
Tumor Growth Remains Refractory to Myc Ablation in Host Macrophages
 , and
, and 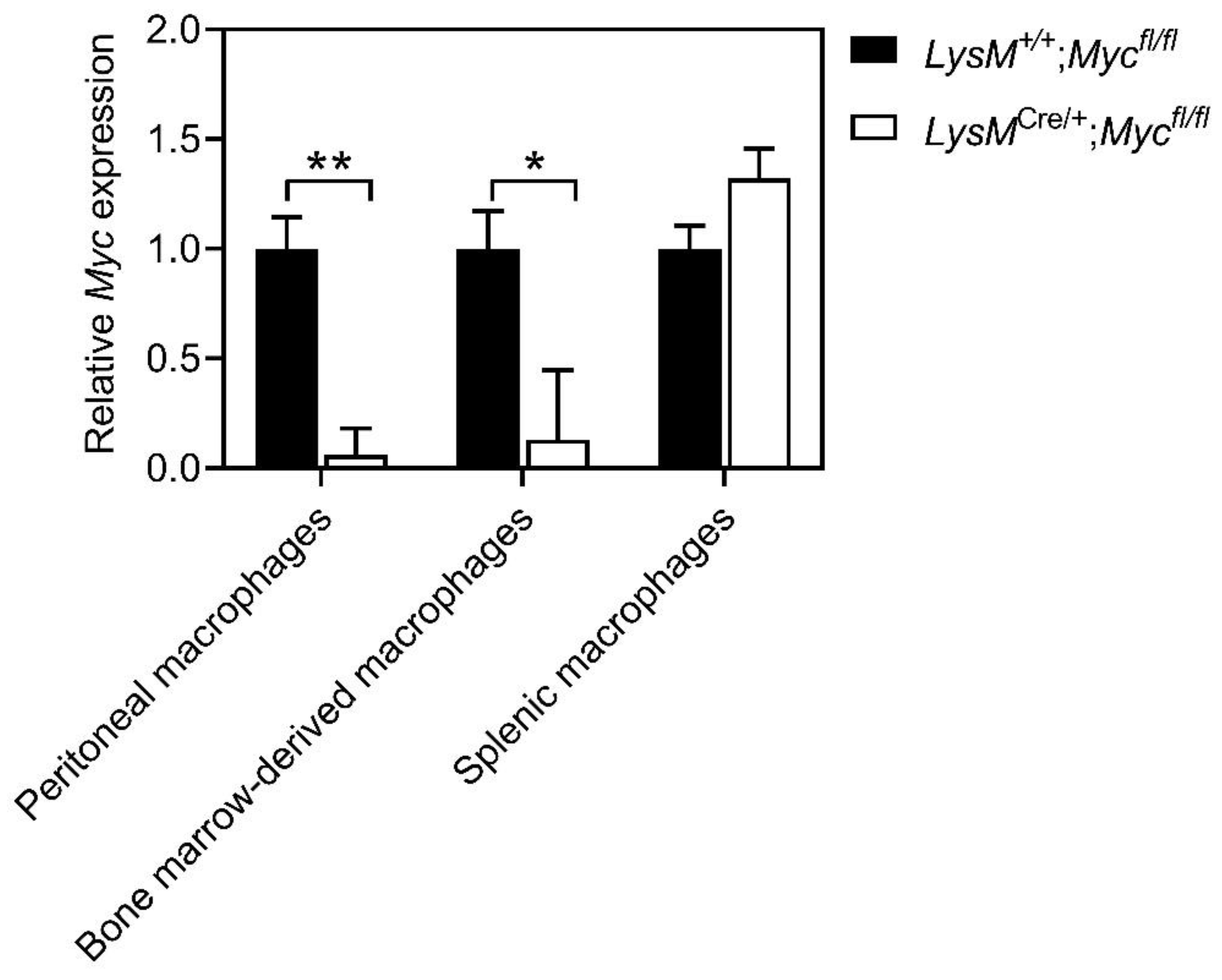{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Tumor Models
2.3. Ablation of Gastric Epithelial Cells via Tamoxifen Treatment
2.4. Immunofluorescence
2.5. Flow Cytometry
2.6. RNA Extraction and qPCR
2.7. Western Blot Analysis
2.8. Isolation of Bone-Marrow-Derived Macrophages
2.9. Isolation of Peritoneal Macrophages
2.10. Isolation of Splenic Macrophages
2.11. Quantification and Statistical Analysis
3. Results
3.1. Myc Expression Is Reduced in Macrophages of LysMCre/+;Mycfl/fl Mice
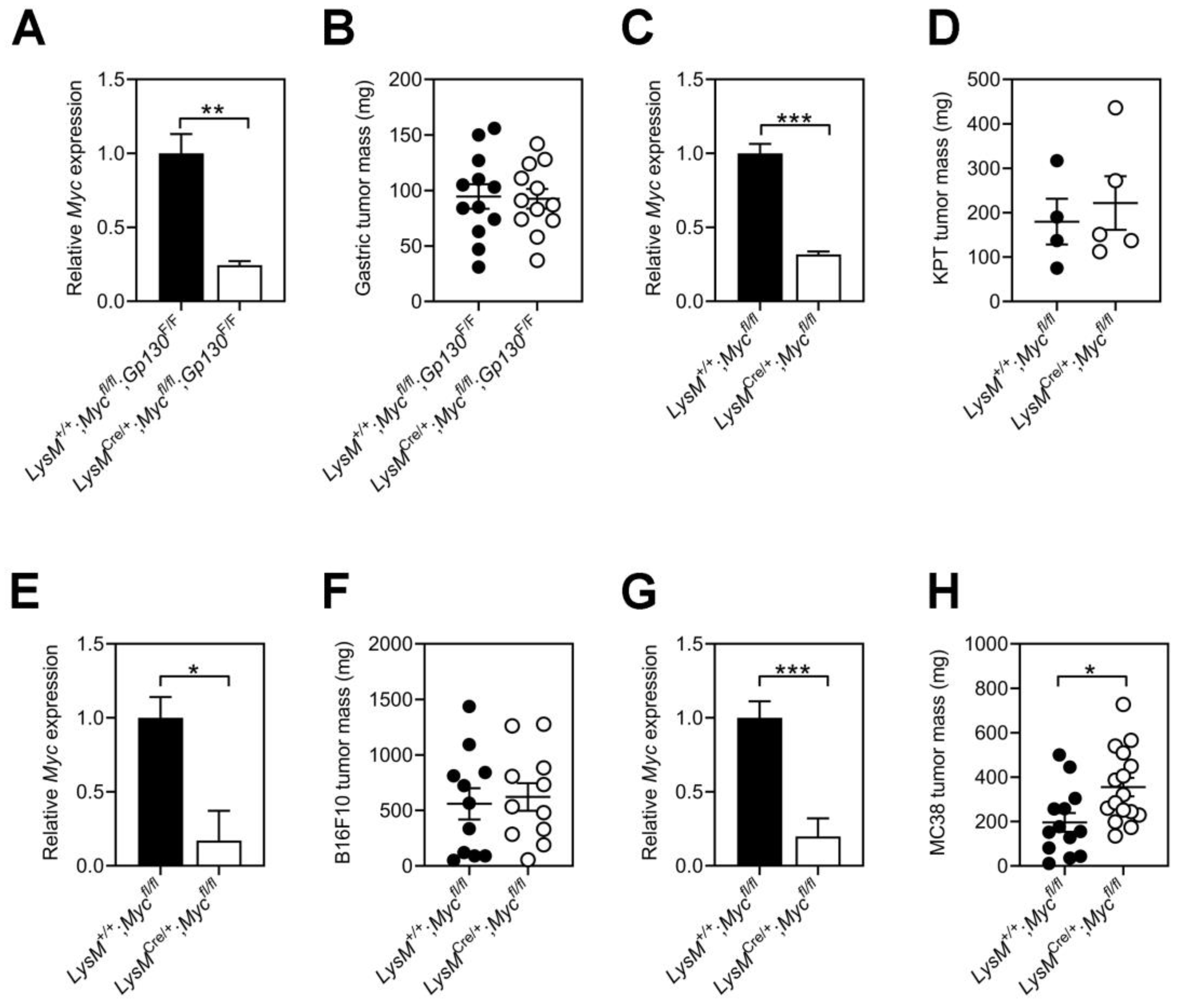
3.2. Tumor Growth Remains Refractory to Myc Ablation in Host Macrophages
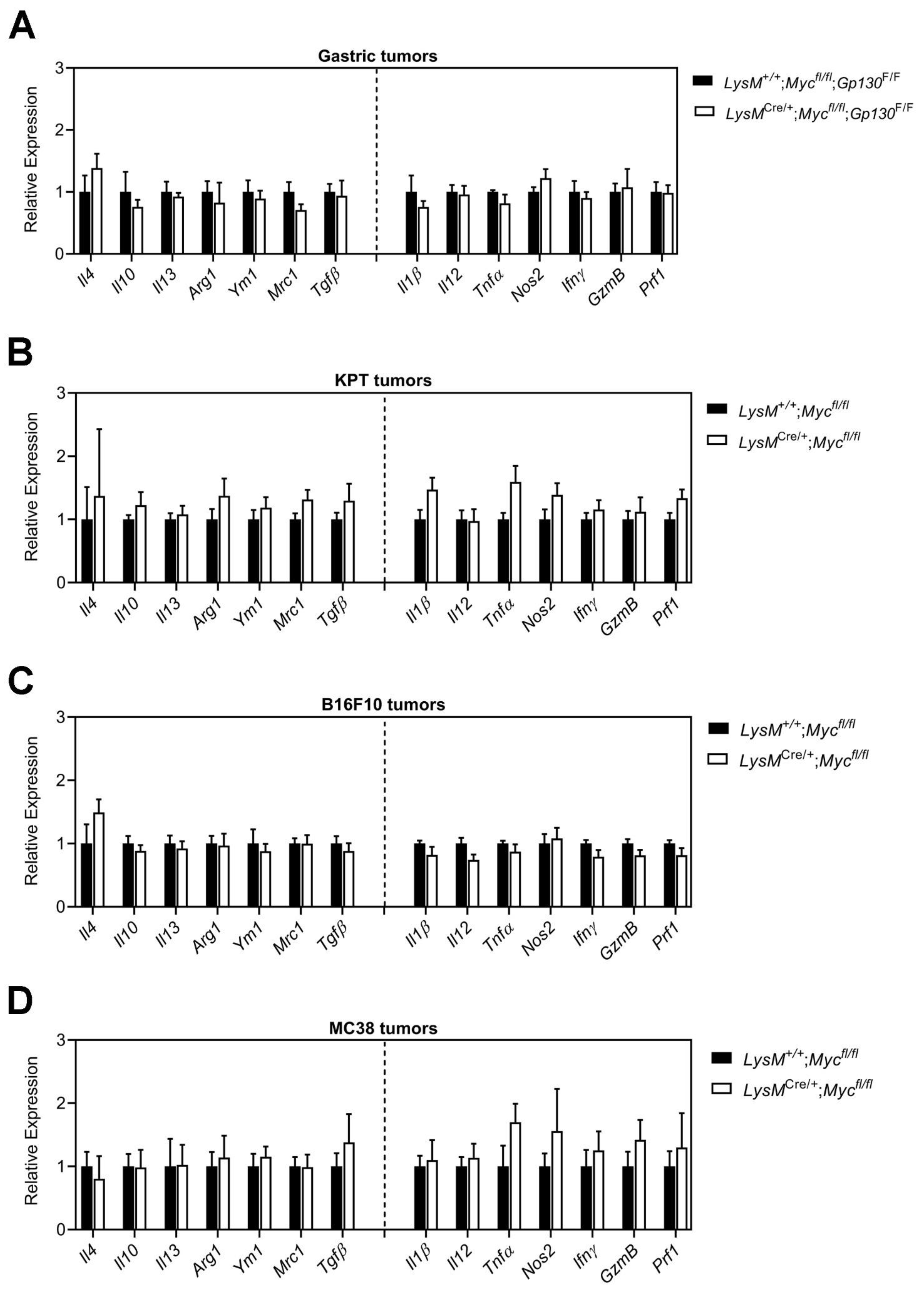
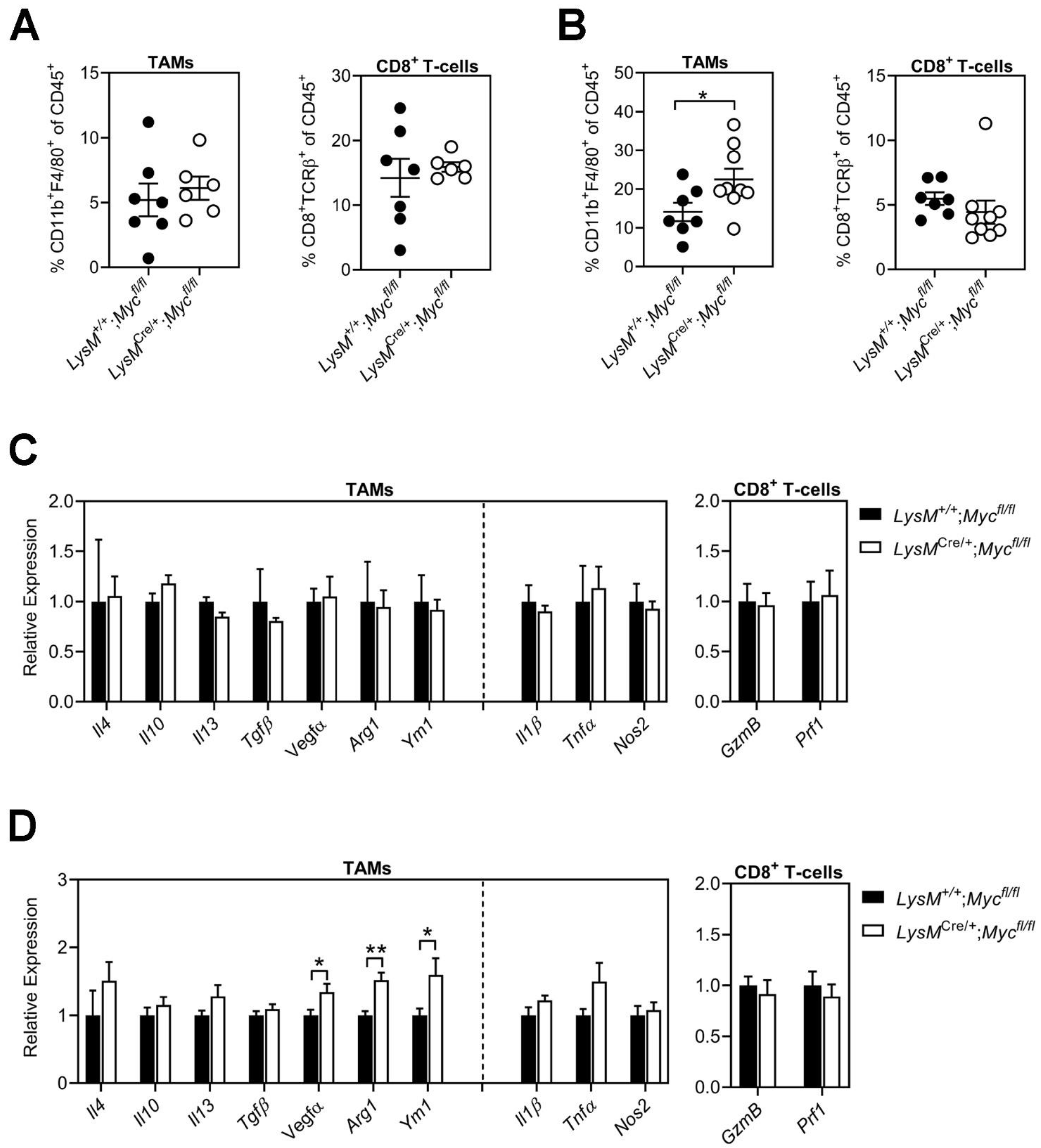
3.3. Conditional Ablation of Myc in TAMs Neither Reduces Tumor Immune Suppression Nor Impairs Alternative Macrophage Polarization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. c-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rathmell, J.C. T Cell Myc-tabolism. Immunity 2011, 35, 845–846. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Felsher, D.W. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer 2010, 1, 597–604. [Google Scholar] [CrossRef]
- Schrier, P.I.; Peltenburg, L.T. Relationship between myc oncogene activation and MHC class I expression. Adv. Cancer Res. 1993, 60, 181–246. [Google Scholar]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene-the grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2021, 19, 23–36. [Google Scholar] [CrossRef]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Milde, R.; Varin, A.; Melgert, B.N.; Draijer, C.; Thomas, B.; Fabbri, M.; Crawshaw, A.; Ho, L.P.; et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: Similarities and differences. Blood 2013, 121, e57–e69. [Google Scholar] [CrossRef] [PubMed]
- Pello, O.M.; De Pizzol, M.; Mirolo, M.; Soucek, L.; Zammataro, L.; Amabile, A.; Doni, A.; Nebuloni, M.; Swigart, L.B.; Evan, G.I.; et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 2012, 119, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pello, O.M.; Chèvre, R.; Laoui, D.; De Juan, A.; Lolo, F.; Andrés-Manzano, M.J.; Serrano, M.; Van Ginderachter, J.; Andrés, V. In Vivo Inhibition of c-MYC in Myeloid Cells Impairs Tumor-Associated Macrophage Maturation and Pro-Tumoral Activities. PLoS ONE 2012, 7, e45399. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, P.S.U.; Lee, Y.; Mun, S.H.; Giannopoulou, E.; Fujii, T.; Lee, K.P.; Violante, S.N.; Cross, J.R.; Park-Min, K.-H. MYC-mediated early glycolysis negatively regulates proinflammatory responses by controlling IRF4 in inflammatory macrophages. Cell Rep. 2021, 35, 109264. [Google Scholar] [CrossRef]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Förster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef]
- de Alboran, I.M.; O’Hagan, R.C.; Gärtner, F.; Malynn, B.; Davidson, L.; Rickert, R.; Rajewsky, K.; DePinho, R.A.; Alt, F.W. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 2001, 2001. 14, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Tebbutt, N.C.; Giraud, A.S.; Inglese, M.; Jenkins, B.; Waring, P.; Clay, F.J.; Malki, S.; Alderman, B.M.; Grail, D.; Hollande, F.; et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 2002, 8, 1089–1097. [Google Scholar] [CrossRef]
- Thiem, S.; Eissmann, M.F.; Stuart, E.; Elzer, J.; Jonas, A.; Buchert, M.; Ernst, M. Inducible gene modification in the gastric epithelium of Tff1-CreERT2, Tff2-rtTA, Tff3-luc mice. Genesis 2016, 54, 626–635. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Schwab, R.H.M.; Tran, B.M.; Phesse, T.J.; Vincan, E. Isolation and Culture of Adult Intestinal, Gastric, and Liver Organoids for Cre-recombinase-Mediated Gene Deletion. Methods Mol. Biol. 2019, 1576, 123–133. [Google Scholar]
- Eissmann, M.F.; Dijkstra, C.; Jarnicki, A.; Phesse, T.; Brunnberg, J.; Poh, A.R.; Etemadi, N.; Tsantikos, E.; Thiem, S.; Huntington, N.D.; et al. IL-33-mediated mast cell activation promotes gastric cancer through macrophage mobilization. Nat. Commun. 2019, 10, 2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allam, A.H.; Russell, S.M. Establishing a multiplex imaging panel to study T cell development in the thymus in mouse. STAR Protoc. 2022, 3, 101472. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Love, C.G.; Chisanga, D.; Steer, J.H.; Baloyan, D.; Chopin, M.; Nutt, S.; Rautela, J.; Huntington, N.D.; Etemadi, N.; et al. Therapeutic inhibition of the SRC-kinase HCK facilitates T cell tumor infiltration and improves response to immunotherapy. Sci. Adv. 2022, 8, eabl7882. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Love, C.G.; Masson, F.; Preaudet, A.; Tsui, C.; Whitehead, L.; Monard, S.; Khakham, Y.; Burstroem, L.; Lessene, G.; et al. Inhibition of Hematopoietic Cell Kinase Activity Suppresses Myeloid Cell-Mediated Colon Cancer Progression. Cancer Cell 2017, 31, 563–575.e5. [Google Scholar] [CrossRef] [Green Version]
- Poh, A.R.; Dwyer, A.R.; Eissmann, M.F.; Chand, A.L.; Baloyan, D.; Boon, L.; Murrey, M.W.; Whitehead, L.; O′Brien, M.; Lowell, C.A.; et al. Inhibition of the SRC Kinase HCK Impairs STAT3-Dependent Gastric Tumor Growth in Mice. Cancer Immunol. Res. 2020, 8, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Abram, C.L.; Roberge, G.L.; Hu, Y.; Lowell, C.A. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 2014, 408, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Poh, A.R.; O’Donoghue, R.J.J.; Ernst, M.; Putoczki, T.L. Mouse models for gastric cancer: Matching models to biological questions. J. Gastroenterol. Hepatol. 2016, 31, 1257–1272. [Google Scholar] [CrossRef]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-β signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar] [CrossRef]
- Meškytė, E.M.; Keskas, S.; Ciribilli, Y. MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. Int. J. Mol. Sci. 2020, 21, 7710. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, Q.; Fan, Y.; Li, J.; Zhang, J.; Wang, W.; Fan, J.; Guo, Y.; Liu, S.; Hao, D.; et al. MYC inhibition reprograms tumor immune microenvironment by recruiting T lymphocytes and activating the CD40/CD40L system in osteosarcoma. Cell Death Discov. 2022, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Chawla, A. Control of macrophage activation and function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Rahman, S.M.; Janssen, R.C.; Choudhury, M.; Baquero, K.C.; Aikens, R.M.; de la Houssaye, B.A.; Friedman, J.E. CCAAT/enhancer-binding protein β (C/EBPβ) expression regulates dietary-induced inflammation in macrophages and adipose tissue in mice. J. Biol. Chem. 2012, 287, 34349–34360. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Myasoedova, V.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 2018, 223, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kγ Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poh, A.R.; O’Brien, M.; Chisanga, D.; He, H.; Baloyan, D.; Traichel, J.; Dijkstra, C.; Chopin, M.; Nutt, S.; Whitehead, L.; et al. Inhibition of HCK in myeloid cells restricts pancreatic tumor growth and metastasis. Cell Rep. 2022, 41, 111479. [Google Scholar] [CrossRef]
- Pello, O.M. Macrophages and c-Myc cross paths. OncoImmunology 2016, 5, e1151991. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α–mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [Green Version]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc Posttranscriptionally Induces HIF1 Protein and Target Gene Expression in Normal and Cancer Cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.-Y.; Wang, X.; Jiang, C. HIF1α-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediat. Inflamm. 2017, 2017, 9029327. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Zhang, Y.; Li, M.; Song, Y.; Liu, D.; Yang, X.; Yang, D.; Qu, H.; Lai, L.; Wang, Q.; et al. E3 ligase FBXW7 restricts M2-like tumor-associated macrophage polarization by targeting c-Myc. Aging 2020, 12, 24394–24423. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, Y.; Lv, H.; Meng, H.; Xiong, G.; Guan, X.; Chen, X.; Bai, Y.; Wang, K. miR-26a and miR-26b inhibit esophageal squamous cancer cell proliferation through suppression of c-MYC pathway. Gene 2017, 625, 1–9. [Google Scholar] [CrossRef]
- Sahu, S.K.; Kumar, M.; Chakraborty, S.; Banerjee, S.K.; Kumar, R.; Gupta, P.; Jana, K.; Gupta, U.D.; Ghosh, Z.; Kundu, M.; et al. MicroRNA 26a (miR-26a)/KLF4 and CREB-C/EBPβ regulate innate immune signaling, the polarization of macrophages and the trafficking of Mycobacterium tuberculosis to lysosomes during infection. PLoS Pathog. 2017, 13, e1006410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Sodir, N.; Wilson, C.; Burkhart, D.L.; Pellegrinet, L.; Swigart, L.B.; Littlewood, T.; Evan, G. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morrow, R.J.; Allam, A.H.; Konecnik, J.; Baloyan, D.; Dijkstra, C.; Eissmann, M.F.; Jacob, S.P.; O’Brien, M.; Poh, A.R.; Ernst, M. Tumor Growth Remains Refractory to Myc Ablation in Host Macrophages. Cells 2022, 11, 4104. https://doi.org/10.3390/cells11244104
Morrow RJ, Allam AH, Konecnik J, Baloyan D, Dijkstra C, Eissmann MF, Jacob SP, O’Brien M, Poh AR, Ernst M. Tumor Growth Remains Refractory to Myc Ablation in Host Macrophages. Cells. 2022; 11(24):4104. https://doi.org/10.3390/cells11244104
Chicago/Turabian StyleMorrow, Riley J., Amr H. Allam, Josh Konecnik, David Baloyan, Christine Dijkstra, Moritz F. Eissmann, Saumya P. Jacob, Megan O’Brien, Ashleigh R. Poh, and Matthias Ernst. 2022. "Tumor Growth Remains Refractory to Myc Ablation in Host Macrophages" Cells 11, no. 24: 4104. https://doi.org/10.3390/cells11244104