
Neutrophil Elastase Increases Vascular Permeability and Leukocyte Transmigration in Cultured Endothelial Cells and Obese Mice

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Experimental Animals
2.4. Evans Blue Vascular Permeability Test
2.5. Isolation of the SVF and FACS Analysis
2.6. Real-Time Quantitative RT-PCR
2.7. Mouse and Human PCR Primers
2.8. Immunofluorescence and F-Actin Phalloidin Staining
2.9. Immunoblotting
2.10. Percoll Gradient Purification of Neutrophils
2.11. Lucifer Yellow Permeability Study
2.12. Monocyte Transmigration
2.13. Statistical Analysis
3. Results
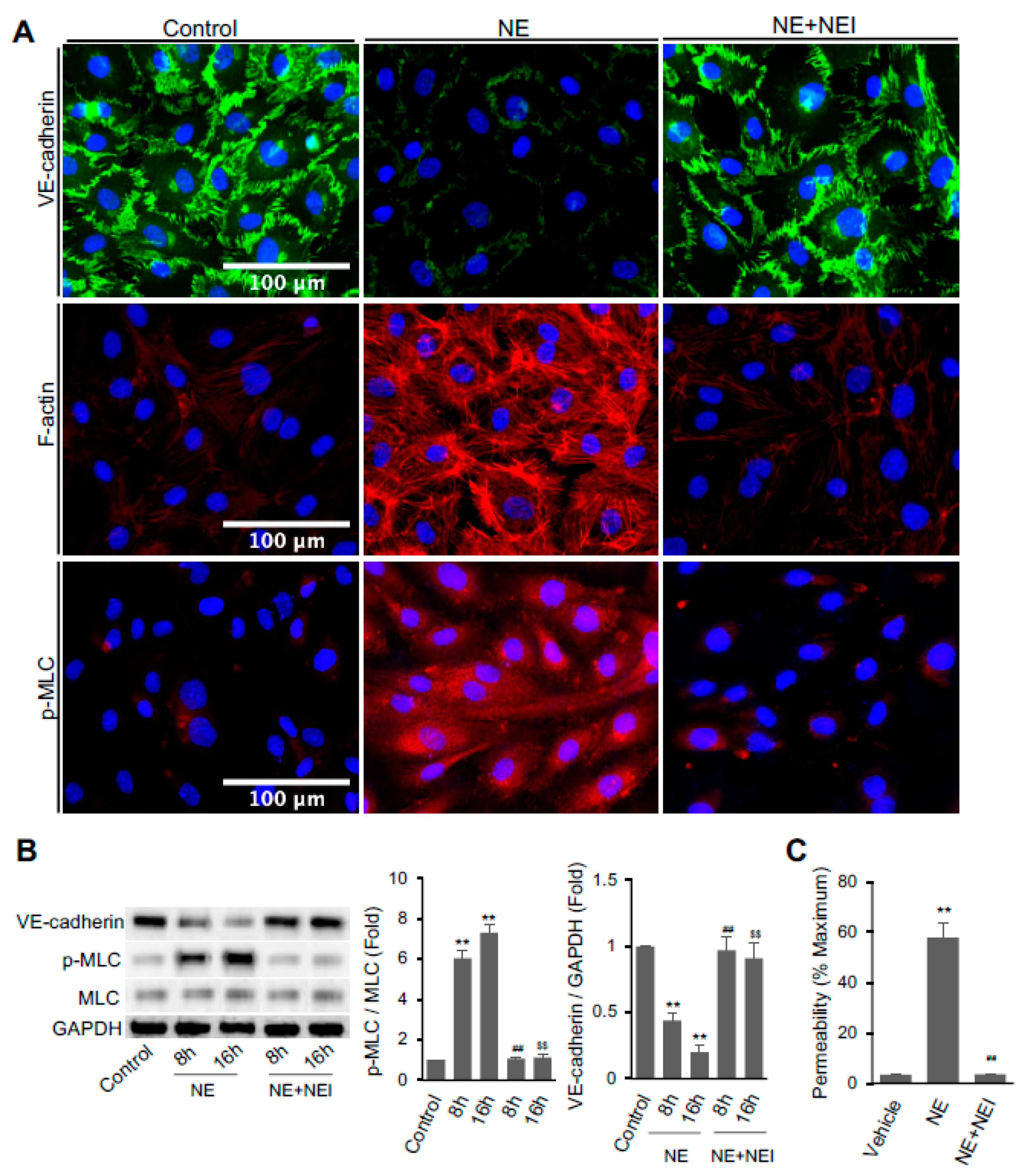
3.1. NE Regulated Vascular Endothelial Cell Permeability, Actomyosin Cytoskeleton, and the Expression of Cell–Cell Junction Molecule VE-Cadherin
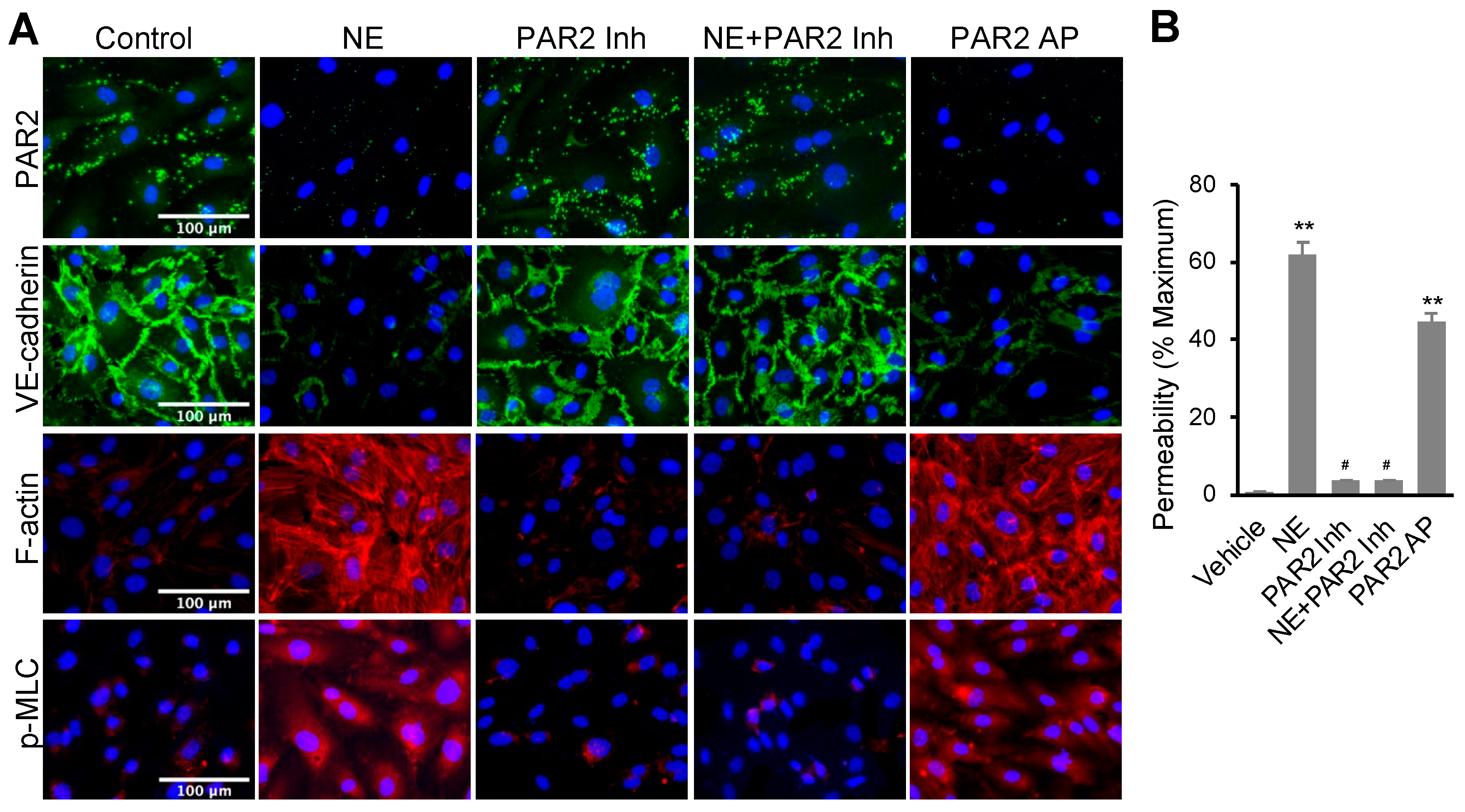
3.2. NE Regulated hEC Permeability through Activating Protease-Activated Receptor 2 (PAR2)
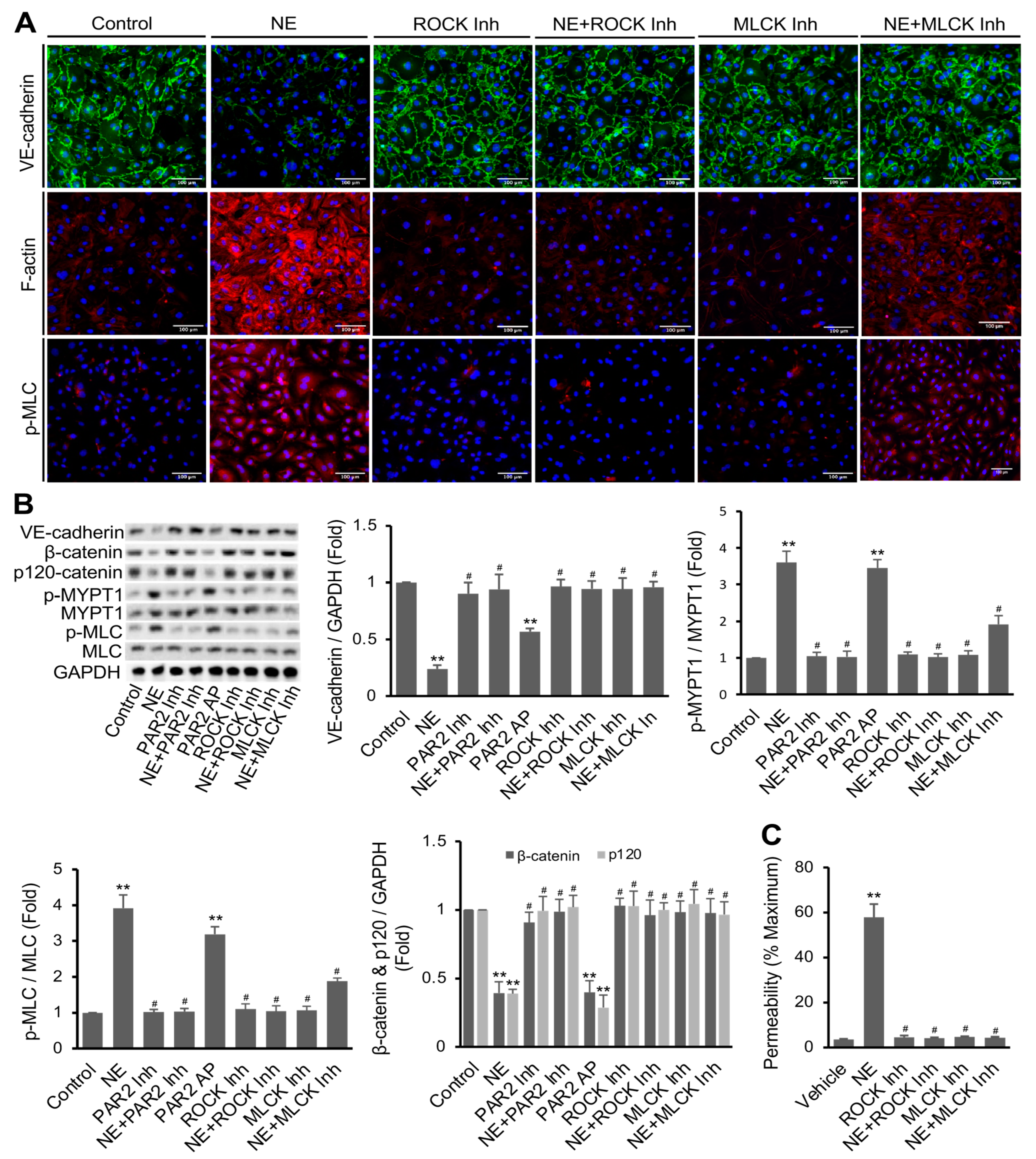
3.3. NE’s Effects on hEC Permeability, F-Actin Formation, MLC Phosphorylation, and VE-Cadherin Expression Were Mediated via PAR2 Downstream Signaling Pathway
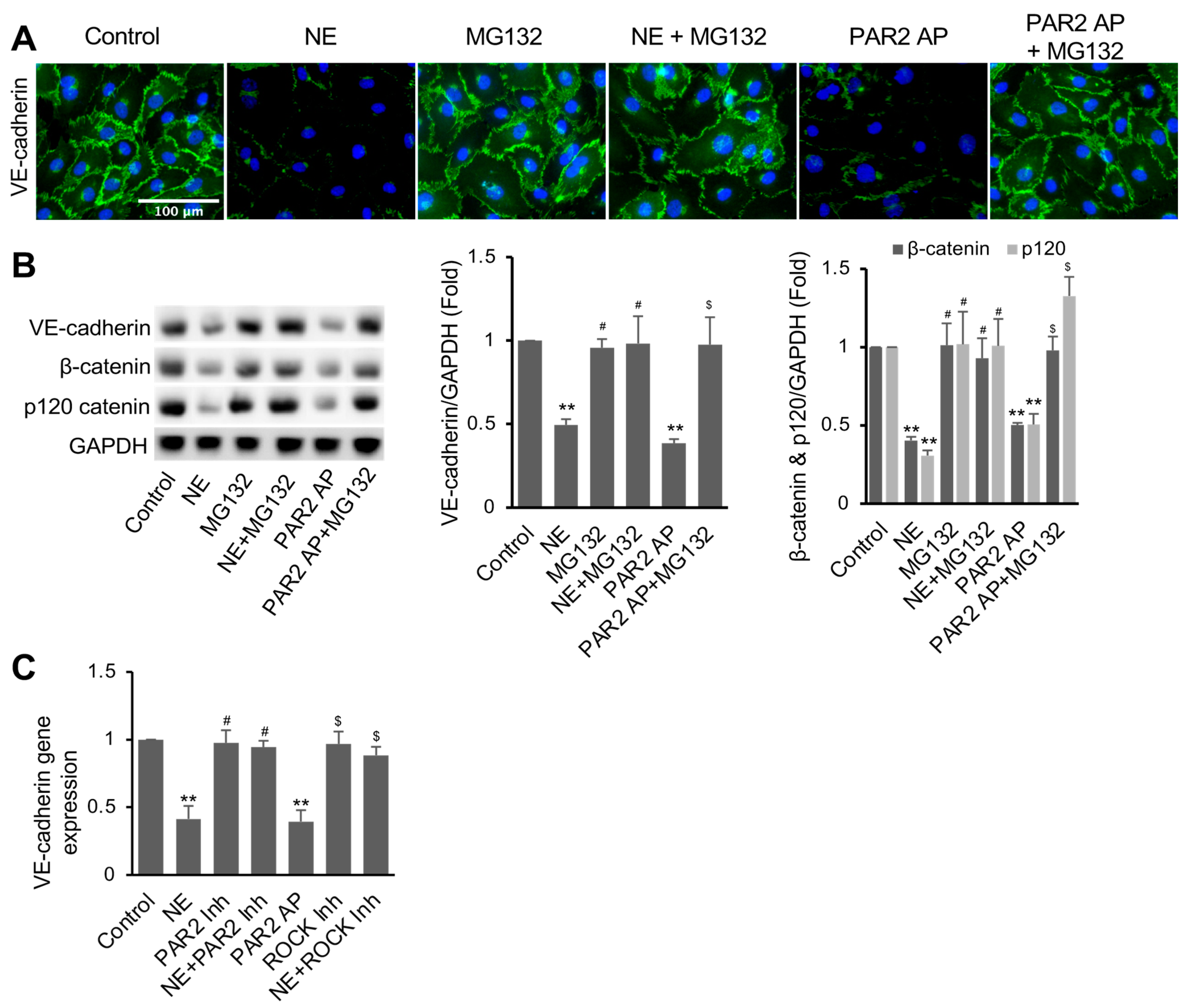
3.4. NE Induced VE-Cadherin Protein Degradation and Repressed VE-Cadherin Gene Expression
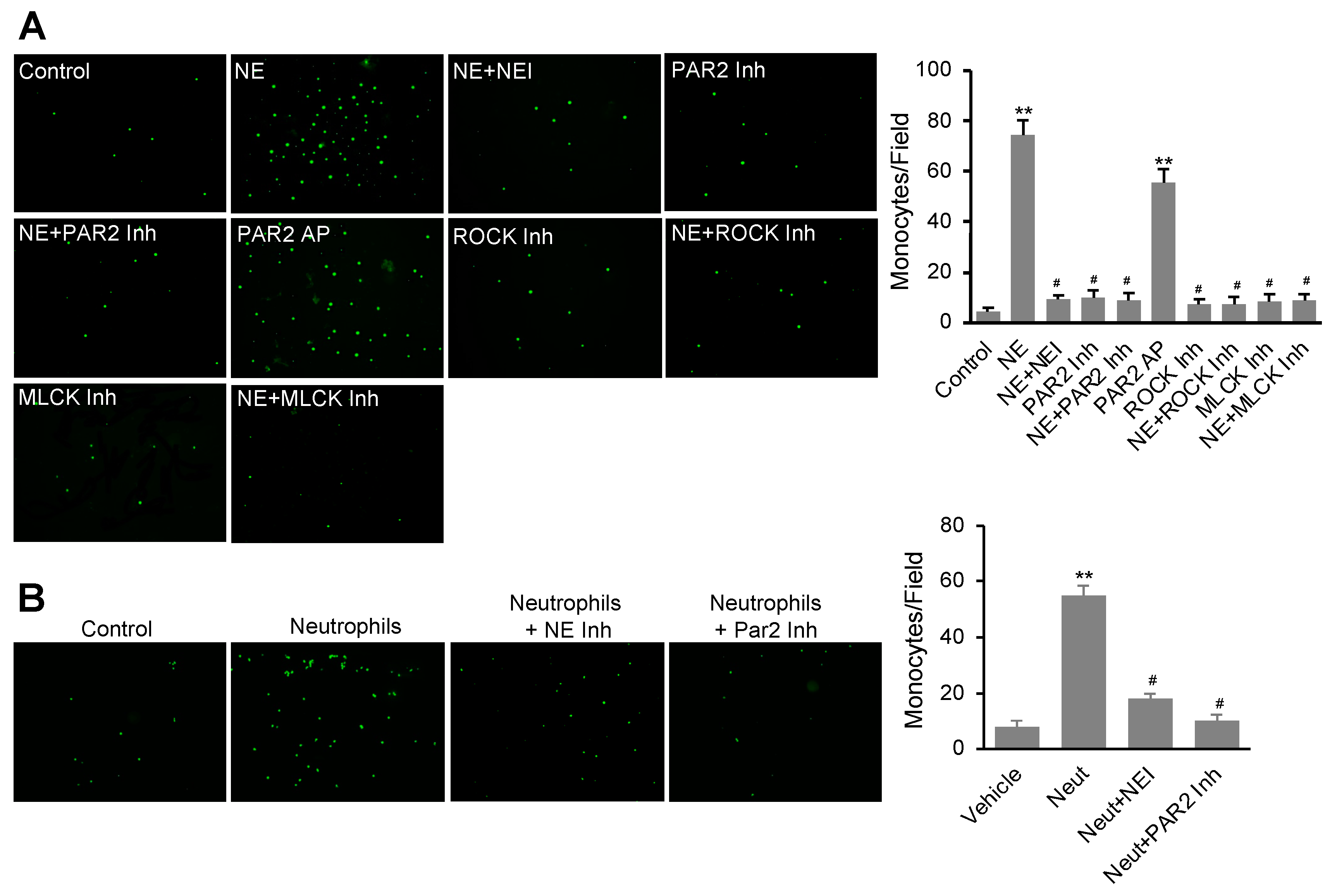
3.5. NE Stimulated Monocyte Transendothelial Migration Dependent on the Activation of the PAR2 Signal Pathway
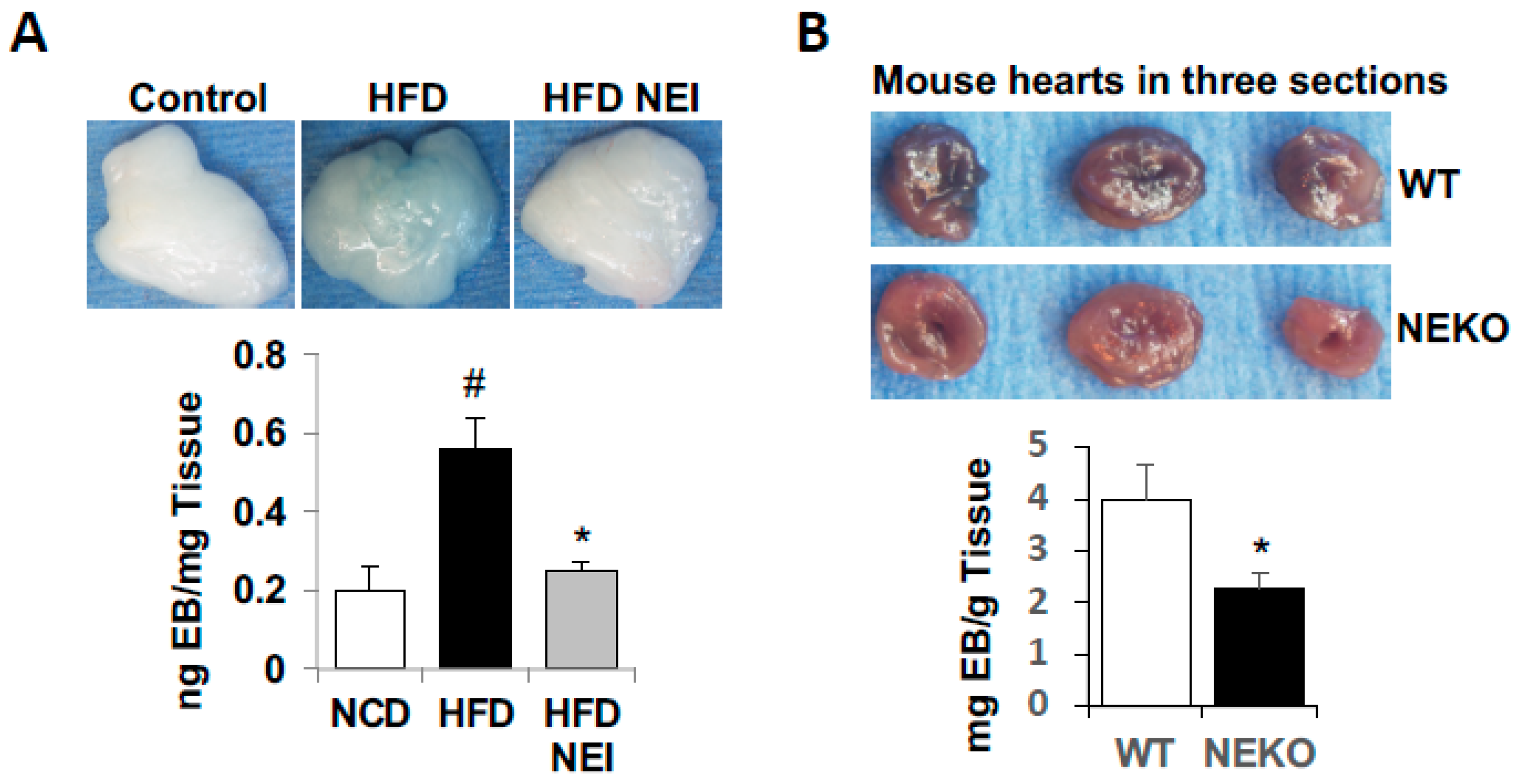
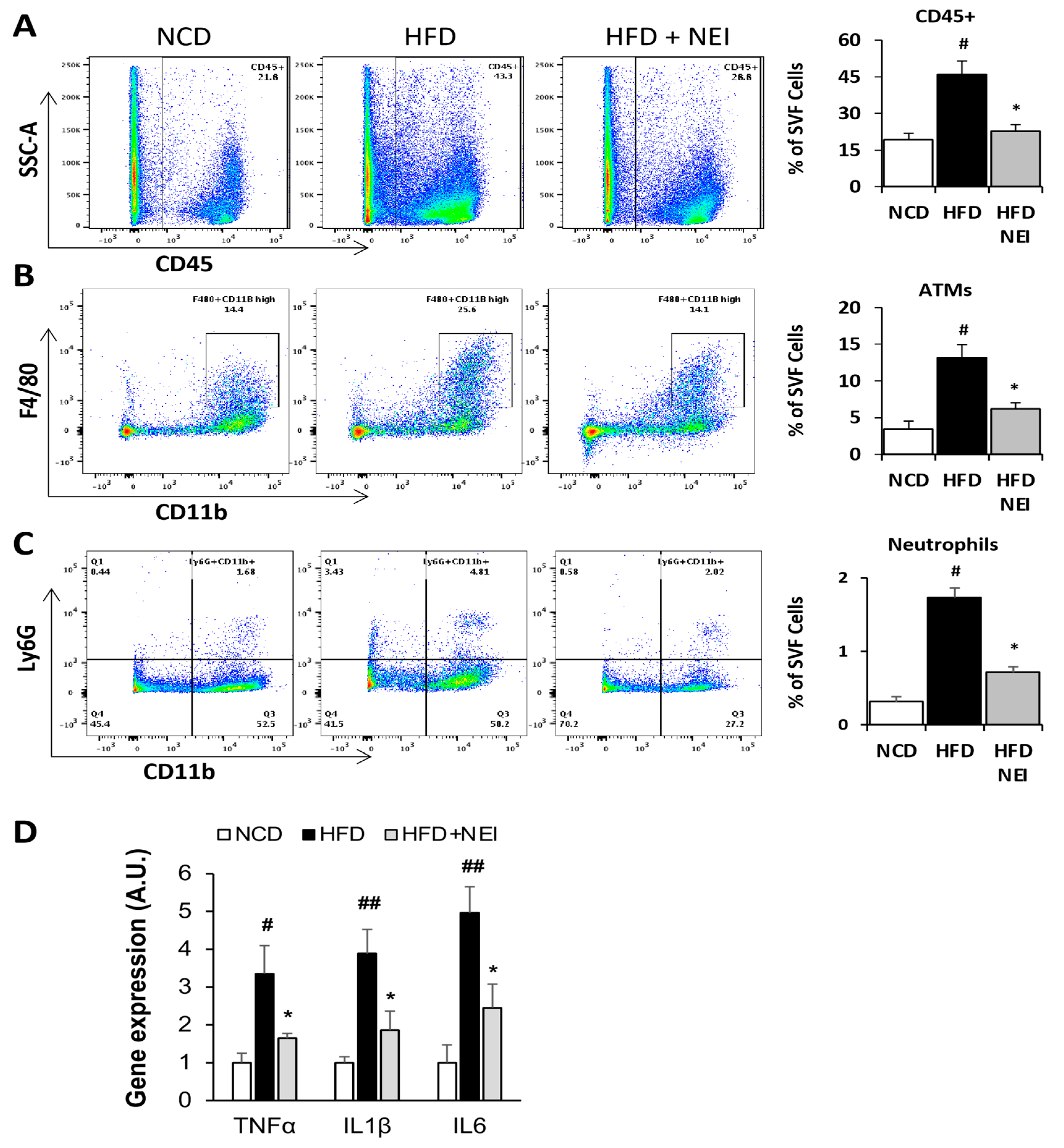
3.6. Inhibition of NE Attenuated HFD-Induced Vascular Leakage in White Adipose Tissue (WAT) and Heart, as Well as Leukocyte Infiltration and Inflammation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2019, 10, 1568. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef]
- Christopher, M.J.; Link, D.C. Regulation of neutrophil homeostasis. Curr. Opin. Hematol. 2007, 14, 3–8. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, X.; Chatterjee, V.; Meegan, J.E.; Beard, R.S., Jr.; Yuan, S.Y. Role of Neutrophil Extracellular Traps and Vesicles in Regulating Vascular Endothelial Permeability. Front. Immunol. 2019, 10, 1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaventura, A.; Vecchie, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuhiro, T.; Ishikawa, A.; Sato, H.; Takita, S.; Yoshikawa, A.; Anzai, R.; Sato, S.; Aoyagi, R.; Arita, M.; Shibuya, T.; et al. Oxidized Phospholipids and Neutrophil Elastase Coordinately Play Critical Roles in N.E.T Formation. Front. Cell Dev. Biol. 2021, 9, 718586. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Wang, Y.; Sano, S.; Oshima, K.; Sano, M.; Watanabe, Y.; Katanasaka, Y.; Yura, Y.; Jung, C.; Anzai, A.; Swirski, F.K.; et al. Wnt5a-Mediated Neutrophil Recruitment Has an Obligatory Role in Pressure Overload-Induced Cardiac Dysfunction. Circulation 2019, 140, 487–499. [Google Scholar] [CrossRef]
- Mansuy-Aubert, V.; Zhou, Q.L.; Xie, X.; Gong, Z.; Huang, J.Y.; Khan, A.R.; Aubert, G.; Candelaria, K.; Thomas, S.; Shin, D.J.; et al. Imbalance between neutrophil elastase and its inhibitor alpha1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab. 2013, 17, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, S.; Oh da, Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.Y.; Zhou, Q.L.; Huang, C.H.; Song, Y.; Sharma, A.G.; Liao, Z.; Zhu, K.; Massidda, M.W.; Jamieson, R.R.; Zhang, J.Y.; et al. Neutrophil Elastase Regulates Emergency Myelopoiesis Preceding Systemic Inflammation in Diet-induced Obesity. J. Biol. Chem. 2017, 292, 4770–4776. [Google Scholar] [CrossRef] [Green Version]
- Elgazar-Carmon, V.; Rudich, A.; Hadad, N.; Levy, R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J. Lipid Res. 2008, 49, 1894–1903. [Google Scholar] [CrossRef] [Green Version]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Vincent, P.A.; Xiao, K.; Buckley, K.M.; Kowalczyk, A.P. VE-cadherin: Adhesion at arm’s length. Am. J. Physiol.-Cell Physiol. 2004, 286, C987–C997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavard, J. Endothelial permeability and VE-cadherin: A wacky comradeship. Cell Adhes. Migr. 2014, 8, 158–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, C.N.; Vestweber, D. Mechanisms Ensuring Endothelial Junction Integrity Beyond VE-Cadherin. Front. Physiol. 2020, 11, 519. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Ferreri, D.M.; DeCocco, N.C.; Minnear, F.L.; Vincent, P.A. VE-cadherin-p120 interaction is required for maintenance of endothelial barrier function. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 286, L1143–L1153. [Google Scholar] [CrossRef]
- Bravi, L.; Dejana, E.; Lampugnani, M.G. VE-cadherin at a glance. Cell Tissue Res. 2014, 355, 515–522. [Google Scholar] [CrossRef]
- Abu Taha, A.; Schnittler, H.J. Dynamics between actin and the VE-cadherin/catenin complex: Novel aspects of the ARP2/3 complex in regulation of endothelial junctions. Cell Adhes. Migr. 2014, 8, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Nanes, B.A.; Grimsley-Myers, C.M.; Cadwell, C.M.; Robinson, B.S.; Lowery, A.M.; Vincent, P.A.; Mosunjac, M.; Fruh, K.; Kowalczyk, A.P. p120-catenin regulates VE-cadherin endocytosis and degradation induced by the Kaposi sarcoma-associated ubiquitin ligase K5. Mol. Biol. Cell 2017, 28, 30–40. [Google Scholar] [CrossRef]
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms regulating endothelial permeability. Pulm. Circ. 2014, 4, 535–551. [Google Scholar] [CrossRef] [Green Version]
- van Nieuw Amerongen, G.P.; van Delft, S.; Vermeer, M.A.; Collard, J.G.; van Hinsbergh, V.W. Activation of RhoA by thrombin in endothelial hyperpermeability: Role of Rho kinase and protein tyrosine kinases. Circ. Res. 2000, 87, 335–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Wu, M.H.; Yuan, S.Y. Nonmuscle myosin light-chain kinase deficiency attenuates atherosclerosis in apolipoprotein E-deficient mice via reduced endothelial barrier dysfunction and monocyte migration. Circulation 2011, 124, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarenbach, S.W.; Chipiuk, A.; Nelson, R.C.; Hollenberg, M.D.; Murray, A.G. Differential actions of PAR2 and PAR1 in stimulating human endothelial cell exocytosis and permeability: The role of Rho-GTPases. Circ. Res. 2003, 92, 272–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, M.C.A.; van Bezu, J.S.M.; van Nieuw Amerongen, G.P.; van Hinsbergh, V.W.M.; Hordijk, P.L. RhoA, RhoB and RhoC differentially regulate endothelial barrier function. Small GTPases 2019, 10, 466–484. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.L.; Song, Y.; Huang, C.H.; Huang, J.Y.; Gong, Z.; Liao, Z.; Sharma, A.G.; Greene, L.; Deng, J.Z.; Rigor, M.C.; et al. Membrane Trafficking Protein CDP138 Regulates Fat Browning and Insulin Sensitivity through Controlling Catecholamine Release. Mol. Cell. Biol. 2018, 38, e00153-17. [Google Scholar] [CrossRef] [Green Version]
- Siemsen, D.W.; Malachowa, N.; Schepetkin, I.A.; Whitney, A.R.; Kirpotina, L.N.; Lei, B.; Deleo, F.R.; Quinn, M.T. Neutrophil isolation from nonhuman species. Methods Mol. Biol. 2014, 1124, 19–37. [Google Scholar]
- Kundumani-Sridharan, V.; Dyukova, E.; Hansen, D.E., 3rd; Rao, G.N. 12/15-Lipoxygenase mediates high-fat diet-induced endothelial tight junction disruption and monocyte transmigration: A new role for 15(S)-hydroxyeicosatetraenoic acid in endothelial cell dysfunction. J. Biol. Chem. 2013, 288, 15830–15842. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, S.J.; Dowle, M.D.; Harrison, L.A.; Shah, P.; Johnson, M.R.; Inglis, G.G.; Clarke, G.D.; Smith, R.A.; Humphreys, D.; Molloy, C.R.; et al. The discovery of a potent, intracellular, orally bioavailable, long duration inhibitor of human neutrophil elastase--GW311616A a development candidate. Bioorganic Med. Chem. Lett. 2001, 11, 895–898. [Google Scholar] [CrossRef]
- Ribeiro-Gomes, F.L.; Moniz-de-Souza, M.C.; Alexandre-Moreira, M.S.; Dias, W.B.; Lopes, M.F.; Nunes, M.P.; Lungarella, G.; DosReis, G.A. Neutrophils activate macrophages for intracellular killing of Leishmania major through recruitment of TLR4 by neutrophil elastase. J. Immunol. 2007, 179, 3988–3994. [Google Scholar] [CrossRef] [Green Version]
- Domon, H.; Nagai, K.; Maekawa, T.; Oda, M.; Yonezawa, D.; Takeda, W.; Hiyoshi, T.; Tamura, H.; Yamaguchi, M.; Kawabata, S.; et al. Neutrophil Elastase Subverts the Immune Response by Cleaving Toll-Like Receptors and Cytokines in Pneumococcal Pneumonia. Front. Immunol. 2018, 9, 732. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Lieu, T.; Barlow, N.; Sostegni, S.; Haerteis, S.; Korbmacher, C.; Liedtke, W.; Jimenez-Vargas, N.N.; Vanner, S.J.; Bunnett, N.W. Neutrophil Elastase Activates Protease-activated Receptor-2 (PAR2) and Transient Receptor Potential Vanilloid 4 (TRPV4) to Cause Inflammation and Pain. J. Biol. Chem. 2015, 290, 13875–13887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.S.; Cho, K.S.; Yoon, H.K.; Moon, H.S.; Park, S.H. Neutrophil elastase causes MUC5AC mucin synthesis via EGF receptor, ERK and NF-kB pathways in A549 cells. Korean J. Intern. Med. 2005, 20, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Mihara, K.; Chung, H.; Renaux, B.; Lau, C.S.; Muruve, D.A.; DeFea, K.A.; Bouvier, M.; Hollenberg, M.D. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J. Biol. Chem. 2011, 286, 24638–24648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Dery, O.; Mullins, R.D.; Bunnett, N.W. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 2000, 148, 1267–1281. [Google Scholar] [CrossRef]
- Rayees, S.; Rochford, I.; Joshi, J.C.; Joshi, B.; Banerjee, S.; Mehta, D. Macrophage TLR4 and PAR2 Signaling: Role in Regulating Vascular Inflammatory Injury and Repair. Front. Immunol. 2020, 11, 2091. [Google Scholar] [CrossRef]
- Jiang, Y.; Yau, M.K.; Lim, J.; Wu, K.C.; Xu, W.; Suen, J.Y.; Fairlie, D.P. A Potent Antagonist of Protease-Activated Receptor 2 That Inhibits Multiple Signaling Functions in Human Cancer Cells. J. Pharmacol. Exp. Ther. 2018, 364, 246–257. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, K.A.; Cunningham, M.R.; Bushell, T.; Plevin, R. The development of proteinase-activated receptor-2 modulators and the challenges involved. Biochem. Soc. Trans. 2020, 48, 2525–2537. [Google Scholar] [CrossRef]
- Sriwai, W.; Mahavadi, S.; Al-Shboul, O.; Grider, J.R.; Murthy, K.S. Distinctive G Protein-Dependent Signaling by Protease-Activated Receptor 2 (PAR2) in Smooth Muscle: Feedback Inhibition of RhoA by cAMP-Independent PKA. PLoS ONE 2013, 8, e66743. [Google Scholar] [CrossRef] [Green Version]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Hebda, J.K.; Leclair, H.M.; Azzi, S.; Roussel, C.; Scott, M.G.; Bidere, N.; Gavard, J. The C-terminus region of beta-arrestin1 modulates VE-cadherin expression and endothelial cell permeability. Cell Commun. Signal. 2013, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Ge, J. Proteinase-Activated Receptor-2 Modulates Ve-Cadherin Expression to Affect Human Vascular Endothelial Barrier Function. J. Cell. Biochem. 2017, 118, 4587–4593. [Google Scholar] [CrossRef]
- DiStasi, M.R.; Ley, K. Opening the flood-gates: How neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009, 30, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Rizo, V.; Martinez-Guzman, M.A.; Iniguez-Gutierrez, L.; Garcia-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef]
- Artemniak-Wojtowicz, D.; Kucharska, A.M.; Pyrzak, B. Obesity and chronic inflammation crosslinking. Cent. Eur. J. Immunol. 2020, 45, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Metcalf, M.; Bunnett, N.W. Biased signaling of protease-activated receptors. Front. Endocrinol. 2014, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Trejo, J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits beta-arrestin-mediated endosomal signaling. J. Biol. Chem. 2013, 288, 11203–11215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dery, O.; Thoma, M.S.; Wong, H.; Grady, E.F.; Bunnett, N.W. Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. beta-Arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 1999, 274, 18524–18535. [Google Scholar] [CrossRef] [Green Version]
- Ricks, T.K.; Trejo, J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J. Biol. Chem. 2009, 284, 34444–34457. [Google Scholar] [CrossRef] [Green Version]
- Stalheim, L.; Ding, Y.; Gullapalli, A.; Paing, M.M.; Wolfe, B.L.; Morris, D.R.; Trejo, J. Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol. Pharmacol. 2005, 67, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.S.; Lee, S.E.; Lee, H.S.; Kim, G.H.; Yoon, C.J.; Han, J.S.; Lee, J.Y.; Sohn, U.D. The signaling of protease-activated receptor-2 activating peptide-induced contraction in cat esophageal smooth muscle cells. Arch. Pharmacal Res. 2017, 40, 1443–1454. [Google Scholar] [CrossRef]
- Sawant, D.A.; Tharakan, B.; Adekanbi, A.; Hunter, F.A.; Smythe, W.R.; Childs, E.W. Inhibition of VE-cadherin proteasomal degradation attenuates microvascular hyperpermeability. Microcirculation 2011, 18, 46–55. [Google Scholar] [CrossRef]
- Xiao, K.; Allison, D.F.; Kottke, M.D.; Summers, S.; Sorescu, G.P.; Faundez, V.; Kowalczyk, A.P. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J. Biol. Chem. 2003, 278, 19199–19208. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ushakumari, C.J.; Zhou, Q.L.; Wang, Y.-H.; Na, S.; Rigor, M.C.; Zhou, C.Y.; Kroll, M.K.; Lin, B.D.; Jiang, Z.Y. Neutrophil Elastase Increases Vascular Permeability and Leukocyte Transmigration in Cultured Endothelial Cells and Obese Mice. Cells 2022, 11, 2288. https://doi.org/10.3390/cells11152288
Ushakumari CJ, Zhou QL, Wang Y-H, Na S, Rigor MC, Zhou CY, Kroll MK, Lin BD, Jiang ZY. Neutrophil Elastase Increases Vascular Permeability and Leukocyte Transmigration in Cultured Endothelial Cells and Obese Mice. Cells. 2022; 11(15):2288. https://doi.org/10.3390/cells11152288
Chicago/Turabian StyleUshakumari, Chinchu Jagadan, Qiong L. Zhou, Yu-Hua Wang, Sijia Na, Michael C. Rigor, Cindy Y. Zhou, Max K. Kroll, Benjamin D. Lin, and Zhen Y. Jiang. 2022. "Neutrophil Elastase Increases Vascular Permeability and Leukocyte Transmigration in Cultured Endothelial Cells and Obese Mice" Cells 11, no. 15: 2288. https://doi.org/10.3390/cells11152288