


MiR-22 Deficiency Fosters Hepatocellular Carcinoma Development in Fatty Liver

 ,
,  ,
,
Abstract
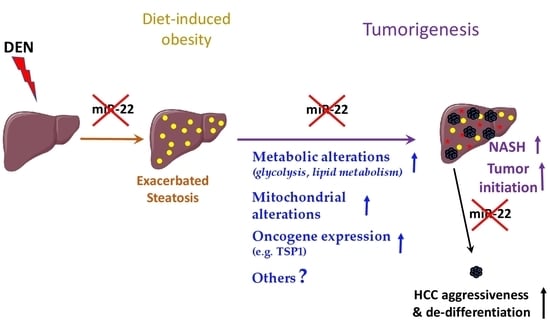
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mouse Studies
2.1.1. Mouse Generation and Housing
2.1.2. Hepatic Tumor Induction
2.1.3. Diet-Induced Steatosis and Obesity in DEN-Treated Mice
2.1.4. Computer Tomography (CT) Analysis
2.1.5. Glucose Tolerance Test
2.1.6. Plasma Analysis
2.2. Cell Culture
2.2.1. Cell Maintenance
2.2.2. Cell Transfection
2.2.3. Cell Migration Assay (Boyden Chamber Assay)
2.2.4. Cell Proliferation
2.2.5. Luciferase-Based Gene Reporter Assay
2.3. Mitochondrial Respiration and Glycolysis Analyses
2.3.1. Long-Chain Fatty acid Oxidation Stress Test
2.3.2. Glycolytic Rate Assay (GlycoRate)
2.4. RT-qPCR Analysis
2.5. Bioinformatic Analysis
2.5.1. miR-22 Expression in GEO Datasets
2.5.2. Survival Analysis
2.5.3. HCC Segregation by Main Mutations
2.5.4. Biological Process Enrichment Analysis
2.5.5. HCC-Associated Target Genes of miR-22
2.6. Statistical Analysis
3. Results
3.1. miR-22 Expression Is Downregulated in Hepatocellular Carcinoma (HCC)
3.2. miR-22 Deficiency Fosters Tumor Development In Vivo
3.3. miR-22 Loss Promotes Histopathological Features of NASH and the Development of More Undifferentiated Tumoral Nodules
3.4. miR-22 Regulates Hepatic Metabolism through Multiple Mechanisms
3.5. Loss of miR-22 Increases the Expression of Key Target Oncogenes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R. Descriptive epidemiology of incidence and mortality of primary liver cancer in 185 countries: Evidence from GLOBOCAN 2018. Jpn. J. Clin. Oncol. 2020, 50, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Dufour, J.-F. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Hepatic Oncol. 2017, 4, 83–98. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Karagozian, R.; Derdák, Z.; Baffy, G. Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism 2014, 63, 607–617. [Google Scholar] [CrossRef]
- Wegermann, K.; Hyun, J.; Diehl, A.M. Molecular Mechanisms Linking Nonalcoholic Steatohepatitis to Cancer. Clin. Liver Dis. 2021, 17, 6–10. [Google Scholar] [CrossRef]
- Lu, Q.; Tian, X.; Wu, H.; Huang, J.; Li, M.; Mei, Z.; Zhou, L.; Xie, H.; Zheng, S. Metabolic Changes of Hepatocytes in NAFLD. Front. Physiol. 2021, 12, 710420. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Peyrou, M.; Bourgoin, L.; Foti, M. PTEN in liver diseases and cancer. World J. Gastroenterol. 2010, 16, 4627–4633. [Google Scholar] [CrossRef]
- Kwabi-Addo, B.; Giri, D.; Schmidt, K.; Podsypanina, K.; Parsons, R.; Greenberg, N.; Ittmann, M. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc. Natl. Acad. Sci. USA 2001, 98, 11563–11568. [Google Scholar] [CrossRef] [Green Version]
- Vinciguerra, M.; Carrozzino, F.; Peyrou, M.; Carlone, S.; Montesano, R.; Benelli, R.; Foti, M. Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN. J. Hepatol. 2009, 50, 1132–1141. [Google Scholar] [CrossRef]
- Peyrou, M.; Bourgoin, L.; Foti, M. PTEN in Non-Alcoholic Fatty Liver Disease/Non-Alcoholic Steatohepatitis and Cancer. Dig. Dis. 2010, 28, 236–246. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Friso, S.; Choi, S.-W. Epigenetics in non-alcoholic fatty liver disease. Mol. Asp. Med. 2017, 54, 78–88. [Google Scholar] [CrossRef]
- Xu, F.; Guo, W. The progress of epigenetics in the development and progression of non-alcoholic fatty liver disease. Liver Res. 2020, 4, 118–123. [Google Scholar] [CrossRef]
- López-Sánchez, G.N.; Dóminguez-Pérez, M.; Uribe, M.; Chávez-Tapia, N.C.; Nuño-Lámbarri, N. Non-alcoholic fatty liver disease and microRNAs expression, how it affects the development and progression of the disease. Ann. Hepatol. 2021, 21, 100212. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; De Sousa, M.C.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef]
- Tessitore, A.; Cicciarelli, G.; Del Vecchio, F.; Gaggiano, A.; Verzella, D.; Fischietti, M.; Mastroiaco, V.; Vetuschi, A.; Sferra, R.; Barnabei, R.; et al. MicroRNA expression analysis in high fat diet-induced NAFLD-NASH-HCC progression: Study on C57BL/6J mice. BMC Cancer 2016, 16, 3. [Google Scholar] [CrossRef]
- Dolicka, D.; Sobolewski, C.; De Sousa, M.C.; Gjorgjieva, M.; Foti, M. mRNA Post-Transcriptional Regulation by AU-Rich Element-Binding Proteins in Liver Inflammation and Cancer. Int. J. Mol. Sci. 2020, 21, 6648. [Google Scholar] [CrossRef]
- Zhang, K.; Barry, A.E.; Lamm, R.; Patel, K.; Schafer, M.; Dang, H. The role of RNA binding proteins in hepatocellular carcinoma. Adv. Drug Deliv. Rev. 2022, 182, 114114. [Google Scholar] [CrossRef]
- Xiao, Z.; Shen, J.; Zhang, L.; Li, M.; Hu, W.; Cho, C. Therapeutic targeting of noncoding RNAs in hepatocellular carcinoma: Recent progress and future prospects (Review). Oncol. Lett. 2018, 15, 3395–3402. [Google Scholar] [CrossRef] [Green Version]
- Roy, B.; Ghose, S.; Biswas, S. Therapeutic strategies for miRNA delivery to reduce hepatocellular carcinoma. Semin. Cell Dev. Biol. 2022, 124, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Van Der Ree, M.H.; Van Der Meer, A.J.; De Bruijne, J.; Maan, R.; Van Vliet, A.; Welzel, T.M.; Zeuzem, S.; Lawitz, E.J.; Rodriguez-Torres, M.; Kupcova, V. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antivir. Res. 2014, 111, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Sobolewski, C.; Ay, A.-S.; Abegg, D.; De Sousa, M.C.; Portius, D.; Berthou, F.; Fournier, M.; Maeder, C.; Rantakari, P.; et al. Genetic Ablation of MiR-22 Fosters Diet-Induced Obesity and NAFLD Development. J. Pers. Med. 2020, 10, 170. [Google Scholar] [CrossRef]
- Wang, G.; Dong, F.; Xu, Z.; Sharma, S.; Hu, X.; Chen, D.; Zhang, L.; Zhang, J.; Dong, Q. MicroRNA profile in HBV-induced infection and hepatocellular carcinoma. BMC Cancer 2017, 17, 805. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.-D.; Yang, J.; Lei, X.-F.; Mi, G.-L.; Li, S.-L.; Li, K.; Xu, C.-Q.; Yang, H.-L. Expression of microRNA-122 and microRNA-22 in HBV-related liver cancer and the correlation with clinical features. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 742–747. [Google Scholar]
- Luo, L.-J.; Zhang, L.-P.; Duan, C.-Y.; Wang, B.; He, N.-N.; Abulimiti, P.; Lin, Y. The inhibition role of miR-22 in hepatocellular carcinoma cell migration and invasion via targeting CD147. Cancer Cell Int. 2017, 17, 17. [Google Scholar] [CrossRef]
- Chen, M.; Hu, W.; Xiong, C.-L.; Qu, Z.; Yin, C.-Q.; Wang, Y.-H.; Luo, C.-L.; Guan, Q.; Yuan, C.-H.; Wang, F.-B. miR-22 targets YWHAZ to inhibit metastasis of hepatocellular carcinoma and its down-regulation predicts a poor survival. Oncotarget 2016, 7, 80751–80764. [Google Scholar] [CrossRef]
- Zeng, Z.; Dong, J.; Li, Y.; Dong, Z.; Liu, Z.; Huang, J.; Wang, Y.; Zhen, Y.; Lu, Y. The expression level and diagnostic value of microRNA-22 in HCC patients. Artif. Cells Nanomed. Biotechnol. 2020, 48, 683–686. [Google Scholar] [CrossRef]
- Zekri, A.-R.N.; El-Sisi, E.R.; Youssef, A.S.E.-D.; Kamel, M.M.; Nassar, A.; Ahmed, O.S.; El Kassas, M.; Barakat, A.B.; El-Motaleb, A.I.A.; Bahnassy, A.A. MicroRNA Signatures for circulating CD133-positive cells in hepatocellular carcinoma with HCV infection. PLoS ONE 2018, 13, e0193709. [Google Scholar] [CrossRef]
- You, Y.; Tan, J.-X.; Dai, H.-S.; Chen, H.-W.; Xu, X.-J.; Yang, A.-G.; Zhang, Y.-J.; Bai, L.-H.; Bie, P. MiRNA-22 inhibits oncogene galectin-1 in hepatocellular carcinoma. Oncotarget 2016, 7, 57099–57116. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; He, J.; Zhang, Y. MicroRNA-22 expression in hepatocellular carcinoma and its correlation with ezrin protein. J. Int. Med. Res. 2013, 41, 1009–1016. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Yang, T.; Liu, Y.; Li, A.; Fu, S.; Wu, M.; Pan, Z.; Zhou, W. microRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br. J. Cancer 2010, 103, 1215–1220. [Google Scholar] [CrossRef]
- Chen, J.; Wu, F.-X.; Luo, H.-L.; Liu, J.-J.; Luo, T.; Bai, T.; Li, L.-Q.; Fan, X.-H. Berberine upregulates miR-22-3p to suppress hepatocellular carcinoma cell proliferation by targeting Sp1. Am. J. Transl. Res. 2016, 8, 4932–4941. [Google Scholar]
- Budd, W.T.; Seashols-Williams, S.; Clark, G.C.; Weaver, D.; Calvert, V.; Petricoin, E.; Dragoescu, E.A.; O’Hanlon, K.; Zehner, Z.E. Dual Action of miR-125b As a Tumor Suppressor and OncomiR-22 Promotes Prostate Cancer Tumorigenesis. PLoS ONE 2015, 10, e0142373. [Google Scholar] [CrossRef]
- Jiang, R.; Deng, L.; Zhao, L.; Li, X.; Zhang, F.; Xia, Y.; Gao, Y.; Wang, X.; Sun, B. miR-22 Promotes HBV-Related Hepatocellular Carcinoma Development in Males. Clin. Cancer Res. 2011, 17, 5593–5603. [Google Scholar] [CrossRef]
- Palacios, F.; De Abreu, C.N.; Prieto, D.; Morande, P.E.; Ruiz, S.; Fernandezcalero, T.; Naya, H.; Libisch, G.; Robello, C.; Landoni, A.I.; et al. Activation of the PI3K/AKT pathway by microRNA-22 results in CLL B-cell proliferation. Leukemia 2014, 29, 115–125. [Google Scholar] [CrossRef]
- Zhang, D.-Y.; Zou, X.-J.; Cao, C.-H.; Zhang, T.; Lei, L.; Qi, X.-L.; Liu, L.; Wu, D.-H. Identification and Functional Characterization of Long Non-coding RNA MIR22HG as a Tumor Suppressor for Hepatocellular Carcinoma. Theranostics 2018, 8, 3751–3765. [Google Scholar] [CrossRef]
- Zhang, L.; Li, C.; Su, X. Emerging impact of the long noncoding RNA MIR22HG on proliferation and apoptosis in multiple human cancers. J. Exp. Clin. Cancer Res. 2020, 39, 271. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Goossens, N.; Sun, X.; Hoshida, Y. Molecular classification of hepatocellular carcinoma: Potential therapeutic implications. Hepatic Oncol. 2015, 2, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Österreicher, C.H.; Takahashi, H.; Karin, M. Dietary and Genetic Obesity Promote Liver Inflammation and Tumorigenesis by Enhancing IL-6 and TNF Expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, A.; Mastroiaco, V.; Vetuschi, A.; Sferra, R.; Pompili, S.; Cicciarelli, G.; Barnabei, R.; Capece, D.; Zazzeroni, F.; Capalbo, C.; et al. Development of hepatocellular cancer induced by long term low fat-high carbohydrate diet in a NAFLD/NASH mouse model. Oncotarget 2017, 8, 53482–53494. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, O.A.; Akinloye, O.; Adaramoye, O.A. Cerium Oxide Nanoparticles Attenuate Oxidative Stress and Inflammation in the Liver of Diethylnitrosamine-Treated Mice. Biol. Trace Elem. Res. 2020, 193, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Velu, P.; Vijayalakshmi, A.; Iyappan, P.; Indumathi, D. Evaluation of antioxidant and stabilizing lipid peroxidation nature of Solanum xanthocarpum leaves in experimentally diethylnitrosamine induced hepatocellular carcinogenesis. Biomed. Pharmacother. 2016, 84, 430–437. [Google Scholar] [CrossRef]
- Al-Rejaie, S.S.; Aleisa, A.M.; Al-Yahya, A.A.; Bakheet, S.A.; Alsheikh, A.; Fatani, A.G.; Al-Shabanah, O.A.; Sayed-Ahmed, M.M. Progression of diethylnitrosamine-induced hepatic carcinogenesis in carnitine-depleted rats. World J. Gastroenterol. 2009, 15, 1373–1380. [Google Scholar] [CrossRef]
- Sánchez-Pérez, Y.; Carrasco-Legleu, C.; García-Cuellar, C.; Pérez-Carreón, J.; Hernández-García, S.; Salcido-Neyoy, M.; Alemán-Lazarini, L.; Villa-Treviño, S. Oxidative stress in carcinogenesis. Correlation between lipid peroxidation and induction of preneoplastic lesions in rat hepatocarcinogenesis. Cancer Lett. 2005, 217, 25–32. [Google Scholar] [CrossRef]
- Li, J.R.; Malhotra, A.; Bi, J. Potential of Ablation Therapy during Hepatocellular Carcinoma. Nutr. Cancer 2019, 71, 881–885. [Google Scholar] [CrossRef]
- McGill, M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar] [CrossRef]
- Liu, R.; Pan, X.; Whitington, P.F. Increased hepatic expression is a major determinant of serum alanine aminotransferase elevation in mice with nonalcoholic steatohepatitis. Liver Int. 2009, 29, 337–343. [Google Scholar] [CrossRef]
- Zhu, W.; Li, H.; Yu, Y.; Chen, J.; Chen, X.; Ren, F.; Ren, Z.; Cui, G. Enolase-1 serves as a biomarker of diagnosis and prognosis in hepatocellular carcinoma patients. Cancer Manag. Res. 2018, 10, 5735–5745. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, C.; Lee, S.; Kim, W.; Klevstig, M.; Harzandi, A.M.; Sikanic, N.; Arif, M.; Ståhlman, M.; Nielsen, J.; et al. Pyruvate kinase L/R is a regulator of lipid metabolism and mitochondrial function. Metab. Eng. 2019, 52, 263–272. [Google Scholar] [CrossRef]
- Liu, W.; Liu, X.; Liu, Y.; Ling, T.; Chen, D.; Otkur, W.; Zhao, H.; Ma, M.; Ma, K.; Dong, B.; et al. PLIN2 promotes HCC cells proliferation by inhibiting the degradation of HIF1α. Exp. Cell Res. 2022, 418, 113244. [Google Scholar] [CrossRef]
- Wang, M.; Liu, X.; Lin, S.; Tian, T.; Guan, F.; Guo, Y.; Li, X.; Deng, Y.; Zheng, Y.; Xu, P.; et al. FABP1 Polymorphisms Contribute to Hepatocellular Carcinoma Susceptibility in Chinese Population with Liver Cirrhosis: A Case-Control Study. J. Cancer 2018, 9, 4294–4300. [Google Scholar] [CrossRef]
- Leung, Z.; Ko, F.C.F.; Tey, S.K.; Kwong, E.M.L.; Mao, X.; Liu, B.H.M.; Ma, A.P.Y.; Fung, Y.M.E.; Che, C.-M.; Wong, D.K.H.; et al. Galectin-1 promotes hepatocellular carcinoma and the combined therapeutic effect of OTX008 galectin-1 inhibitor and sorafenib in tumor cells. J. Exp. Clin. Cancer Res. 2019, 38, 423. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Chen, X.; Yang, Z. Polymeric immunoglobulin receptor (PIGR) exerts oncogenic functions via activating ribosome pathway in hepatocellular carcinoma. Int. J. Med. Sci. 2021, 18, 364–371. [Google Scholar] [CrossRef]
- Kang, H.J.; Jung, S.K.; Kim, S.J.; Chung, S.J. Structure of human alpha-enolase (hENO1), a multifunctional glycolytic enzyme. Acta Crystallogr. D Biol. Crystallogr. 2008, 64 Pt 6, 651–657. [Google Scholar] [CrossRef]
- Aisaki, K.; Aizawa, S.; Fujii, H.; Kanno, J.; Kanno, H. Glycolytic inhibition by mutation of pyruvate kinase gene increases oxidative stress and causes apoptosis of a pyruvate kinase deficient cell line. Exp. Hematol. 2007, 35, 1190–1200. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef]
- Orlicky, D.J.; Libby, A.E.; Bales, E.S.; Mcmahan, R.H.; Monks, J.; La Rosa, F.G.; McManaman, J.L. Perilipin-2 promotes obesity and progressive fatty liver disease in mice through mechanistically distinct hepatocyte and extra-hepatocyte actions. J. Physiol. 2019, 597, 1565–1584. [Google Scholar] [CrossRef]
- Morishita, A.; Oura, K.; Tadokoro, T.; Fujita, K.; Tani, J.; Masaki, T. MicroRNAs in the Pathogenesis of Hepatocellular Carcinoma: A Review. Cancers 2021, 13, 514. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Yagi, S.; Ito, T.; Lowenstein, C.J. MicroRNA-22 Regulates Hypoxia Signaling in Colon Cancer Cells. PLoS ONE 2011, 6, e20291. [Google Scholar] [CrossRef]
- Ali, M.I.; Li, L.; Li, L.; Yao, L.; Liu, J.; Gu, W.; Huang, S.; Wang, B.; Liu, G. The tissue specific regulation of miR22 expression in the lung and brain by ribosomal protein L29. Sci. Rep. 2020, 10, 16242. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.-S.; Mugiyanto, E.; Chang, W.-C.; Wan, Y.-J.Y. MiR-22 as a metabolic silencer and liver tumor suppressor. Liver Res. 2020, 4, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, H.M.; Muiwo, P.; Muthuswami, R.; Bhattacharya, A. FosB regulates expression of miR-22 during PMA induced differentiation of K562 cells to megakaryocytes. Biochimie 2017, 133, 1–6. [Google Scholar] [CrossRef]
- Gu, W.; Zhan, H.; Zhou, X.Y.; Yao, L.; Yan, M.; Chen, A.; Liu, J.; Ren, X.; Zhang, X.; Liu, J.X.; et al. MicroRNA-22 regulates inflammation and angiogenesis via targeting VE-cadherin. FEBS Lett. 2017, 591, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Ashraf, U.; Ye, J.; Duan, X.; Zohaib, A.; Wang, W.; Chen, Z.; Zhu, B.; Li, Y.; Chen, H.; et al. MicroRNA-22 negatively regulates poly(I:C)-triggered type I interferon and inflammatory cytokine production via targeting mitochondrial antiviral signaling protein (MAVS). Oncotarget 2016, 7, 76667–76683. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Liu, Q. Catalpol inhibits cell proliferation, invasion and migration through regulating miR-22-3p/MTA3 signalling in hepatocellular carcinoma. Exp. Mol. Pathol. 2019, 109, 51–60. [Google Scholar] [CrossRef]
- Yang, F.; Hu, Y.; Liu, H.-X.; Wan, Y.-J.Y. MiR-22-silenced Cyclin A Expression in Colon and Liver Cancer Cells is Regulated by Bile Acid Receptor. J. Biol. Chem. 2015, 290, 6507–6515. [Google Scholar] [CrossRef]
- Pant, K.; Yadav, A.K.; Gupta, P.; Islam, R.; Saraya, A.; Venugopal, S.K. Butyrate induces ROS-mediated apoptosis by modulating miR-22/SIRT-1 pathway in hepatic cancer cells. Redox Biol. 2017, 12, 340–349. [Google Scholar] [CrossRef]
- Xu, D.; Guo, Y.; Liu, T.; Li, S.; Sun, Y. miR-22 contributes to endosulfan-induced endothelial dysfunction by targeting SRF in HUVECs. Toxicol. Lett. 2017, 269, 33–40. [Google Scholar] [CrossRef]
- Hu, Y.; French, S.W.; Chau, T.; Liu, H.-X.; Sheng, L.; Wei, F.; Stondell, J.; Garcia, J.C.; Du, Y.; Bowlus, C.L.; et al. RARβ acts as both an upstream regulator and downstream effector of miR-22, which epigenetically regulates NUR77 to induce apoptosis of colon cancer cells. FASEB J. 2018, 33, 2314–2326. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, H.-X.; Jena, P.K.; Sheng, L.; Ali, M.R.; Wan, Y.-J.Y. miR-22 inhibition reduces hepatic steatosis via FGF21 and FGFR1 induction. JHEP Rep. 2020, 2, 100093. [Google Scholar] [CrossRef]
- He, J.-S.; Lian, C.-W.; Fang, Y.-L.; Wu, J.-Z.; Ye, X.-L.; Zhu, S.-B. Influence and significance of intervening diabetes microRNA expression profile of NOD mice with exendin-4. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4322–4327. [Google Scholar]
- Gidlöf, O.; Sathanoori, R.; Magistri, M.; Faghihi, M.A.; Wahlestedt, C.; Olde, B.; Erlinge, D. Extracellular Uridine Triphosphate and Adenosine Triphosphate Attenuate Endothelial Inflammation through miR-22-Mediated ICAM-1 Inhibition. J. Vasc. Res. 2015, 52, 71–80. [Google Scholar] [CrossRef]
- Lu, W.; You, R.; Yuan, X.; Yang, T.; Samuel, E.L.; Marcano, D.C.; Sikkema, W.K.; Tour, J.M.; Rodriguez, A. The microRNA miR-22 inhibits the histone deacetylase HDAC4 to promote T(H)17 cell-dependent emphysema. Nat. Immunol. 2015, 16, 1185–1194. [Google Scholar] [CrossRef]
- Lin, J.; Huo, R.; Xiao, L.; Zhu, X.; Xie, J.; Sun, S.; He, Y.; Zhang, J.; Sun, Y.; Zhou, Z.; et al. A novel p53/microRNA-22/Cyr61 axis in synovial cells regulates inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 49–59. [Google Scholar] [CrossRef]
- Lee, J.-H.; Park, S.-J.; Kim, S.W.; Hariharasudhan, G.; Jung, S.-M.; Jun, S.; Yong, J.; You, H.J. c-Fos-dependent miR-22 targets MDC1 and regulates DNA repair in terminally differentiated cells. Oncotarget 2017, 8, 48204–48221. [Google Scholar] [CrossRef]
- Shen, C.; Chen, M.-T.; Zhang, X.-H.; Yin, X.-L.; Ning, H.-M.; Su, R.; Lin, H.-S.; Song, L.; Wang, F.; Ma, Y.-N.; et al. The PU.1-Modulated MicroRNA-22 is a Regulator of Monocyte/Macrophage Differentiation and Acute Myeloid Leukemia. PLoS Genet. 2016, 12, e1006259. [Google Scholar] [CrossRef]
- Sibbesen, N.A.; Kopp, K.L.; Litvinov, I.V.; Jønson, L.; Willerslev-Olsen, A.; Fredholm, S.; Petersen, D.L.; Nastasi, C.; Krejsgaard, T.; Lindahl, L.M.; et al. Jak3, STAT3, and STAT5 inhibit expression of miR-22, a novel tumor suppressor microRNA, in cutaneous T-Cell lymphoma. Oncotarget 2015, 6, 20555–20569. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Wang, M.; Hu, S.; Ru, X.; Ren, Y.; Zhang, Z.; Yu, S.; Zhang, Y. Oncogenic Activation of Nrf2, Though as a Master Antioxidant Transcription Factor, Liberated by Specific Knockout of the Full-Length Nrf1α that Acts as a Dominant Tumor Repressor. Cancers 2018, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Bar, N.; Dikstein, R. miR-22 Forms a Regulatory Loop in PTEN/AKT Pathway and Modulates Signaling Kinetics. PLoS ONE 2010, 5, e10859. [Google Scholar] [CrossRef]
- Delic, D.; Grosser, C.; Dkhil, M.; Al-Quraishy, S.; Wunderlich, F. Testosterone-induced upregulation of miRNAs in the female mouse liver. Steroids 2010, 75, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Moheimani, F.; Koops, J.; Williams, T.; Reid, A.T.; Hansbro, P.M.; Wark, P.A.; Knight, D.A. Influenza A virus infection dysregulates the expression of microRNA-22 and its targets; CD147 and HDAC4, in epithelium of asthmatics. Respir. Res. 2018, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49 (Suppl. 1), 59–69. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Erhard, F.; Haas, J.; Lieber, D.; Malterer, G.; Jaskiewicz, L.; Zavolan, M.; Dölken, L.; Zimmer, R. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014, 24, 906–919. [Google Scholar] [CrossRef]
- Zapalska-Sozoniuk, M.; Chrobak, L.; Kowalczyk, K.; Kankofer, M. Is it useful to use several “omics” for obtaining valuable results? Mol. Biol. Rep. 2019, 46, 3597–3606. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Luque-Garcia, J.L.; Zhou, G.; Spellman, D.S.; Sun, T.-T.; Neubert, T.A. Analysis of Electroblotted Proteins by Mass Spectrometry: Protein Identification after Western Blotting. Mol. Cell. Proteom. 2008, 7, 308–314. [Google Scholar] [CrossRef]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2021, 12, 558–580. [Google Scholar] [CrossRef]
- De Matteis, S.; Ragusa, A.; Marisi, G.; De Domenico, S.; Casadei Gardini, A.; Bonafè, M.; Giudetti, A.M. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxid. Med. Cell Longev. 2018, 2018, 7512159. [Google Scholar] [CrossRef] [Green Version]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef]
- Lincet, H.; Icard, P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 2015, 34, 3751–3759. [Google Scholar] [CrossRef]
- Lhan, M. Non-metabolic functions of Pyruvate kinase M2: PKM2 in tumorigenesis and therapy resistance. Neoplasma 2022, 69, 747–754. [Google Scholar]
- Huang, C.K.; Sun, Y.; Lv, L.; Ping, Y. ENO1 and Cancer. Mol. Ther. Oncol. 2022, 24, 288–298. [Google Scholar] [CrossRef]
- Gurung, S.; Chung, K.P.S.; Lee, T.K.-W. Emerging role of fatty acid binding proteins in cancer pathogenesis. Histol. Histopathol. 2019, 34, 1–12. [Google Scholar]
- Conte, M.; Franceschi, C.; Sandri, M.; Salvioli, S. Perilipin 2 and Age-Related Metabolic Diseases: A New Perspective. Trends Endocrinol. Metab. 2016, 27, 893–903. [Google Scholar] [CrossRef]
- Miyata, Y.; Sakai, H. Thrombospondin-1 in Urological Cancer: Pathological Role, Clinical Significance, and Therapeutic Prospects. Int. J. Mol. Sci. 2013, 14, 12249–12272. [Google Scholar] [CrossRef]
- Lawler, P.; Lawler, J. Molecular Basis for the Regulation of Angiogenesis by Thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.T.; Chung, K.K.; Cheung, S.T.; Lau, C.P.; Tong, S.W.; Leung, K.L.; Yu, W.C.; Tuszynski, G.P.; Fan, S.T. Clinical Significance of Thrombospondin 1 Expression in Hepatocellular Carcinoma. Clin. Cancer Res. 2004, 10, 4150–4157. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Turpin, C.P.; Wang, S. Role of thrombospondin 1 in liver diseases. Hepatol. Res. 2017, 47, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lawler, J. Thrombospondin-based antiangiogenic therapy. Microvasc. Res. 2007, 74, 90–99. [Google Scholar] [CrossRef]
- Chen, R.; Tan, Y.; Wang, M.; Wang, F.; Yao, Z.; Dong, L.; Ye, M.; Wang, H.; Zou, H. Development of Glycoprotein Capture-Based Label-Free Method for the High-throughput Screening of Differential Glycoproteins in Hepatocellular Carcinoma. Mol. Cell. Proteom. 2011, 10, 06445. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Tapia, N.C.; Rosso, N.; Tiribelli, C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012, 12, 20. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, P.; Wang, J.; Liu, Q.; Wang, T.; Wang, Y.; Lin, F. MiR-22 regulated T cell differentiation and hepatocellular carcinoma growth by directly targeting Jarid2. Am. J. Cancer Res. 2021, 11, 2159–2173. [Google Scholar]
- Mah, S.M.; Buske, C.; Humphries, R.K.; Kuchenbauer, F. miRNA*: A passenger stranded in RNA-induced silencing complex? Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 141–148. [Google Scholar] [CrossRef]
- Calo, N.; Ramadori, P.; Sobolewski, C.; Romero, Y.; Maeder, C.; Fournier, M.; Rantakari, P.; Zhang, F.P.; Poutanen, M.; Dufour, J.F.; et al. Stress-activated miR-21/miR-21* in hepatocytes promotes lipid and glucose metabolic disorders associated with high-fat diet consumption. Gut 2016, 65, 1871–1881. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gjorgjieva, M.; Ay, A.-S.; Correia de Sousa, M.; Delangre, E.; Dolicka, D.; Sobolewski, C.; Maeder, C.; Fournier, M.; Sempoux, C.; Foti, M. MiR-22 Deficiency Fosters Hepatocellular Carcinoma Development in Fatty Liver. Cells 2022, 11, 2860. https://doi.org/10.3390/cells11182860
Gjorgjieva M, Ay A-S, Correia de Sousa M, Delangre E, Dolicka D, Sobolewski C, Maeder C, Fournier M, Sempoux C, Foti M. MiR-22 Deficiency Fosters Hepatocellular Carcinoma Development in Fatty Liver. Cells. 2022; 11(18):2860. https://doi.org/10.3390/cells11182860
Chicago/Turabian StyleGjorgjieva, Monika, Anne-Sophie Ay, Marta Correia de Sousa, Etienne Delangre, Dobrochna Dolicka, Cyril Sobolewski, Christine Maeder, Margot Fournier, Christine Sempoux, and Michelangelo Foti. 2022. "MiR-22 Deficiency Fosters Hepatocellular Carcinoma Development in Fatty Liver" Cells 11, no. 18: 2860. https://doi.org/10.3390/cells11182860