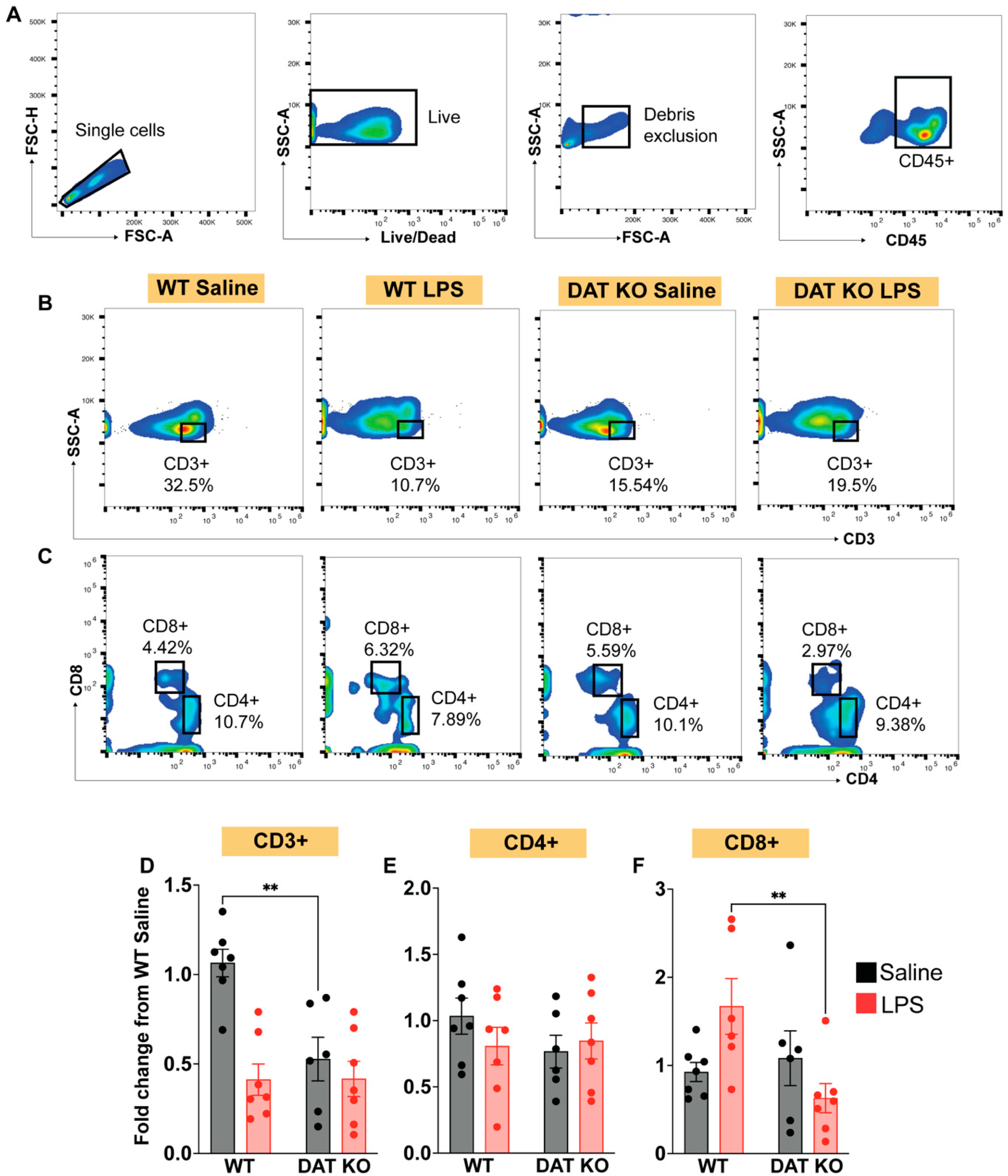
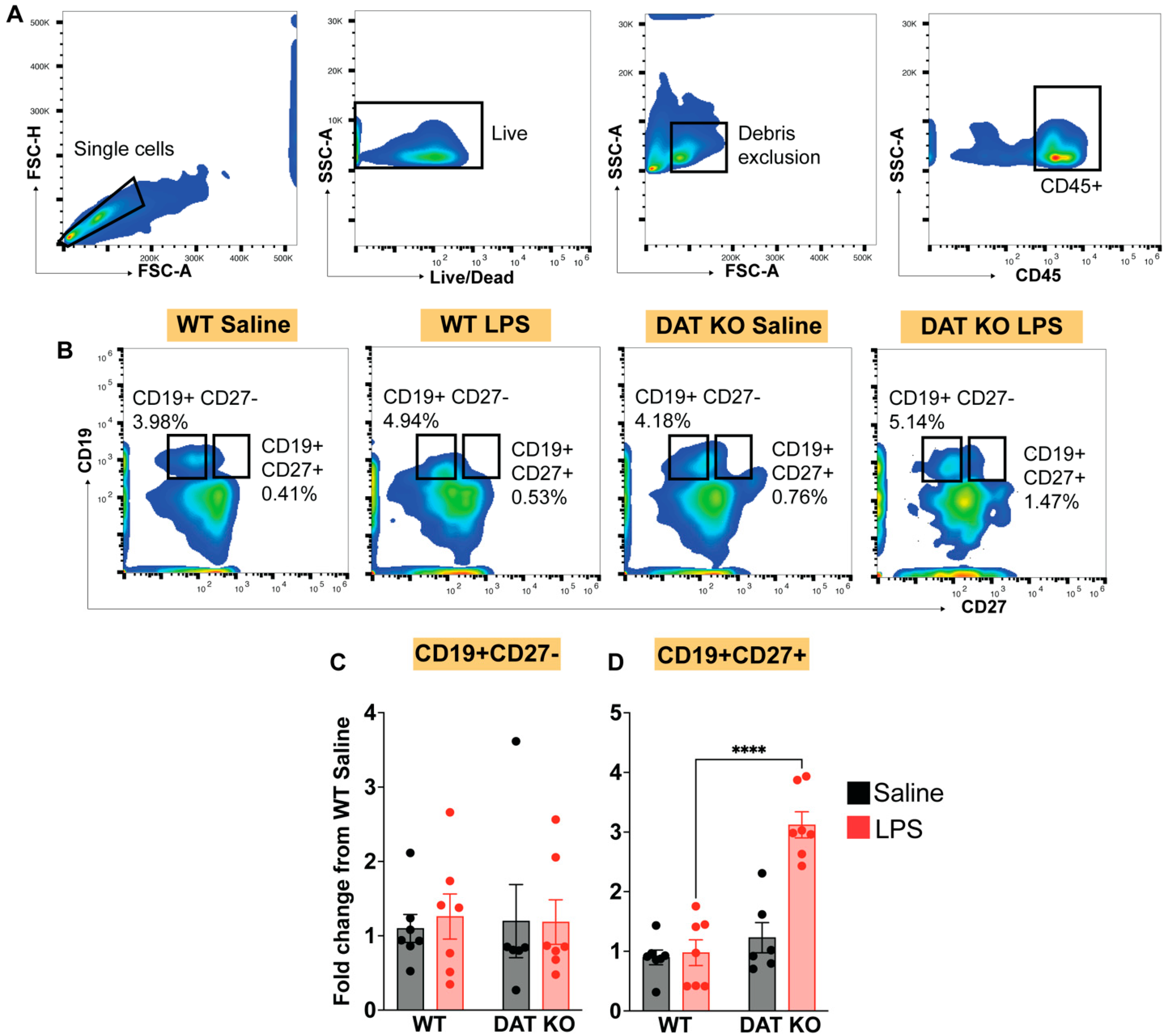
3.4. Loss of DAT Has Divergent Effects on the Circulating Lymphoid Compartment
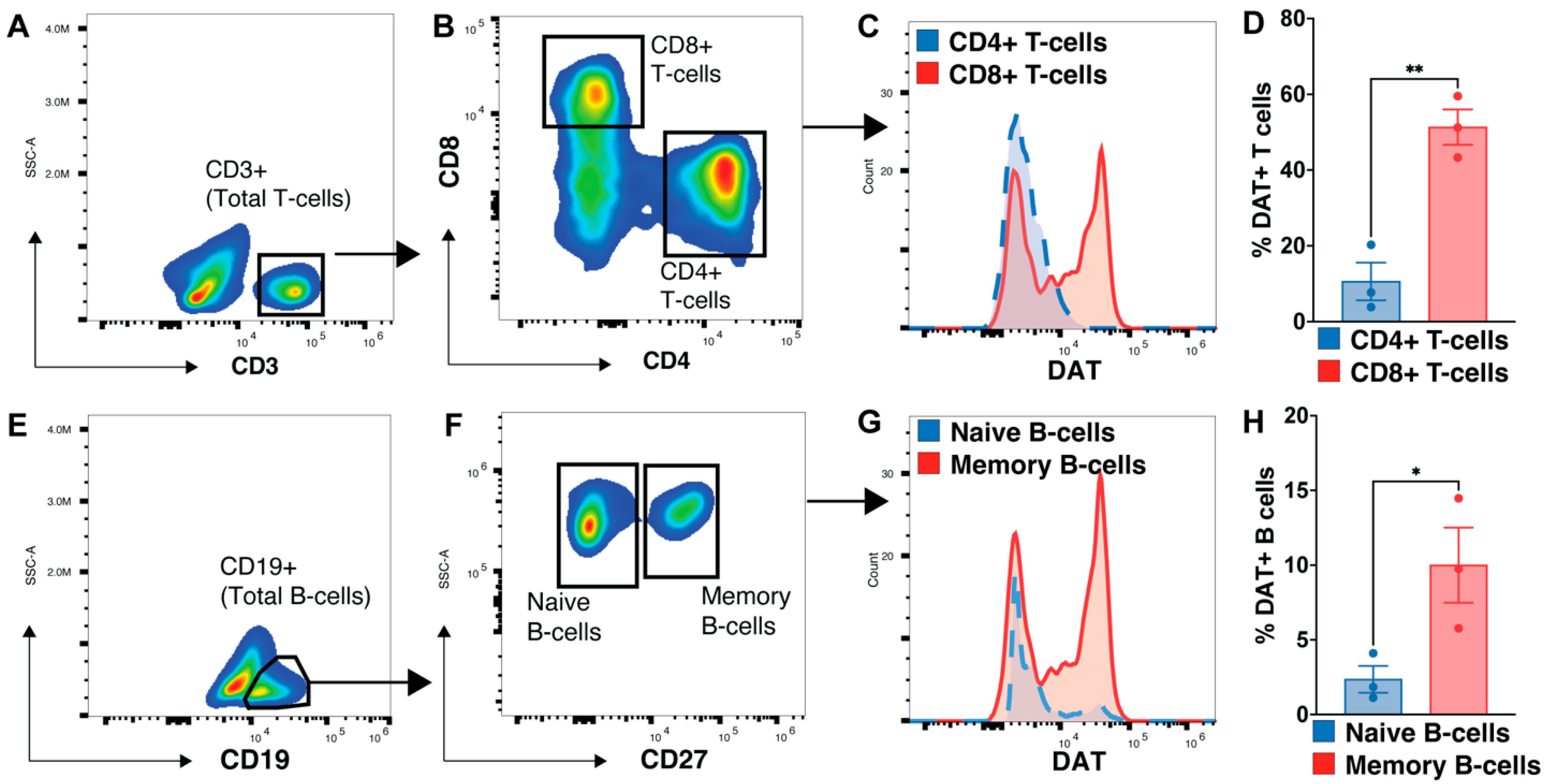
3.5. Memory B-Cell and CD8 T-Cells Express More DAT
3.6. Loss of DAT Alters the Peripheral Cytokine Profile
4. Discussion, Broader Biological Implications, and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arreola, R.; Alvarez-Herrera, S.; Pérez-Sánchez, G.; Becerril-Villanueva, E.; Cruz-Fuentes, C.; Flores-Gutierrez, E.O.; Garcés-Alvarez, M.E.; de la Cruz-Aguilera, D.L.; Medina-Rivero, E.; Hurtado-Alvarado, G.; et al. Immunomodulatory Effects Mediated by Dopamine. J. Immunol. Res. 2016, 2016, 3160486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farino, Z.J.; Morgenstern, T.J.; Maffei, A.; Quick, M.; De Solis, A.J.; Wiriyasermkul, P.; Freyberg, R.J.; Aslanoglou, D.; Sorisio, D.; Inbar, B.P.; et al. New roles for dopamine D2 and D3 receptors in pancreatic beta cell insulin secretion. Mol. Psychiatry 2019, 25, 2070–2085. [Google Scholar] [CrossRef] [PubMed]
- Harms, R.; Morsey, B.; Boyer, C.W.; Fox, H.S.; Sarvetnick, N. Methamphetamine administration targets multiple immune subsets and induces phenotypic alterations suggestive of immunosuppression. PLoS ONE 2012, 7, e49897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herisson, F.; Frodermann, V.; Courties, G.; Rohde, D.; Sun, Y.; Vandoorne, K.; Wojtkiewicz, G.R.; Masson, G.S.; Vinegoni, C.; Kim, J.; et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat. Neurosci. 2018, 21, 1209–1217. [Google Scholar] [CrossRef]
- Liu, L.; Yuan, G.; Cheng, Z.; Zhang, G.; Liu, X.; Zhang, H. Identification of the mRNA expression status of the dopamine D2 receptor and dopamine transporter in peripheral blood lymphocytes of schizophrenia patients. PLoS ONE 2013, 8, e75259. [Google Scholar] [CrossRef] [Green Version]
- Marazziti, D.; Dell’osso, M.C.; Baroni, S.; Masala, I.; Dell’Osso, B.; Consoli, G.; Giannaccini, G.; Betti, L.; Lucacchini, A. Alterations of the dopamine transporter in resting lymphocytes of patients with different psychotic disorders. Psychiatry Res. 2010, 175, 54–57. [Google Scholar] [CrossRef]
- Gaskill, P.J.; Carvallo, L.; Eugenin, E.A.; Berman, J.W. Characterization and function of the human macrophage dopaminergic system: Implications for CNS disease and drug abuse. J. Neuroinflamm. 2012, 9, 203. [Google Scholar] [CrossRef] [Green Version]
- Mackie, P.; Lebowitz, J.; Saadatpour, L.; Nickoloff, E.; Gaskill, P.; Khoshbouei, H. The dopamine transporter: An unrecognized nexus for dysfunctional peripheral immunity and signaling in Parkinson’s Disease. Brain Behav. Immun. 2018, 70, 21–35. [Google Scholar] [CrossRef]
- Mackie, P.M.; Gopinath, A.; Montas, D.M.; Nielsen, A.; Smith, A.; Nolan, R.A.; Runner, K.; Matt, S.M.; McNamee, J.; Riklan, J.E.; et al. Functional characterization of the biogenic amine transporters on human macrophages. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Tsao, C.W.; Lin, Y.S.; Cheng, J.T. Inhibition of immune cell proliferation with haloperidol and relationship of tyrosine hydroxylase expression to immune cell growth. Life Sci. 1998, 62, 335–344. [Google Scholar] [CrossRef]
- Caronti, B.; Tanda, G.; Colosimo, C.; Ruggieri, S.; Calderaro, C.; Palladini, G.; Pontieri, F.E.; Di Chiara, G. Reduced dopamine in peripheral blood lymphocytes in Parkinson’s disease. Neuroreport 1999, 10, 2907–2910. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.M.; Gaskill, P.J. Where Is Dopamine and how do Immune Cells See it?: Dopamine-Mediated Immune Cell Function in Health and Disease. J. Neuroimmune Pharmacol. 2020, 15, 114–164. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, E.; Bergquist, J.; Ekman, R.; Tarkowski, A. Catecholamines are synthesized by mouse lymphocytes and regulate function of these cells by induction of apoptosis. Immunology 1996, 88, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Musso, N.R.; Brenci, S.; Setti, M.; Indiveri, F.; Lotti, G. Catecholamine content and in vitro catecholamine synthesis in peripheral human lymphocytes. J. Clin. Endocrinol. Metab. 1996, 81, 3553–3557. [Google Scholar] [CrossRef] [PubMed]
- Bergquist, J.; Silberring, J. Identification of catecholamines in the immune system by electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1998, 12, 683–688. [Google Scholar] [CrossRef]
- Amenta, F.; Bronzetti, E.; Cantalamessa, F.; El-Assouad, D.; Felici, L.; Ricci, A.; Tayebati, S.K. Identification of dopamine plasma membrane and vesicular transporters in human peripheral blood lymphocytes. J. Neuroimmunol. 2001, 117, 133–142. [Google Scholar] [CrossRef]
- Cosentino, M.; Fietta, A.M.; Ferrari, M.; Rasini, E.; Bombelli, R.; Carcano, E.; Saporiti, F.; Meloni, F.; Marino, F.; Lecchini, S. Human CD4+CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood 2007, 109, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Faraj, B.A.; Olkowski, Z.L.; Jackson, R.T. Binding of [3H]-dopamine to human lymphocytes: Possible relationship to neurotransmitter uptake sites. Pharmacology 1991, 42, 135–141. [Google Scholar] [CrossRef]
- Papa, I.; Saliba, D.; Ponzoni, M.; Bustamante, S.; Canete, P.F.; Gonzalez-Figueroa, P.; McNamara, H.A.; Valvo, S.; Grimbaldeston, M.; Sweet, R.A.; et al. T(FH)-derived dopamine accelerates productive synapses in germinal centres. Nature 2017, 547, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Mignini, F.; Traini, E.; Tomassoni, D.; Amenta, F. Dopamine plasma membrane transporter (DAT) in rat thymus and spleen: An immunochemical and immunohistochemical study. Auton. Autacoid Pharmacol. 2006, 26, 183–189. [Google Scholar] [CrossRef]
- Mignini, F.; Tomassoni, D.; Traini, E.; Amenta, F. Dopamine, vesicular transporters and dopamine receptor expression and localization in rat thymus and spleen. J. Neuroimmunol. 2009, 206, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Mignini, F.; Sabbatini, M.; D’Andrea, V.; Cavallotti, C. Intrinsic innervation and dopaminergic markers after experimental denervation in rat thymus. Eur. J. Histochem. 2010, 54, e17. [Google Scholar] [CrossRef] [Green Version]
- Assis, M.A.; Valdomero, A.; García-Keller, C.; Sotomayor, C.; Cancela, L.M. Decrease of lymphoproliferative response by amphetamine is mediated by dopamine from the nucleus accumbens: Influence on splenic met-enkephalin levels. Brain Behav. Immun. 2011, 25, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Mignini, F.; Sabbatini, M.; Capacchietti, M.; Amantini, C.; Bianchi, E.; Artico, M.; Tammaro, A. T-cell subpopulations express a different pattern of dopaminergic markers in intra- and extra-thymic compartments. J. Biol. Regul. Homeost. Agents 2013, 27, 463–475. [Google Scholar]
- Gopinath, A.; Mackie, P.; Hashimi, B.; Buchanan, A.M.; Smith, A.R.; Bouchard, R.; Shaw, G.; Badov, M.; Saadatpour, L.; Gittis, A.; et al. DAT and TH expression marks human Parkinson’s disease in peripheral immune cells. NPJ Park. Dis. 2022, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Dittel, B.N. Isolation of mouse peritoneal cavity cells. J. Vis. Exp. 2010, 28, e1488. [Google Scholar] [CrossRef]
- Ruddle, N.H.; Akirav, E.M. Secondary lymphoid organs: Responding to genetic and environmental cues in ontogeny and the immune response. J. Immunol. 2009, 183, 2205–2212. [Google Scholar] [CrossRef] [Green Version]
- Drayton, D.L.; Liao, S.; Mounzer, R.H.; Ruddle, N.H. Lymphoid organ development: From ontogeny to neogenesis. Nat. Immunol. 2006, 7, 344–353. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Jones, S.R.; Caron, M.G. Functional hyperdopaminergia in dopamine transporter knock-out mice. Biol. Psychiatry 1999, 46, 303–311. [Google Scholar] [CrossRef]
- Khan, N.; Kaur, S.; Knuth, C.M.; Jeschke, M.G. CNS-Spleen Axis—A Close Interplay in Mediating Inflammatory Responses in Burn Patients and a Key to Novel Burn Therapeutics. Front. Immunol. 2021, 12, 720221. [Google Scholar] [CrossRef]
- Lori, A.; Perrotta, M.; Lembo, G.; Carnevale, D. The Spleen: A Hub Connecting Nervous and Immune Systems in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2017, 18, 1216. [Google Scholar] [CrossRef]
- Chhatar, S.; Lal, G. Role of adrenergic receptor signalling in neuroimmune communication. Curr. Res. Immunol. 2021, 2, 202–217. [Google Scholar] [CrossRef]
- Lewis, A.J.; Seymour, C.W.; Rosengart, M.R. Current Murine Models of Sepsis. Surg. Infect. 2016, 17, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, S.L.; Gold, M.R.; DeFranco, A.L. Bacterial lipopolysaccharide stimulates protein tyrosine phosphorylation in macrophages. Proc. Natl. Acad. Sci. USA 1991, 88, 4148–4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollenweber, H.W.; Morrison, D.C. Synthesis and biochemical characterization of a photoactivatable, iodinatable, cleavable bacterial lipopolysaccharide derivative. J. Biol. Chem. 1985, 260, 15068–15074. [Google Scholar] [CrossRef] [PubMed]
- Schumak, B.; Klocke, K.; Kuepper, J.M.; Biswas, A.; Djie-Maletz, A.; Limmer, A.; van Rooijen, N.; Mack, M.; Hoerauf, A.; Dunay, I.R. Specific depletion of Ly6C(hi) inflammatory monocytes prevents immunopathology in experimental cerebral malaria. PLoS ONE 2015, 10, e0124080. [Google Scholar] [CrossRef]
- Fischer, M.A.; Davies, M.L.; Reider, I.E.; Heipertz, E.L.; Epler, M.R.; Sei, J.J.; Ingersoll, M.A.; Rooijen, N.V.; Randolph, G.J.; Norbury, C.C. CD11b⁺, Ly6G⁺ cells produce type I interferon and exhibit tissue protective properties following peripheral virus infection. PLoS Pathog. 2011, 7, e1002374. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Misharin, A.; Perlman, H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytom. A 2012, 81, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Zigmond, E.; Varol, C.; Farache, J.; Elmaliah, E.; Satpathy, A.T.; Friedlander, G.; Mack, M.; Shpigel, N.; Boneca, I.G.; Murphy, K.M.; et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 2012, 37, 1076–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopinath, A.; Badov, M.; Francis, M.; Shaw, G.; Collins, A.; Miller, D.R.; Hansen, C.A.; Mackie, P.; Tansey, M.G.; Dagra, A.; et al. TNFα increases tyrosine hydroxylase expression in human monocytes. npj Park. Dis. 2021, 7, 62. [Google Scholar] [CrossRef]
- Ghosn, E.E.; Cassado, A.A.; Govoni, G.R.; Fukuhara, T.; Yang, Y.; Monack, D.M.; Bortoluci, K.R.; Almeida, S.R.; Herzenberg, L.A.; Herzenberg, L.A. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl. Acad. Sci. USA 2010, 107, 2568–2573. [Google Scholar] [CrossRef]
- Ados, A.C.; Lima, M.R.D.; Bortoluci, K.R. Revisiting mouse peritoneal macrophages: Heterogeneity, development, and function. Front. Immunol. 2015, 6, 225. [Google Scholar] [CrossRef] [Green Version]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Bö, L.; Mörk, S.; Kong, P.A.; Nyland, H.; Pardo, C.A.; Trapp, B.D. Detection of MHC class II-antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis lesions. J. Neuroimmunol. 1994, 51, 135–146. [Google Scholar] [CrossRef]
- Choi, S.H.; Gonen, A.; Diehl, C.J.; Kim, J.; Almazan, F.; Witztum, J.L.; Miller, Y.I. SYK regulates macrophage MHC-II expression via activation of autophagy in response to oxidized LDL. Autophagy 2015, 11, 785–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Marin, D.; Jarvis, C.; Nelius, T.; Filleur, S. Assessment of phagocytic activity in live macrophages-tumor cells co-cultures by Confocal and Nomarski Microscopy. Biol. Methods Protoc. 2017, 2, bpx002. [Google Scholar] [CrossRef] [PubMed]
- de Lima, T.M.; Sampaio, S.C.; Petroni, R.; Brigatte, P.; Velasco, I.T.; Soriano, F.G. Phagocytic activity of LPS tolerant macrophages. Mol. Immunol. 2014, 60, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. GM-CSF in inflammation. J. Exp. Med. 2020, 217, e20190945. [Google Scholar] [CrossRef] [Green Version]
- Bassler, K.; Schulte-Schrepping, J.; Warnat-Herresthal, S.; Aschenbrenner, A.C.; Schultze, J.L. The Myeloid Cell Compartment-Cell by Cell. Annu. Rev. Immunol. 2019, 37, 269–293. [Google Scholar] [CrossRef]
- Prinz, M.; Erny, D.; Hagemeyer, N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 2017, 18, 385–392. [Google Scholar] [CrossRef]
- Hey, Y.Y.; Quah, B.; O’Neill, H.C. Antigen presenting capacity of murine splenic myeloid cells. BMC Immunol. 2017, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.W.; Tam, J.W.; Okan, N.A.; Mena, P.; Furie, M.B.; Thanassi, D.G.; Benach, J.L.; van der Velden, A.W. Phenotypic, morphological, and functional heterogeneity of splenic immature myeloid cells in the host response to tularemia. Infect. Immun. 2012, 80, 2371–2381. [Google Scholar] [CrossRef] [Green Version]
- Hey, Y.Y.; Tan, J.K.; O’Neill, H.C. Redefining Myeloid Cell Subsets in Murine Spleen. Front. Immunol. 2015, 6, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hey, Y.Y.; O’Neill, H.C. Murine spleen contains a diversity of myeloid and dendritic cells distinct in antigen presenting function. J. Cell. Mol. Med. 2012, 16, 2611–2619. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hua, Q.; Zheng, L. Generation of Myeloid Cells in Cancer: The Spleen Matters. Front. Immunol. 2020, 11, 1126. [Google Scholar] [CrossRef]
- Germain, R.N. T-cell development and the CD4-CD8 lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef]
- Liu, W.; Menoret, A.; Vella, A.T. Responses to LPS boost effector CD8 T-cell accumulation outside of signals 1 and 2. Cell. Mol. Immunol. 2017, 14, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Sueyoshi, K.; Ledderose, C.; Shen, Y.; Lee, A.H.; Shapiro, N.I.; Junger, W.G. Lipopolysaccharide suppresses T cells by generating extracellular ATP that impairs their mitochondrial function via P2Y11 receptors. J. Biol. Chem. 2019, 294, 6283–6293. [Google Scholar] [CrossRef] [PubMed]
- Born, W.K.; Yin, Z.; Hahn, Y.S.; Sun, D.; O’Brien, R.L. Analysis of gamma delta T cell functions in the mouse. J. Immunol. 2010, 184, 4055–4061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venet, F.; Bohé, J.; Debard, A.L.; Bienvenu, J.; Lepape, A.; Monneret, G. Both percentage of gammadelta T lymphocytes and CD3 expression are reduced during septic shock. Crit. Care Med. 2005, 33, 2836–2840. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Cyster, J.G.; Allen, C.D.C. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaghan, S.A.; Peterson, L.C.; James, C.; Marszalek, L.; Khoong, A.; Bachta, D.J.; Karpus, W.J.; Goolsby, C.L. Pan B-cell markers are not redundant in analysis of chronic lymphocytic leukemia (CLL). Cytom. B Clin. Cytom. 2003, 56, 30–42. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Avery, D.T.; Ellyard, J.I.; Mackay, F.; Corcoran, L.M.; Hodgkin, P.D.; Tangye, S.G. Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. J. Immunol. 2005, 174, 4034–4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahota, S.S.; Babbage, G.; Weston-Bell, N.J. CD27 in defining memory B-cell origins in Waldenström’s macroglobulinemia. Clin. Lymphoma Myeloma 2009, 9, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Stow, J.L.; Low, P.C.; Offenhäuser, C.; Sangermani, D. Cytokine secretion in macrophages and other cells: Pathways and mediators. Immunobiology 2009, 214, 601–612. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Sendra, V.G.; Liadi, I.; Ramesh, B.; Romain, G.; Haymaker, C.; Martinez-Paniagua, M.; Lu, Y.; Radvanyi, L.G.; Roysam, B.; et al. Single-cell profiling of dynamic cytokine secretion and the phenotype of immune cells. PLoS ONE 2017, 12, e0181904. [Google Scholar] [CrossRef] [Green Version]
- Duque, G.A.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Pradeep, S.P.; Venkatesh, P.H.; Manchala, N.R.; Veedu, A.V.; Basavaraju, R.K.; Selvasundari, L.; Ramakrishna, M.; Chandrakiran, Y.; Krishnamurthy, V.; Holigi, S.; et al. Innate Immune Cytokine Profiling and Biomarker Identification for Outcome in Dengue Patients. Front. Immunol. 2021, 12, 677874. [Google Scholar] [CrossRef]
- Romagnani, S. Th1 and Th2 in human diseases. Clin. Immunol. Immunopathol. 1996, 80, 225–235. [Google Scholar] [CrossRef]
- Paredes, J.; Zabaleta, J.; Garai, J.; Ji, P.; Imtiaz, S.; Spagnardi, M.; Alvarado, J.; Li, L.; Akadri, M.; Barrera, K.; et al. Immune-Related Gene Expression and Cytokine Secretion Is Reduced Among African American Colon Cancer Patients. Front. Oncol. 2020, 10, 1498. [Google Scholar] [CrossRef]
- Mera, S.; Tatulescu, D.; Cismaru, C.; Bondor, C.; Slavcovici, A.; Zanc, V.; Carstina, D.; Oltean, M. Multiplex cytokine profiling in patients with sepsis. Apmis 2011, 119, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Bozza, F.A.; Salluh, J.I.; Japiassu, A.M.; Soares, M.; Assis, E.F.; Gomes, R.N.; Bozza, M.T.; Castro-Faria-Neto, H.C.; Bozza, P.T. Cytokine profiles as markers of disease severity in sepsis: A multiplex analysis. Crit. Care 2007, 11, R49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; Deitch, E.A.; Ulloa, L. Novel insights for systemic inflammation in sepsis and hemorrhage. Mediat. Inflamm. 2010, 2010, 642462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Zhu, L.; Tian, H.; Sun, H.X.; Wang, R.; Zhang, L.; Zhao, Y. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell 2018, 9, 1027–1038. [Google Scholar] [CrossRef]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Roberts, A.I.; Liu, C.; Ren, G.; Xu, G.; Zhang, L.; Devadas, S.; Shi, Y. A novel subset of helper T cells promotes immune responses by secreting GM-CSF. Cell Death Differ. 2013, 20, 1731–1741. [Google Scholar] [CrossRef]
- Simmons, A.D.; Li, B.; Gonzalez-Edick, M.; Lin, C.; Moskalenko, M.; Du, T.; Creson, J.; VanRoey, M.J.; Jooss, K. GM-CSF-secreting cancer immunotherapies: Preclinical analysis of the mechanism of action. Cancer Immunol. Immunother. 2007, 56, 1653–1665. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Thiruppathi, M.; Elshabrawy, H.A.; Alharshawi, K.; Kumar, P.; Prabhakar, B.S. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine 2015, 75, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Lew, A.M.; Chopin, M. The Pleiotropic Effects of the GM-CSF Rheostat on Myeloid Cell Differentiation and Function: More Than a Numbers Game. Front. Immunol. 2019, 10, 2679. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Rautela, J.; Delconte, R.B.; Souza-Fonseca-Guimaraes, F.; Carrington, E.M.; Schenk, R.L.; Herold, M.J.; Huntington, N.D.; Lew, A.M.; Xu, Y.; et al. GM-CSF Quantity Has a Selective Effect on Granulocytic vs. Monocytic Myeloid Development and Function. Front. Immunol. 2018, 9, 1922. [Google Scholar] [CrossRef] [PubMed]
- Verreck, F.A.; de Boer, T.; Langenberg, D.M.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Diny, N.L.; Ong, S.; Barin, J.G.; Hou, X.; Rose, N.R.; Talor, M.V.; Čiháková, D. Pathogenic IL-23 signaling is required to initiate GM-CSF-driven autoimmune myocarditis in mice. Eur. J. Immunol. 2016, 46, 582–592. [Google Scholar] [CrossRef]
- Hirata, Y.; Egea, L.; Dann, S.M.; Eckmann, L.; Kagnoff, M.F. GM-CSF-facilitated dendritic cell recruitment and survival govern the intestinal mucosal response to a mouse enteric bacterial pathogen. Cell Host Microbe 2010, 7, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Saba, S.; Soong, G.; Greenberg, S.; Prince, A. Bacterial stimulation of epithelial G-CSF and GM-CSF expression promotes PMN survival in CF airways. Am. J. Respir. Cell Mol. Biol. 2002, 27, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, T.A.; Zhao, C.; Pavicic, P.G., Jr.; Datta, S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front. Immunol. 2014, 5, 554. [Google Scholar] [CrossRef] [Green Version]
- Schon, M.P.; Erpenbeck, L. The Interleukin-23/Interleukin-17 Axis Links Adaptive and Innate Immunity in Psoriasis. Front. Immunol. 2018, 9, 1323. [Google Scholar] [CrossRef] [Green Version]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar] [CrossRef]
- Sorimachi, K.; Akimoto, K.; Hattori, Y.; Ieiri, T.; Niwa, A. Activation of macrophages by lactoferrin: Secretion of TNF-alpha, IL-8 and NO. Biochem. Mol. Biol. Int. 1997, 43, 79–87. [Google Scholar] [CrossRef]
- Sorimachi, K.; Akimoto, K.; Tsuru, K.; Nagashima, S.; Ieiri, T.; Niwa, A. Secretion of TNF-alpha from macrophages following induction with a lignin derivative. Cell Biol. Int. 1995, 19, 833–838. [Google Scholar] [CrossRef]
- Zganiacz, A.; Santosuosso, M.; Wang, J.; Yang, T.; Chen, L.; Anzulovic, M.; Alexander, S.; Gicquel, B.; Wan, Y.; Bramson, J.; et al. TNF-alpha is a critical negative regulator of type 1 immune activation during intracellular bacterial infection. J. Clin. Investig. 2004, 113, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeham, J.; Harkness, R.; Xing, Z. Macrophages are a significant source of type 1 cytokines during mycobacterial infection. J. Clin. Investig. 1999, 103, 1023–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, B.A.; Fumagalli, F.; Gainetdinov, R.R.; Jones, S.R.; Ator, R.; Giros, B.; Miller, G.W.; Caron, M.G. Cocaine self-administration in dopamine-transporter knockout mice. Nat. Neurosci. 1998, 1, 132–137. [Google Scholar] [CrossRef]
- Meredith, E.J.; Holder, M.J.; Rosén, A.; Lee, A.D.; Dyer, M.J.; Barnes, N.M.; Gordon, J. Dopamine targets cycling B cells independent of receptors/transporter for oxidative attack: Implications for non-Hodgkin’s lymphoma. Proc. Natl. Acad. Sci. USA 2006, 103, 13485–13490. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shaanan, T.L.; Azulay-Debby, H.; Dubovik, T.; Starosvetsky, E.; Korin, B.; Schiller, M.; Green, N.L.; Admon, Y.; Hakim, F.; Shen-Orr, S.S.; et al. Activation of the reward system boosts innate and adaptive immunity. Nat. Med. 2016, 22, 940–944. [Google Scholar] [CrossRef]
- Ben-Shaanan, T.L.; Schiller, M.; Azulay-Debby, H.; Korin, B.; Boshnak, N.; Koren, T.; Krot, M.; Shakya, J.; Rahat, M.A.; Hakim, F.; et al. Modulation of anti-tumor immunity by the brain’s reward system. Nat. Commun. 2018, 9, 2723. [Google Scholar] [CrossRef] [Green Version]
- Buller, K.M.; Dayas, C.V.; Day, T.A. Descending pathways from the paraventricular nucleus contribute to the recruitment of brainstem nuclei following a systemic immune challenge. Neuroscience 2003, 118, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.C.; Cox, S.; King, C.; Becker, H.; Reichel, C.M. Oxytocin and Rodent Models of Addiction. Int. Rev. Neurobiol. 2018, 140, 201–247. [Google Scholar] [CrossRef] [PubMed]
- Peris, J.; MacFadyen, K.; Smith, J.A.; de Kloet, A.D.; Wang, L.; Krause, E.G. Oxytocin receptors are expressed on dopamine and glutamate neurons in the mouse ventral tegmental area that project to nucleus accumbens and other mesolimbic targets. J. Comp. Neurol. 2017, 525, 1094–1108. [Google Scholar] [CrossRef]
- Ogundele, O.M.; Lee, C.C.; Francis, J. Thalamic dopaminergic neurons projects to the paraventricular nucleus-rostral ventrolateral medulla/C1 neural circuit. Anat. Rec. 2017, 300, 1307–1314. [Google Scholar] [CrossRef]
- Chen, K.K.; Chan, J.Y.; Chang, L.S. Dopaminergic neurotransmission at the paraventricular nucleus of hypothalamus in central regulation of penile erection in the rat. J. Urol. 1999, 162, 237–242. [Google Scholar] [CrossRef]
- Gaskill, P.J.; Khoshbouei, H. Dopamine and norepinephrine are embracing their immune side and so should we. Curr. Opin. Neurobiol. 2022, 77, 102626. [Google Scholar] [CrossRef] [PubMed]
- Pinoli, M.; Marino, F.; Cosentino, M. Dopaminergic Regulation of Innate Immunity: A Review. J. Neuroimmune Pharmacol. 2017, 12, 602–623. [Google Scholar] [CrossRef]
- Marino, F.; Pinoli, M.; Rasini, E.; Martini, S.; Luini, A.; Pulze, L.; Gasperina, D.D.; Grossi, P.; Legnaro, M.; Ferrari, M.; et al. Dopaminergic inhibition of human neutrophils is exerted through D1-like receptors and affected by bacterial infection. Immunology 2022, 167, 508–527. [Google Scholar] [CrossRef]
- Kustrimovic, N.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Comi, C.; Mauri, M.; Minafra, B.; Riboldazzi, G.; et al. Dopaminergic Receptors on CD4+ T Naive and Memory Lymphocytes Correlate with Motor Impairment in Patients with Parkinson’s Disease. Sci. Rep. 2016, 6, 33738. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavelaars, A.; Cobelens, P.M.; Teunis, M.A.; Heijnen, C.J. Changes in innate and acquired immune responses in mice with targeted deletion of the dopamine transporter gene. J. Neuroimmunol. 2005, 161, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Mondal, A.C.; Majumder, J.; Basu, S.; Dasgupta, P.S. Physiological concentrations of dopamine inhibit the proliferation and cytotoxicity of human CD4+ and CD8+ T cells in vitro: A receptor-mediated mechanism. Neuroimmunomodulation 2001, 9, 23–33. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gopinath, A.; Mackie, P.M.; Phan, L.T.; Mirabel, R.; Smith, A.R.; Miller, E.; Franks, S.; Syed, O.; Riaz, T.; Law, B.K.; et al. Who Knew? Dopamine Transporter Activity Is Critical in Innate and Adaptive Immune Responses. Cells 2023, 12, 269. https://doi.org/10.3390/cells12020269
Gopinath A, Mackie PM, Phan LT, Mirabel R, Smith AR, Miller E, Franks S, Syed O, Riaz T, Law BK, et al. Who Knew? Dopamine Transporter Activity Is Critical in Innate and Adaptive Immune Responses. Cells. 2023; 12(2):269. https://doi.org/10.3390/cells12020269
Chicago/Turabian StyleGopinath, Adithya, Phillip M. Mackie, Leah T. Phan, Rosa Mirabel, Aidan R. Smith, Emily Miller, Stephen Franks, Ohee Syed, Tabish Riaz, Brian K. Law, and et al. 2023. "Who Knew? Dopamine Transporter Activity Is Critical in Innate and Adaptive Immune Responses" Cells 12, no. 2: 269. https://doi.org/10.3390/cells12020269