
Effects of Epigenetic Modification of PGC-1α by a Chemical Chaperon on Mitochondria Biogenesis and Visual Function in Retinitis Pigmentosa

 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
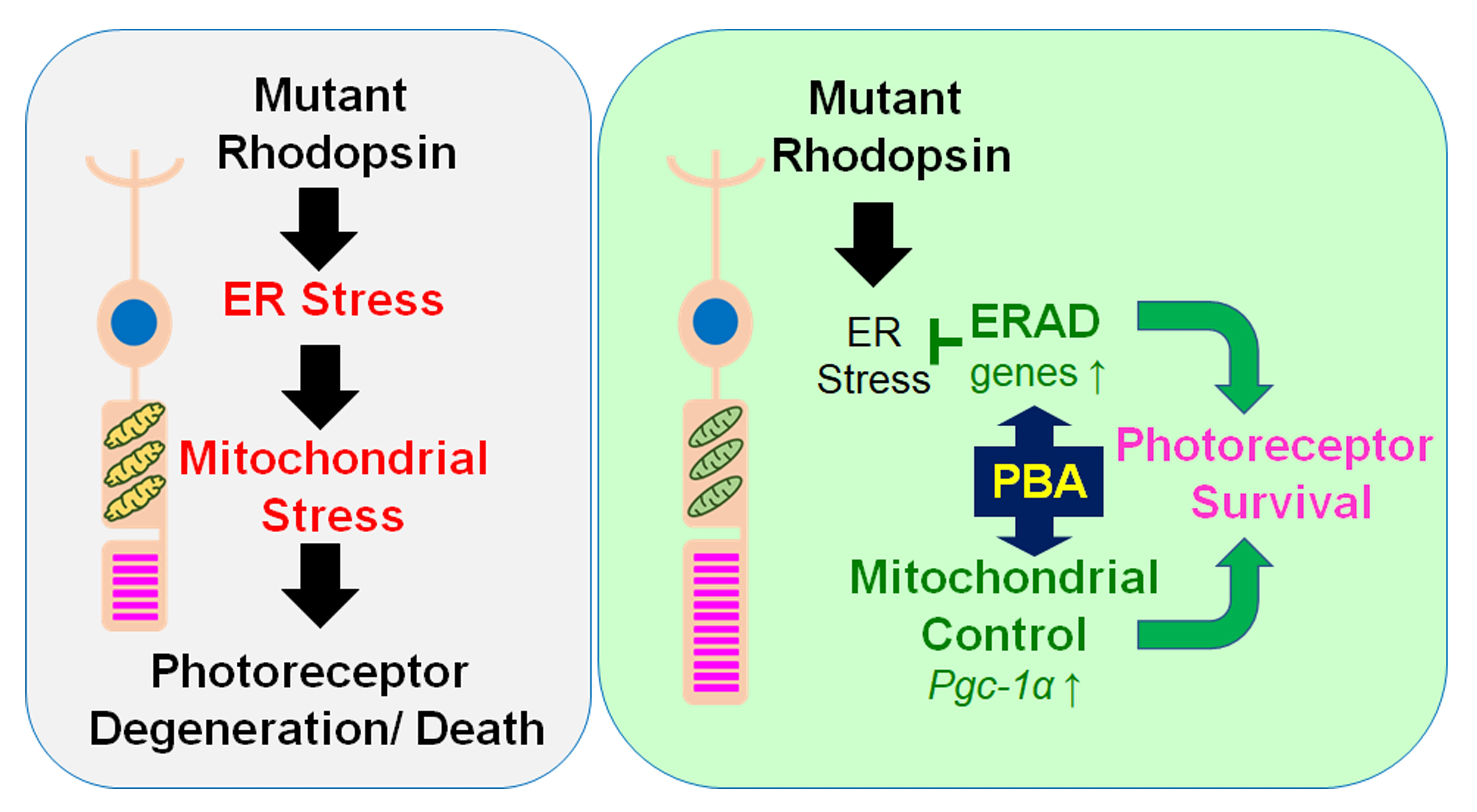
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Histological Analyses
2.3. Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.4. Electroretinography (ERG) Recordings
2.5. Cell Culture
2.6. Mitochondrial Membrane Potential Measurement
2.7. Cytochrome c Oxidase (CcO) Activity Measurement
2.8. ATP Measurement
2.9. Chromatin Immunoprecipitation Quantitative Real-Time PCR (ChIP-qPCR)
2.10. Statistical Analysis
3. Results
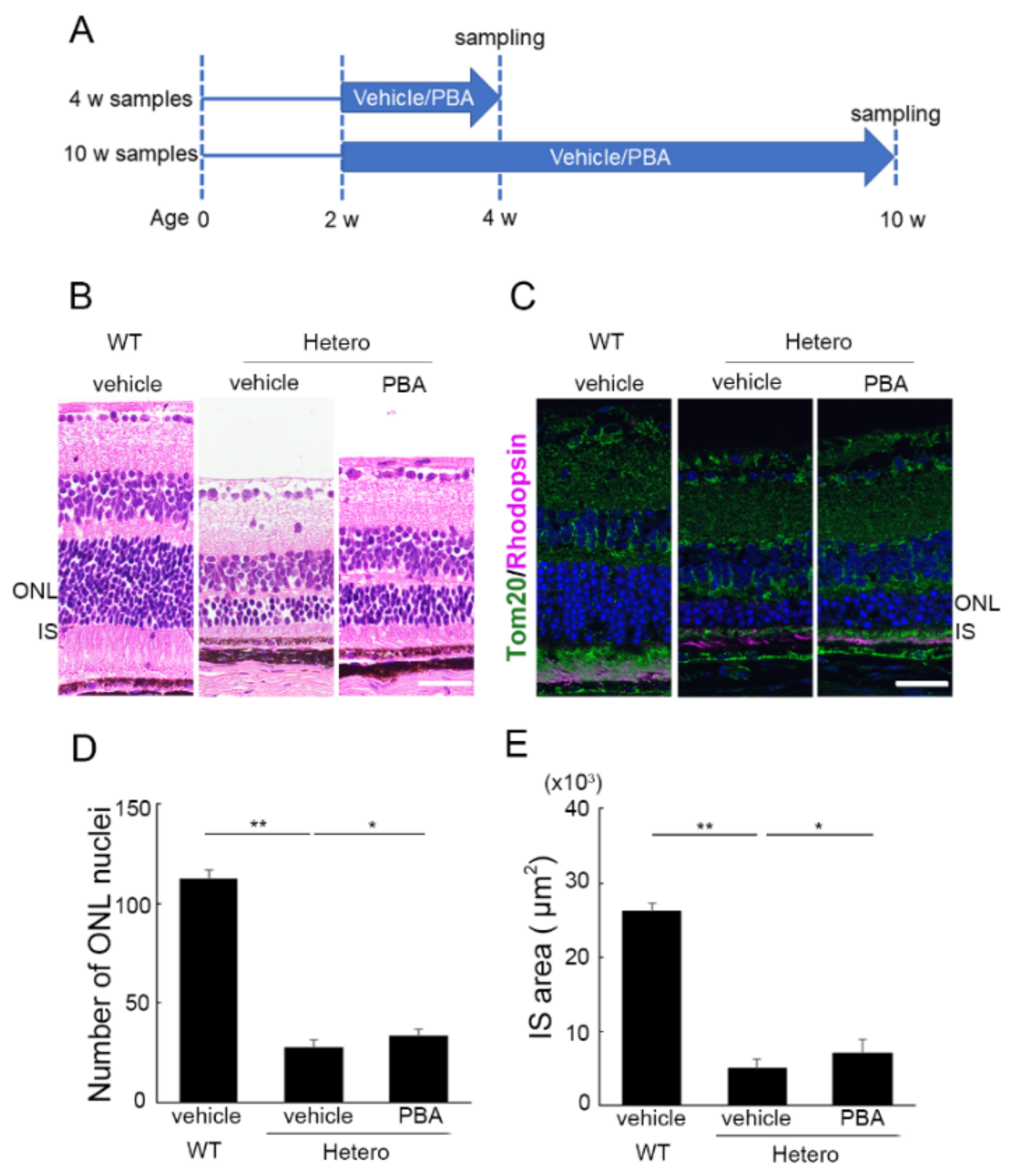
3.1. PBA Promoted Photoreceptor Survival in P23H Knock-In Heterozygotes (P23H RP Models)
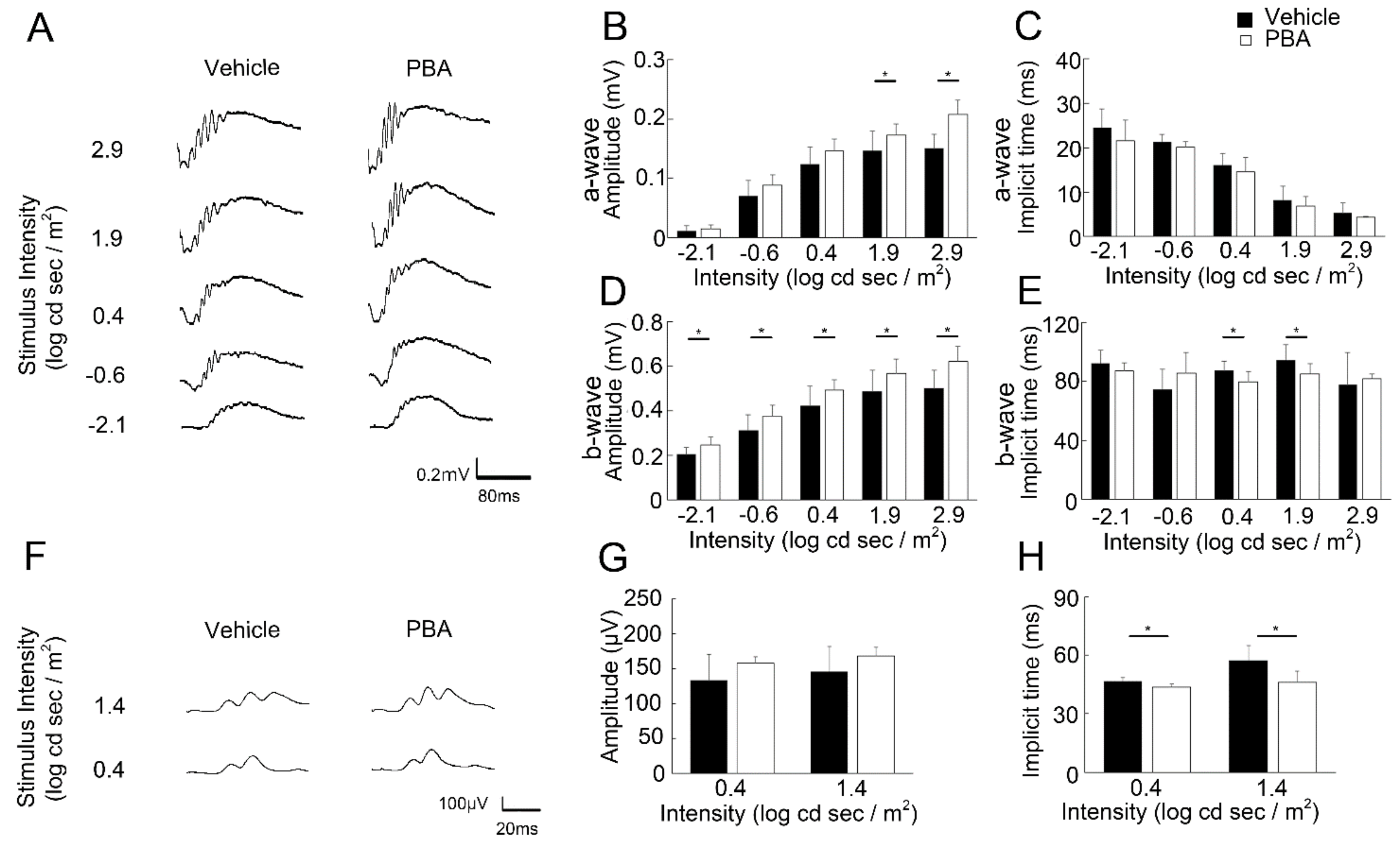
3.2. PBA Preserved Visual Function in the P23H RP Models
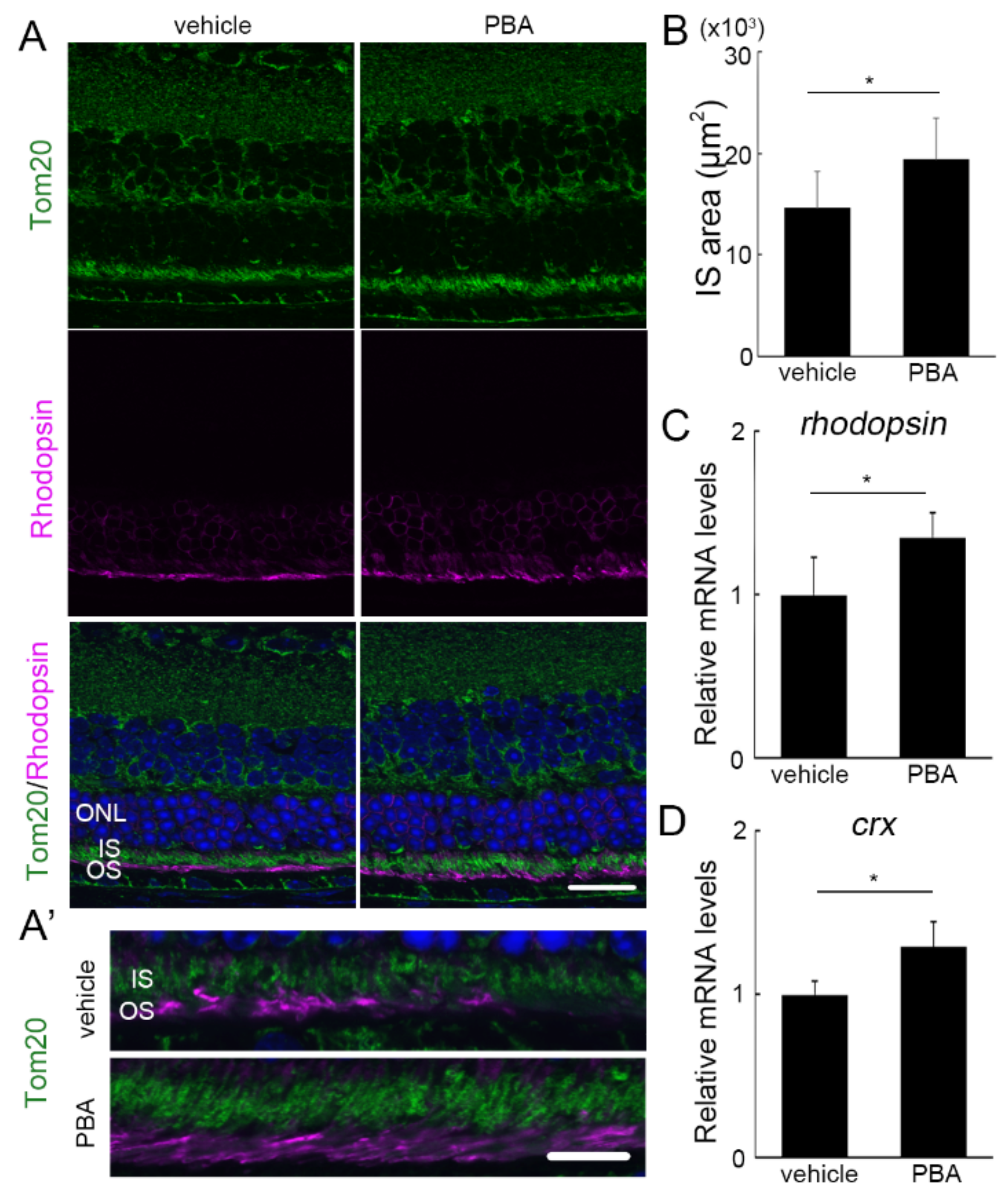
3.3. The Protective Effect of PBA Was Detected in the Photoreceptors of P23H RP Models before Photoreceptor Loss
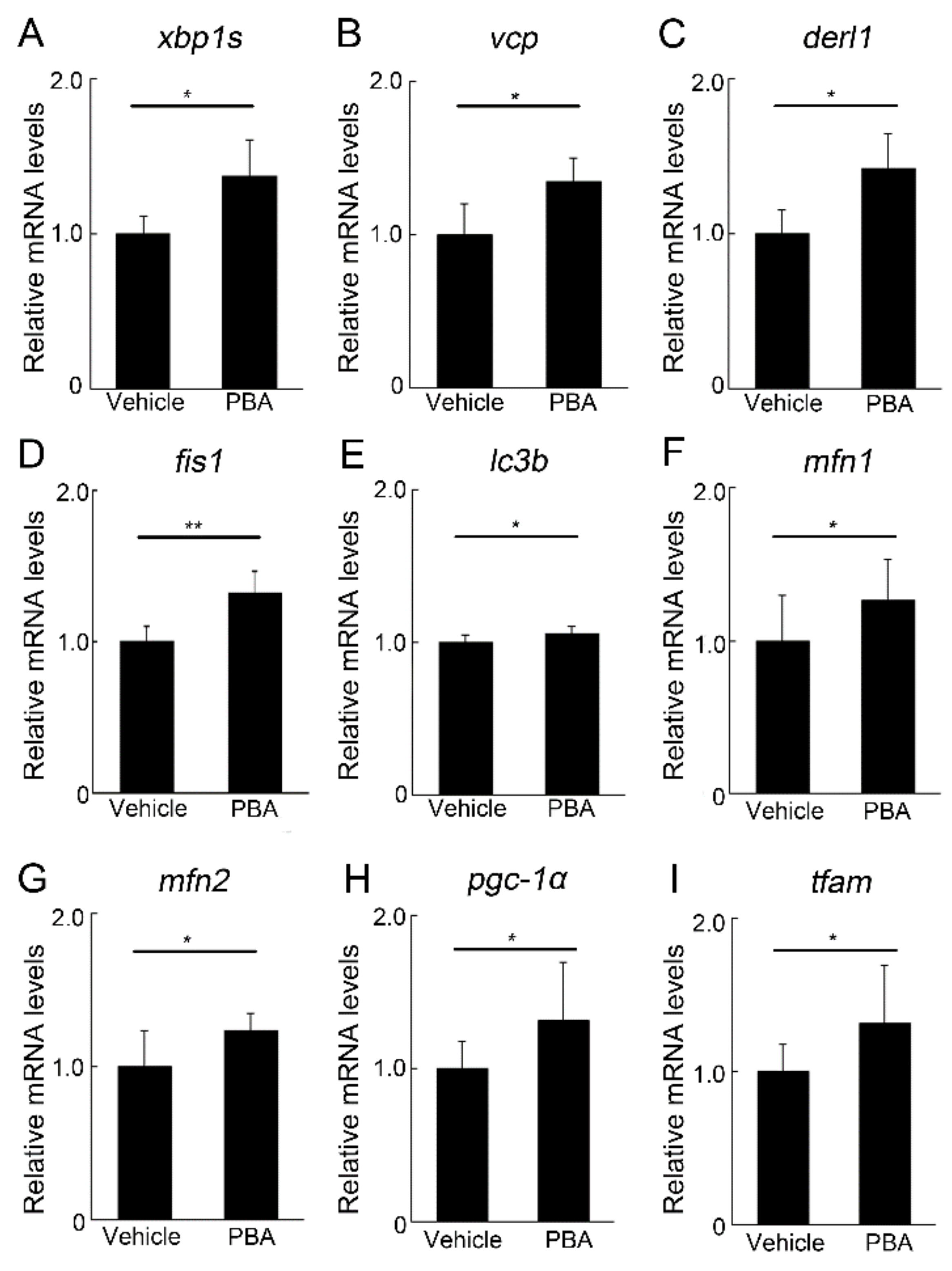
3.4. PBA Induced ERAD and Mitochondrial Markers in the Retina of P23H RP Models
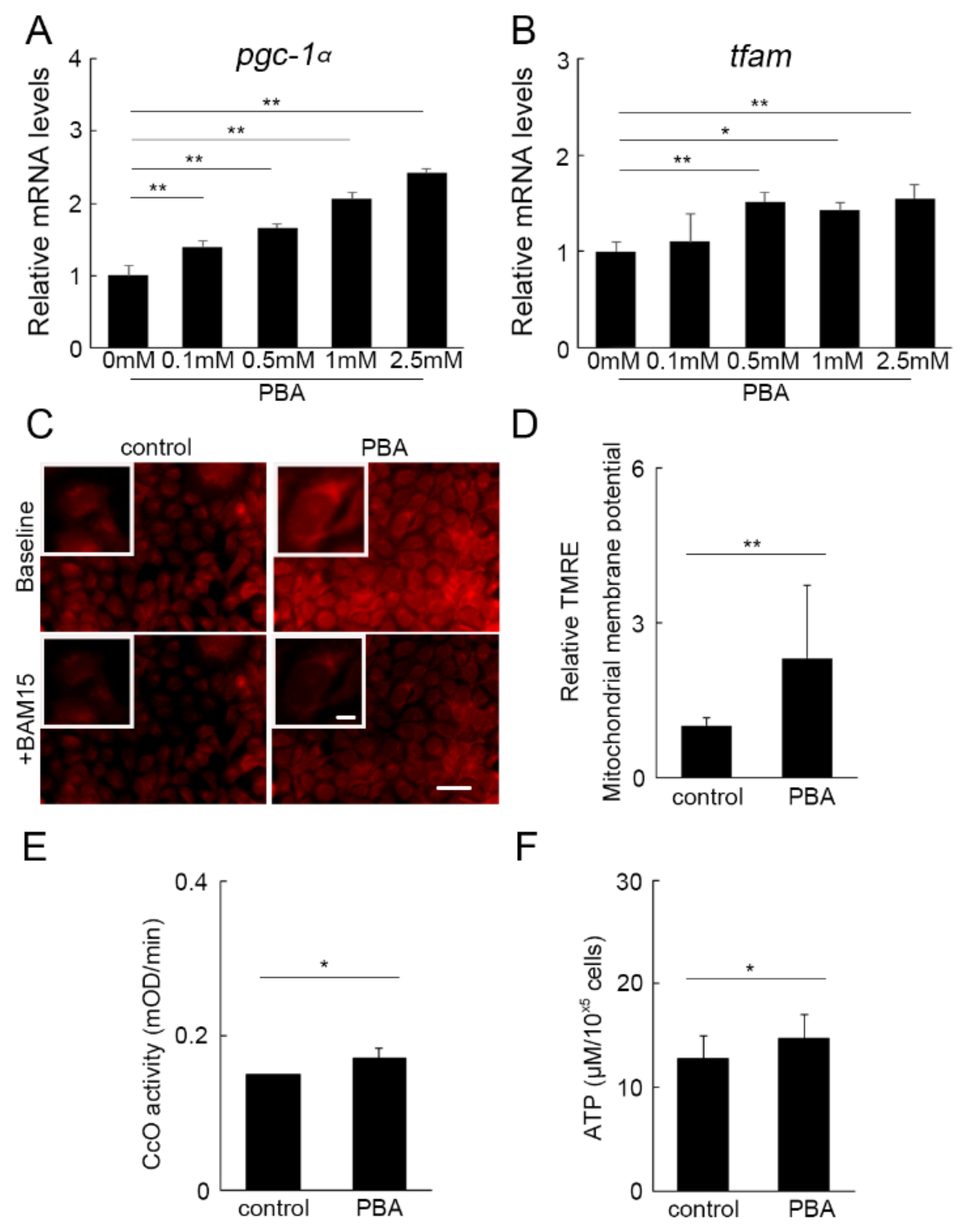
3.5. PBA Activated Oxidative Phosphorylation (OXPHOS)
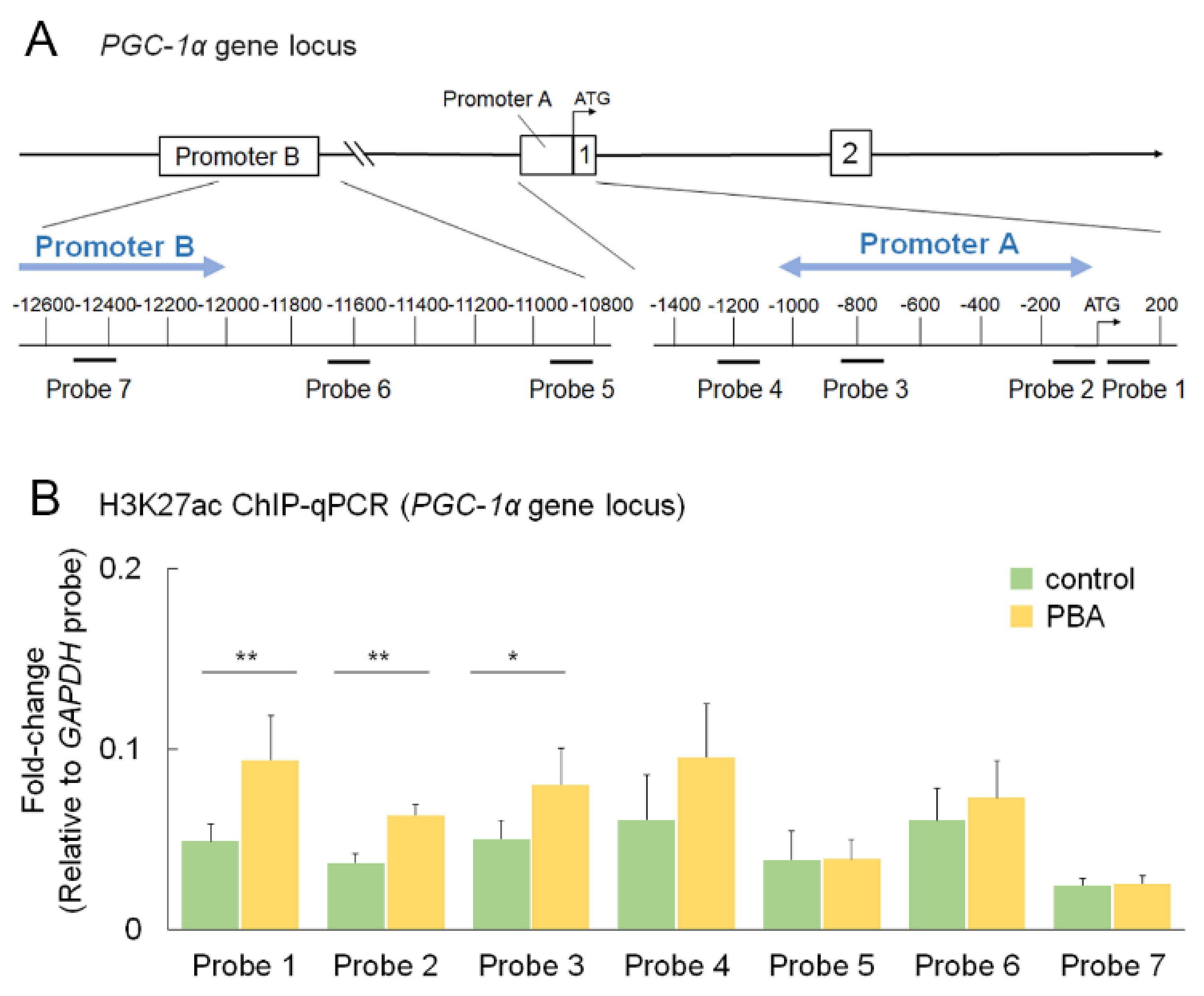
3.6. PBA Increased Histone Acetylation of the PGC-1α Promoter
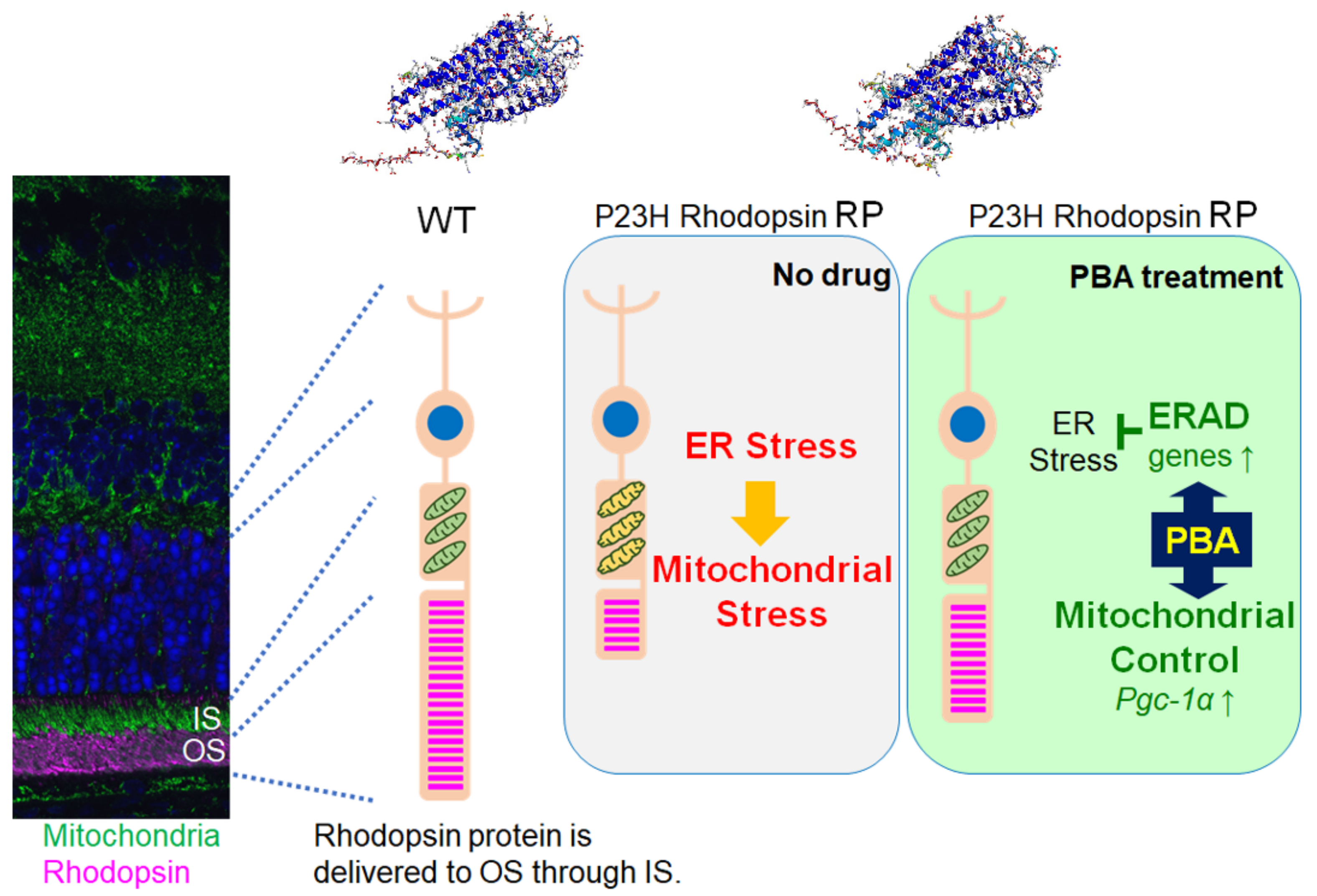
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morizane, Y.; Morimoto, N.; Fujiwara, A.; Kawasaki, R.; Yamashita, H.; Ogura, Y.; Shiraga, F. Incidence and causes of visual impairment in Japan: The first nation-wide complete enumeration survey of newly certified visually impaired individuals. Jpn. J. Ophthalmol. 2019, 63, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, S. Effect of rapamycin on the fate of P23H opsin associated with retinitis pigmentosa (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc. 2006, 104, 517–529. [Google Scholar] [PubMed]
- Yau, K.W.; Hardie, R.C. Phototransduction motifs and variations. Cell 2009, 139, 246–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, H.F.; van der Spuy, J.; Chapple, J.P.; Cheetham, M.E. Mechanisms of cell death in rhodopsin retinitis pigmentosa: Implications for therapy. Trends Mol. Med. 2005, 11, 177–185. [Google Scholar] [CrossRef]
- Yoshida, T.; Ozawa, Y.; Suzuki, K.; Yuki, K.; Ohyama, M.; Akamatsu, W.; Matsuzaki, Y.; Shimmura, S.; Mitani, K.; Tsubota, K.; et al. The use of induced pluripotent stem cells to reveal pathogenic gene mutations and explore treatments for retinitis pigmentosa. Mol. Brain 2014, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Behnen, P.; Felline, A.; Comitato, A.; Di Salvo, M.T.; Raimondi, F.; Gulati, S.; Kahremany, S.; Palczewski, K.; Marigo, V.; Fanelli, F. A Small Chaperone Improves Folding and Routing of Rhodopsin Mutants Linked to Inherited Blindness. iScience 2018, 4, 1–19. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Knox, T.; LaVail, M.M.; Gorbatyuk, O.S.; Noorwez, S.M.; Hauswirth, W.W.; Lin, J.H.; Muzyczka, N.; Lewin, A.S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. USA 2010, 107, 5961–5966. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Komarya, S.K.; Jena, G. Phenylbutyrate and beta-cell function: Contribution of histone deacetylases and ER stress inhibition. Epigenomics 2017, 9, 711–720. [Google Scholar] [CrossRef]
- Kolb, P.S.; Ayaub, E.A.; Zhou, W.; Yum, V.; Dickhout, J.G.; Ask, K. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int. J. Biochem. Cell Biol. 2015, 61, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Yao, J.; Jia, L.; Thompson, D.A.; Zacks, D.N. Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: A novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 2019, 10, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, R.C.; Egan, M.E.; Zeitlin, P.L. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J. Clin. Investig. 1997, 100, 2457–2465. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.M.; Chen, Y.J.; Ouyang, H.J. Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem. J. 2011, 433, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Ye, Y. Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nat. Struct. Mol. Biol. 2014, 21, 325–335. [Google Scholar] [CrossRef]
- Claessen, J.H.; Kundrat, L.; Ploegh, H.L. Protein quality control in the ER: Balancing the ubiquitin checkbook. Trends Cell Biol. 2012, 22, 22–32. [Google Scholar] [CrossRef]
- Guzman Mendoza, N.A.; Homma, K.; Osada, H.; Toda, E.; Ban, N.; Nagai, N.; Negishi, K.; Tsubota, K.; Ozawa, Y. Neuroprotective Effect of 4-Phenylbutyric Acid against Photo-Stress in the Retina. Antioxidants 2021, 10, 1147. [Google Scholar] [CrossRef]
- Chiang, W.C.; Kroeger, H.; Sakami, S.; Messah, C.; Yasumura, D.; Matthes, M.T.; Coppinger, J.A.; Palczewski, K.; LaVail, M.M.; Lin, J.H. Robust Endoplasmic Reticulum-Associated Degradation of Rhodopsin Precedes Retinal Degeneration. Mol. Neurobiol. 2015, 52, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Sakami, S.; Maeda, T.; Bereta, G.; Okano, K.; Golczak, M.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G.; Palczewski, K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem. 2011, 286, 10551–10567. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, H.; Ozawa, Y.; Toda, E.; Homma, K.; Osada, H.; Narimatsu, T.; Nagai, N.; Tsubota, K. Neuroprotective and vision-protective effect of preserving ATP levels by AMPK activator. FASEB J. 2020, 34, 5016–5026. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, T.; Ozawa, Y.; Miyake, S.; Kubota, S.; Yuki, K.; Nagai, N.; Tsubota, K. Biological effects of blocking blue and other visible light on the mouse retina. Clin. Exp. Ophthalmol. 2014, 42, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narimatsu, T.; Ozawa, Y.; Miyake, S.; Nagai, N.; Tsubota, K. Angiotensin II type 1 receptor blockade suppresses light-induced neural damage in the mouse retina. Free Radic. Biol. Med. 2014, 71, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, H.; Okamoto, T.; Kawashima, H.; Toda, E.; Miyake, S.; Nagai, N.; Kobayashi, S.; Tsubota, K.; Ozawa, Y. Neuroprotective effect of bilberry extract in a murine model of photo-stressed retina. PLoS ONE 2017, 12, e0178627. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Kawashima, H.; Toda, E.; Homma, K.; Osada, H.; Guzman, N.A.; Shibata, S.; Uchiyama, Y.; Okano, H.; Tsubota, K.; et al. Renin-angiotensin system impairs macrophage lipid metabolism to promote age-related macular degeneration in mouse models. Commun. Biol. 2020, 3, 767. [Google Scholar] [CrossRef] [PubMed]
- Osada, H.; Toda, E.; Homma, K.; Guzman, N.A.; Nagai, N.; Ogawa, M.; Negishi, K.; Arita, M.; Tsubota, K.; Ozawa, Y. ADIPOR1 deficiency-induced suppression of retinal ELOVL2 and docosahexaenoic acid levels during photoreceptor degeneration and visual loss. Cell Death Dis. 2021, 12, 458. [Google Scholar] [CrossRef]
- Ozawa, Y.; Toda, E.; Kawashima, H.; Homma, K.; Osada, H.; Nagai, N.; Abe, Y.; Yasui, M.; Tsubota, K. Aquaporin 4 Suppresses Neural Hyperactivity and Synaptic Fatigue and Fine-Tunes Neurotransmission to Regulate Visual Function in the Mouse Retina. Mol. Neurobiol. 2019, 56, 8124–8135. [Google Scholar] [CrossRef] [Green Version]
- Gorbatyuk, M.S.; Gorbatyuk, O.S.; LaVail, M.M.; Lin, J.H.; Hauswirth, W.W.; Lewin, A.S. Functional rescue of P23H rhodopsin photoreceptors by gene delivery. Adv. Exp. Med. Biol. 2012, 723, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, H.; Messah, C.; Ahern, K.; Gee, J.; Joseph, V.; Matthes, M.T.; Yasumura, D.; Gorbatyuk, M.S.; Chiang, W.C.; LaVail, M.M.; et al. Induction of endoplasmic reticulum stress genes, BiP and chop, in genetic and environmental models of retinal degeneration. Invest. Ophthalmol. Vis. Sci. 2012, 53, 7590–7599. [Google Scholar] [CrossRef] [Green Version]
- Ohgane, K.; Dodo, K.; Hashimoto, Y. Retinobenzaldehydes as proper-trafficking inducers of folding-defective P23H rhodopsin mutant responsible for retinitis pigmentosa. Bioorg. Med. Chem. 2010, 18, 7022–7028. [Google Scholar] [CrossRef]
- Gragg, M.; Park, P.S. Misfolded rhodopsin mutants display variable aggregation properties. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Harlen, K.M.; Roush, E.C.; Clayton, J.E.; Martinka, S.; Hughes, T.E. Live-Cell Assays for Cell Stress Responses Reveal New Patterns of Cell Signaling Caused by Mutations in Rhodopsin, alpha-Synuclein and TDP-43. Front. Cell Neurosci. 2019, 13, 535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfitt, D.A.; Aguila, M.; McCulley, C.H.; Bevilacqua, D.; Mendes, H.F.; Athanasiou, D.; Novoselov, S.S.; Kanuga, N.; Munro, P.M.; Coffey, P.J.; et al. The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis. 2014, 5, e1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenech, E.J.; Lari, F.; Charles, P.D.; Fischer, R.; Laetitia-Thezenas, M.; Bagola, K.; Paton, A.W.; Paton, J.C.; Gyrd-Hansen, M.; Kessler, B.M.; et al. Interaction mapping of endoplasmic reticulum ubiquitin ligases identifies modulators of innate immune signalling. eLife 2020, 9, e57306. [Google Scholar] [CrossRef] [PubMed]
- Ravanelli, S.; den Brave, F.; Hoppe, T. Mitochondrial Quality Control Governed by Ubiquitin. Front. Cell Dev. Biol. 2020, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.; Kevei, E.; Hoppe, T. Double-edged alliance: Mitochondrial surveillance by the UPS and autophagy. Curr. Opin. Cell Biol. 2015, 37, 18–27. [Google Scholar] [CrossRef]
- Farmer, T.; Naslavsky, N.; Caplan, S. Tying trafficking to fusion and fission at the mighty mitochondria. Traffic 2018, 19, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnaune-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef]
- Zhou, H.; Ren, J.; Toan, S.; Mui, D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res. Rev. 2021, 66, 101250. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, Y. Oxidative stress in the light-exposed retina and its implication in age-related macular degeneration. Redox. Biol. 2020, 37, 101779. [Google Scholar] [CrossRef] [PubMed]
- Hata, M.; Ikeda, H.O. Modulation of valosin-containing protein by Kyoto University Substances (KUS) as a novel therapeutic strategy for ischemic neuronal diseases. Neural. Regen. Res. 2017, 12, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Lochmann, T.L.; Thomas, R.R.; Bennett, J.P., Jr.; Taylor, S.M. Epigenetic Modifications of the PGC-1alpha Promoter during Exercise Induced Expression in Mice. PLoS ONE 2015, 10, e0129647. [Google Scholar] [CrossRef]
- Tam, B.M.; Moritz, O.L. Characterization of rhodopsin P23H-induced retinal degeneration in a Xenopus laevis model of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2006, 47, 3234–3241. [Google Scholar] [CrossRef] [Green Version]
- Athanasiou, D.; Aguila, M.; Opefi, C.A.; South, K.; Bellingham, J.; Bevilacqua, D.; Munro, P.M.; Kanuga, N.; Mackenzie, F.E.; Dubis, A.M.; et al. Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor degeneration. Hum. Mol. Genet. 2017, 26, 305–319. [Google Scholar] [CrossRef]
- Adekeye, A.; Haeri, M.; Solessio, E.; Knox, B.E. Ablation of the proapoptotic genes CHOP or Ask1 does not prevent or delay loss of visual function in a P23H transgenic mouse model of retinitis pigmentosa. PLoS ONE 2014, 9, e83871. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Kanuga, N.; Adamson, P.; Cheetham, M.E. The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2017, 26, 4896–4905. [Google Scholar] [CrossRef] [Green Version]
- Comitato, A.; Schiroli, D.; Montanari, M.; Marigo, V. Calpain Activation Is the Major Cause of Cell Death in Photoreceptors Expressing a Rhodopsin Misfolding Mutation. Mol. Neurobiol. 2020, 57, 589–599. [Google Scholar] [CrossRef]
- Thangaraj, A.; Sil, S.; Tripathi, A.; Chivero, E.T.; Periyasamy, P.; Buch, S. Targeting endoplasmic reticulum stress and autophagy as therapeutic approaches for neurological diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 285–325. [Google Scholar] [CrossRef]
- Wen, R.H.; Loewen, A.D.; Vent-Schmidt, R.Y.J.; Moritz, O.L. Autophagy Induction by HDAC Inhibitors Is Unlikely to be the Mechanism of Efficacy in Prevention of Retinal Degeneration Caused by P23H Rhodopsin. Adv. Exp. Med. Biol. 2019, 1185, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschlager, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Roux, M.J.; Merl, J.; Giangrande, A.; Hauck, S.M.; Aron, L.; Ueffing, M. Proteomic survey reveals altered energetic patterns and metabolic failure prior to retinal degeneration. J. Neurosci. 2014, 34, 2797–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, N.; Ikeda, H.O.; Hasegawa, T.; Muraoka, Y.; Iwai, S.; Tsuruyama, T.; Nakano, M.; Fuchigami, T.; Shudo, T.; Kakizuka, A.; et al. Neuroprotective effects of VCP modulators in mouse models of glaucoma. Heliyon 2016, 2, e00096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, M.; Ikeda, H.O.; Kikkawa, C.; Iwai, S.; Muraoka, Y.; Hasegawa, T.; Kakizuka, A.; Yoshimura, N. KUS121, a VCP modulator, attenuates ischemic retinal cell death via suppressing endoplasmic reticulum stress. Sci. Rep. 2017, 7, 44873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef] [Green Version]
- Woo, Y.; Lee, H.J.; Jung, Y.M.; Jung, Y.J. mTOR-Mediated Antioxidant Activation in Solid Tumor Radioresistance. J. Oncol. 2019, 2019, 5956867. [Google Scholar] [CrossRef] [Green Version]
- Parra, M. Class IIa HDACs-new insights into their functions in physiology and pathology. FEBS J. 2015, 282, 1736–1744. [Google Scholar] [CrossRef]
- Czubryt, M.P.; McAnally, J.; Fishman, G.I.; Olson, E.N. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc. Natl. Acad. Sci. USA 2003, 100, 1711–1716. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Sancho-Pelluz, J.; Alavi, M.V.; Sahaboglu, A.; Kustermann, S.; Farinelli, P.; Azadi, S.; van Veen, T.; Romero, F.J.; Paquet-Durand, F.; Ekstrom, P. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 2010, 1, e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leveillard, T.; Mohand-Said, S.; Lorentz, O.; Hicks, D.; Fintz, A.C.; Clerin, E.; Simonutti, M.; Forster, V.; Cavusoglu, N.; Chalmel, F.; et al. Identification and characterization of rod-derived cone viability factor. Nat. Genet. 2004, 36, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Ait-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clerin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridlich, R.; Delalande, F.; Jaillard, C.; Lu, J.; Poidevin, L.; Cronin, T.; Perrocheau, L.; Millet-Puel, G.; Niepon, M.L.; Poch, O.; et al. The thioredoxin-like protein rod-derived cone viability factor (RdCVFL) interacts with TAU and inhibits its phosphorylation in the retina. Mol. Cell Proteom. 2009, 8, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Mohand-Said, S.; Danan, A.; Simonutti, M.; Fontaine, V.; Clerin, E.; Picaud, S.; Leveillard, T.; Sahel, J.A. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol. Ther. 2009, 17, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Aron, L.; Ueffing, M. ER stress in retinal degeneration: A target for rational therapy? Trends Mol. Med. 2011, 17, 442–451. [Google Scholar] [CrossRef]
- Sankrityayan, H.; Oza, M.J.; Kulkarni, Y.A.; Mulay, S.R.; Gaikwad, A.B. ER stress response mediates diabetic microvascular complications. Drug Discov. Today 2019, 24, 2247–2257. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozawa, Y.; Toda, E.; Homma, K.; Osada, H.; Nagai, N.; Tsubota, K.; Okano, H. Effects of Epigenetic Modification of PGC-1α by a Chemical Chaperon on Mitochondria Biogenesis and Visual Function in Retinitis Pigmentosa. Cells 2022, 11, 1497. https://doi.org/10.3390/cells11091497
Ozawa Y, Toda E, Homma K, Osada H, Nagai N, Tsubota K, Okano H. Effects of Epigenetic Modification of PGC-1α by a Chemical Chaperon on Mitochondria Biogenesis and Visual Function in Retinitis Pigmentosa. Cells. 2022; 11(9):1497. https://doi.org/10.3390/cells11091497
Chicago/Turabian StyleOzawa, Yoko, Eriko Toda, Kohei Homma, Hideto Osada, Norihiro Nagai, Kazuo Tsubota, and Hideyuki Okano. 2022. "Effects of Epigenetic Modification of PGC-1α by a Chemical Chaperon on Mitochondria Biogenesis and Visual Function in Retinitis Pigmentosa" Cells 11, no. 9: 1497. https://doi.org/10.3390/cells11091497