
RIP3 Translocation into Mitochondria Promotes Mitofilin Degradation to Increase Inflammation and Kidney Injury after Renal Ischemia–Reperfusion



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Kidney Ischemia Reperfusion Injury
2.3. Immunohistochemical Staining and Injury Assessment
2.4. Mitochondrial Isolation
2.5. Mitochondrial ROS Measurement
2.6. Ca2+-Induced Mitochondrial Permeability Transition
2.7. Mitochondrial Membrane Potential
2.8. Cell Culture and siRNA Transfection
2.9. RIP3 and Mitofilin Plasmid Transfection
2.10. Transmission Electron Microscopy
2.11. Immunoprecipitation
2.12. Western Blot
2.13. Immunofluorescence Staining
2.14. mtDNA Isolation
2.15. RNA Isolation and Real-Time PCR
2.16. Statistical Analysis
3. Results
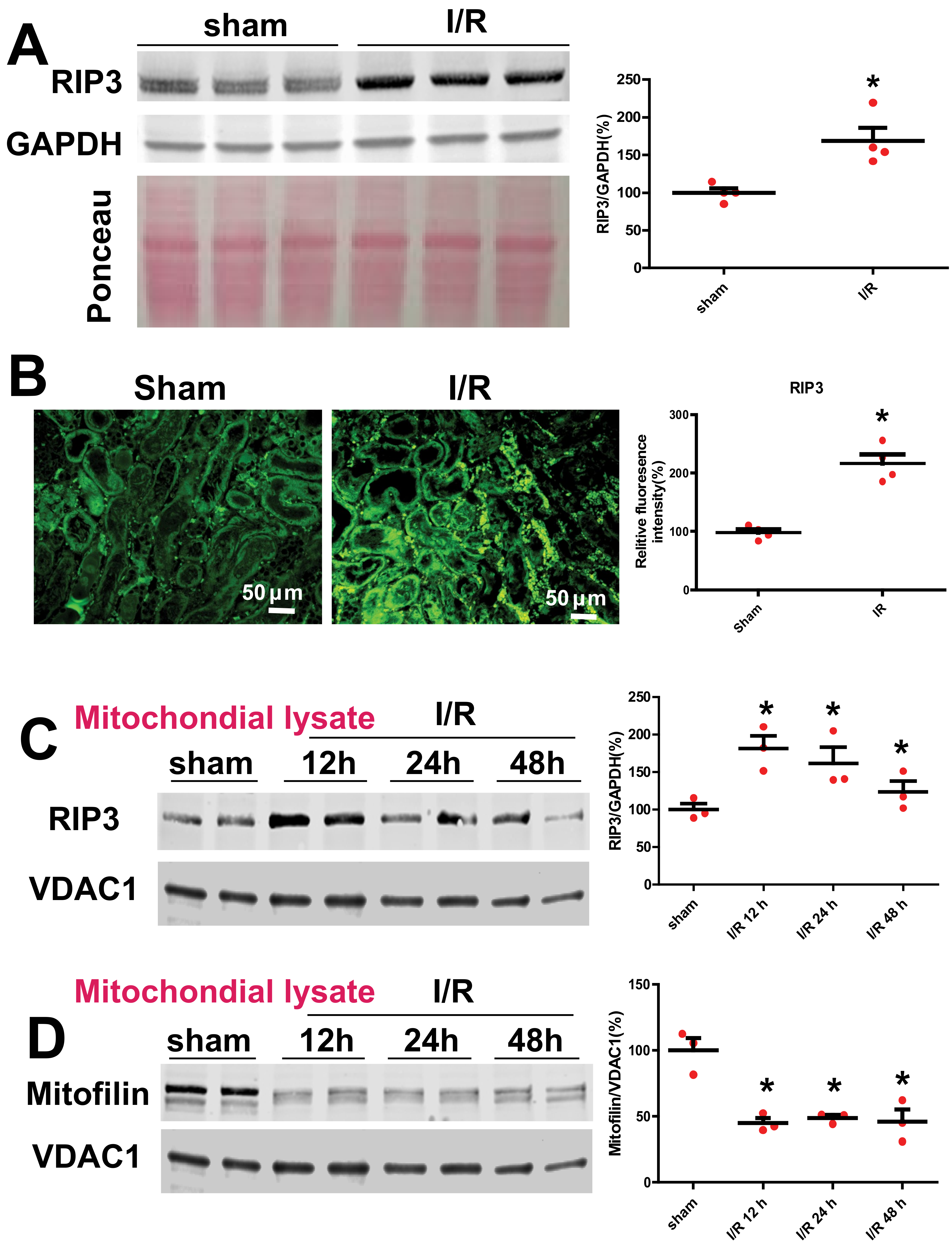
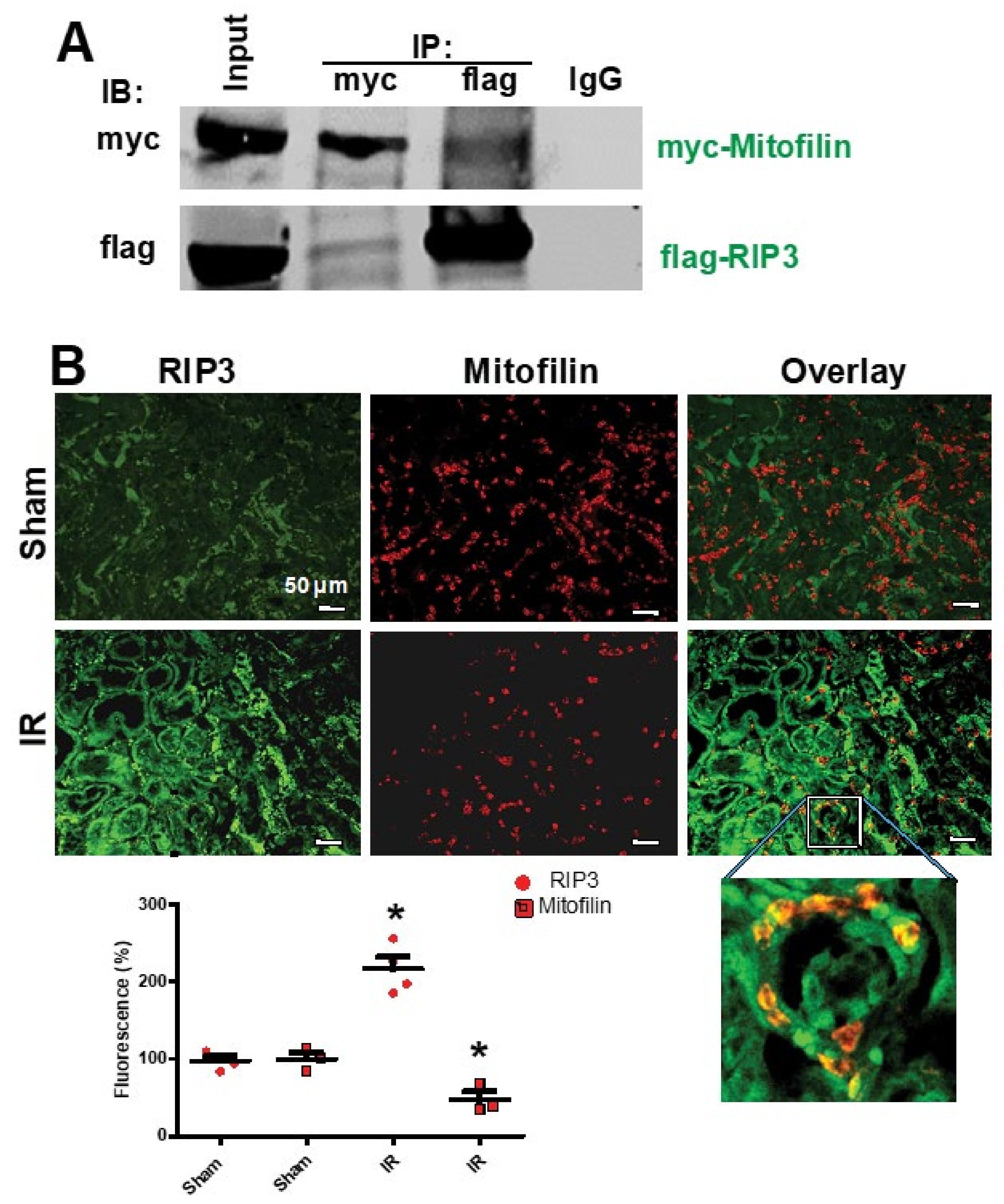
3.1. Renal I/R Injury Increases RIP3 Levels and Promotes Its Translocation in Mitochondria Where It Interacts with Mitofilin
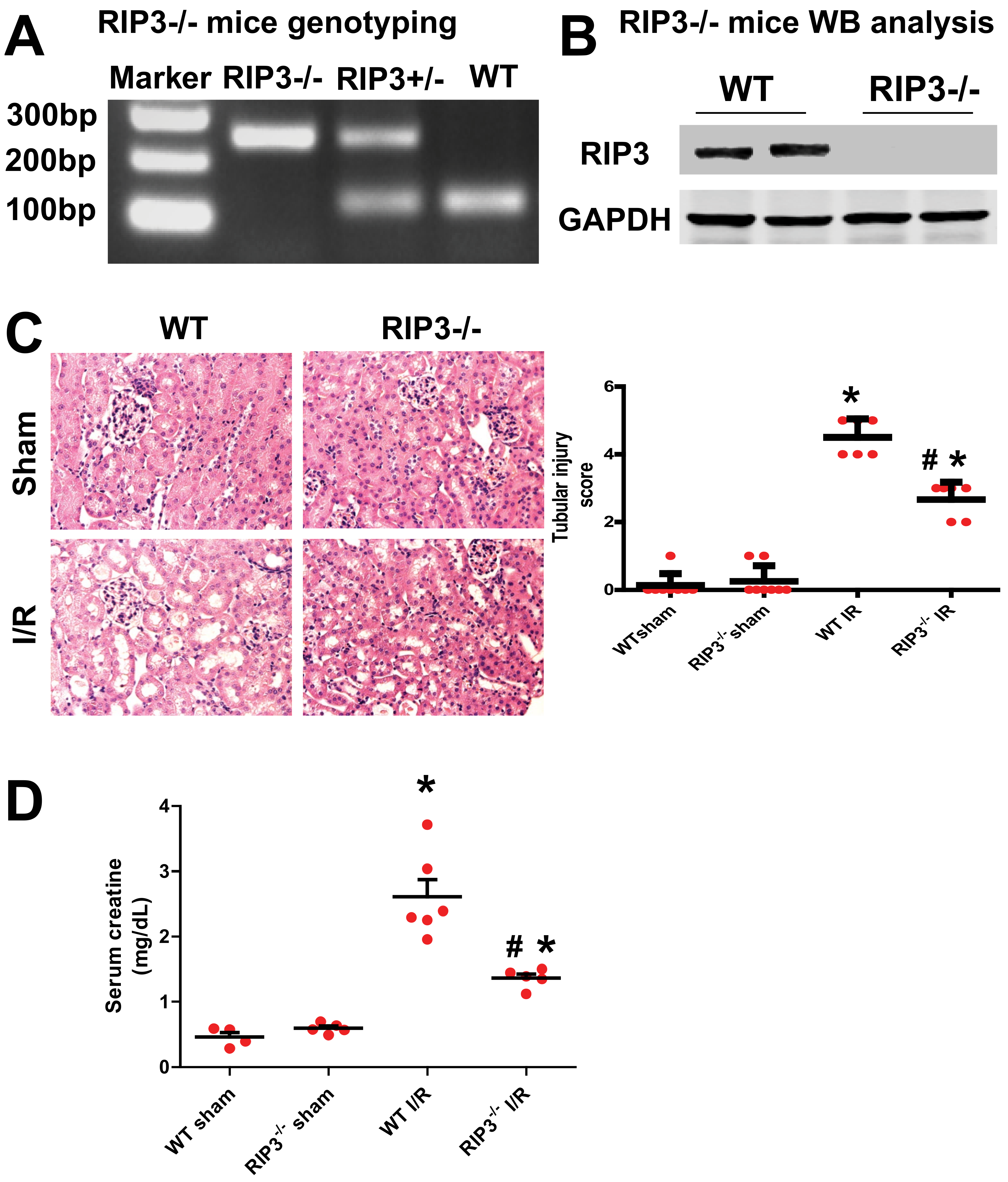
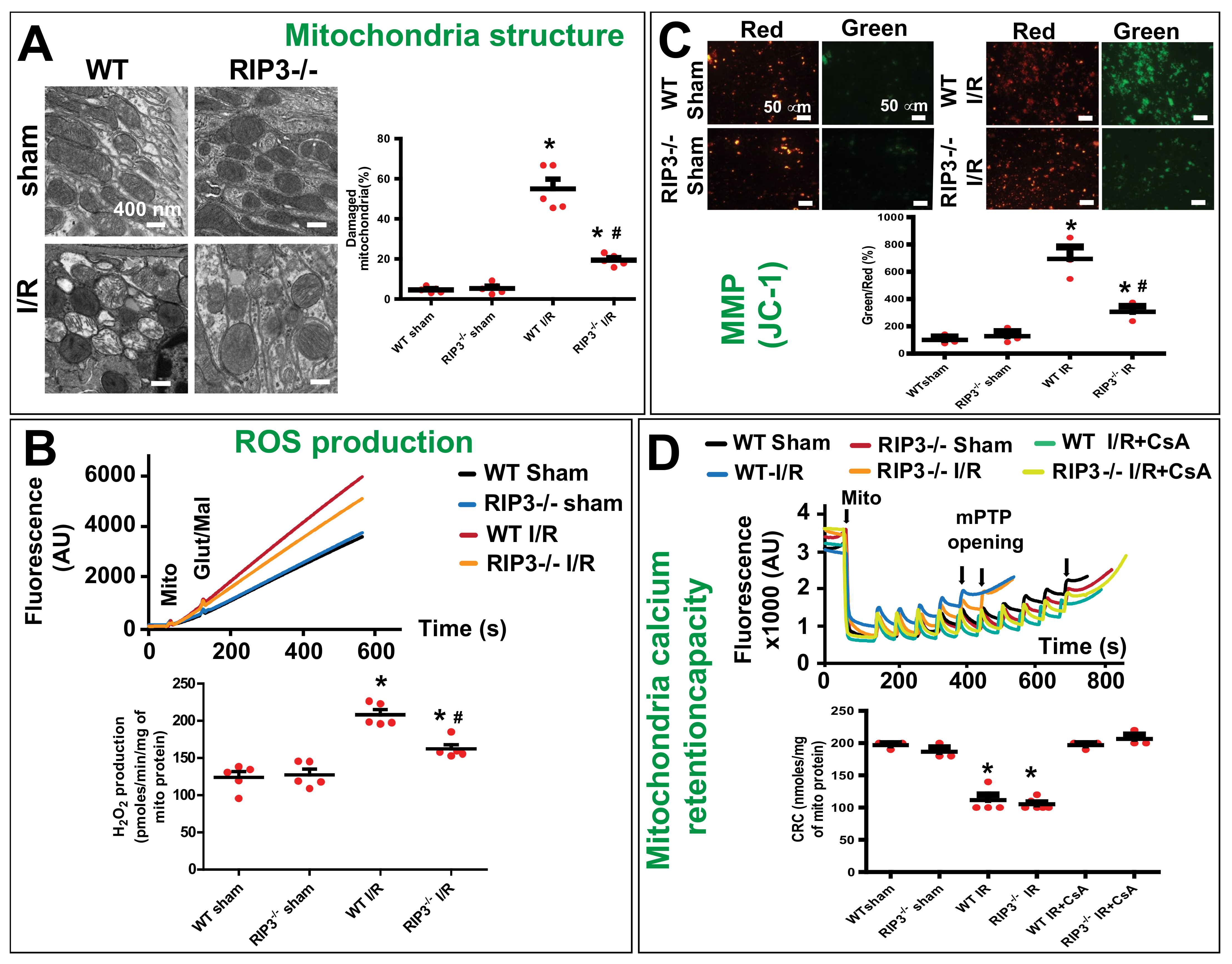
3.2. RIP3 Knockout Mice Display Decrease in Kidney Injury after Renal IR
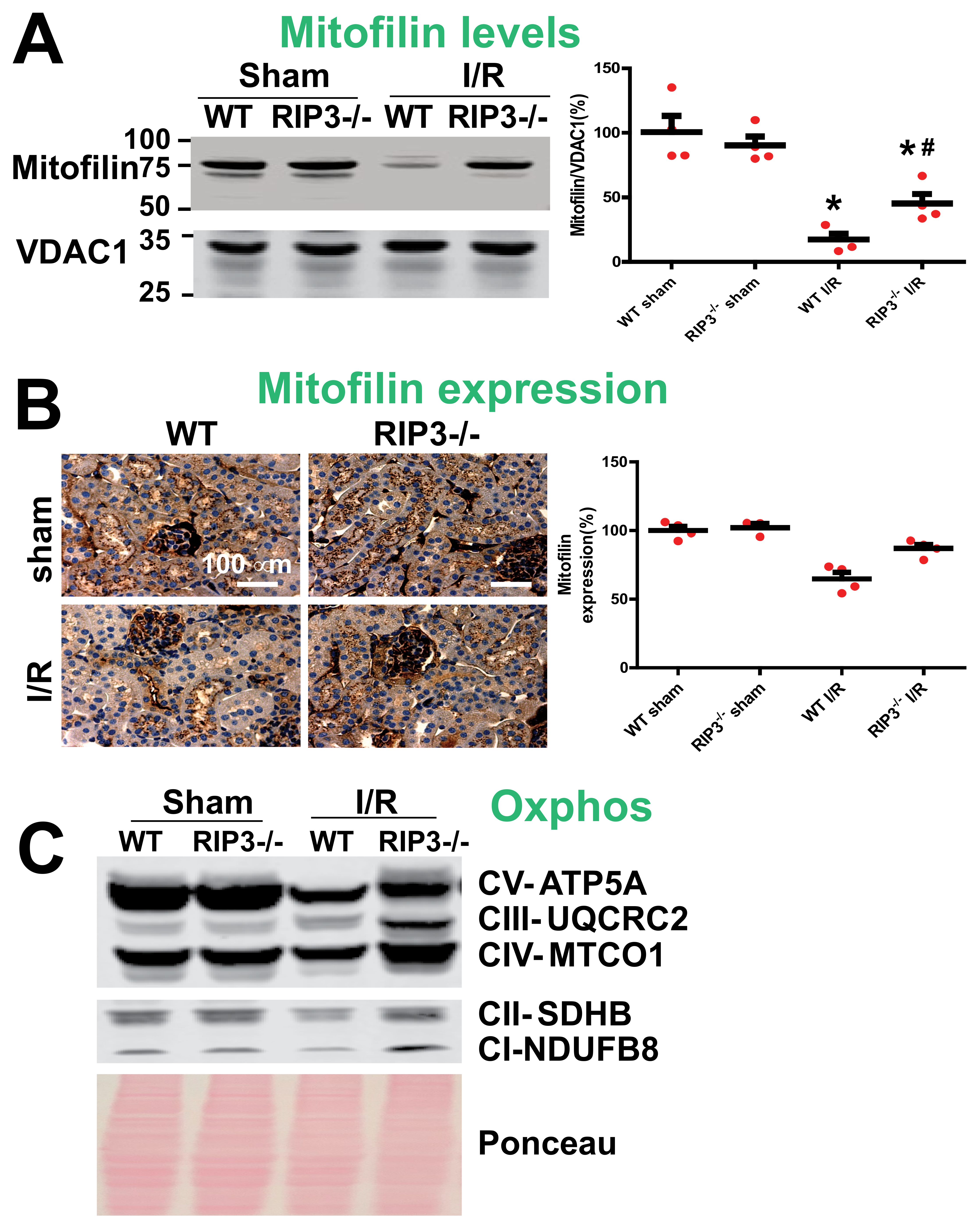
3.3. RIP3 Knockdown Reduces the Mitofilin Degradation and Protects Mitochondrial Damage after Renal I/R
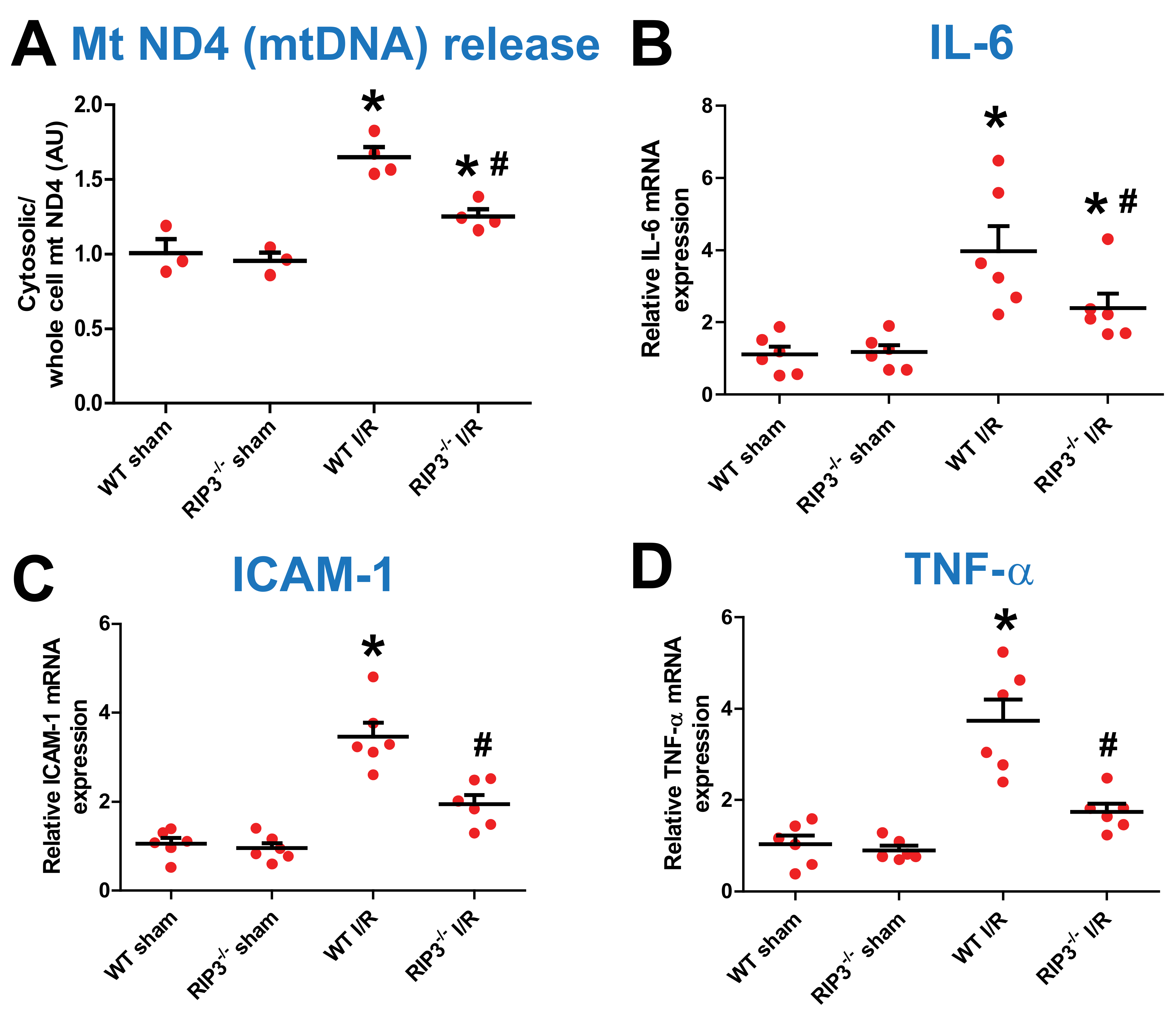
3.4. RIP3 Knockdown Reduces Mitochondrial DNA Release and Inflammatory Factors after Kidney IR Injury
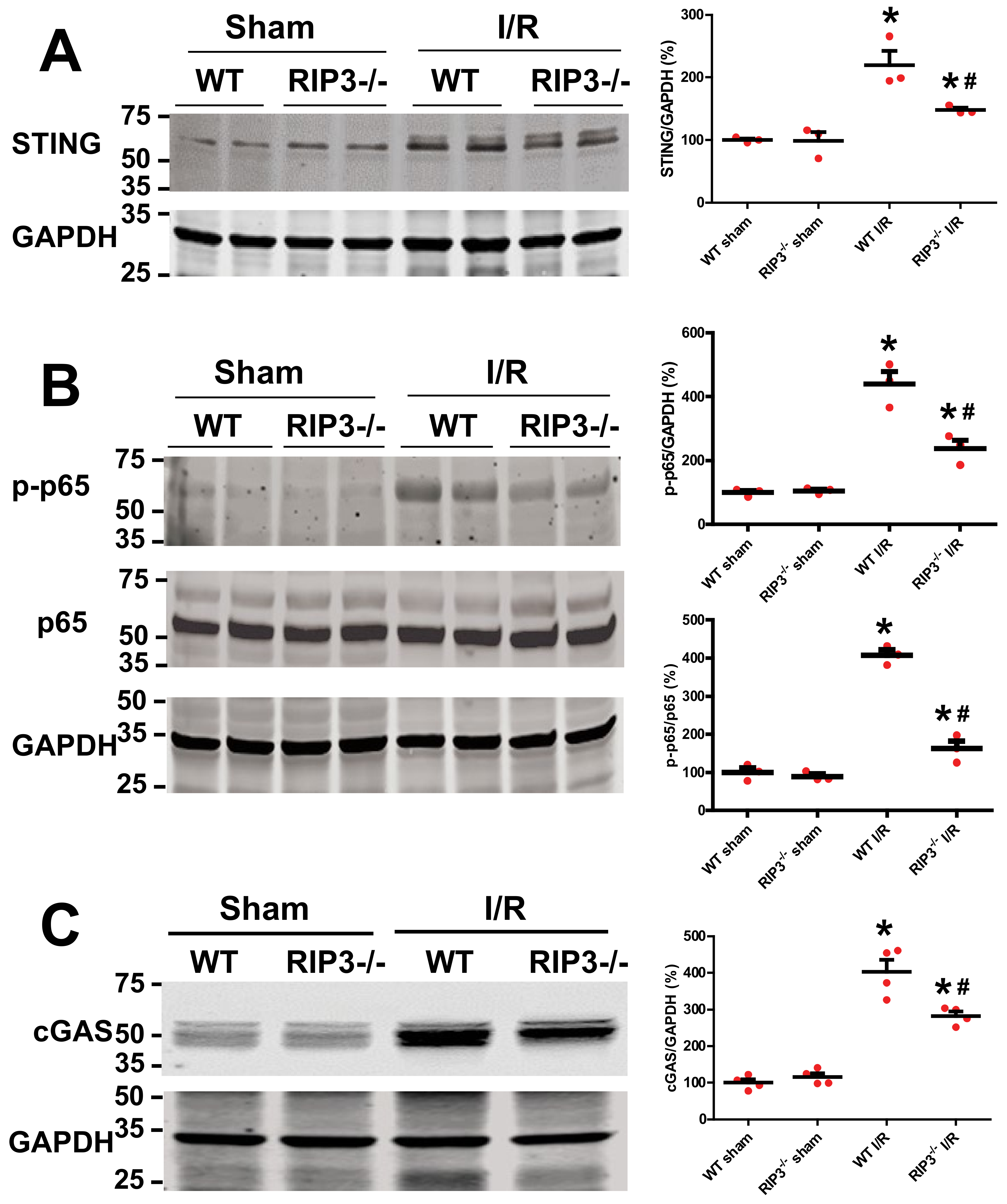
3.5. RIP3 Knockout Deactivates cGAS–STING–P-p65 Pathway after Kidney I/R Injury
3.6. RIP3 Overexpression in HK-2 Cells Activates the cGAS–STING–p65 Pathway
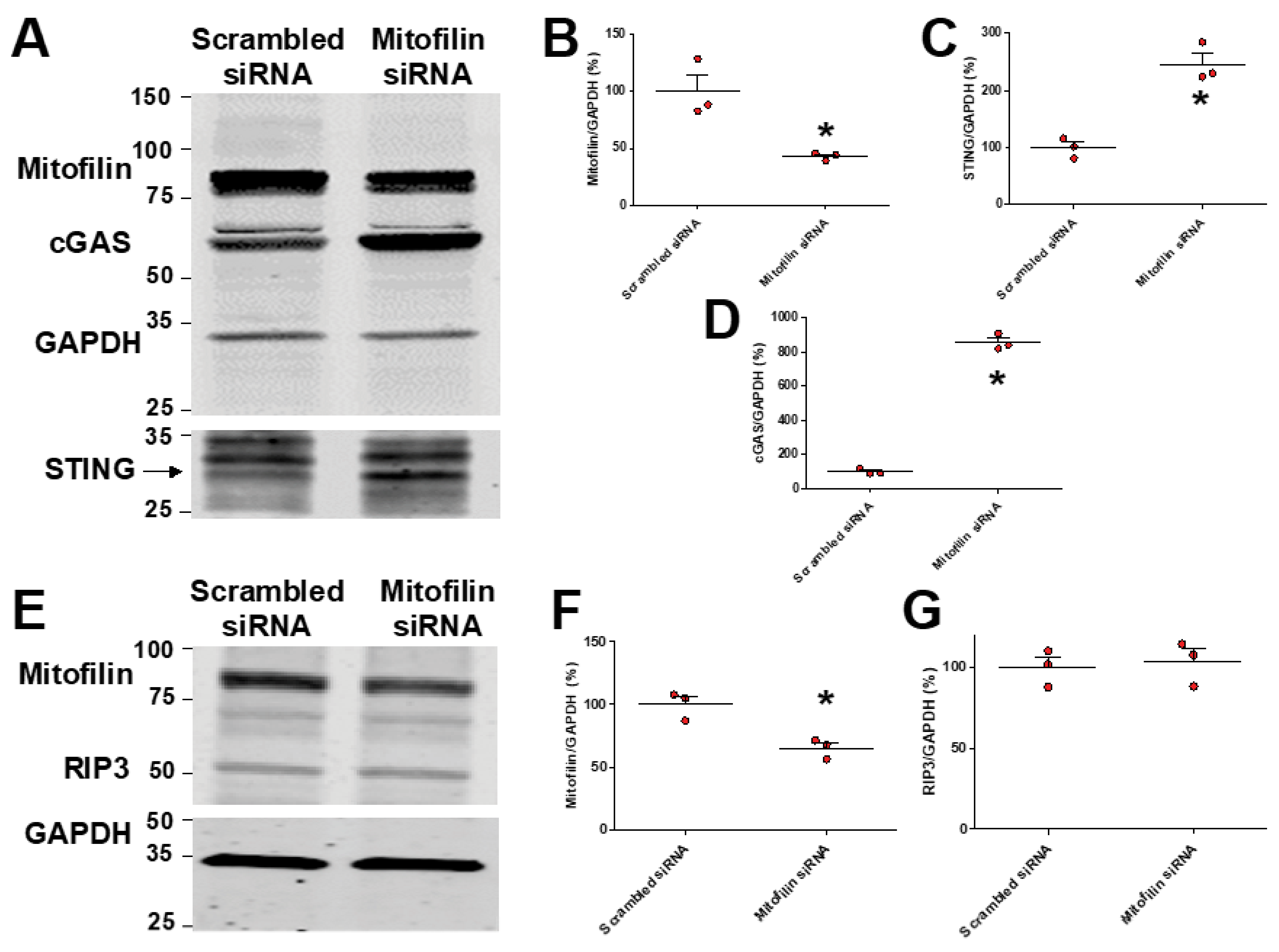
3.7. Mitofilin Knockdown in HK-2 Cells Activates cGAS–STING–p65 Pathway
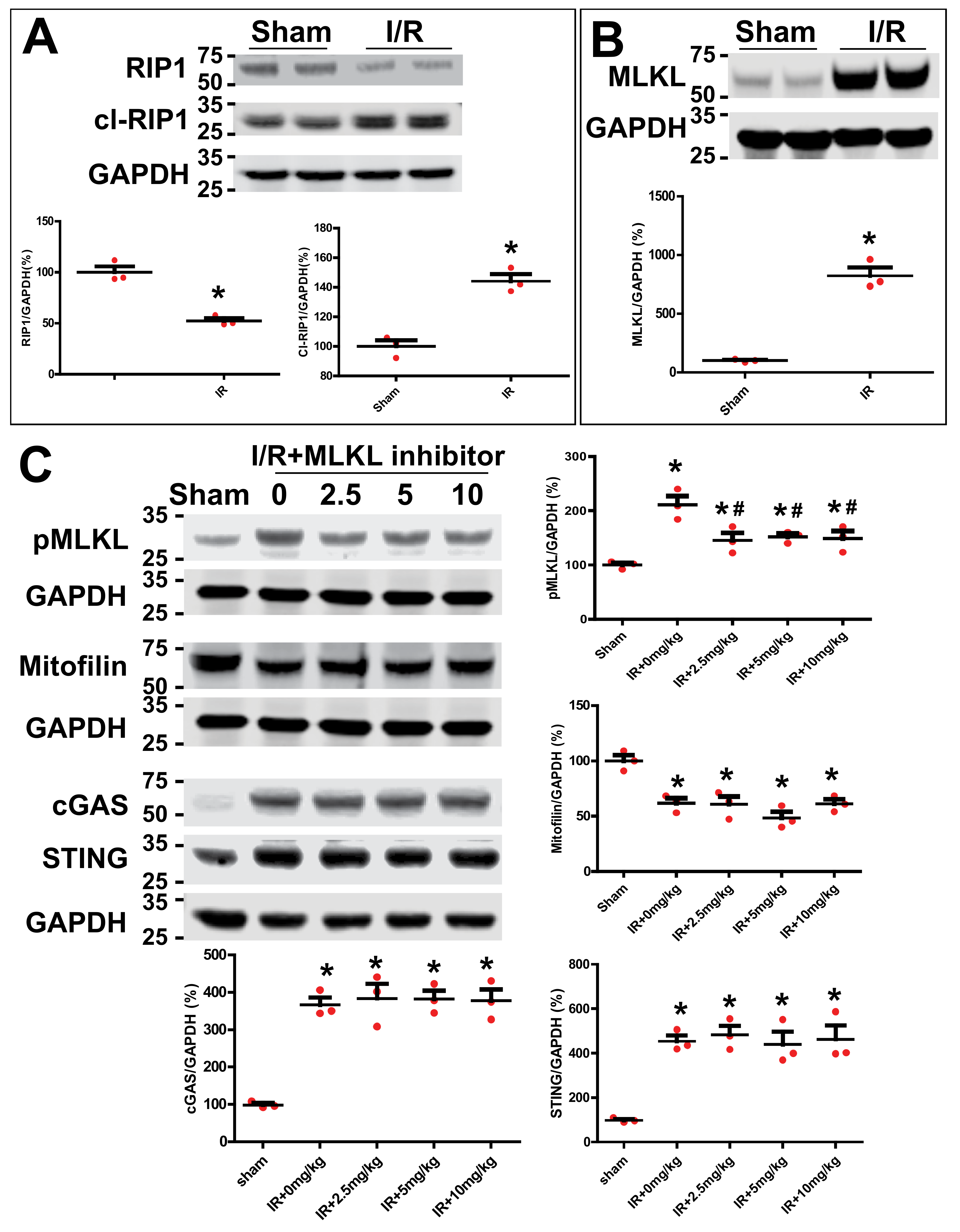
3.8. Inhibition of MLKL Does Not Affect Mitofilin Levels and cGAS–STING Pathway after Kidney IR Injury
4. Discussion
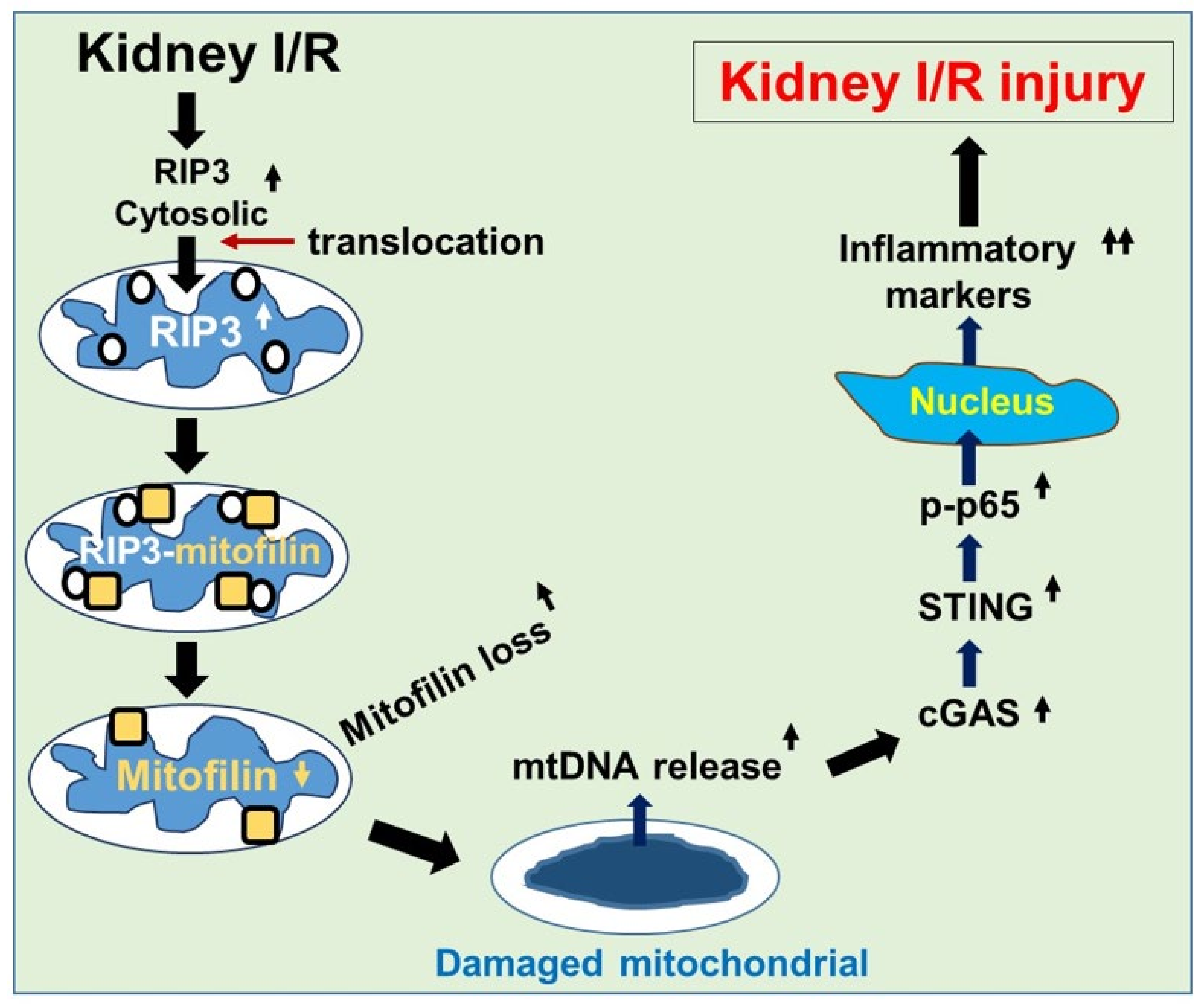
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IMMT, Mic60, Mitofilin | inner mitochondrial membrane protein |
| RIP3/RIPK3 | receptor-interacting protein kinase 3 |
| MICOS | mitochondrial contact site and cristae organizing system |
| MINOS | mitochondrial inner membrane organizing system |
| IMM | inner mitochondrial membrane |
| MLKL | mixed lineage kinase domain-like pseudokinase |
| mtDNA | mitochondrial deoxyribonucleic acid |
| ROS | reactive oxygen species |
| I/R | ischemia–reperfusion |
| mPTP | mitochondrial permeability transition pore |
| CRC | calcium retention capacity |
| MMP | mitochondrial membrane potential |
| CypD | cyclophilin D |
| cGAS | cyclic GMP–AMP synthase |
| STING | stimulator of interferon genes |
| IL-6 | interleukin 6 |
| ICAM | intercellular adhesion molecule 1 |
| p-p65 | phospho-p65 (P65 is one of the five components that form the NF-kB nuclear factor kappa-light-chain-enhancer of activated B cells. |
| AIM2 | absent in melanoma 2 |
| NLRP3 | NACHT, LRR, and PYD domains-containing protein 3 |
| TLR9 | Toll-like receptor 9 |
| ATP | adenosine triphosphate |
| HK-2 | human kidney 2 |
| CaMKII | calcium/calmodulin-dependent protein kinase II |
| H2O2 | hydrogen peroxide |
| EDTA | ethylenediaminetetraacetic acid is an aminopolycarboxylic acid |
| EGTA | ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| cDNA | complementary DNA |
References
- Moriwaki, K.; Chan, F.K. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013, 27, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.W.; Huang, B.C.; Shen, M.; Quast, J.; Chan, E.; Xu, X.; Nolan, G.P.; Payan, D.G.; Luo, Y. Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkappaB. Curr. Biol. 1999, 9, 539–542. [Google Scholar] [CrossRef]
- Weber, K.; Roelandt, R.; Bruggeman, I.; Estornes, Y.; Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 2018, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Miller, G. Molecular Pathways: The Necrosome-A Target for Cancer Therapy. Clin. Cancer. Res. 2017, 23, 1132–1136. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Chen, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Patino, E.; Ma, K.C.; Laursen, K.; Finkelsztein, E.J.; Akchurin, O.; Muthukumar, T.; Ryter, S.W.; Gudas, L.; Choi, A.M.K.; et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight 2018, 3, e98411. [Google Scholar] [CrossRef]
- Jennings, R.B. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ. Res. 2013, 113, 428–438. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Renal Inj. Prev. 2015, 4, 20–27. [Google Scholar] [PubMed]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Baetz, D.; Ovize, M. Cyclophilin D and myocardial ischemia-reperfusion injury: A fresh perspective. J. Mol. Cell Cardiol. 2015, 78, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Madungwe, N.B.; Bopassa, J.C. Mitochondrial inner membrane protein, Mic60/mitofilin in mammalian organ protection. J. Cell Physiol. 2019, 234, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Adachi, Y.; Fukaya, M.; Iijima, M.; Sesaki, H. Dynamin-Related Protein 1 Deficiency Leads to Receptor-Interacting Protein Kinase 3-Mediated Necroptotic Neurodegeneration. Am. J. Pathol. 2016, 186, 2798–2802. [Google Scholar] [CrossRef] [PubMed]
- Odgren, P.R.; Toukatly, G.; Bangs, P.L.; Gilmore, R.; Fey, E.G. Molecular characterization of mitofilin (HMP), a mitochondria-associated protein with predicted coiled coil and intermembrane space targeting domains. J. Cell Sci. 1996, 109, 2253–2264. [Google Scholar] [CrossRef]
- Icho, T.; Ikeda, T.; Matsumoto, Y.; Hanaoka, F.; Kaji, K.; Tsuchida, N. A novel human gene that is preferentially transcribed in heart muscle. Gene 1994, 144, 301–306. [Google Scholar]
- Zerbes, R.M.; van der Klei, I.J.; Veenhuis, M.; Pfanner, N.; van der Laan, M.; Bohnert, M. Mitofilin complexes: Conserved organizers of mitochondrial membrane architecture. Biol. Chem. 2012, 393, 1247–1261. [Google Scholar] [CrossRef]
- Von der Malsburg, K.; Muller, J.M.; Bohnert, M.; Oeljeklaus, S.; Kwiatkowska, P.; Becker, T.; Loniewska-Lwowska, A.; Wiese, S.; Rao, S.; Milenkovic, D.; et al. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell 2011, 21, 694–707. [Google Scholar] [CrossRef]
- Yang, R.F.; Zhao, G.W.; Liang, S.T.; Zhang, Y.; Sun, L.H.; Chen, H.Z.; Liu, D.P. Mitofilin regulates cytochrome c release during apoptosis by controlling mitochondrial cristae remodeling. Biochem. Biophys. Res. Commun. 2012, 428, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Thapa, D.; Nichols, C.E.; Lewis, S.E.; Shepherd, D.L.; Jagannathan, R.; Croston, T.L.; Tveter, K.J.; Holden, A.A.; Baseler, W.A.; Hollander, J.M. Transgenic overexpression of mitofilin attenuates diabetes mellitus-associated cardiac and mitochondria dysfunction. J. Mol. Cell Cardiol. 2015, 79, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Luo, Y.X.; An, X.Z.; Zhang, R.; Liu, G.; Li, H.; Chen, H.Z.; Liu, D.P. Overexpression of mitofilin in the mouse heart promotes cardiac hypertrophy in response to hypertrophic stimuli. Antioxid. Redox Signal. 2014, 21, 1693–1707. [Google Scholar] [CrossRef]
- Xue, R.Q.; Zhao, M.; Wu, Q.; Yang, S.; Cui, Y.L.; Yu, X.J.; Liu, J.; Zang, W.J. Regulation of mitochondrial cristae remodelling by acetylcholine alleviates palmitate-induced cardiomyocyte hypertrophy. Free Radic. Biol. Med. 2019, 145, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, D.; Gentili, V.; Caselli, E.; Sicolo, M.; Soffritti, I.; D’Accolti, M.; Barao, I.; Rotola, A.; Di Luca, D.; Rizzo, R. DNA Sensors’ Signaling in NK Cells During HHV-6A, HHV-6B and HHV-7 Infection. Front. Microbiol. 2020, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zen, K. Toll-Like Receptors Regulate the Development and Progression of Renal Diseases. Kidney Dis. 2021, 7, 14–23. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Lee, Y.L.; Obiako, B.; Gorodnya, O.M.; Ruchko, M.V.; Kuck, J.L.; Pastukh, V.M.; Wilson, G.L.; Simmons, J.D.; Gillespie, M.N. Mitochondrial DNA Damage Initiates Acute Lung Injury and Multi-Organ System Failure Evoked in Rats by Intra-Tracheal Pseudomonas Aeruginosa. Shock 2017, 48, 54–60. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Ren, H.; Wu, J.; Wu, X.; Liu, S.; Wang, G.; Gu, G.; Guo, K.; Li, J. Urinary Mitochondrial DNA Identifies Renal Dysfunction and Mitochondrial Damage in Sepsis-Induced Acute Kidney Injury. Oxid. Med. Cell Longev. 2018, 2018, 8074936. [Google Scholar] [CrossRef]
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201. [Google Scholar] [CrossRef] [PubMed]
- Van der Slikke, E.C.; Star, B.S.; van Meurs, M.; Henning, R.H.; Moser, J.; Bouma, H.R. Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-AKI. Crit. Care 2021, 25, 36. [Google Scholar] [CrossRef] [PubMed]
- Elefantova, K.; Lakatos, B.; Kubickova, J.; Sulova, Z.; Breier, A. Detection of the Mitochondrial Membrane Potential by the Cationic Dye JC-1 in L1210 Cells with Massive Overexpression of the Plasma Membrane ABCB1 Drug Transporter. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Imam Aliagan, A.D.; Ahwazi, M.D.; Tombo, N.; Feng, Y.; Bopassa, J.C. Parkin interacts with Mitofilin to increase dopaminergic neuron death in response to Parkinson’s disease-related stressors. Am. J. Transl. Res. 2020, 12, 7542–7564. [Google Scholar]
- Kabir, M.E.; Singh, H.; Lu, R.; Olde, B.; Leeb-Lundberg, L.M.; Bopassa, J.C. G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3beta Pathway after Ischemia/Reperfusion. PLoS ONE 2015, 10, e0135988. [Google Scholar] [CrossRef]
- Chen, H.; Fang, Y.; Wu, J.; Chen, H.; Zou, Z.; Zhang, X.; Shao, J.; Xu, Y. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. 2018, 9, 878. [Google Scholar] [CrossRef]
- Lau, A.; Wang, S.; Jiang, J.; Haig, A.; Pavlosky, A.; Linkermann, A.; Zhang, Z.X.; Jevnikar, A.M. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant 2013, 13, 2805–2818. [Google Scholar] [CrossRef]
- Madungwe, N.B.; Feng, Y.; Lie, M.; Tombo, N.; Liu, L.; Kaya, F.; Bopassa, J.C. Mitochondrial inner membrane protein (mitofilin) knockdown induces cell death by apoptosis via an AIF-PARP-dependent mechanism and cell cycle arrest. Am. J. Physiol. Cell Physiol. 2018, 315, C28–C43. [Google Scholar] [CrossRef]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct mPTP activation mechanisms in ischaemia-reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar] [CrossRef]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef]
- Pefanis, A.; Ierino, F.L.; Murphy, J.M.; Cowan, P.J. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. 2019, 96, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, J.N.; He, X.Y.; Suo, X.G.; Li, C.; Ni, W.J.; Cai, Y.T.; He, Y.; Fang, X.Y.; Dong, Y.H.; et al. Stratifin promotes renal dysfunction in ischemic and nephrotoxic AKI mouse models via enhancing RIPK3-mediated necroptosis. Acta Pharmacol. Sin. 2022, 43, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Coupienne, I.; Fettweis, G.; Rubio, N.; Agostinis, P.; Piette, J. 5-ALA-PDT induces RIP3-dependent necrosis in glioblastoma. Photochem. Photobiol. Sci. 2011, 10, 1868–1878. [Google Scholar] [CrossRef]
- Hockenbery, D.M.; Giedt, C.D.; O’Neill, J.W.; Manion, M.K.; Banker, D.E. Mitochondria and apoptosis: New therapeutic targets. Adv. Cancer Res. 2002, 85, 203–242. [Google Scholar]
- Rayamajhi, M.; Miao, E.A. The RIP1-RIP3 complex initiates mitochondrial fission to fuel NLRP3. Nat. Immunol. 2014, 15, 1100–1102. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, W.; Yan, Y.; Gong, T.; Han, J.; Tian, Z.; Zhou, R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014, 15, 1126–1133. [Google Scholar] [CrossRef]
- Alkhaja, A.K.; Jans, D.C.; Nikolov, M.; Vukotic, M.; Lytovchenko, O.; Ludewig, F.; Schliebs, W.; Riedel, D.; Urlaub, H.; Jakobs, S.; et al. MINOS1 is a conserved component of mitofilin complexes and required for mitochondrial function and cristae organization. Mol. Biol. Cell 2012, 23, 247–257. [Google Scholar] [CrossRef]
- Tombo, N.; Imam Aliagan, A.D.; Feng, Y.; Singh, H.; Bopassa, J.C. Cardiac ischemia/reperfusion stress reduces inner mitochondrial membrane protein (mitofilin) levels during early reperfusion. Free Radic. Biol. Med. 2020, 158, 181–194. [Google Scholar] [CrossRef]
- Magi, B.; Ettorre, A.; Liberatori, S.; Bini, L.; Andreassi, M.; Frosali, S.; Neri, P.; Pallini, V.; Di Stefano, A. Selectivity of protein carbonylation in the apoptotic response to oxidative stress associated with photodynamic therapy: A cell biochemical and proteomic investigation. Cell Death Differ. 2004, 11, 842–852. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Guo, J.; Hu, N.; Wang, S.; Li, D.; Hu, S.; Ren, J.; Cao, F.; Chen, Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017, 13, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, Y.; Zhang, Y. Mitochondrial reactive oxygen species cause major oxidative mitochondrial DNA damages and repair pathways. J. Biosci. 2020, 45, 84. [Google Scholar] [CrossRef]
- Khalifa, A.A.; Rashad, R.M.; El-Hadidy, W.F. Thymoquinone protects against cardiac mitochondrial DNA loss, oxidative stress, inflammation and apoptosis in isoproterenol-induced myocardial infarction in rats. Heliyon 2021, 7, e07561. [Google Scholar] [CrossRef]
- Qin, C.Y.; Zhang, H.W.; Gu, J.; Xu, F.; Liang, H.M.; Fan, K.J.; Shen, J.Y.; Xiao, Z.H.; Zhang, E.Y.; Hu, J. Mitochondrial DNAinduced inflammatory damage contributes to myocardial ischemia reperfusion injury in rats: Cardioprotective role of epigallocatechin. Mol. Med. Rep. 2017, 16, 7569–7576. [Google Scholar] [CrossRef]
- Feoktistova, M.; Leverkus, M. Programmed necrosis and necroptosis signalling. FEBS J. 2015, 282, 19–31. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, L.; Ma, X.; Wang, J.; Yang, J.; Zhou, X.; Yang, Y.; Liu, H. RIP1/RIP3/MLKL-mediated necroptosis contributes to vinblastine-induced myocardial damage. Mol. Cell Biochem. 2021, 476, 1233–1243. [Google Scholar] [CrossRef]
- Johnston, A.; Wang, Z. Necroptosis: MLKL Polymerization. J. Nat. Sci. 2018, 4, e513. [Google Scholar]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Y.; Imam Aliagan, A.; Tombo, N.; Draeger, D.; Bopassa, J.C. RIP3 Translocation into Mitochondria Promotes Mitofilin Degradation to Increase Inflammation and Kidney Injury after Renal Ischemia–Reperfusion. Cells 2022, 11, 1894. https://doi.org/10.3390/cells11121894
Feng Y, Imam Aliagan A, Tombo N, Draeger D, Bopassa JC. RIP3 Translocation into Mitochondria Promotes Mitofilin Degradation to Increase Inflammation and Kidney Injury after Renal Ischemia–Reperfusion. Cells. 2022; 11(12):1894. https://doi.org/10.3390/cells11121894
Chicago/Turabian StyleFeng, Yansheng, Abdulhafiz Imam Aliagan, Nathalie Tombo, Derrick Draeger, and Jean C. Bopassa. 2022. "RIP3 Translocation into Mitochondria Promotes Mitofilin Degradation to Increase Inflammation and Kidney Injury after Renal Ischemia–Reperfusion" Cells 11, no. 12: 1894. https://doi.org/10.3390/cells11121894