
TRIP13 Participates in Immediate-Early Sensing of DNA Strand Breaks and ATM Signaling Amplification through MRE11

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Neutral COMET Assay
2.3. Sister Chromatid Exchange (SCE) Assay
2.4. Plasmids
2.5. Antibodies
2.6. Regulation of TRIP13 Expression and MRE11 Exonuclease Activity
2.7. DSB Induction
2.8. Immunofluorescence Imaging
2.9. Proteomics Experiments with SILAC
2.10. Western Blot Analysis
2.11. In Vitro Pulldown Assay
2.12. Immunoprecipitation
2.13. Colony Formation Assay
2.14. I-SceI-Induced Assay
2.15. DNA End Resection Assay
2.16. MRN Endonuclease Activity Assay
3. Results
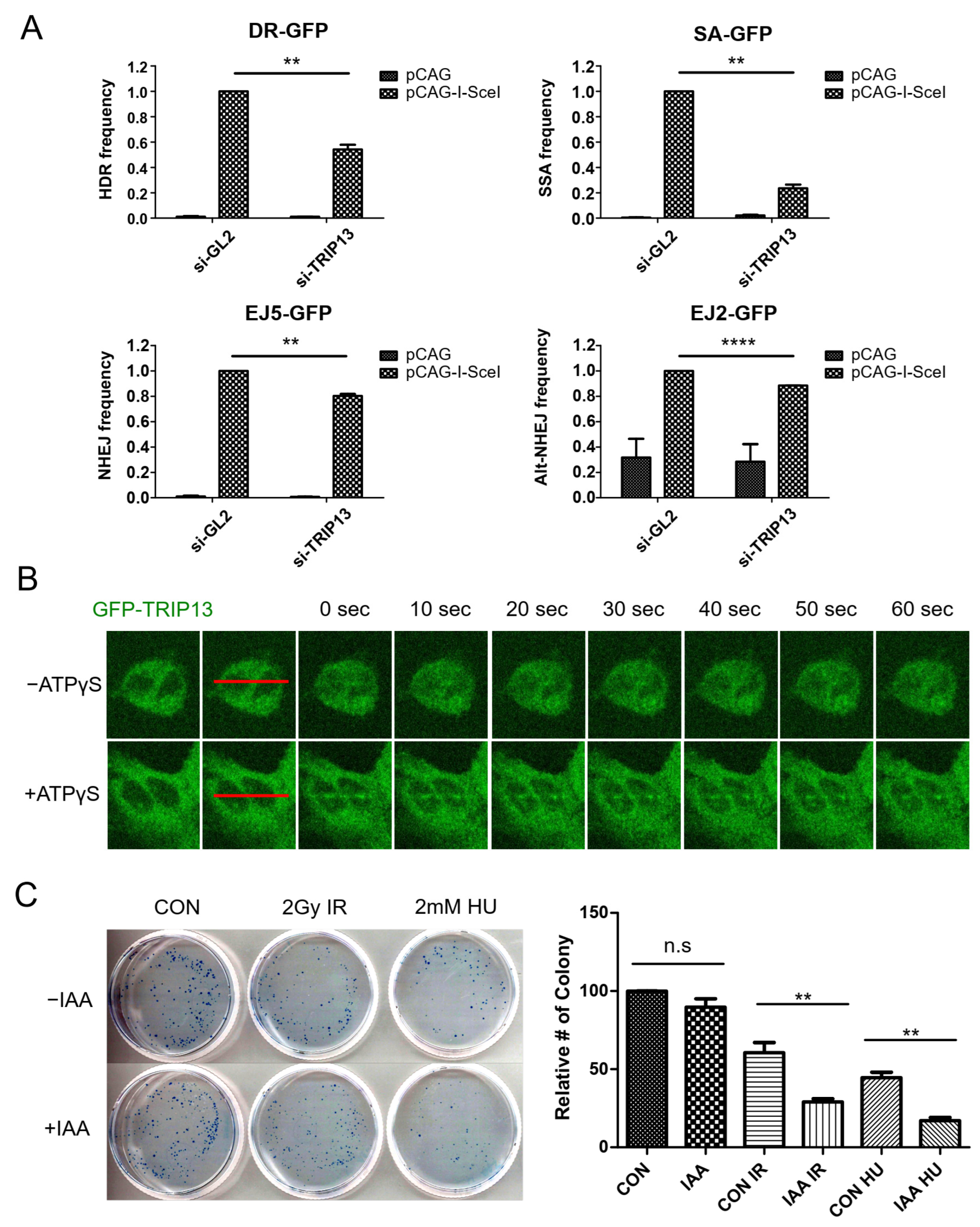
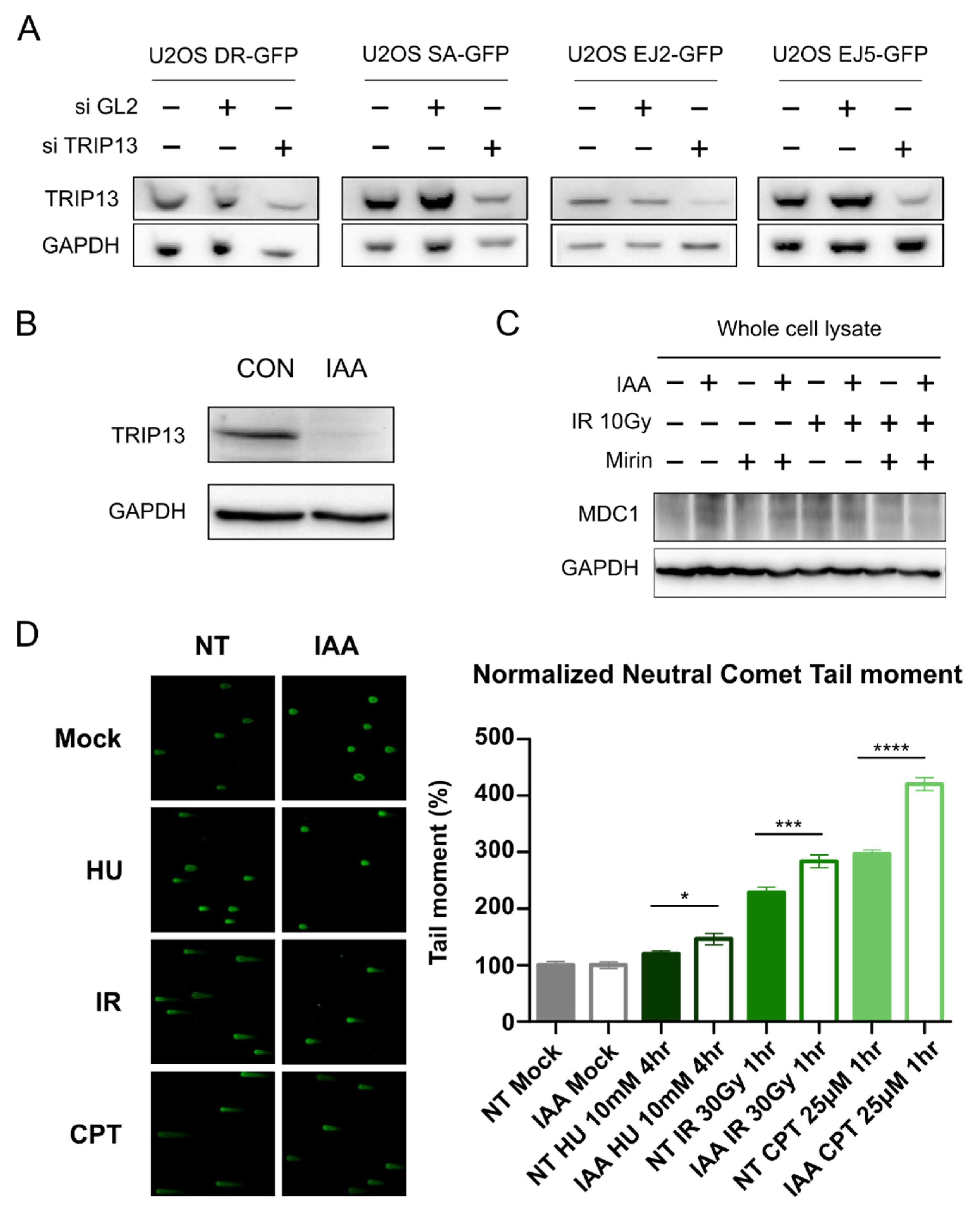
3.1. TRIP13 Participates in the DNA Damage Response
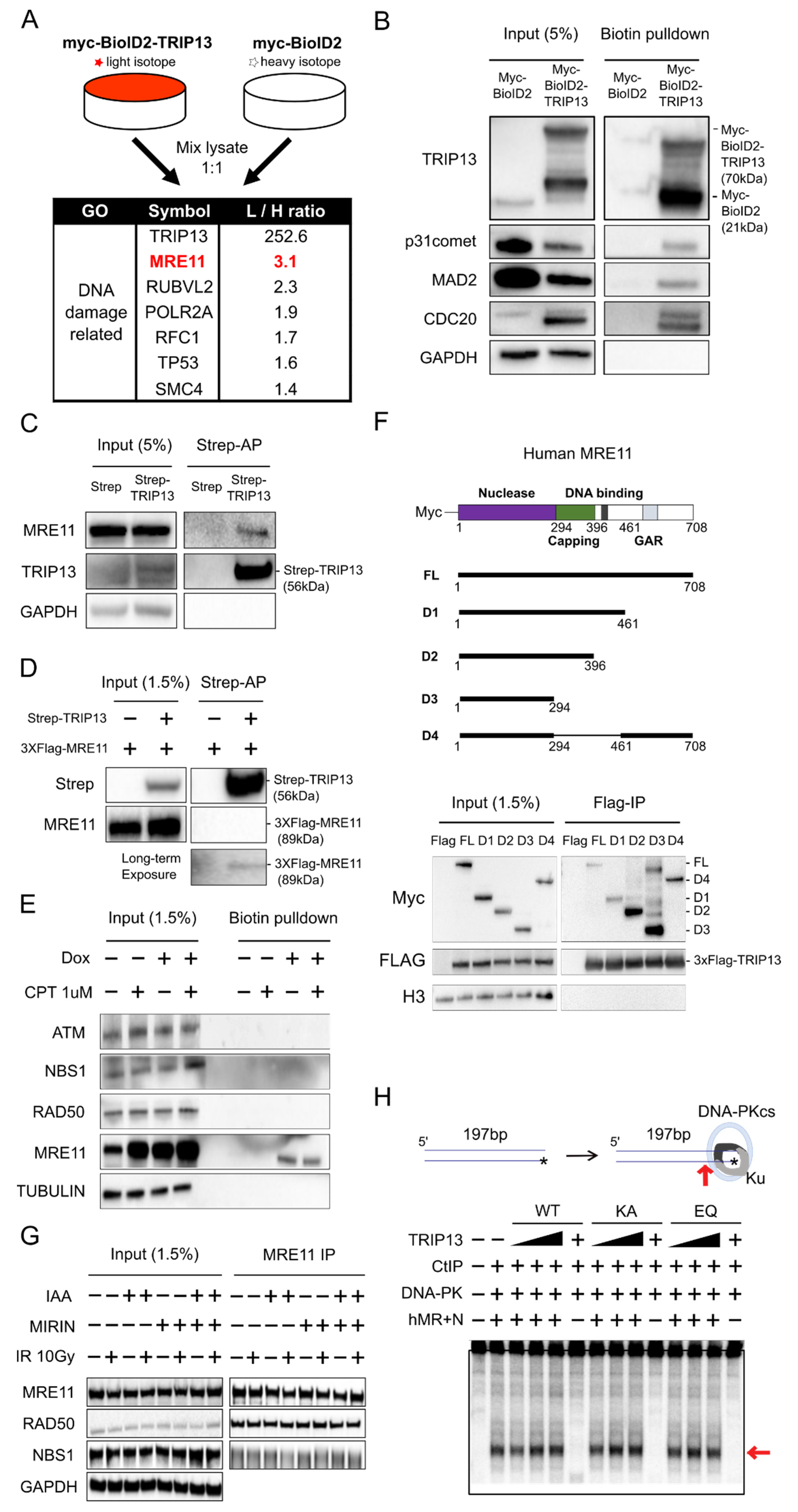
3.2. MRE11 Is a Novel Interaction Partner of TRIP13, but this Interaction Is Independent of the Conformation and Nuclease Activity of the MRN Complex
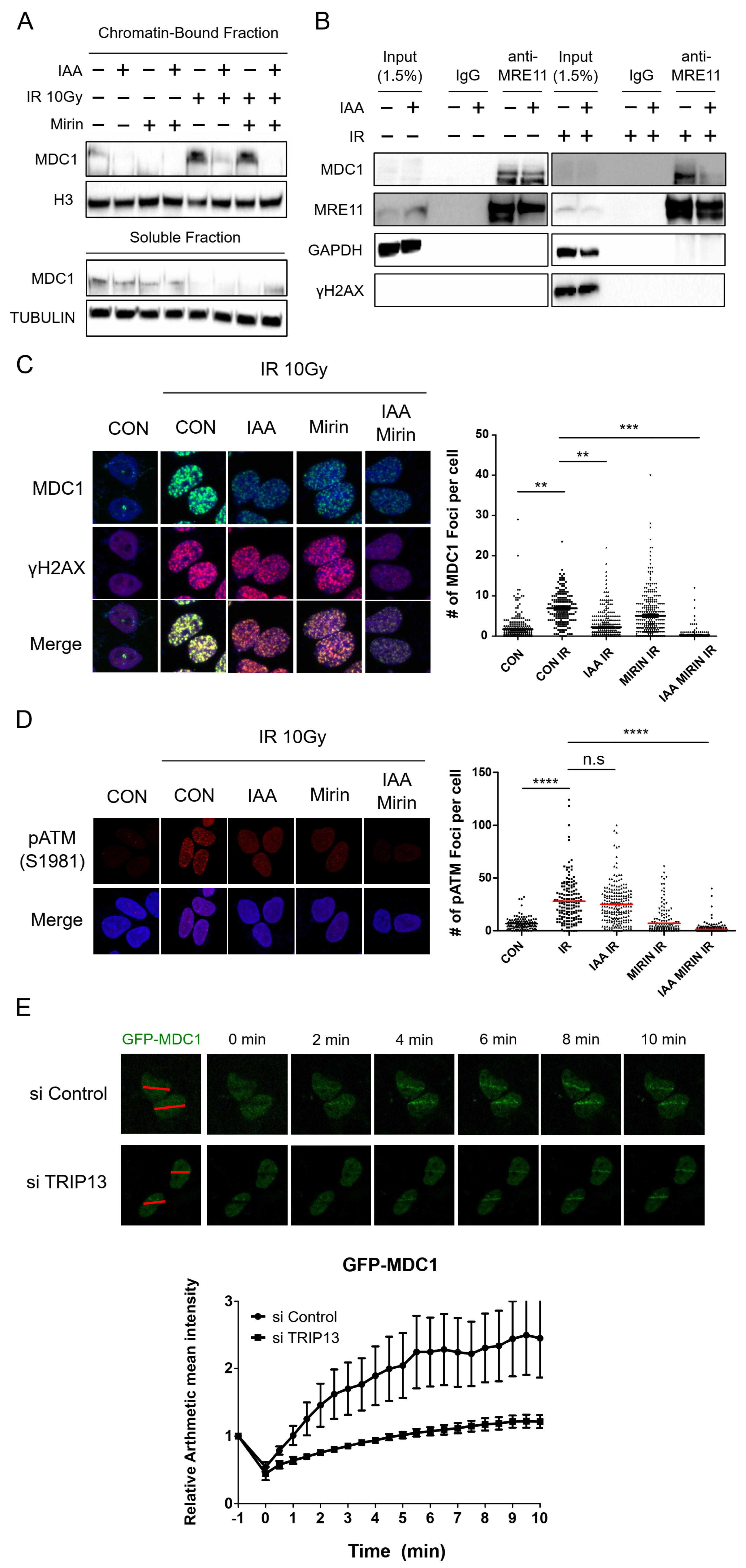
3.3. TRIP13 Depletion Inhibits the Physical Interaction of MDC1 with MRE11 and Reduces the Recruitment of MDC1 to DNA Damage Sites
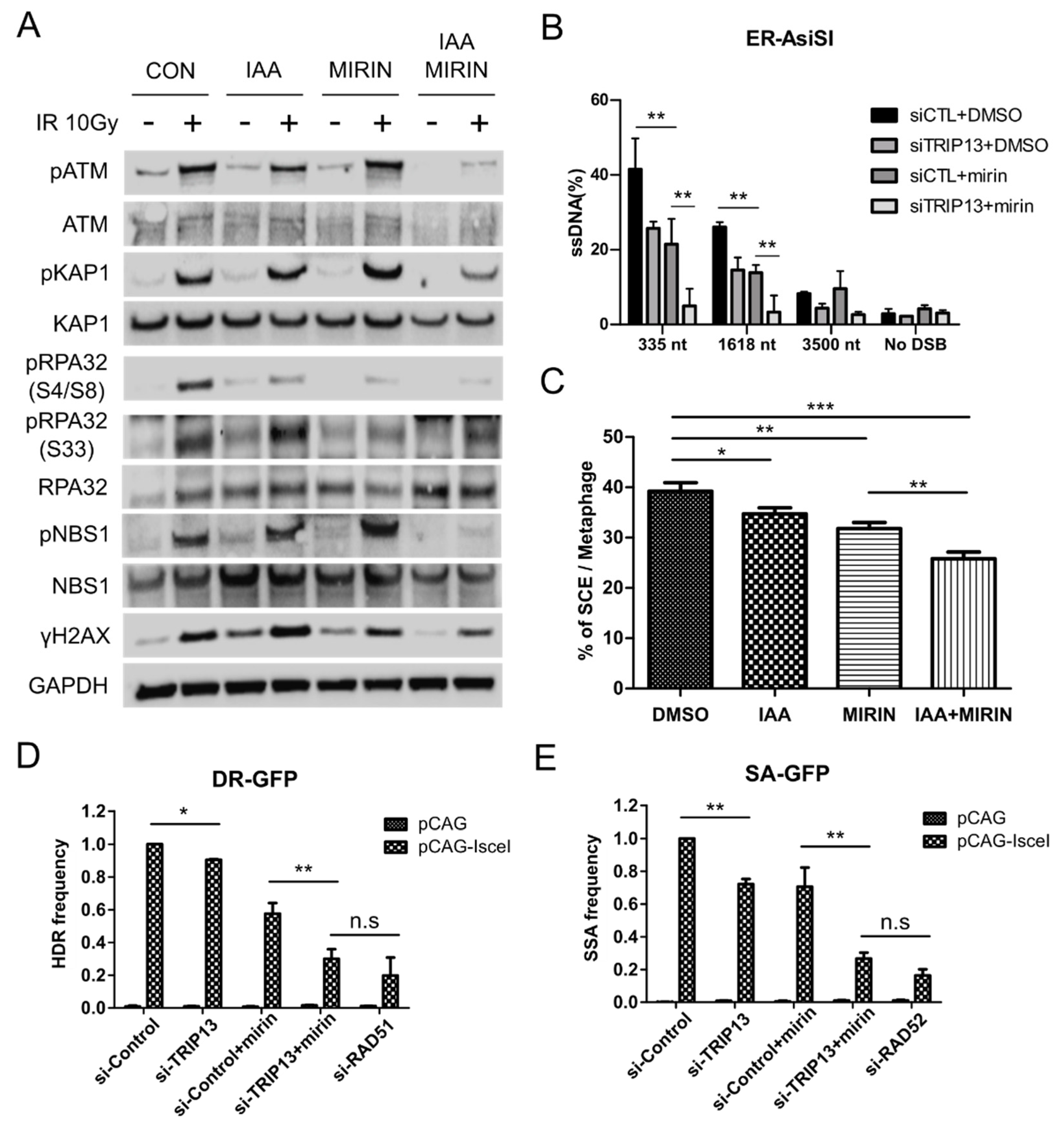
3.4. TRIP13 Enhances DNA Resection and Homologous Recombination by Regulating ATM Downstream Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Aravind, L.; Koonin, E.V. The HORMA Domain: A Common Structural Denominator in Mitotic Checkpoints, Chromosome Synapsis and DNA Repair. Trends Biochem. Sci. 1998, 23, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.C.; Corbett, K.D. The Multifaceted Roles of the HORMA Domain in Cellular Signaling. J. Cell Biol. 2015, 211, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 Regulates DNA Repair Pathway Choice through REV7 Conformational Change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Wojtasz, L.; Daniel, K.; Roig, I.; Bolcun-Filas, E.; Xu, H.; Boonsanay, V.; Eckmann, C.R.; Cooke, H.J.; Jasin, M.; Keeney, S.; et al. Mouse HORMAD1 and HORMAD2, Two Conserved Meiotic Chromosomal Proteins, Are Depleted from Synapsed Chromosome Axes with the Help of TRIP13 AAA-ATPase. PLoS Genet. 2009, 5, e1000702. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [Green Version]
- Yost, S.; de Wolf, B.; Hanks, S.; Zachariou, A.; Marcozzi, C.; Clarke, M.; de Voer, R.M.; Etemad, B.; Uijttewaal, E.; Ramsay, E.; et al. Biallelic TRIP13 Mutations Predispose to Wilms Tumor and Chromosome Missegregation. Nat. Genet. 2017, 49, 1148–1151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Zhu, Q.; Fu, G.; Hou, J.; Hu, X.; Cao, J.; Peng, W.; Wang, X.; Chen, F.; Cui, H. TRIP13 Promotes the Cell Proliferation, Migration and Invasion of Glioblastoma through the FBXW7/c-MYC Axis. Br. J. Cancer 2019, 121, 1069–1078. [Google Scholar] [CrossRef]
- Banerjee, R.; Russo, N.; Liu, M.; Basrur, V.; Bellile, E.; Palanisamy, N.; Scanlon, C.S.; van Tubergen, E.; Inglehart, R.C.; Metwally, T.; et al. TRIP13 Promotes Error-Prone Nonhomologous End Joining and Induces Chemoresistance in Head and Neck Cancer. Nat. Commun. 2014, 5, 4527. [Google Scholar] [CrossRef] [Green Version]
- Sarangi, P.; Clairmont, C.S.; Galli, L.D.; Moreau, L.A.; D’Andrea, A.D. P31comet Promotes Homologous Recombination by Inactivating REV7 through the TRIP13 ATPase. Proc. Natl. Acad. Sci. USA 2020, 117, 26795–26803. [Google Scholar] [CrossRef]
- Venkatesan, K.; Rual, J.-F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.-I.; et al. An Empirical Framework for Binary Interactome Mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef]
- Ho, H.-C.; Burgess, S.M. Pch2 Acts through Xrs2 and Tel1/ATM to Modulate Interhomolog Bias and Checkpoint Function during Meiosis. PLoS Genet. 2011, 7, e1002351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar-Fernández, M.A.; da Silva, R.C.; Firlej, M.; Pan, D.; Weir, E.; Sarembe, A.; Raina, V.B.; Bange, T.; Weir, J.R.; Vader, G. Biochemical and Functional Characterization of a Meiosis-Specific Pch2/ORC AAA+ Assembly. Life Sci. Alliance 2020, 3, e201900630. [Google Scholar] [CrossRef]
- Zhou, Y.; Caron, P.; Legube, G.; Paull, T.T. Quantitation of DNA Double-Strand Break Resection Intermediates in Human Cells. Nucleic Acids Res. 2014, 42, e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Han, J.S.; Ly, P.; Ye, Q.; McMahon, M.A.; Myung, K.; Corbett, K.D.; Cleveland, D.W. TRIP13 and APC15 Drive Mitotic Exit by Turnover of Interphase- and Unattached Kinetochore-Produced MCC. Nat. Commun. 2018, 9, 4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennardo, N.; Gunn, A.; Cheng, A.; Hasty, P.; Stark, J.M. Limiting the Persistence of a Chromosome Break Diminishes Its Mutagenic Potential. PLoS Genet. 2009, 5, e1000683. [Google Scholar] [CrossRef]
- Motegi, A.; Liaw, H.-J.; Lee, K.-Y.; Roest, H.P.; Maas, A.; Wu, X.; Moinova, H.; Markowitz, S.D.; Ding, H.; Hoeijmakers, J.H.J.; et al. Polyubiquitination of Proliferating Cell Nuclear Antigen by HLTF and SHPRH Prevents Genomic Instability from Stalled Replication Forks. Proc. Natl. Acad. Sci. USA 2008, 105, 12411–12416. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-G.; Kim, N.; Kim, S.-M.; Park, I.B.; Kim, H.; Kim, S.; Kim, B.-G.; Hwang, J.M.; Baek, I.-J.; Gartner, A.; et al. Ewing Sarcoma Protein Promotes Dissociation of Poly(ADP-Ribose) Polymerase 1 from Chromatin. EMBO Rep. 2020, 21, e48676. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Paull, T.T.; Gellert, M. The 3′ to 5′ Exonuclease Activity of Mre11 Facilitates Repair of DNA Double-Strand Breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- de Krijger, I.; Föhr, B.; Pérez, S.H.; Vincendeau, E.; Serrat, J.; Thouin, A.M.; Susvirkar, V.; Lescale, C.; Paniagua, I.; Hoekman, L.; et al. MAD2L2 Dimerization and TRIP13 Control Shieldin Activity in DNA Repair. Nat. Commun. 2021, 12, 5421. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA Damage Response Factors from Diverse Pathways, Including DNA Crosslink Repair, Mediate Alternative End Joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochan, J.A.; Desclos, E.C.B.; Bosch, R.; Meister, L.; Vriend, L.E.M.; van Attikum, H.; Krawczyk, P.M. Meta-Analysis of DNA Double-Strand Break Response Kinetics. Nucleic Acids Res. 2017, 45, 12625–12637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsume, T.; Kiyomitsu, T.; Saga, Y.; Kanemaki, M.T. Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep. 2016, 15, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An Improved Smaller Biotin Ligase for BioID Proximity Labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.-E.; Mann, M. A Practical Recipe for Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC). Nat. Protoc. 2006, 1, 2650–2660. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. Activation and Regulation of ATM Kinase Activity in Response to DNA Double-Strand Breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a Super Complex of BRCA1-Associated Proteins Involved in the Recognition and Repair of Aberrant DNA Structures. Genes Dev. 2000, 14, 927–939. [Google Scholar] [CrossRef]
- Haber, J.E. The Many Interfaces of Mre11. Cell 1998, 95, 583–586. [Google Scholar] [CrossRef]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 Directly Binds Phosphorylated Histone H2AX to Regulate Cellular Responses to DNA Double-Strand Breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.R.; Jackson, S.P. Phospho-Dependent Interactions between NBS1 and MDC1 Mediate Chromatin Retention of the MRN Complex at Sites of DNA Damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spycher, C.; Miller, E.S.; Townsend, K.; Pavic, L.; Morrice, N.A.; Janscak, P.; Stewart, G.S.; Stucki, M. Constitutive Phosphorylation of MDC1 Physically Links the MRE11-RAD50-NBS1 Complex to Damaged Chromatin. J. Cell Biol. 2008, 181, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Luo, K.; Lou, Z.; Chen, J. MDC1 Regulates Intra-S-Phase Checkpoint by Targeting NBS1 to DNA Double-Strand Breaks. Proc. Natl. Acad. Sci. USA 2008, 105, 11200–11205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Edwards, R.A.; Thede, G.L.; Glover, J.N.M. Structure of the BRCT Repeat Domain of MDC1 and Its Specificity for the Free COOH-Terminal End of the Gamma-H2AX Histone Tail. J. Biol. Chem. 2005, 280, 32053–32056. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.R.; Elledge, S.J. MDC1 Is a Mediator of the Mammalian DNA Damage Checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Mand, M.R.; Deshpande, R.A.; Kinoshita, E.; Yang, S.-H.; Wyman, C.; Paull, T.T. Ataxia Telangiectasia-Mutated (ATM) Kinase Activity Is Regulated by ATP-Driven Conformational Changes in the Mre11/Rad50/Nbs1 (MRN) Complex. J. Biol. Chem. 2013, 288, 12840–12851. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Paull, T.T. Direct Measurement of Single-Stranded DNA Intermediates in Mammalian Cells by Quantitative Polymerase Chain Reaction. Anal. Biochem. 2015, 479, 48–50. [Google Scholar] [CrossRef]
- Crossen, P.E.; Drets, M.E.; Arrighi, F.E.; Johnston, D.A. Aanlysis of the Frequency and Distribution of Sister Chromatid Exchanges in Cultured Human Lymphocytes. Hum. Genet. 1977, 35, 345–352. [Google Scholar] [CrossRef]
- Galloway, S.M.; Evans, H.J. Sister Chromatid Exchange in Human Chromosomes from Normal Individuals and Patients with Ataxia Telangiectasia. Cytogenet. Cell Genet. 1975, 15, 17–29. [Google Scholar] [CrossRef]
- Sonoda, E.; Sasaki, M.S.; Morrison, C.; Yamaguchi-Iwai, Y.; Takata, M.; Takeda, S. Sister Chromatid Exchanges Are Mediated by Homologous Recombination in Vertebrate Cells. Mol. Cell. Biol. 1999, 19, 5166–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieffer, S.R.; Lowndes, N.F. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front. Genet. 2022, 13, 793884. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. Direct Activation of the ATM Protein Kinase by the Mre11/Rad50/Nbs1 Complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Lee, J.-H. The Mre11/Rad50/Nbs1 Complex and Its Role as a DNA Double-Strand Break Sensor for ATM. Cell Cycle 2005, 4, 737–740. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Limbo, O.; Yamada, Y.; Russell, P. Mre11-Rad50-Dependent Activity of ATM/Tel1 at DNA Breaks and Telomeres in the Absence of Nbs1. Mol. Biol. Cell 2018, 29, 1389–1399. [Google Scholar] [CrossRef]
- Drew, K.; Wallingford, J.B.; Marcotte, E.M. Hu.MAP 2.0: Integration of over 15,000 Proteomic Experiments Builds a Global Compendium of Human Multiprotein Assemblies. Mol. Syst. Biol. 2021, 17, e10016. [Google Scholar] [CrossRef]
- Yasugi, T.; Vidal, M.; Sakai, H.; Howley, P.M.; Benson, J.D. Two Classes of Human Papillomavirus Type 16 E1 Mutants Suggest Pleiotropic Conformational Constraints Affecting E1 Multimerization, E2 Interaction, and Interaction with Cellular Proteins. J. Virol. 1997, 71, 5942–5951. [Google Scholar] [CrossRef] [Green Version]
- Eytan, E.; Wang, K.; Miniowitz-Shemtov, S.; Sitry-Shevah, D.; Kaisari, S.; Yen, T.J.; Liu, S.-T.; Hershko, A. Disassembly of Mitotic Checkpoint Complexes by the Joint Action of the AAA-ATPase TRIP13 and P31(Comet). Proc. Natl. Acad. Sci. USA 2014, 111, 12019–12024. [Google Scholar] [CrossRef] [Green Version]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef]
- Nowsheen, S.; Aziz, K.; Aziz, A.; Deng, M.; Qin, B.; Luo, K.; Jeganathan, K.B.; Zhang, H.; Liu, T.; Yu, J.; et al. L3MBTL2 Orchestrates Ubiquitin Signalling by Dictating the Sequential Recruitment of RNF8 and RNF168 after DNA Damage. Nat. Cell Biol. 2018, 20, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 Couples Initiation and Amplification of Ubiquitin Signalling after DNA Damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.R.; Barral, P.; Vannier, J.-B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 Regulates DSB Repair Using Rif1 to Control 5′ End Resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, I.M.; Jowsey, P.A.; Toth, R.; Rouse, J. Phospho-Epitope Binding by the BRCT Domains of HPTIP Controls Multiple Aspects of the Cellular Response to DNA Damage. Nucleic Acids Res. 2007, 35, 5312–5322. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The Shieldin Complex Mediates 53BP1-Dependent DNA Repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [Green Version]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 Controls DNA Repair at Telomeres and DNA Breaks by Inhibiting 5′ End Resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Species | Dilution (WB) | Dilution (IF) | Company | Cat# |
|---|---|---|---|---|---|
| MRE11 | Rabbit | 1:2000 | - | NOVUS, St. Louis, MO, USA | NB100-142 |
| GAPDH | Rabbit | 1:5000 | - | Santa Cruz, Dallas, TX, USA | sc-32233 |
| Myc | Mouse | 1:1000 | - | Santa Cruz, Dallas, TX, USA | sc-40 |
| TRIP13 | Rabbit | 1:1000 | - | Bethyl, Montgomery, TX, USA | A303-605A |
| Strep | Rabbit | 1:1000 | - | Abcam, Cambridge, UK | ab76949 |
| ATM | Rabbit | 1:1000 | - | Abcam, Cambridge, UK | ab32420 |
| NBS1 | Mouse | 1:1000 | - | Cell Signaling Tech, Danvers, MA, USA | 14956S |
| RAD50 | Mouse | 1:1000 | - | Santa Cruz, Dallas, TX, USA | sc-74460 |
| CtIP | Goat | 1:1000 | - | Santa Cruz, Dallas, TX, USA | sc5970 |
| Tubulin | Mouse | 1:10000 | - | Sigma, St. Louis, MO, USA | T-5168 |
| MDC1 | Rabbit | 1:1000 | 1:200 | Abcam, Cambridge, UK | ab11171 |
| H3 | Rabbit | 1:1000 | - | Santa Cruz, Dallas, TX, USA | sc-8654R |
| γH2AX | Rabbit | 1:1000 | 1:200 | GeneTex, Irvine, CA, USA | GTX127340 |
| pATM(S1981) | Rabbit | 1:1000 | - | Abcam, Cambridge, UK | ab81292 |
| pKAP1 | Mouse | 1:1000 | - | Bethyl, Montgomery, TX, USA | A300-767A |
| KAP1 | Mouse | 1:1000 | - | Bethyl, Montgomery, TX, USA | A300-274A |
| pRPA32(S4/8) | Rabbit | 1:1000 | - | Bethyl, Montgomery, TX, USA | A300-245A |
| pRPA32(S33) | Rabbit | 1:1000 | - | Bethyl, Montgomery, TX, USA | A300-246A |
| RPA32 | Rabbit | 1:1000 | - | Bethyl, Montgomery, TX, USA | A300-244A |
| pNBS1 | Rabbit | 1:1000 | - | Cell Signaling Tech, Danvers, MA, USA | 3002S |
| Primer Name | Primer Sequence | |
|---|---|---|
| DSB1-335 bp | Primer FW | 5′-GAATCGGATGTATGCGACTGATC-3′ |
| Primer REV | 5′-TTCCAAAGTTATTCCAACCCGAT-3′ | |
| Probe | 5′-6FAM-CACAGCTTGCCCATCCTTGCAAACC-TAMRA-3′ | |
| DSB1-1618 bp | Primer FW | 5′-TGAGGAGGTGACATTAGAACTCAGA-3′ |
| Primer REV | 5′-AGGACTCACTTACACGGCCTTT-3′ | |
| Probe | 5′-6FAM-TTGCAAGGCTGCTTCCTTACCATTCAA-TAMRA-3′ | |
| DSB1-3500 bp | Primer FW | 5′-TCCTAGCCAGATAATAATAGCTATACAAACA-3′ |
| Primer REV | 5′-TGAATAGACAGACAACAGATAAATGAGACA-3′ | |
| Probe | 5′-6FAM-ACCCTGATCAGCCTTTCCATGGGTTAAG-TAMRA-3′ | |
| No DSB | Primer FW | 5′-ATTGGGTATCTGCGTCTAGTGAGG-3′ |
| Primer REV | 5′-GACTCAATTACATCCCTGCAGCT-3′ | |
| Probe | 5′-6FAM-TCTCTGCACAGACCGGCTTCCCTTC-TAMRA-3′ | |
| siRNA Name | siRNA Sequence |
|---|---|
| TRIP13 mRNA-specific-sense | 5′-GCA AAU CAC UGG GUU CUA C-3′ |
| TRIP13 mRNA-specific-antisense | 5′-G UAG AAC CCA GUG AUU UGC-3′ |
| GL2 | 5′-CGUACGCGGAAUACUUCGA=UU-3′ 5′-UCGAAGUAUUCCGCGUACG=UU-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.; Wie, M.; Baek, I.-J.; Sohn, G.; Um, S.-H.; Lee, S.-G.; Seo, Y.; Ra, J.; Lee, E.A.; Kim, S.; et al. TRIP13 Participates in Immediate-Early Sensing of DNA Strand Breaks and ATM Signaling Amplification through MRE11. Cells 2022, 11, 4095. https://doi.org/10.3390/cells11244095
Jeong H, Wie M, Baek I-J, Sohn G, Um S-H, Lee S-G, Seo Y, Ra J, Lee EA, Kim S, et al. TRIP13 Participates in Immediate-Early Sensing of DNA Strand Breaks and ATM Signaling Amplification through MRE11. Cells. 2022; 11(24):4095. https://doi.org/10.3390/cells11244095
Chicago/Turabian StyleJeong, Hyeongsun, Minwoo Wie, In-Joon Baek, Gyuwon Sohn, Si-Hyeon Um, Seon-Gyeong Lee, Yuri Seo, Jaesun Ra, Eun A Lee, Shinseog Kim, and et al. 2022. "TRIP13 Participates in Immediate-Early Sensing of DNA Strand Breaks and ATM Signaling Amplification through MRE11" Cells 11, no. 24: 4095. https://doi.org/10.3390/cells11244095