
Smurf1 Suppression Enhances Temozolomide Chemosensitivity in Glioblastoma by Facilitating PTEN Nuclear Translocation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfections
2.2. MTT Assay
2.3. Plasmids, si-RNA, and Transfections
2.4. Mice
2.5. Western Blotting
2.6. Immunofluorescence
2.7. Cytoplasmic and Nuclear Protein Extraction
2.8. Immunoprecipitation
3. Results
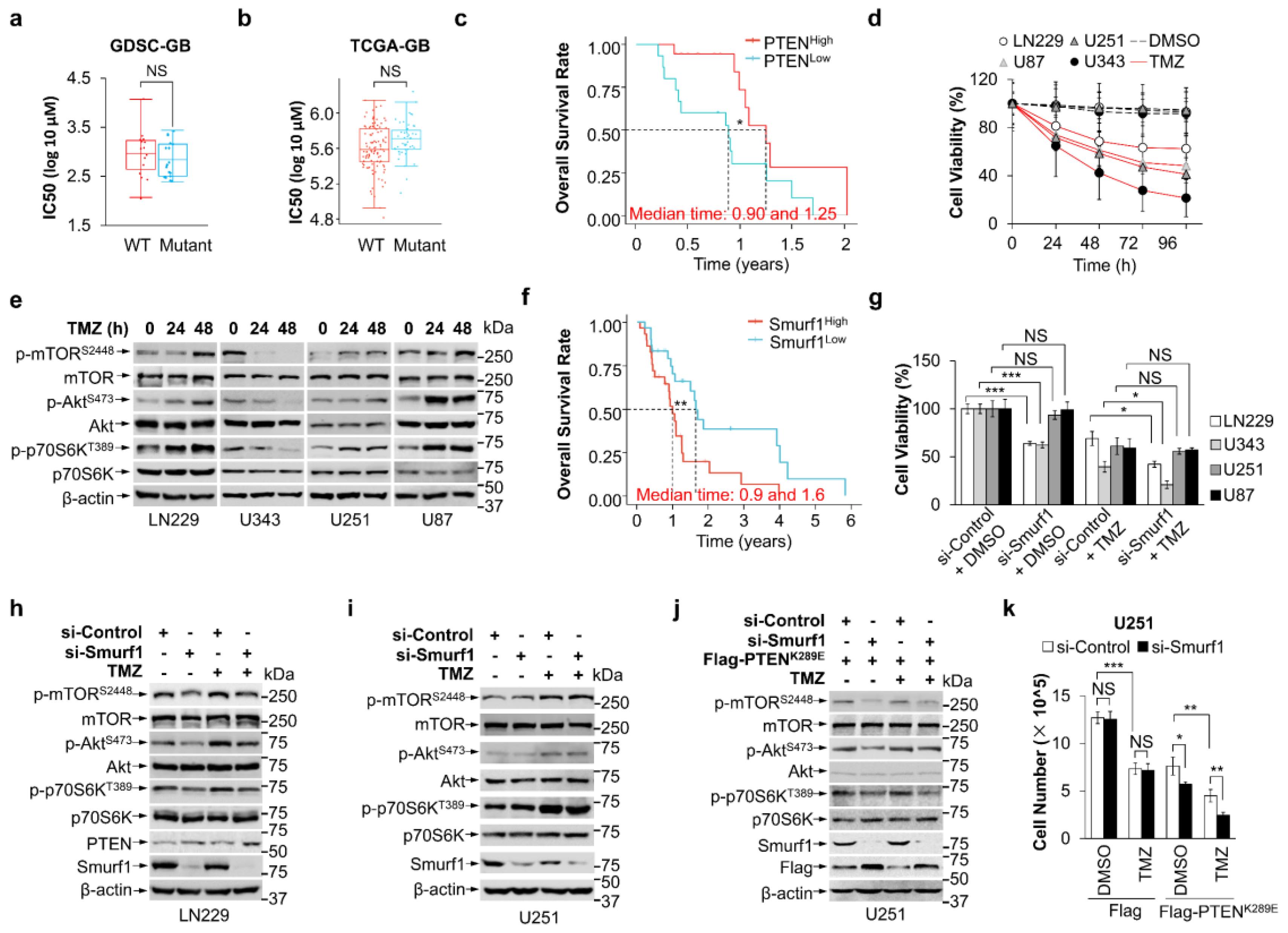
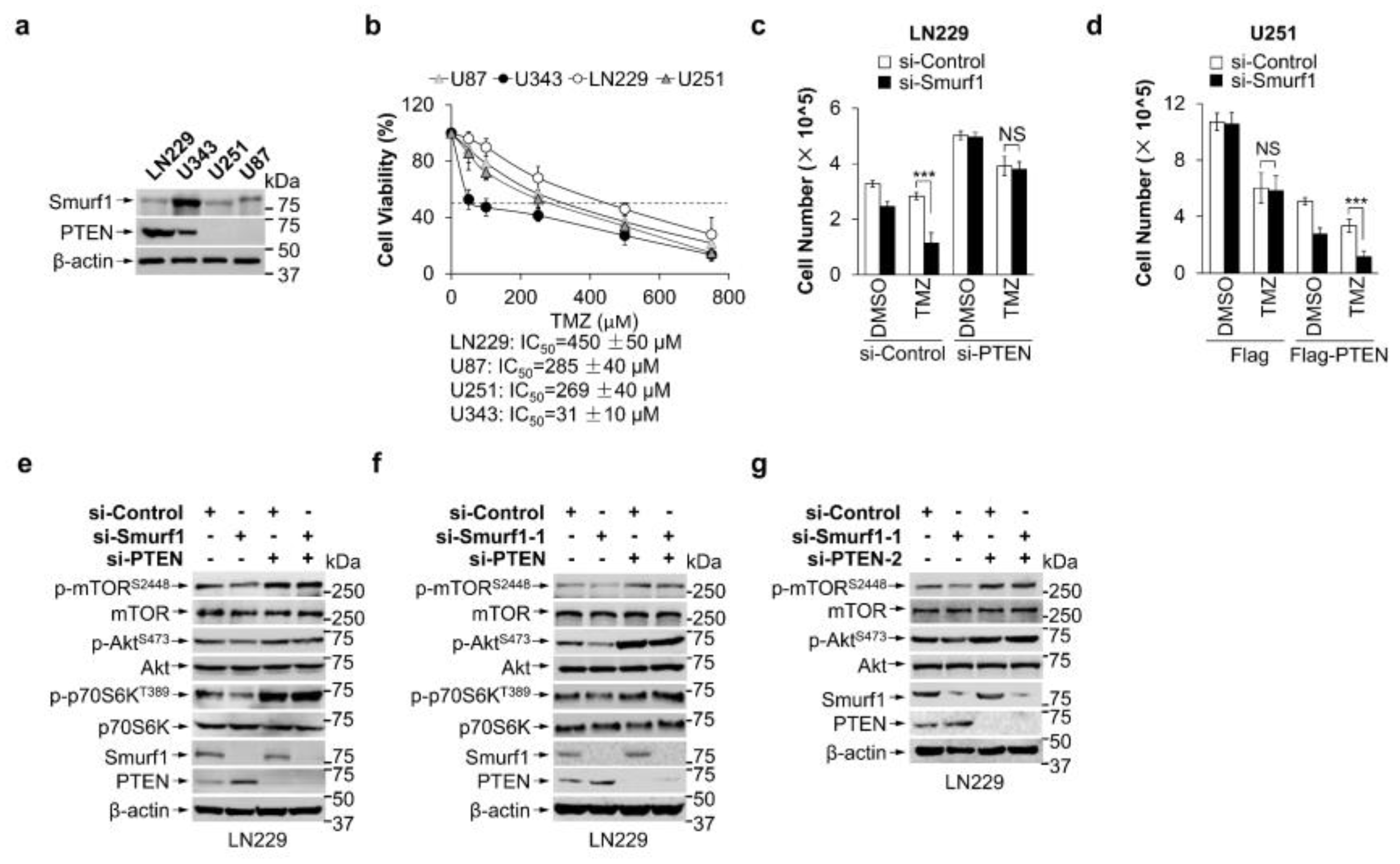
3.1. Combination of Smurf1 Suppression with TMZ Synthetically Inhibits PI3K/Akt Pathway
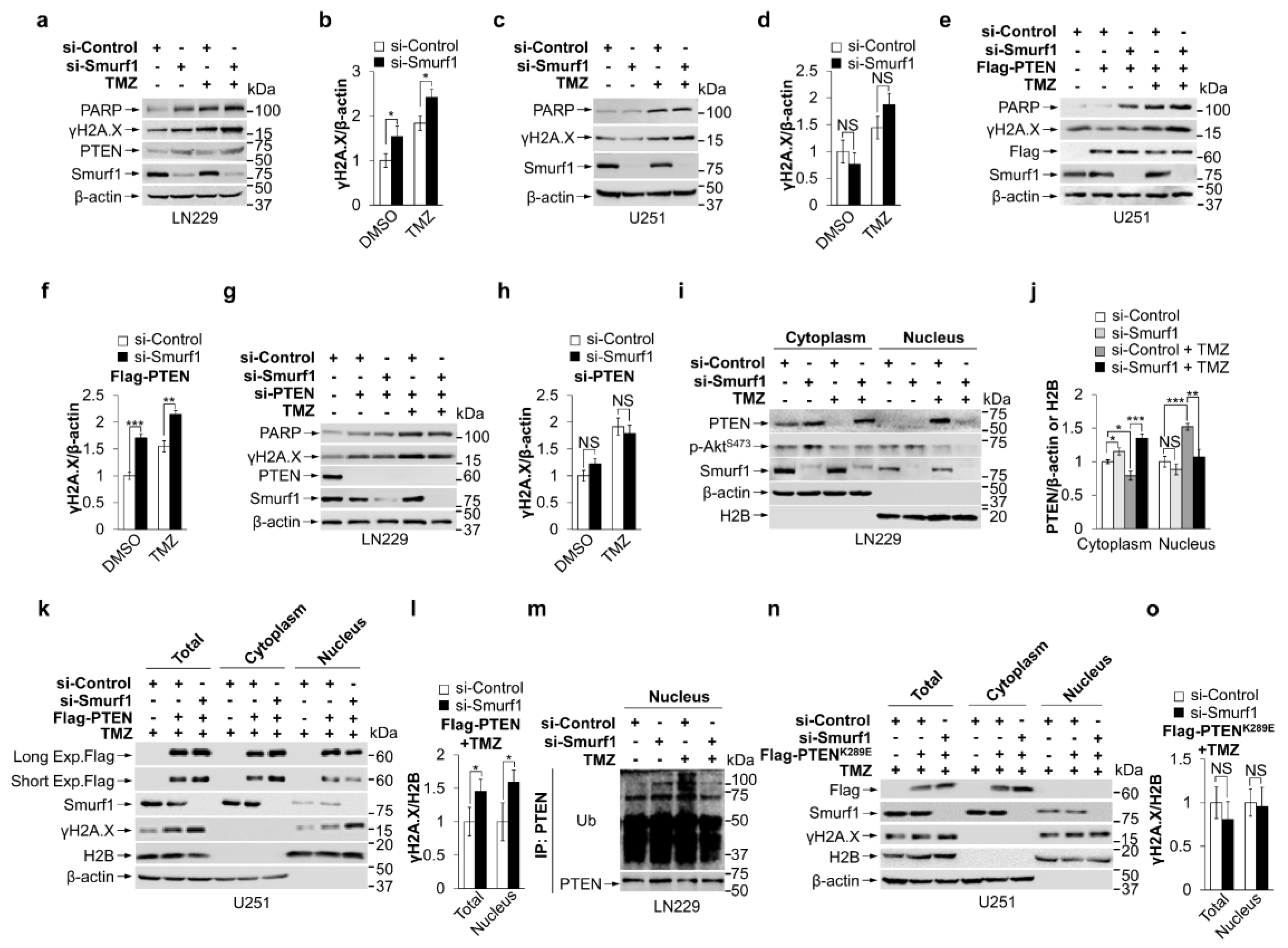
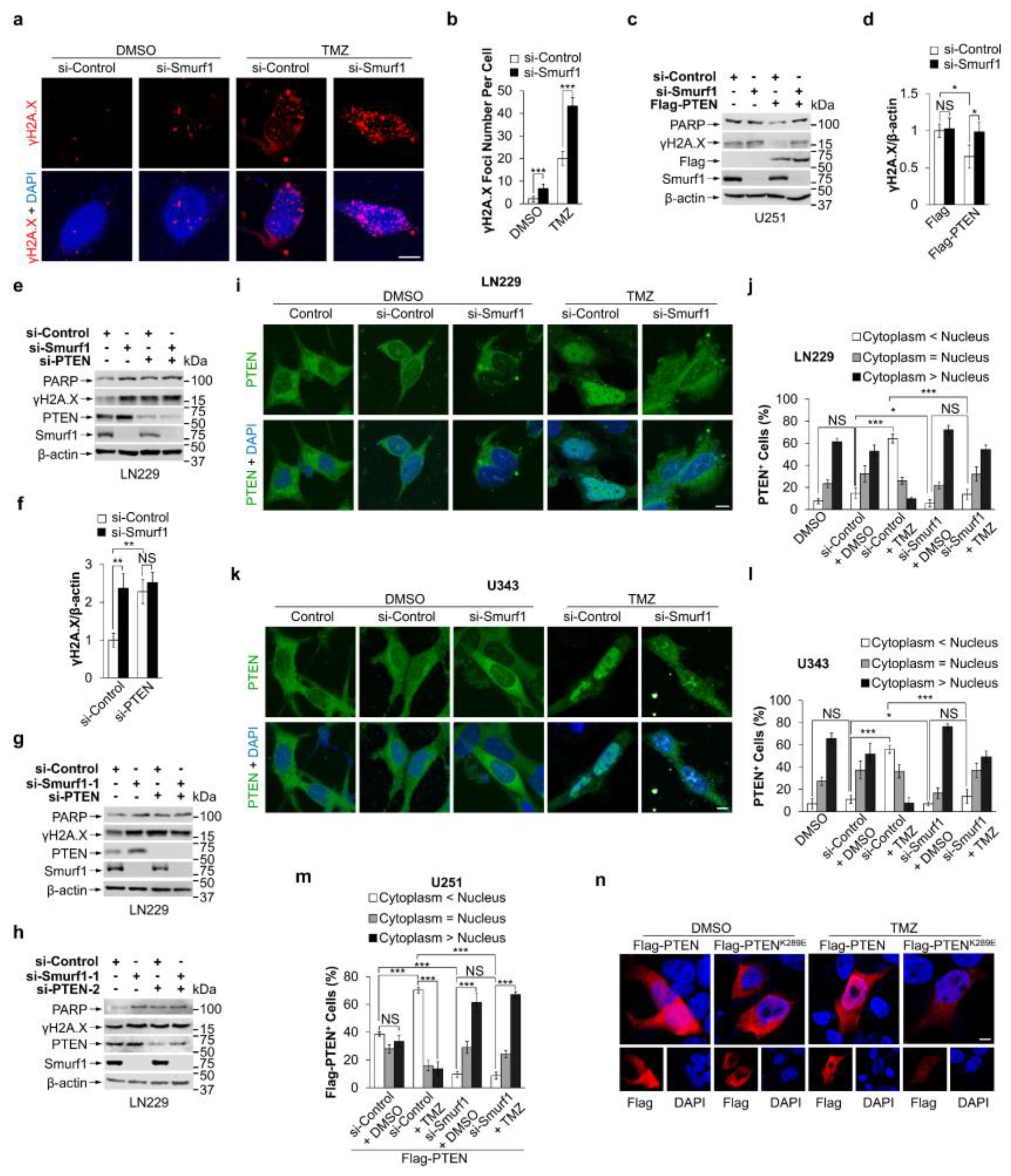
3.2. Smurf1 Facilitates PTEN Nuclear Translocation to Repair DNA Damage under TMZ Treatment
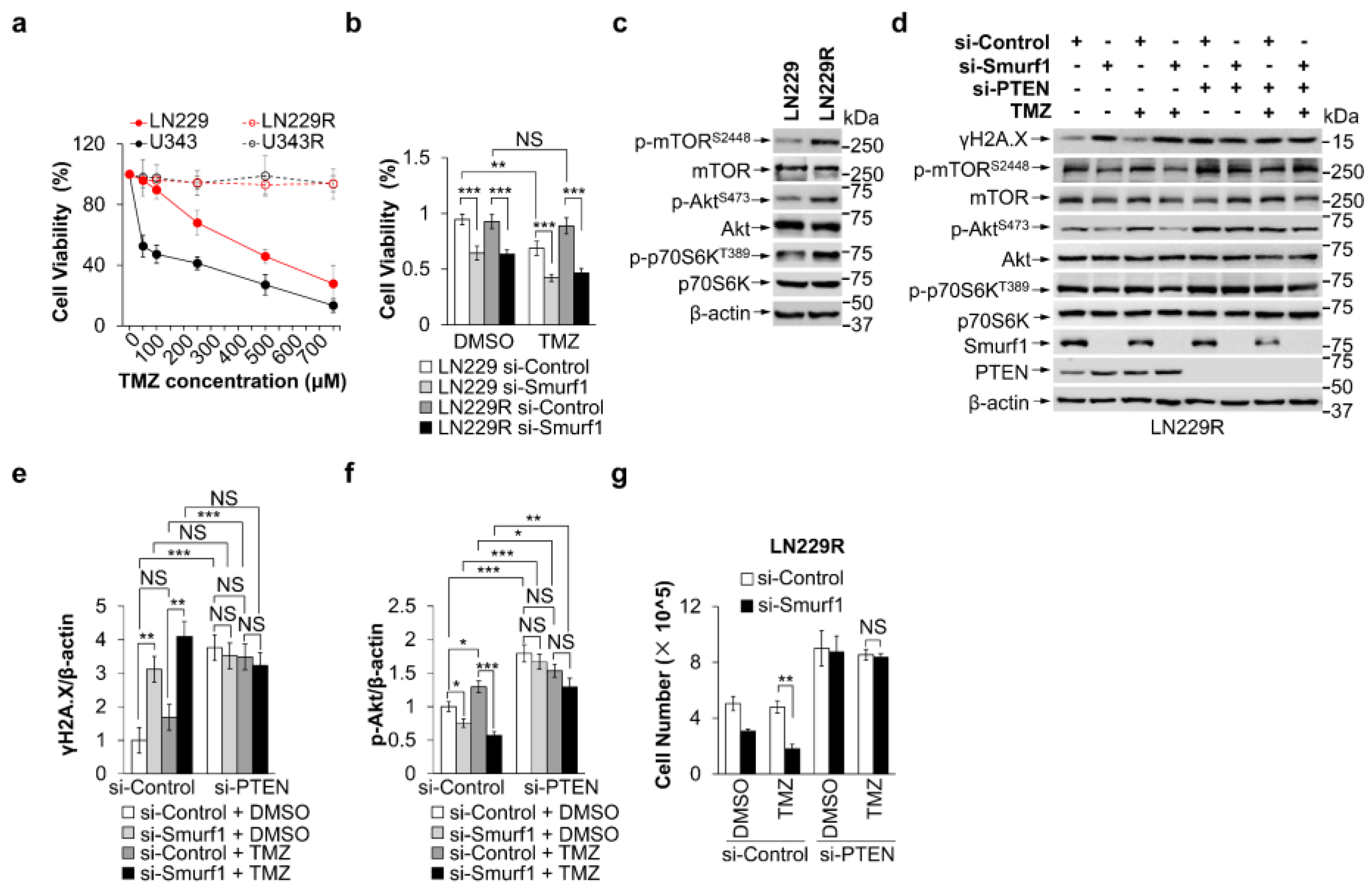
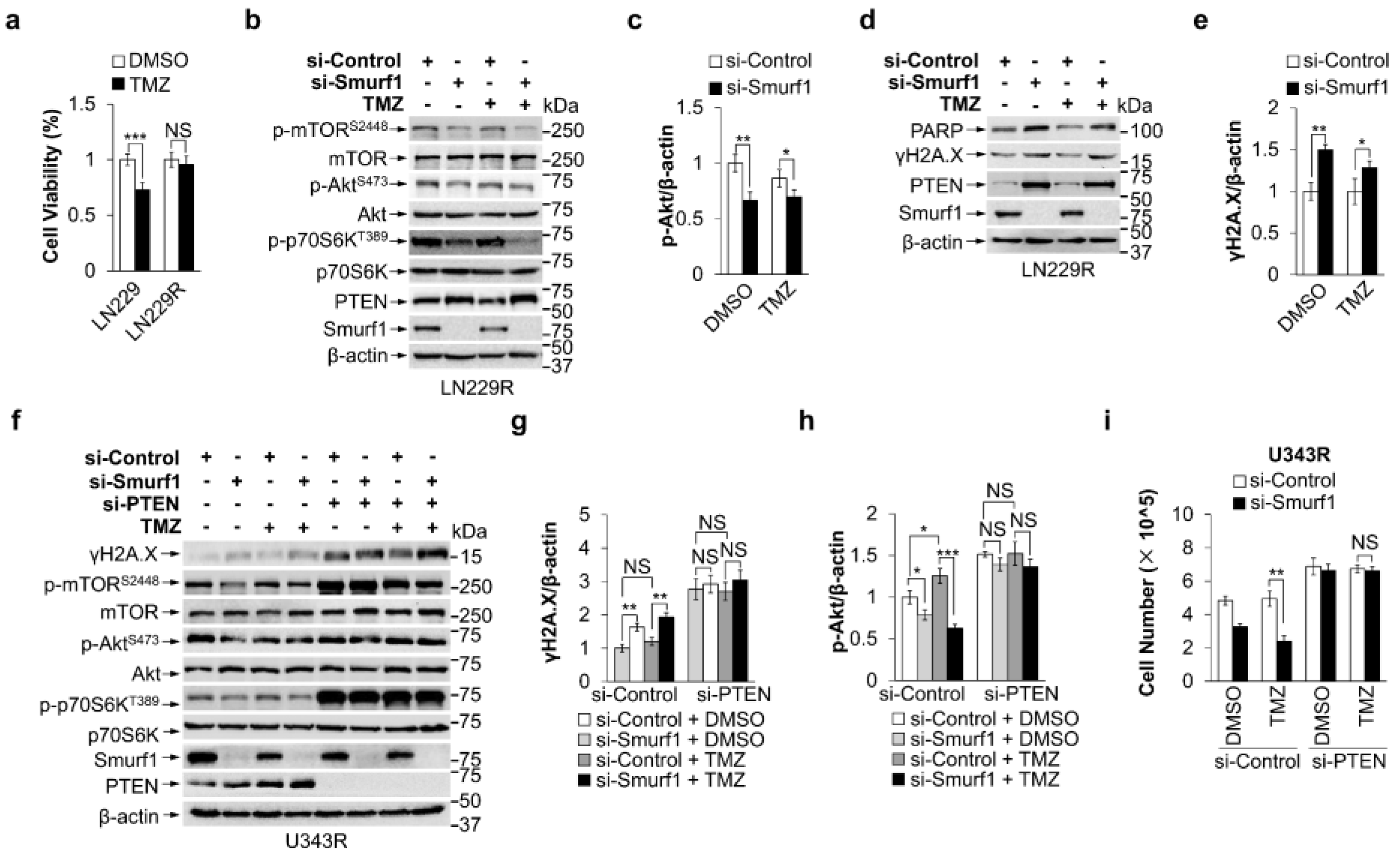
3.3. Smurf1 Suppression Restores the Sensitivity of TMZ-Resistant GB Cells
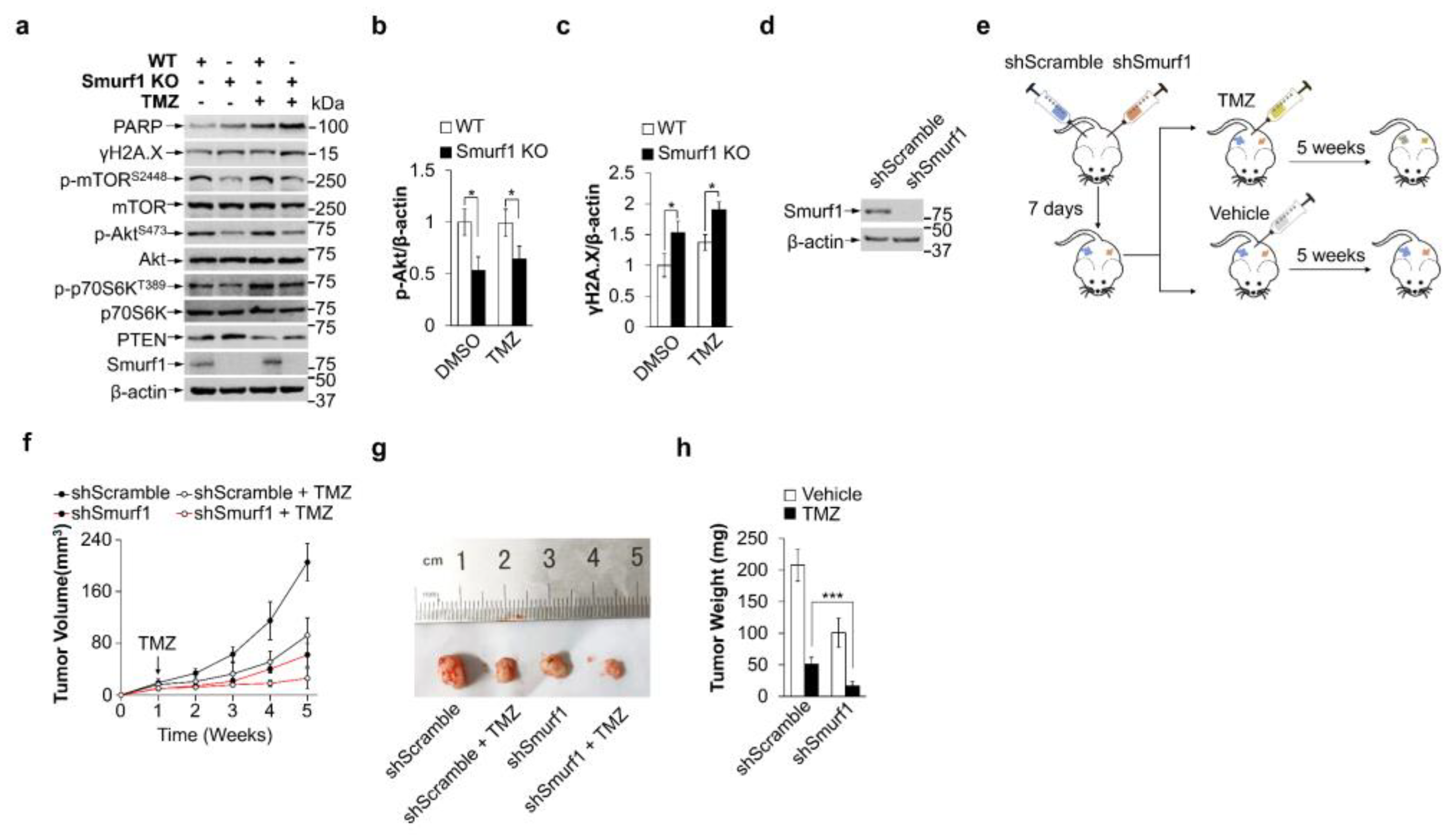
3.4. Smurf1 Knockout Restores TMZ Sensitivity of PTEN Wild-Type GB Cells In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lu, C.; Zhou, P.; Zhao, L.; Lyu, X.; Yin, J.; Shi, Z.; You, Y. EIF4A3-induced circular RNA ASAP1 promotes tumorigenesis and temozolomide resistance of glioblastoma via NRAS/MEK1/ERK1-2 signaling. Neuro Oncol. 2021, 23, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.H.; Liu, W.L.; Cao, H.; Wen, C.; Chen, L.; Jiang, G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013, 4, e876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Zhou, W.; Yang, M.; Ding, Y.; Wang, Q.; Hu, R. Fasudil increases temozolomide sensitivity and suppresses temozolomide-resistant glioma growth via inhibiting ROCK2/ABCG2. Cell Death Dis. 2018, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 2021, 23, 920–931. [Google Scholar] [CrossRef]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.; Kroon, P.; Zhang, Y.; Galán-Ganga, M.; Bao, Z.; Wang, Z.; Liu, H.; Sa, J.K.; et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 2020, 11, 3883. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Rabkin, S.D.; Martuza, R.L. Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J. Immunother. Cancer 2020, 8, e000345. [Google Scholar] [CrossRef]
- Tang, J.B.; Svilar, D.; Trivedi, R.N.; Wang, X.H.; Goellner, E.M.; Moore, B.; Hamilton, R.L.; Banze, L.A.; Brown, A.R.; Sobol, R.W. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol. 2011, 13, 471–486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, T.; Yang, M.; Du, Q.; Wang, R.; Fu, B.; Tan, Y.; Cao, M.; Chen, Y.; Wang, Q.; et al. Acquired temozolomide resistance in MGMT(low) gliomas is associated with regulation of homologous recombination repair by ROCK2. Cell Death Dis. 2022, 13, 138. [Google Scholar] [CrossRef]
- Vengoji, R.; Atri, P.; Macha, M.A.; Seshacharyulu, P.; Perumal, N.; Mallya, K.; Liu, Y.; Smith, L.M.; Rachagani, S.; Mahapatra, S.; et al. Differential gene expression-based connectivity mapping identified novel drug candidate and improved Temozolomide efficacy for Glioblastoma. J. Exp. Clin. Cancer Res. 2021, 40, 335. [Google Scholar] [CrossRef]
- Han, B.; Meng, X.; Wu, P.; Li, Z.; Li, S.; Zhang, Y.; Zha, C.; Ye, Q.; Jiang, C.; Cai, J.; et al. ATRX/EZH2 complex epigenetically regulates FADD/PARP1 axis, contributing to TMZ resistance in glioma. Theranostics 2020, 10, 3351–3365. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Prados, M.D.; Chang, S.M.; Butowski, N.; DeBoer, R.; Parvataneni, R.; Carliner, H.; Kabuubi, P.; Ayers-Ringler, J.; Rabbitt, J.; Page, M.; et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J. Clin. Oncol. 2009, 27, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Cominelli, M.; Grisanti, S.; Mazzoleni, S.; Branca, C.; Buttolo, L.; Furlan, D.; Liserre, B.; Bonetti, M.F.; Medicina, D.; Pellegrini, V.; et al. EGFR amplified and overexpressing glioblastomas and association with better response to adjuvant metronomic temozolomide. J. Natl. Cancer Inst. 2015, 107, djv041. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Ye, H.; Liu, X.J.; Hui, Y.; Liang, Y.H.; Li, C.H.; Wan, Q. Downregulation of MicroRNA-222 Reduces Insulin Resistance in Rats with PCOS by Inhibiting Activation of the MAPK/ERK Pathway via Pten. Mol. Ther. Nucleic Acids 2020, 22, 733–741. [Google Scholar] [CrossRef]
- Mukherjee, R.; Vanaja, K.G.; Boyer, J.A.; Gadal, S.; Solomon, H.; Chandarlapaty, S.; Levchenko, A.; Rosen, N. Regulation of PTEN translation by PI3K signaling maintains pathway homeostasis. Mol. Cell 2021, 81, 708–723.e705. [Google Scholar] [CrossRef]
- Bian, X.; Gao, J.; Luo, F.; Rui, C.; Zheng, T.; Wang, D.; Wang, Y.; Roberts, T.M.; Liu, P.; Zhao, J.J.; et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 2018, 37, 341–351. [Google Scholar] [CrossRef]
- Puc, J.; Keniry, M.; Li, H.S.; Pandita, T.K.; Choudhury, A.D.; Memeo, L.; Mansukhani, M.; Murty, V.V.; Gaciong, Z.; Meek, S.E.; et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 2005, 7, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Carracedo, A.; Salmena, L.; Song, S.J.; Egia, A.; Malumbres, M.; Pandolfi, P.P. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 2011, 144, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Salmena, L.; Carracedo, A.; Egia, A.; Lo-Coco, F.; Teruya-Feldstein, J.; Pandolfi, P.P. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008, 455, 813–817. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Liu, J.; Li, Y.L.; Li, J.P.; Zhang, R. Ubiquitination/de-ubiquitination: A promising therapeutic target for PTEN reactivation in cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188723. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Fouladkou, F.; Landry, T.; Kawabe, H.; Neeb, A.; Lu, C.; Brose, N.; Stambolic, V.; Rotin, D. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc. Natl. Acad. Sci. USA 2008, 105, 8585–8590. [Google Scholar] [CrossRef]
- Chen, J.H.; Zhang, P.; Chen, W.D.; Li, D.D.; Wu, X.Q.; Deng, R.; Jiao, L.; Li, X.; Ji, J.; Feng, G.K.; et al. ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 2015, 11, 239–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, P.; Peng, Z.; Chen, Y.; Li, H.; Du, M.; Tan, Y.; Zhang, X.; Lu, Z.; Cui, C.P.; Liu, C.H.; et al. Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res. 2021, 31, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013, 341, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Benitez, J.A.; Li, J.; Miki, S.; Ponte de Albuquerque, C.; Galatro, T.; Orellana, L.; Zanca, C.; Reed, R.; Boyer, A.; et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019, 35, 504–518.e507. [Google Scholar] [CrossRef] [Green Version]
- Fenton, T.R.; Nathanson, D.; Ponte de Albuquerque, C.; Kuga, D.; Iwanami, A.; Dang, J.; Yang, H.; Tanaka, K.; Oba-Shinjo, S.M.; Uno, M.; et al. Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240. Proc. Natl. Acad. Sci. USA 2012, 109, 14164–14169. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, D.; Liu, B.; Jin, X.; Wang, X.; Pan, J.; Tu, W.; Shao, Y. IMP3 accelerates the progression of prostate cancer through inhibiting PTEN expression in a SMURF1-dependent way. J. Exp. Clin. Cancer Res. CR 2020, 39, 190. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, Q.; Liu, W.; Zhu, Q.; Cui, C.P.; Xu, L.; Guo, X.; Wang, P.; Liu, J.; Dong, G.; et al. Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control tumor growth. Nat. Commun. 2018, 9, 2464. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Jia, Y.; Zhang, Y.; Ma, F.; Zhu, Y.; Hong, X.; Zhou, Q.; He, R.; Zhang, H.; Jin, J.; et al. Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 2019, 15, 1130–1149. [Google Scholar] [CrossRef]
- Yang, H.; Yu, N.; Xu, J.; Ding, X.; Deng, W.; Wu, G.; Li, X.; Hou, Y.; Liu, Z.; Zhao, Y.; et al. SMURF1 facilitates estrogen receptor ɑ signaling in breast cancer cells. J. Exp. Clin. Cancer Res. CR 2018, 37, 24. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Li, G.; Wen, C.; Zeng, T.; Fan, Y.; Liu, C.; Fu, G.F.; Xie, C.; Lin, Q.; Xie, L.; et al. Monoubiquitination of p120-catenin is essential for TGFβ-induced epithelial-mesenchymal transition and tumor metastasis. Sci. Adv. 2020, 6, eaay9819. [Google Scholar] [CrossRef]
- Wei, X.; Wang, X.; Zhan, J.; Chen, Y.; Fang, W.; Zhang, L.; Zhang, H. Smurf1 inhibits integrin activation by controlling Kindlin-2 ubiquitination and degradation. J. Cell Biol. 2017, 216, 1455–1471. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Li, W.; Ali, S.; Xu, M.; Li, Y.; Li, S.; Meng, X.; Liu, L.; Dong, L. Smurf1 silencing restores PTEN expression that ameliorates progression of human glioblastoma and sensitizes tumor cells to mTORC1/C2 inhibitor Torin1. iScience 2021, 24, 103528. [Google Scholar] [CrossRef]
- Xia, Q.; Zhang, H.; Zhang, P.; Li, Y.; Xu, M.; Li, X.; Li, X.; Dong, L. Oncogenic Smurf1 promotes PTEN wild-type glioblastoma growth by mediating PTEN ubiquitylation. Oncogene 2020, 39, 5902–5915. [Google Scholar] [CrossRef]
- Gaspar, N.; Marshall, L.; Perryman, L.; Bax, D.A.; Little, S.E.; Viana-Pereira, M.; Sharp, S.Y.; Vassal, G.; Pearson, A.D.; Reis, R.M.; et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010, 70, 9243–9252. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Chen, X.; Yan, Y.; Ren, X.; Wang, X.; Peng, B.; Cai, Y.; Liang, Q.; Xu, Z.; Peng, J. Aberrant Expression of ADARB1 Facilitates Temozolomide Chemoresistance and Immune Infiltration in Glioblastoma. Front. Pharm. 2022, 13, 768743. [Google Scholar] [CrossRef]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Lin, F.; de Gooijer, M.C.; Roig, E.M.; Buil, L.C.; Christner, S.M.; Beumer, J.H.; Würdinger, T.; Beijnen, J.H.; van Tellingen, O. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin. Cancer Res. 2014, 20, 2703–2713. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Paluch, B.E.; Wang, X.; Jiang, X. PTEN at a glance. J. Cell Sci. 2012, 125, 4687–4692. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef]
- Karlowee, V.; Amatya, V.J.; Hirano, H.; Takayasu, T.; Nosaka, R.; Kolakshyapati, M.; Yoshihiro, M.; Takeshima, Y.; Sugiyama, K.; Arita, K.; et al. Multicentric Glioma Develops via a Mutant IDH1-Independent Pathway: Immunohistochemical Study of Multicentric Glioma. Pathobiology 2017, 84, 99–107. [Google Scholar] [CrossRef]
- Dai, C.; Wu, B.; Chen, Y.; Li, X.; Bai, Y.; Du, Y.; Pang, Y.; Wang, Y.T.; Dong, Z. Aagab acts as a novel regulator of NEDD4-1-mediated Pten nuclear translocation to promote neurological recovery following hypoxic-ischemic brain damage. Cell Death Differ. 2021, 28, 2367–2384. [Google Scholar] [CrossRef]
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 2007, 128, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Yu, Z.; Xie, G.; Zhou, G.; Cheng, Y.; Zhang, G.; Yao, G.; Chen, Y.; Li, Y.; Zhao, G. NVP-BEZ235, a novel dual PI3K-mTOR inhibitor displays anti-glioma activity and reduces chemoresistance to temozolomide in human glioma cells. Cancer Lett. 2015, 367, 58–68. [Google Scholar] [CrossRef]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef]
- Brown, K.K.; Montaser-Kouhsari, L.; Beck, A.H.; Toker, A. MERIT40 Is an Akt Substrate that Promotes Resolution of DNA Damage Induced by Chemotherapy. Cell Rep. 2015, 11, 1358–1366. [Google Scholar] [CrossRef] [Green Version]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef]
- Hu, H.L.; Shiflett, L.A.; Kobayashi, M.; Chao, M.V.; Wilson, A.C.; Mohr, I.; Huang, T.T. TOP2β-Dependent Nuclear DNA Damage Shapes Extracellular Growth Factor Responses via Dynamic AKT Phosphorylation to Control Virus Latency. Mol. Cell 2019, 74, 466–480.e464. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Harding, S.M.; Zhao, H.; Coackley, C.; Durocher, D.; Bristow, R.G. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle 2011, 10, 2218–2232. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Kanai, F.; Stehn, J.; Xu, J.; Sarbassova, D.; Frangioni, J.V.; Dalal, S.N.; DeCaprio, J.A.; Greenberg, M.E.; Yaffe, M.B. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J. Cell Biol. 2002, 156, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Guo, C.; Xie, A.; Gao, D.; Guo, J.; Zhang, J.; Willis, N.; Su, A.; Asara, J.M.; et al. Akt-mediated phosphorylation of XLF impairs non-homologous end-joining DNA repair. Mol. Cell 2015, 57, 648–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alao, J.P.; Johansson-Sjölander, J.; Rallis, C.; Sunnerhagen, P. Caffeine Stabilises Fission Yeast Wee1 in a Rad24-Dependent Manner but Attenuates Its Expression in Response to DNA Damage. Microorganisms 2020, 8, 1512. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, A.J.; Golding, S.E.; Khalil, A.; Valerie, K. DNA double-strand break-induced pro-survival signaling. Radiother. Oncol. 2011, 101, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Drinjakovic, J.; Jung, H.; Campbell, D.S.; Strochlic, L.; Dwivedy, A.; Holt, C.E. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 2010, 65, 341–357. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Choi, S.; Wen, T.; Chen, C.; Thapa, N.; Lee, J.H.; Cryns, V.L.; Anderson, R.A. A p53-phosphoinositide signalosome regulates nuclear AKT activation. Nat. Cell Biol. 2022, 24, 1099–1113. [Google Scholar] [CrossRef]
- Wu, P.; Cai, J.; Chen, Q.; Han, B.; Meng, X.; Li, Y.; Li, Z.; Wang, R.; Lin, L.; Duan, C.; et al. Lnc-TALC promotes O(6)-methylguanine-DNA methyltransferase expression via regulating the c-Met pathway by competitively binding with miR-20b-3p. Nat. Commun. 2019, 10, 2045. [Google Scholar] [CrossRef]
- Huang, W.; Zhong, Z.; Luo, C.; Xiao, Y.; Li, L.; Zhang, X.; Yang, L.; Xiao, K.; Ning, Y.; Chen, L.; et al. The miR-26a/AP-2α/Nanog signaling axis mediates stem cell self-renewal and temozolomide resistance in glioma. Theranostics 2019, 9, 5497–5516. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, L.; Li, Y.; Liu, L.; Meng, X.; Li, S.; Han, D.; Xiao, Z.; Xia, Q. Smurf1 Suppression Enhances Temozolomide Chemosensitivity in Glioblastoma by Facilitating PTEN Nuclear Translocation. Cells 2022, 11, 3302. https://doi.org/10.3390/cells11203302
Dong L, Li Y, Liu L, Meng X, Li S, Han D, Xiao Z, Xia Q. Smurf1 Suppression Enhances Temozolomide Chemosensitivity in Glioblastoma by Facilitating PTEN Nuclear Translocation. Cells. 2022; 11(20):3302. https://doi.org/10.3390/cells11203302
Chicago/Turabian StyleDong, Lei, Yang Li, Liqun Liu, Xinyi Meng, Shengzhen Li, Da Han, Zhenyu Xiao, and Qin Xia. 2022. "Smurf1 Suppression Enhances Temozolomide Chemosensitivity in Glioblastoma by Facilitating PTEN Nuclear Translocation" Cells 11, no. 20: 3302. https://doi.org/10.3390/cells11203302