
Melatonin Treatment Triggers Metabolic and Intracellular pH Imbalance in Glioblastoma
 , , , ,
, , , ,
Abstract
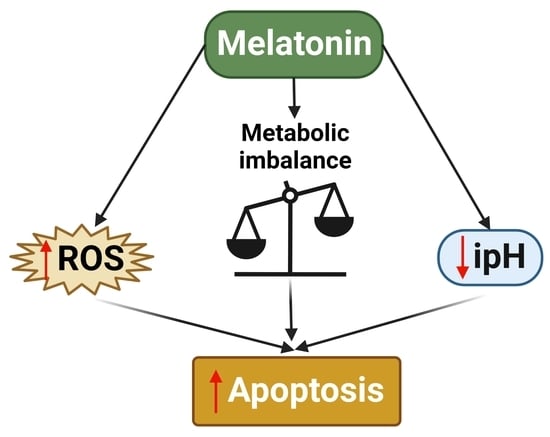
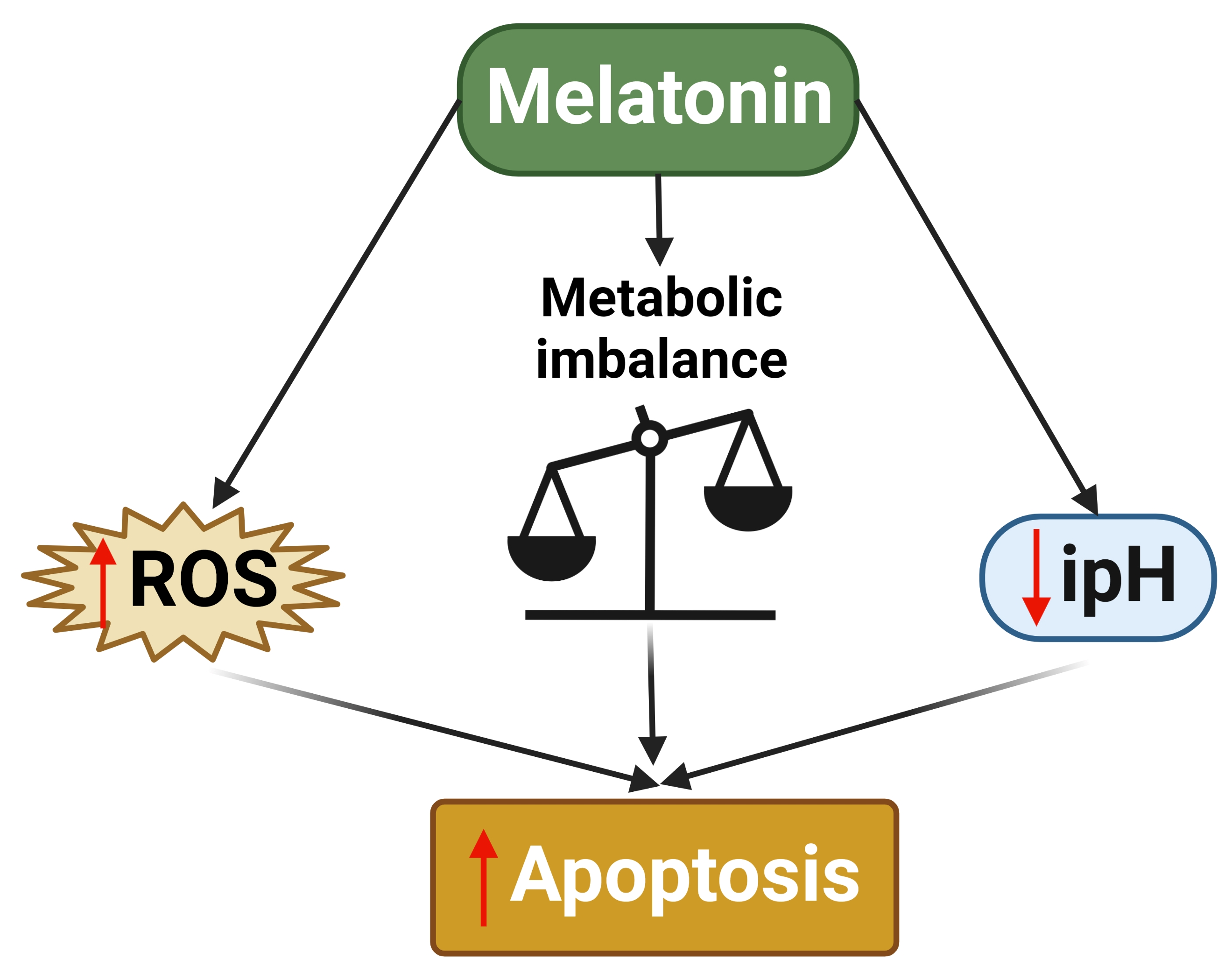
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Cell Proliferation and Viability
2.3. Cell Cycle Assay
2.4. Apoptosis
2.5. Clonogenic Assay
2.6. Synergy Calculation
2.7. Limiting Dilution Assay
2.8. Oxygen Consumption Rate
2.9. Pyruvate Concentration
2.10. Western Blotting
2.11. ROS Production
2.12. Glucose-6-Phosphate Dehydrogenase Activity
2.13. Substrate Oxidation
2.14. ATP Detection
2.15. pH
2.16. Lactate Concentration
2.17. Glucose Availability
2.18. In Vivo Subcutaneous Xenografts
2.19. Histologic Analysis
2.20. Statistical Analysis
3. Results
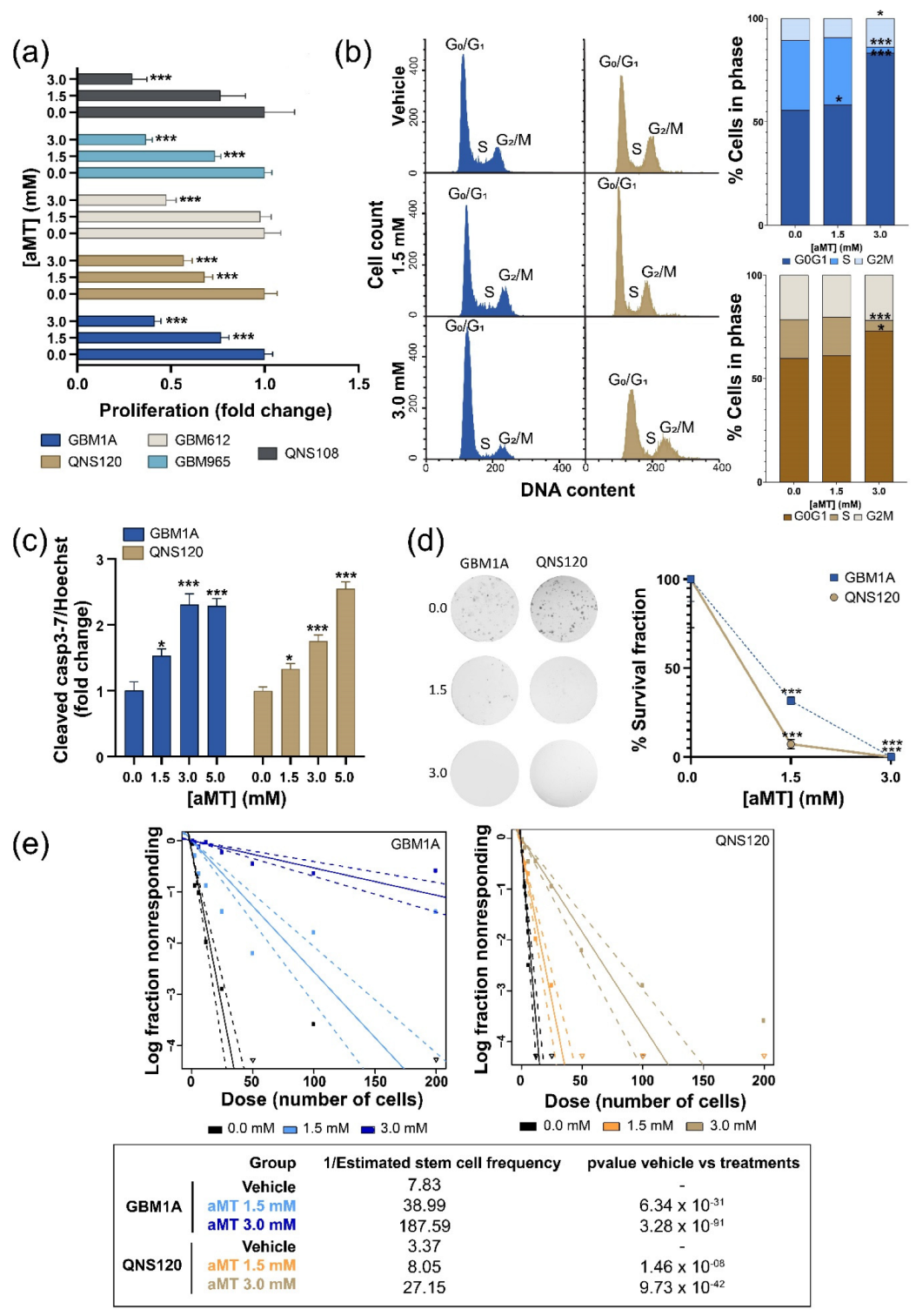
3.1. Melatonin Decreases GBM Viability and Stemness, Inducing Cell Death
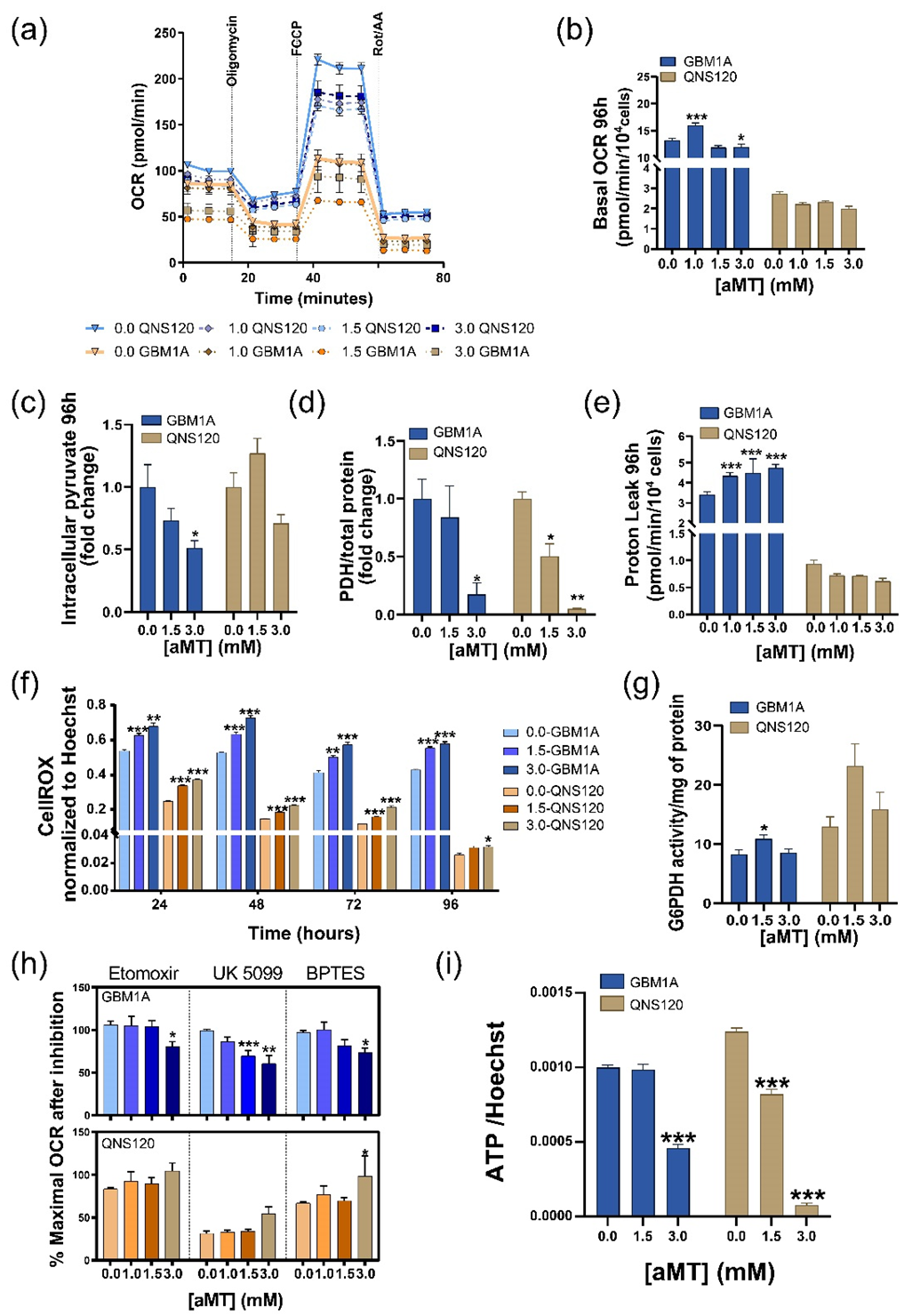
3.2. Melatonin Differentially Disturbs GBM Mitochondrial Metabolism
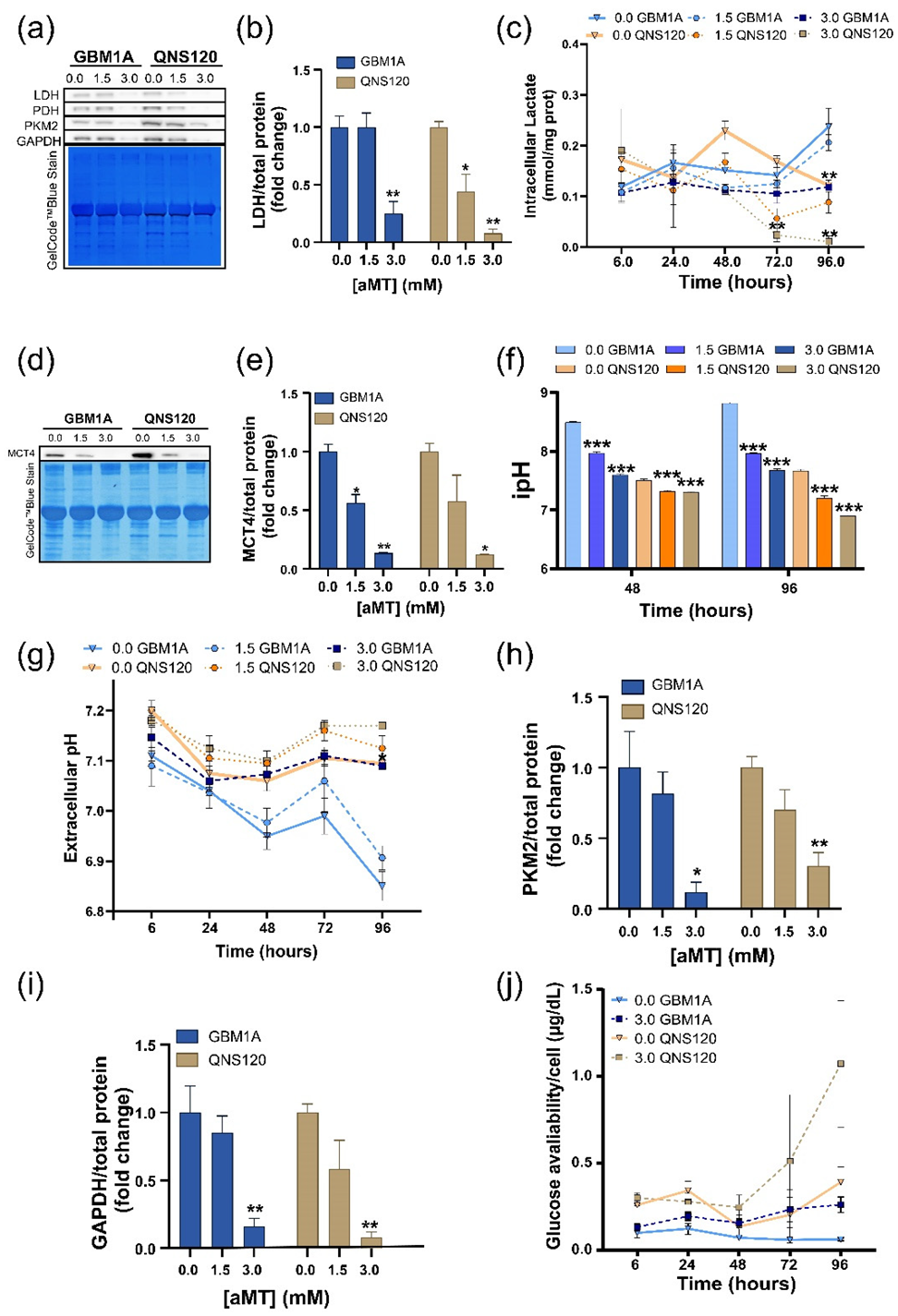
3.3. Melatonin Disrupts pH Balance in GBM while Downregulating Glycolysis
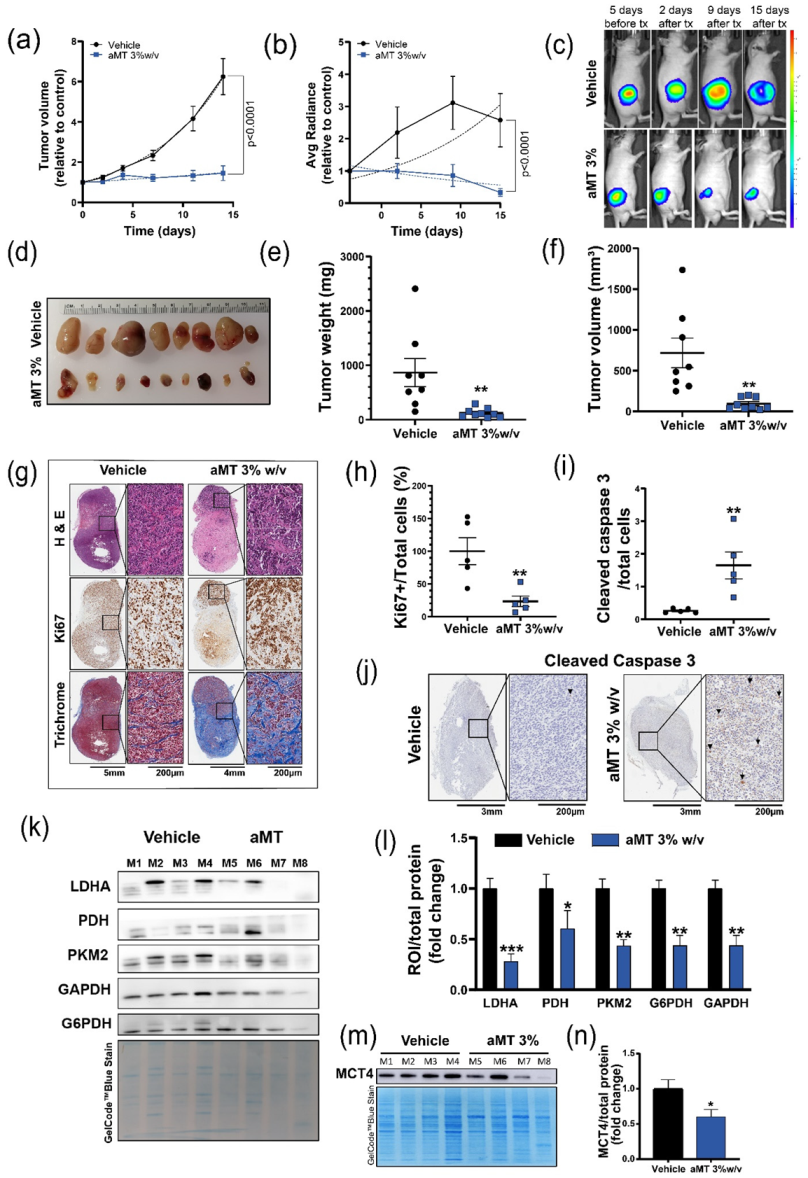
3.4. Intratumoral Melatonin Treatment Decreases Tumor Growth In Vivo
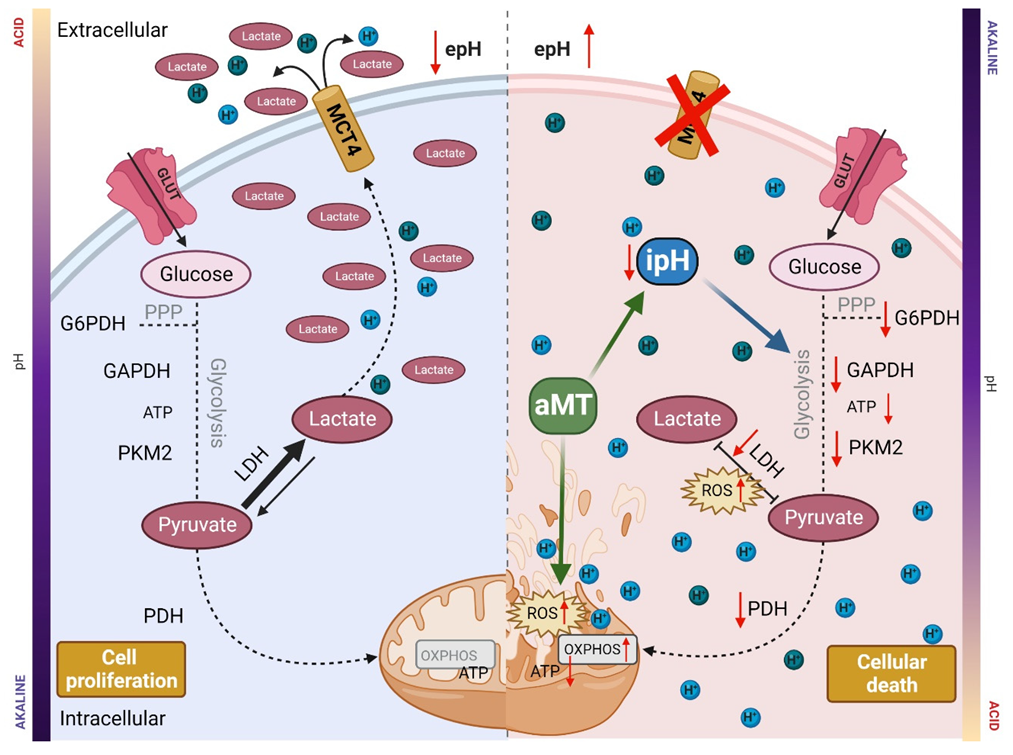
4. Discussion
5. Limitations of This Study and Future Directions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Koltai, T. The Ph paradigm in cancer. Eur. J. Clin. Nutr. 2020, 74, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Quinones-Hinojosa, A.; Rao, R. Emerging links between endosomal pH and cancer. Cancer Metastasis Rev. 2020, 39, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, K.C.; Llongueras, J.P.; Capilla-Gonzalez, V.; Prasad, H.; Hack, A.; Smith, C.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Rao, R. A leak pathway for luminal protons in endosomes drives oncogenic signalling in glioblastoma. Nat. Commun. 2015, 6, 6289. [Google Scholar] [CrossRef] [Green Version]
- Talib, W.H.; Alsayed, A.R.; Abuawad, A.; Daoud, S.; Mahmod, A.I. Melatonin in Cancer Treatment: Current Knowledge and Future Opportunities. Molecules 2021, 26, 2506. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Zhang, H.M.; Zhang, Y. Melatonin: A well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 2014, 57, 131–146. [Google Scholar] [CrossRef]
- Florido, J.; Rodriguez-Santana, C.; Martinez-Ruiz, L.; Lopez-Rodriguez, A.; Acuna-Castroviejo, D.; Rusanova, I.; Escames, G. Understanding the Mechanism of Action of Melatonin, Which Induces ROS Production in Cancer Cells. Antioxidants 2022, 11, 1621. [Google Scholar] [CrossRef]
- Florido, J.; Martinez-Ruiz, L.; Rodriguez-Santana, C.; López-Rodríguez, A.; Hidalgo-Gutiérrez, A.; Cottet-Rousselle, C.; Lamarche, F.; Schlattner, U.; Guerra-Librero, A.; Aranda-Martínez, P.; et al. Melatonin drives apoptosis in head and neck cancer by increasing mitochondrial ROS generated via reverse electron transport. J. Pineal Res. 2022. [Google Scholar] [CrossRef]
- Guerra-Librero, A.; Fernandez-Gil, B.I.; Florido, J.; Martinez-Ruiz, L.; Rodriguez-Santana, C.; Shen, Y.Q.; Garcia-Verdugo, J.M.; Lopez-Rodriguez, A.; Rusanova, I.; Quinones-Hinojosa, A.; et al. Melatonin Targets Metabolism in Head and Neck Cancer Cells by Regulating Mitochondrial Structure and Function. Antioxidants 2021, 10, 603. [Google Scholar] [CrossRef]
- Aust, S.; Brucker, B.; Graf, J.; Klimpfinger, M.; Thalhammer, T. Melatonin modulates acid/base transport in human pancreatic carcinoma cells. Cell Physiol. Biochem. 2006, 18, 91–102. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.A.; Bhargav, A.G.; Brooks, M.; Suarez-Meade, P.; Mondal, S.K.; Zarco, N.; ReFaey, K.; Jentoft, M.; Middlebrooks, E.H.; Snuderl, M.; et al. Functional Characterization of Brain Tumor-Initiating Cells and Establishment of GBM Preclinical Models that Incorporate Heterogeneity, Therapy, and Sex Differences. Mol. Cancer Ther. 2021, 20, 2585–2597. [Google Scholar] [CrossRef]
- Lara-Velazquez, M.; Zarco, N.; Carrano, A.; Phillipps, J.; Norton, E.S.; Schiapparelli, P.; Al-Kharboosh, R.; Rincon-Torroella, J.; Jeanneret, S.; Corona, T.; et al. Alpha 1-antichymotrypsin contributes to stem cell characteristics and enhances tumorigenicity of glioblastoma. Neuro Oncol. 2021, 23, 599–610. [Google Scholar] [CrossRef]
- Shen, Y.Q.; Guerra-Librero, A.; Fernandez-Gil, B.I.; Florido, J.; Garcia-Lopez, S.; Martinez-Ruiz, L.; Mendivil-Perez, M.; Soto-Mercado, V.; Acuna-Castroviejo, D.; Ortega-Arellano, H.; et al. Combination of melatonin and rapamycin for head and neck cancer therapy: Suppression of AKT/mTOR pathway activation, and activation of mitophagy and apoptosis via mitochondrial function regulation. J. Pineal Res. 2018, 64, e12461. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Pang, B.; Gu, G.; Gao, T.; Zhang, R.; Pang, Q.; Liu, Q. Melatonin Inhibits Glioblastoma Stem-like cells through Suppression of EZH2-NOTCH1 Signaling Axis. Int. J. Biol. Sci. 2017, 13, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, N.; Wei, Z.X.; Qi, Z.X. Inhibition of autophagy triggers melatonin-induced apoptosis in glioblastoma cells. BMC Neurosci. 2019, 20, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Canaveras, J.C.; Chen, L.; Rabinowitz, J.D. The Tumor Metabolic Microenvironment: Lessons from Lactate. Cancer Res. 2019, 79, 3155–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; DiPaola, R.S.; Mathew, R.; White, E. Metabolic catastrophe as a means to cancer cell death. J. Cell Sci. 2007, 120, 379–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Wilk, A.; Wyczechowska, D.; Zapata, A.; Dean, M.; Mullinax, J.; Marrero, L.; Parsons, C.; Peruzzi, F.; Culicchia, F.; Ochoa, A.; et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol. Cell Biol. 2015, 35, 182–198. [Google Scholar] [CrossRef] [Green Version]
- Hevia, D.; Gonzalez-Menendez, P.; Fernandez-Fernandez, M.; Cueto, S.; Rodriguez-Gonzalez, P.; Garcia-Alonso, J.I.; Mayo, J.C.; Sainz, R.M. Melatonin Decreases Glucose Metabolism in Prostate Cancer Cells: A (13)C Stable Isotope-Resolved Metabolomic Study. Int. J. Mol. Sci. 2017, 18, 1620. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Sanchez, A.M.; Antolin, I.; Puente-Moncada, N.; Suarez, S.; Gomez-Lobo, M.; Rodriguez, C.; Martin, V. Melatonin Cytotoxicity Is Associated to Warburg Effect Inhibition in Ewing Sarcoma Cells. PLoS ONE 2015, 10, e0135420. [Google Scholar] [CrossRef]
- Hu, X.; Chao, M.; Wu, H. Central role of lactate and proton in cancer cell resistance to glucose deprivation and its clinical translation. Signal Transduct. Target. Ther. 2017, 2, 16047. [Google Scholar] [CrossRef]
- Xie, J.; Wu, H.; Dai, C.; Pan, Q.; Ding, Z.; Hu, D.; Ji, B.; Luo, Y.; Hu, X. Beyond Warburg effect-dual metabolic nature of cancer cells. Sci. Rep. 2014, 4, 4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; White, K.A.; Barber, D.L. Intracellular pH Regulates Cancer and Stem Cell Behaviors: A Protein Dynamics Perspective. Front. Oncol. 2020, 10, 1401. [Google Scholar] [CrossRef] [PubMed]
- Reshkin, S.J.; Greco, M.R.; Cardone, R.A. Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordway, B.; Tomaszewski, M.; Byrne, S.; Abrahams, D.; Swietach, P.; Gillies, R.J.; Damaghi, M. Targeting of Evolutionarily Acquired Cancer Cell Phenotype by Exploiting pHi-Metabolic Vulnerabilities. Cancers 2020, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Man, C.H.; Mercier, F.E.; Liu, N.; Dong, W.; Stephanopoulos, G.; Jiang, L.; Jung, Y.; Lin, C.P.; Leung, A.Y.H.; Scadden, D.T. Proton export alkalinizes intracellular pH and reprograms carbon metabolism to drive normal and malignant cell growth. Blood 2022, 139, 502–522. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A., 3rd. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase: Pyruvate Dehydrogenase Axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Luo, B.; Li, X.; Lu, W.; Yang, J.; Hu, Y.; Huang, P.; Wen, S. Inhibition of cancer growth in vitro and in vivo by a novel ROS-modulating agent with ability to eliminate stem-like cancer cells. Cell Death Dis. 2017, 8, e2887. [Google Scholar] [CrossRef]
- Flinck, M.; Kramer, S.H.; Pedersen, S.F. Roles of pH in control of cell proliferation. Acta Physiol. 2018, 223, e13068. [Google Scholar] [CrossRef]
- Sergeeva, T.F.; Shirmanova, M.V.; Zlobovskaya, O.A.; Gavrina, A.I.; Dudenkova, V.V.; Lukina, M.M.; Lukyanov, K.A.; Zagaynova, E.V. Relationship between intracellular pH, metabolic co-factors and caspase-3 activation in cancer cells during apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 604–611. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, S.; Li, Y.; Liu, Z.; Mi, L.; Cai, Y.; Wang, X.; Chen, L.; Ran, H.; Xiao, D.; et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat. Commun. 2021, 12, 3720. [Google Scholar] [CrossRef]
- Chien, C.H.; Hsueh, W.T.; Chuang, J.Y.; Chang, K.Y. Dissecting the mechanism of temozolomide resistance and its association with the regulatory roles of intracellular reactive oxygen species in glioblastoma. J. Biomed. Sci. 2021, 28, 18. [Google Scholar] [CrossRef]
- Gupta, K.; Vuckovic, I.; Zhang, S.; Xiong, Y.; Carlson, B.L.; Jacobs, J.; Olson, I.; Petterson, X.M.; Macura, S.I.; Sarkaria, J.; et al. Radiation Induced Metabolic Alterations Associate with Tumor Aggressiveness and Poor Outcome in Glioblastoma. Front. Oncol. 2020, 10, 535. [Google Scholar] [CrossRef]
- Oliva, C.R.; Moellering, D.R.; Gillespie, G.Y.; Griguer, C.E. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS ONE 2011, 6, e24665. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.; Nelson, D.S.; Roman, J.; Putt, M.E.; Lee, S.C.; Leeper, D.B.; Glickson, J.D. Effect of Lonidamine on Systemic Therapy of DB-1 Human Melanoma Xenografts with Temozolomide. Anticancer Res. 2017, 37, 3413–3421. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.; Basit, F.; Swarts, H.G.; Forkink, M.; Oliveira, P.J.; Willems, P.; Koopman, W.J.H. Extracellular acidification induces ROS- and mPTP-mediated death in HEK293 cells. Redox Biol. 2018, 15, 394–404. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Ahmed, S.B.M.; Ahmed, A.; Elliott, R.L.; Ibrahim, M.E.; Ali, H.S.; Wales, C.C.; Nourwali, I.; Aljarbou, A.N.; Bashir, A.H.H.; et al. The Interplay of Dysregulated pH and Electrolyte Imbalance in Cancer. Cancers 2020, 12, 898. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Sex | Age | Molecular Subtype |
|---|---|---|---|
| GBM1A | M | - | Classical/Proneural |
| QNS120 | M | 59 | Classical |
| GBM612 | F | 56 | Proneural |
| GBM965 | F | 61 | Classical |
| QNS108 | M | 63 | Classical |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Gil, B.I.; Otamendi-Lopez, A.; Bechtle, A.; Vazquez-Ramos, C.A.; Qosja, N.; Suarez-Meade, P.; Sarabia-Estrada, R.; Jentoft, M.E.; Guerrero-Cázares, H.; Escames, G.; et al. Melatonin Treatment Triggers Metabolic and Intracellular pH Imbalance in Glioblastoma. Cells 2022, 11, 3467. https://doi.org/10.3390/cells11213467
Fernandez-Gil BI, Otamendi-Lopez A, Bechtle A, Vazquez-Ramos CA, Qosja N, Suarez-Meade P, Sarabia-Estrada R, Jentoft ME, Guerrero-Cázares H, Escames G, et al. Melatonin Treatment Triggers Metabolic and Intracellular pH Imbalance in Glioblastoma. Cells. 2022; 11(21):3467. https://doi.org/10.3390/cells11213467
Chicago/Turabian StyleFernandez-Gil, Beatriz I., Andrea Otamendi-Lopez, Alexandra Bechtle, Carla A. Vazquez-Ramos, Neda Qosja, Paola Suarez-Meade, Rachel Sarabia-Estrada, Mark E. Jentoft, Hugo Guerrero-Cázares, Germaine Escames, and et al. 2022. "Melatonin Treatment Triggers Metabolic and Intracellular pH Imbalance in Glioblastoma" Cells 11, no. 21: 3467. https://doi.org/10.3390/cells11213467