
Inhibitory Neurotransmission Is Sex-Dependently Affected by Tat Expression in Transgenic Mice and Suppressed by the Fatty Acid Amide Hydrolase Enzyme Inhibitor PF3845 via Cannabinoid Type-1 Receptor Mechanisms

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Slice Electrophysiology Ex Vivo
2.2.1. Prefrontal Cortex (PFC) Slices
2.2.2. Electrophysiological Recordings
2.2.3. Acquisition and Analysis of Synaptic Currents
2.3. Treatments and Drugs
2.4. Western Blot Analysis
2.5. Analysis of Endocannabinoids (eCBs) and Other Lipids
2.6. Statistical Analysis
3. Results
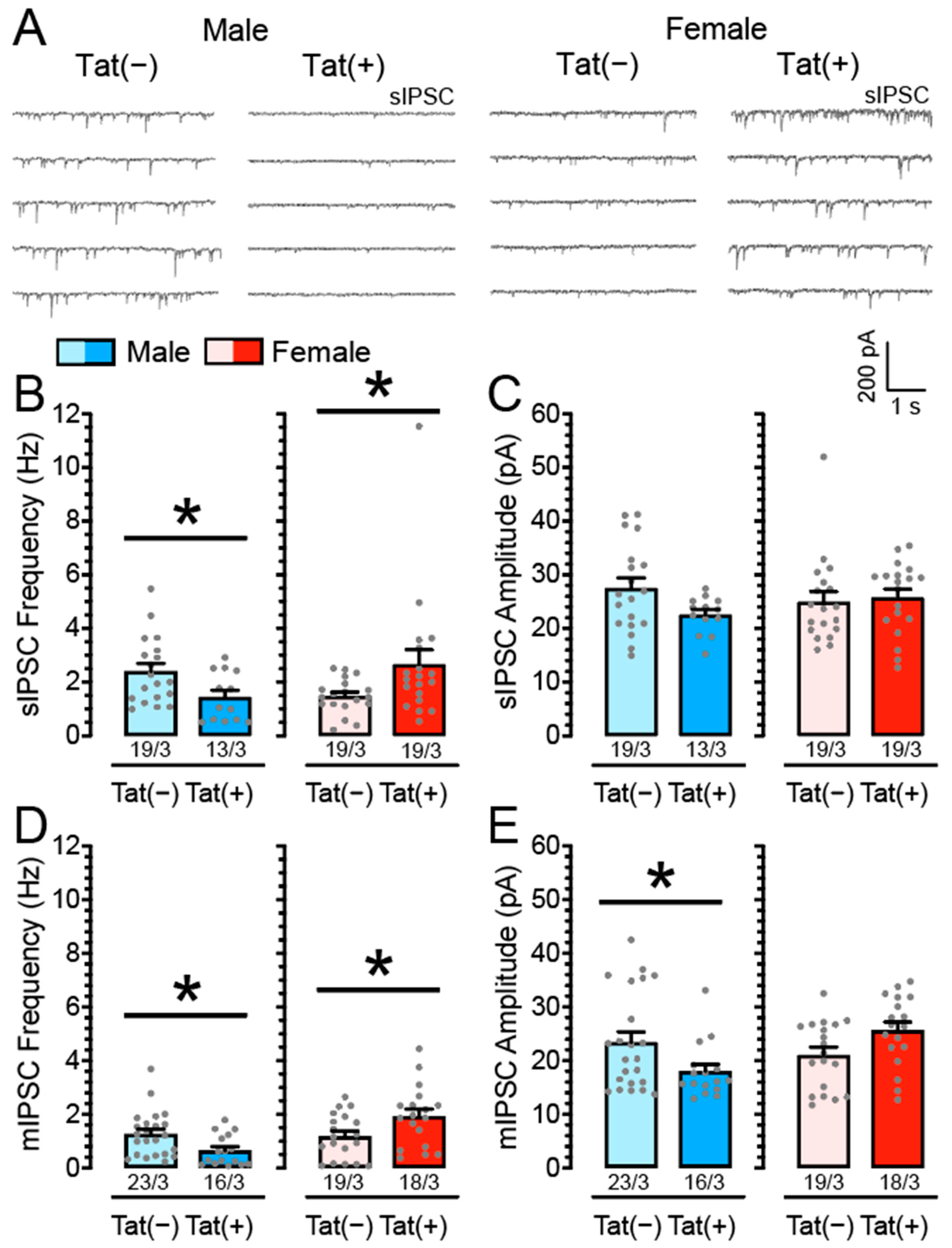
3.1. Inhibitory GABAergic Neurotransmission Is Altered in Tat Transgenic mPFC Brain Slices in a Sex-Dependent Manner
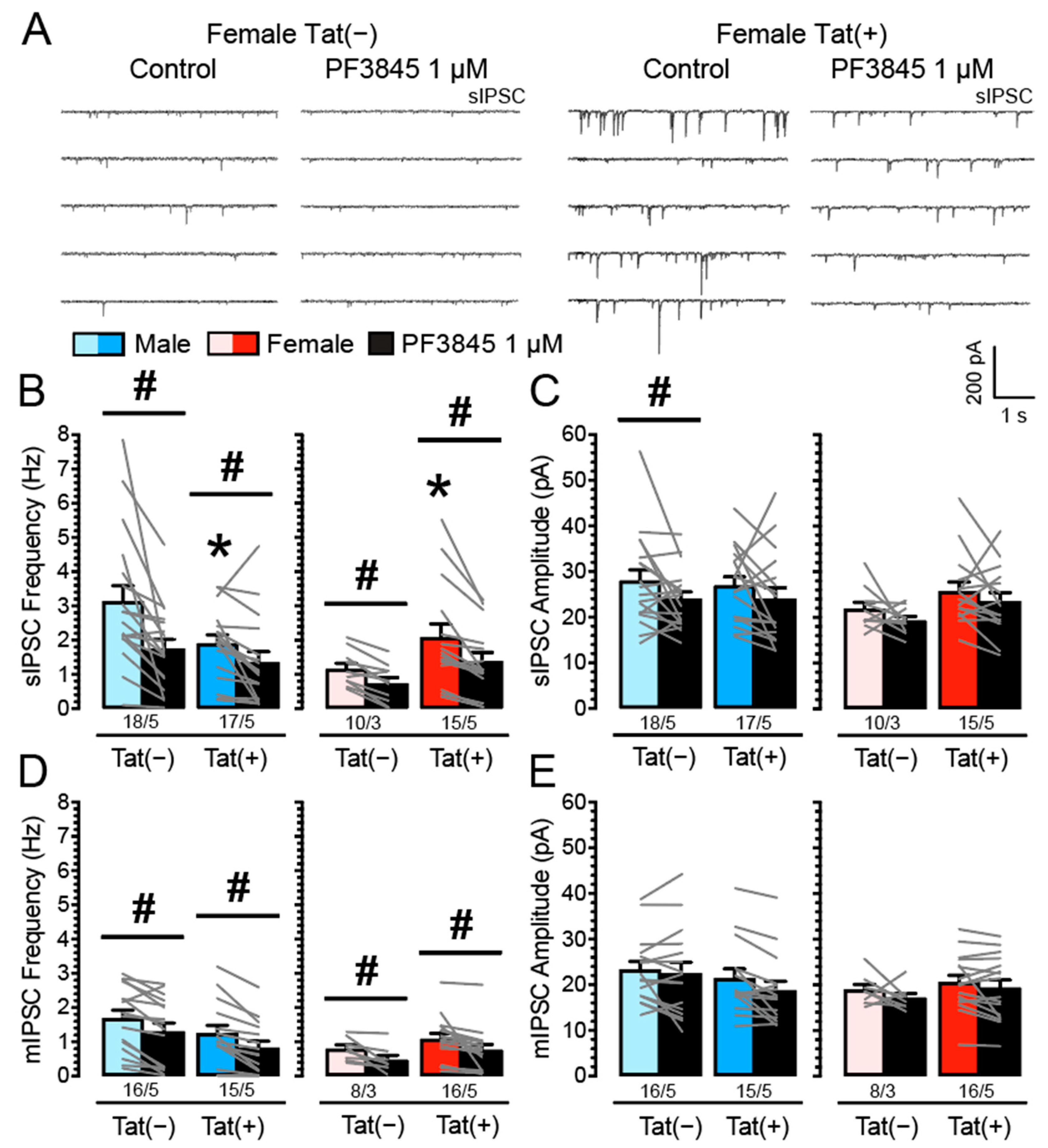
3.2. PF3845 Decreases Inhibitory GABAergic Neurotransmission Independent of Tat Induction and Sex
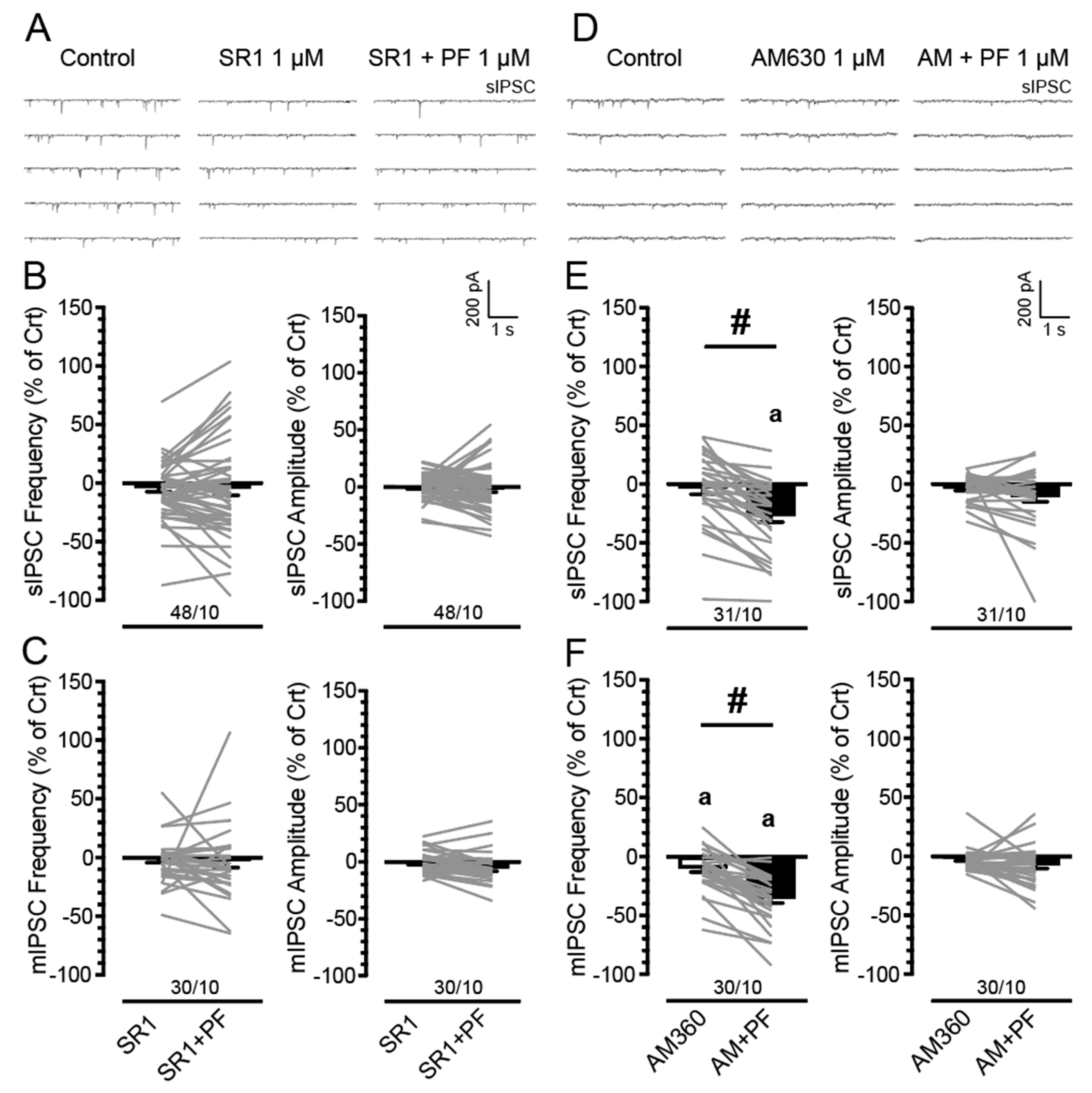
3.3. PF3845′s Inhibitory Effects on GABAergic Neurotransmission Are Mediated by CB1Rs but Not CB2Rs
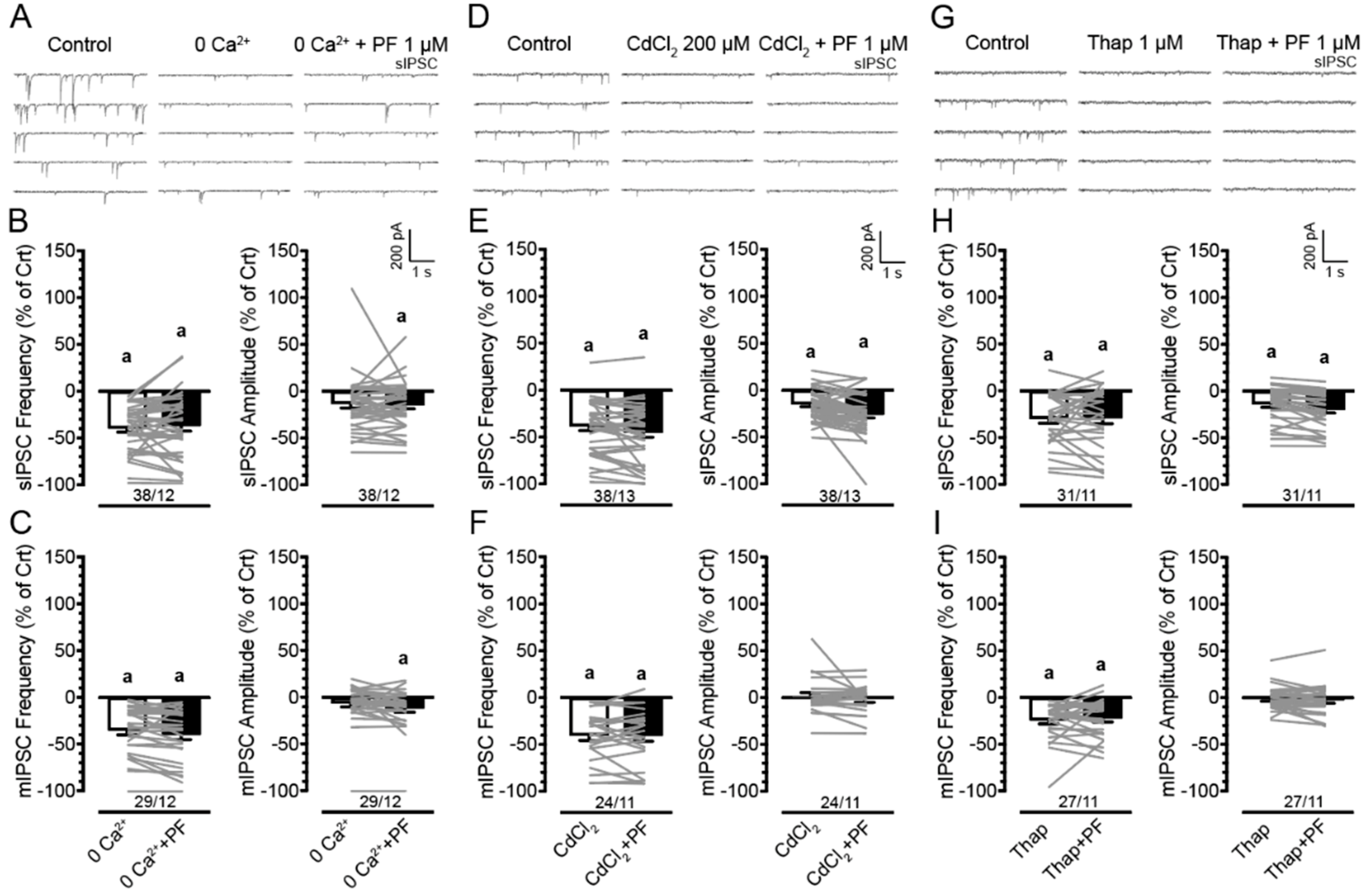
3.4. Effects of PF3845 on Inhibitory GABAergic Neurotransmission Involve the Presence of Extracellular and Intracellular Calcium
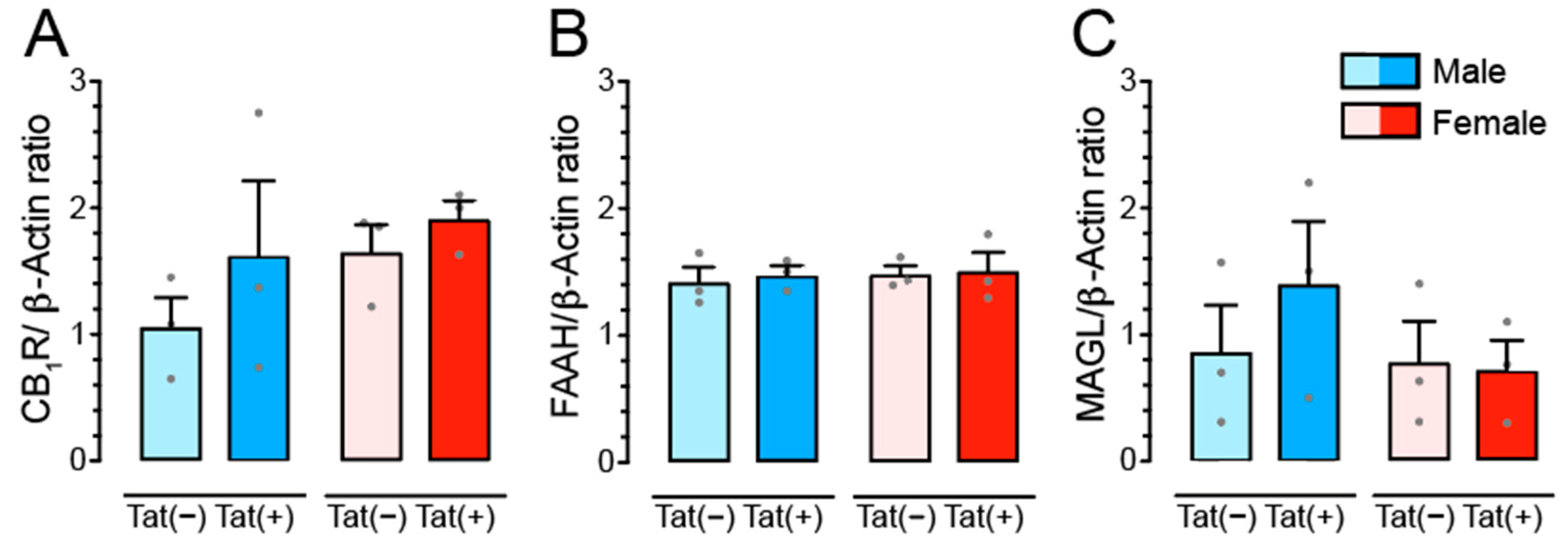
3.5. Tat Transgenic Mice Display No Changes in CB1R, FAAH, and MAGL Protein Expression
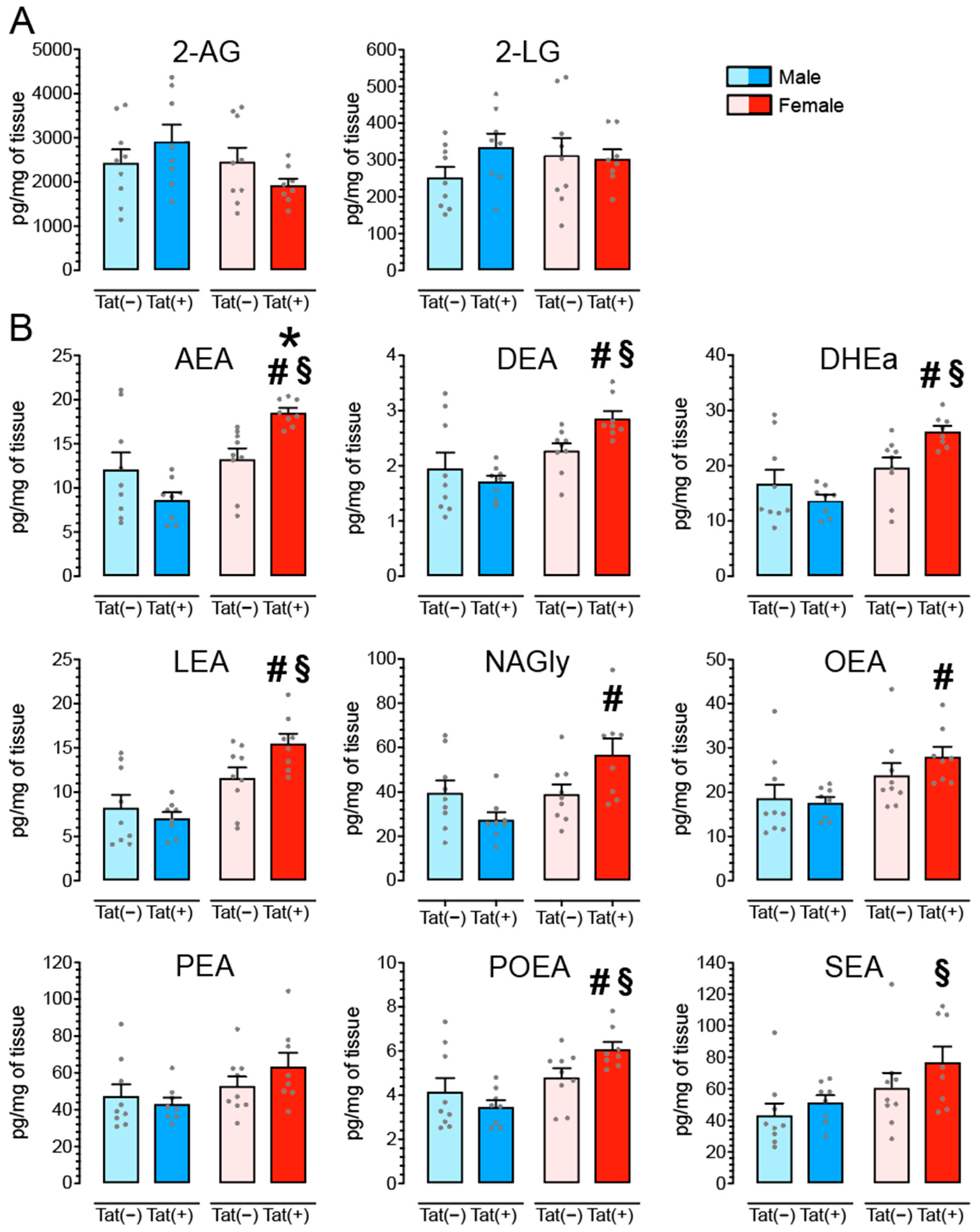
3.6. Tat Transgenic Mice Display Altered Levels in AEA and Related Non-eCB Lipids in the PFC in a Sex and/or Tat-Dependent Manner
4. Discussion
4.1. Inhibitory Neurotransmission Is Sex-Dependently Affected by Tat Exposure
4.2. PF3845 Decreases GABAergic Neurotransmission Independent of Sex and Genotype via CB1R-Related Mechanisms
4.3. Tat Exposure Significantly Alters AEA and Related Non-eCB Lipids in the PFC of Female Mice but Not Male Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrison, K.M.; Song, R.; Zhang, X. Life expectancy after HIV diagnosis based on national HIV surveillance data from 25 states, United States. J. Acquir. Immune Defic. Syndr. 2010, 53, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, M.T.; Sterne, J.A.; Costagliola, D.; Sabin, C.A.; Phillips, A.N.; Justice, A.C.; Dabis, F.; Gill, J.; Lundgren, J.; Hogg, R.S.; et al. HIV treatment response and prognosis in Europe and North America in the first decade of highly active antiretroviral therapy: A collaborative analysis. Lancet 2006, 368, 451–458. [Google Scholar] [PubMed]
- Marcus, J.L.; Chao, C.R.; Leyden, W.A.; Xu, L.; Quesenberry, C.P., Jr.; Klein, D.B.; Towner, W.J.; Horberg, M.A.; Silverberg, M.J. Narrowing the Gap in Life Expectancy Between HIV-Infected and HIV-Uninfected Individuals With Access to Care. J. Acquir. Immune Defic. Syndr. 2016, 73, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannon, P.; Khan, M.Z.; Kolson, D.L. Current understanding of HIV-associated neurocognitive disorders pathogenesis. Curr. Opin. Neurol. 2011, 24, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Antinori, A.; Arendt, G.; Becker, J.T.; Brew, B.J.; Byrd, D.A.; Cherner, M.; Clifford, D.B.; Cinque, P.; Epstein, L.G.; Goodkin, K.; et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef] [Green Version]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Sacktor, N.; McDermott, M.P.; Marder, K.; Schifitto, G.; Selnes, O.A.; McArthur, J.C.; Stern, Y.; Albert, S.; Palumbo, D.; Kieburtz, K.; et al. HIV-associated cognitive impairment before and after the advent of combination therapy. J. Neurovirol. 2002, 8, 136–142. [Google Scholar] [CrossRef]
- Dore, G.J.; Correll, P.K.; Li, Y.; Kaldor, J.M.; Cooper, D.A.; Brew, B.J. Changes to AIDS dementia complex in the era of highly active antiretroviral therapy. AIDS 1999, 13, 1249–1253. [Google Scholar] [CrossRef]
- Ellis, R.; Langford, D.; Masliah, E. HIV and antiretroviral therapy in the brain: Neuronal injury and repair. Nat. Rev. Neurosci. 2007, 8, 33–44. [Google Scholar] [CrossRef]
- Garvey, L.J.; Yerrakalva, D.; Winston, A. Correlations between computerized battery testing and a memory questionnaire for identification of neurocognitive impairment in HIV type 1-infected subjects on stable antiretroviral therapy. AIDS Res. Hum. Retrovir. 2009, 25, 765–769. [Google Scholar] [CrossRef]
- Scott, J.C.; Woods, S.P.; Carey, C.L.; Weber, E.; Bondi, M.W.; Grant, I.; Group HIVNRC. Neurocognitive consequences of HIV infection in older adults: An evaluation of the “cortical” hypothesis. AIDS Behav. 2011, 15, 1187–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cysique, L.A.; Maruff, P.; Brew, B.J. Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: A combined study of two cohorts. J. Neurovirol. 2004, 10, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.J.; Vance, D.E. The neuropsychology of HIV/AIDS in older adults. Neuropsychol. Rev. 2009, 19, 263–272. [Google Scholar] [CrossRef]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brailoiu, G.C.; Brailoiu, E.; Chang, J.K.; Dun, N.J. Excitatory effects of human immunodeficiency virus 1 Tat on cultured rat cerebral cortical neurons. Neuroscience 2008, 151, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musante, V.; Summa, M.; Neri, E.; Puliti, A.; Godowicz, T.T.; Severi, P.; Battaglia, G.; Raiteri, M.; Pittaluga, A. The HIV-1 viral protein Tat increases glutamate and decreases GABA exocytosis from human and mouse neocortical nerve endings. Cereb. Cortex 2010, 20, 1974–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, K.A.; Lyddon, E.; Thayer, S.A. HIV-1 Tat activates a RhoA signaling pathway to reduce NMDA-evoked calcium responses in hippocampal neurons via an actin-dependent mechanism. J. Neurochem. 2015, 132, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Martemyanov, K.A.; Thayer, S.A. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J. Neurosci. 2008, 28, 12604–12613. [Google Scholar] [CrossRef] [Green Version]
- Hermes, D.J.; Xu, C.; Poklis, J.L.; Niphakis, M.J.; Cravatt, B.F.; Mackie, K.; Lichtman, A.H.; Ignatowska-Jankowska, B.M.; Fitting, S. Neuroprotective effects of fatty acid amide hydrolase catabolic enzyme inhibition in a HIV-1 Tat model of neuroAIDS. Neuropharmacology 2018, 141, 55–65. [Google Scholar] [CrossRef]
- Xu, C.; Hermes, D.J.; Nwanguma, B.; Jacobs, I.R.; Mackie, K.; Mukhopadhyay, S.; Lichtman, A.H.; Ignatowska-Jankowska, B.; Fitting, S. Endocannabinoids exert CB1 receptor-mediated neuroprotective effects in models of neuronal damage induced by HIV-1 Tat protein. Mol. Cell Neurosci. 2017, 83, 92–102. [Google Scholar] [CrossRef]
- Nath, A.; Conant, K.; Chen, P.; Scott, C.; Major, E.O. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J. Biol. Chem. 1999, 274, 17098–17102. [Google Scholar] [CrossRef] [Green Version]
- El-Hage, N.; Gurwell, J.A.; Singh, I.N.; Knapp, P.E.; Nath, A.; Hauser, K.F. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia 2005, 50, 91–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, J.J.; Singh, H.D.; Carey, A.N.; McLaughlin, J.P. Exposure to HIV-1 Tat in brain impairs sensorimotor gating and activates microglia in limbic and extralimbic brain regions of male mice. Behav. Brain Res. 2015, 291, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, J.J.; Liere, P.; Kim, S.; Mahdi, F.; Buchanan, M.E.; Nass, S.R.; Qrareya, A.N.; Salahuddin, M.F.; Pianos, A.; Fernandez, N.; et al. Pregnane steroidogenesis is altered by HIV-1 Tat and morphine: Physiological allopregnanolone is protective against neurotoxic and psychomotor effects. Neurobiol. Stress 2020, 12, 100211. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.O.; Liu, Y.; Ruan, Y.; Xu, Z.C.; Schantz, L.; He, J.J. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am. J. Pathol. 2003, 162, 1693–1707. [Google Scholar] [CrossRef] [Green Version]
- Carey, A.N.; Liu, X.; Mintzopoulos, D.; Paris, J.J.; Muschamp, J.W.; McLaughlin, J.P.; Kaufman, M.J. Conditional Tat protein expression in the GT-tg bigenic mouse brain induces gray matter density reductions. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 43, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Carey, A.N.; Liu, X.; Mintzopoulos, D.; Paris, J.J.; McLaughlin, J.P.; Kaufman, M.J. Conditional Tat protein brain expression in the GT-tg bigenic mouse induces cerebral fractional anisotropy abnormalities. Curr. HIV Res. 2015, 13, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Fitting, S.; Ignatowska-Jankowska, B.M.; Bull, C.; Skoff, R.P.; Lichtman, A.H.; Wise, L.E.; Fox, M.A.; Su, J.; Medina, A.E.; Krahe, T.E.; et al. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol. Psychiatry 2013, 73, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Dickens, A.M.; Yoo, S.W.; Chin, A.C.; Xu, J.; Johnson, T.P.; Trout, A.L.; Hauser, K.F.; Haughey, N.J. Chronic low-level expression of HIV-1 Tat promotes a neurodegenerative phenotype with aging. Sci. Rep. 2017, 7, 7748. [Google Scholar] [CrossRef]
- Hahn, Y.K.; Podhaizer, E.M.; Farris, S.P.; Miles, M.F.; Hauser, K.F.; Knapp, P.E. Effects of chronic HIV-1 Tat exposure in the CNS: Heightened vulnerability of males versus females to changes in cell numbers, synaptic integrity, and behavior. Brain Struct. Funct. 2015, 220, 605–623. [Google Scholar] [CrossRef] [Green Version]
- Marks, W.D.; Paris, J.J.; Schier, C.J.; Denton, M.D.; Fitting, S.; McQuiston, A.R.; Knapp, P.E.; Hauser, K.F. HIV-1 Tat causes cognitive deficits and selective loss of parvalbumin, somatostatin, and neuronal nitric oxide synthase expressing hippocampal CA1 interneuron subpopulations. J. Neurovirol. 2016, 22, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.R.; Xu, C.; Hermes, D.J.; League, A.F.; Xu, C.; Nath, B.; Jiang, W.; Niphakis, M.J.; Cravatt, B.F.; Mackie, K.; et al. Inhibitory Control Deficits Associated with Upregulation of CB1R in the HIV-1 Tat Transgenic Mouse Model of HAND. J. Neuroimmune Pharmacol. 2019, 14, 661–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Garcia-Mesa, Y.; Knapp, P.E.; Hauser, K.F.; Karn, J. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019, 15, e1008249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, A.N.; Sypek, E.I.; Singh, H.D.; Kaufman, M.J.; McLaughlin, J.P. Expression of HIV-Tat protein is associated with learning and memory deficits in the mouse. Behav. Brain Res. 2012, 229, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, J.J.; Singh, H.D.; Ganno, M.L.; Jackson, P.; McLaughlin, J.P. Anxiety-like behavior of mice produced by conditional central expression of the HIV-1 regulatory protein, Tat. Psychopharmacology 2014, 231, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, J.J.; Fenwick, J.; McLaughlin, J.P. Progesterone protects normative anxiety-like responding among ovariectomized female mice that conditionally express the HIV-1 regulatory protein, Tat, in the CNS. Horm. Behav. 2014, 65, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Kesby, J.P.; Markou, A.; Semenova, S. The effects of HIV-1 regulatory TAT protein expression on brain reward function, response to psychostimulants and delay-dependent memory in mice. Neuropharmacology 2016, 109, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Hahn, Y.K.; Paris, J.J.; Lichtman, A.H.; Hauser, K.F.; Sim-Selley, L.J.; Selley, D.E.; Knapp, P.E. Central HIV-1 Tat exposure elevates anxiety and fear conditioned responses of male mice concurrent with altered mu-opioid receptor-mediated G-protein activation and beta-arrestin 2 activity in the forebrain. Neurobiol. Dis. 2016, 92, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Bagdas, D.; Paris, J.J.; Carper, M.; Wodarski, R.; Rice, A.S.C.; Knapp, P.E.; Hauser, K.F.; Damaj, M.I. Conditional expression of HIV-1 Tat in the mouse alters the onset and progression of tonic, inflammatory, and neuropathic hypersensitivity in a sex-dependent manner. Eur. J. Pain 2020, 24, 1609–1623. [Google Scholar] [CrossRef]
- Barbour, A.J.; Hauser, K.F.; McQuiston, A.R.; Knapp, P.E. HIV and opiates dysregulate K(+)-Cl(−) cotransporter 2 (KCC2) to cause GABAergic dysfunction in primary human neurons and Tat-transgenic mice. Neurobiol. Dis. 2020, 141, 104878. [Google Scholar] [CrossRef]
- Behnisch, T.; Francesconi, W.; Sanna, P.P. HIV secreted protein Tat prevents long-term potentiation in the hippocampal CA1 region. Brain Res. 2004, 1012, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Wayman, W.N.; Chen, L.; Persons, A.L.; Napier, T.C. Cortical consequences of HIV-1 Tat exposure in rats are enhanced by chronic cocaine. Curr. HIV Res. 2015, 13, 80–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nass, S.R.; Hahn, Y.K.; McLane, V.D.; Varshneya, N.B.; Damaj, M.I.; Knapp, P.E.; Hauser, K.F. Chronic HIV-1 Tat exposure alters anterior cingulate cortico-basal ganglia-thalamocortical synaptic circuitry, associated behavioral control, and immune regulation in male mice. Brain Behav. Immun. Health 2020, 5, 100077. [Google Scholar] [CrossRef] [PubMed]
- Hargus, N.J.; Thayer, S.A. Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J. Neurosci. 2013, 33, 17908–17920. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Hermes, D.J.; Mackie, K.; Lichtman, A.H.; Ignatowska-Jankowska, B.M.; Fitting, S. Cannabinoids Occlude the HIV-1 Tat-Induced Decrease in GABAergic Neurotransmission in Prefrontal Cortex Slices. J. Neuroimmune Pharmacol. 2016, 11, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Fitting, S. Inhibition of GABAergic Neurotransmission by HIV-1 Tat and Opioid Treatment in the Striatum Involves mu-Opioid Receptors. Front. Neurosci. 2016, 10, 497. [Google Scholar] [CrossRef]
- Gelman, B.B.; Chen, T.; Lisinicchia, J.G.; Soukup, V.M.; Carmical, J.R.; Starkey, J.M.; Masliah, E.; Commins, D.L.; Brandt, D.; Grant, I.; et al. The National NeuroAIDS Tissue Consortium brain gene array: Two types of HIV-associated neurocognitive impairment. PLoS ONE 2012, 7, e46178. [Google Scholar] [CrossRef]
- Wu, M.M.; Thayer, S.A. HIV Tat Protein Selectively Impairs CB1 Receptor-Mediated Presynaptic Inhibition at Excitatory But Not Inhibitory Synapses. eNeuro 2020, 7, 1–13. [Google Scholar] [CrossRef]
- Buzhdygan, T.; Lisinicchia, J.; Patel, V.; Johnson, K.; Neugebauer, V.; Paessler, S.; Jennings, K.; Gelman, B. Neuropsychological, Neurovirological and Neuroimmune Aspects of Abnormal GABAergic Transmission in HIV Infection. J. Neuroimmune Pharmacol. 2016, 11, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Harezlak, J.; Buchthal, S.; Taylor, M.; Schifitto, G.; Zhong, J.; Daar, E.; Alger, J.; Singer, E.; Campbell, T.; Yiannoutsos, C.; et al. Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS 2011, 25, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E.; Straube, E.; et al. HIV-1-associated neurocognitive disorder: Epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 2017, 264, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Ru, W.; Tang, S.J. HIV-associated synaptic degeneration. Mol. Brain 2017, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Zhang, X.; Asher, M.J.; Thayer, S.A. Druggable targets of the endocannabinoid system: Implications for the treatment of HIV-associated neurocognitive disorder. Brain Res. 2019, 1724, 146467. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Rapaka, R.S.; Rutter, J. Cannabinoid receptor-2 and HIV-associated neurocognitive disorders. J. Neuroimmune Pharmacol. 2014, 9, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Yadav-Samudrala, B.J.; Fitting, S. Mini-review: The therapeutic role of cannabinoids in neuroHIV. Neurosci. Lett. 2021, 750, 135717. [Google Scholar] [CrossRef]
- Towe, S.L.; Meade, C.S.; Cloak, C.C.; Bell, R.P.; Baptiste, J.; Chang, L. Reciprocal Influences of HIV and Cannabinoids on the Brain and Cognitive Function. J. Neuroimmune Pharmacol. 2020, 15, 765–779. [Google Scholar] [CrossRef]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef]
- Ahn, K.; McKinney, M.K.; Cravatt, B.F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008, 108, 1687–1707. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, A.H.; Blankman, J.L.; Cravatt, B.F. Endocannabinoid overload. Mol. Pharmacol. 2010, 78, 993–995. [Google Scholar] [CrossRef] [Green Version]
- Petrosino, S.; Di Marzo, V. FAAH and MAGL inhibitors: Therapeutic opportunities from regulating endocannabinoid levels. Curr. Opin. Investig. Drugs 2010, 11, 51–62. [Google Scholar]
- Ignatowska-Jankowska, B.; Wilkerson, J.L.; Mustafa, M.; Abdullah, R.; Niphakis, M.; Wiley, J.L.; Cravatt, B.F.; Lichtman, A.H. Selective monoacylglycerol lipase inhibitors: Antinociceptive versus cannabimimetic effects in mice. J. Pharmacol. Exp. Ther. 2015, 353, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatowska-Jankowska, B.M.; Ghosh, S.; Crowe, M.S.; Kinsey, S.G.; Niphakis, M.J.; Abdullah, R.A.; Tao, Q.; O’Neal, S.T.; Walentiny, D.M.; Wiley, J.L.; et al. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: Antinociceptive activity without cannabimimetic side effects. Br. J. Pharmacol. 2014, 171, 1392–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertwee, R.G. Elevating endocannabinoid levels: Pharmacological strategies and potential therapeutic applications. Proc. Nutr. Soc. 2014, 73, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidoo, V.; Nikas, S.P.; Karanian, D.A.; Hwang, J.; Zhao, J.; Wood, J.T.; Alapafuja, S.O.; Vadivel, S.K.; Butler, D.; Makriyannis, A.; et al. A new generation fatty acid amide hydrolase inhibitor protects against kainate-induced excitotoxicity. J. Mol. Neurosci. 2011, 43, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef]
- Zhang, X.; Thayer, S.A. Monoacylglycerol lipase inhibitor JZL184 prevents HIV-1 gp120-induced synapse loss by altering endocannabinoid signaling. Neuropharmacology 2018, 128, 269–281. [Google Scholar] [CrossRef]
- Avraham, H.K.; Jiang, S.; Fu, Y.; Rockenstein, E.; Makriyannis, A.; Wood, J.; Wang, L.; Masliah, E.; Avraham, S. Impaired neurogenesis by HIV-1-Gp120 is rescued by genetic deletion of fatty acid amide hydrolase enzyme. Br. J. Pharmacol. 2015, 172, 4603–4614. [Google Scholar] [CrossRef] [Green Version]
- Nasirinezhad, F.; Jergova, S.; Pearson, J.P.; Sagen, J. Attenuation of persistent pain-related behavior by fatty acid amide hydrolase (FAAH) inhibitors in a rat model of HIV sensory neuropathy. Neuropharmacology 2015, 95, 100–109. [Google Scholar] [CrossRef] [Green Version]
- League, A.F.; Gorman, B.L.; Hermes, D.J.; Johnson, C.T.; Jacobs, I.R.; Yadav-Samudrala, B.J.; Poklis, J.L.; Niphakis, M.J.; Cravatt, B.F.; Lichtman, A.H.; et al. Monoacylglycerol Lipase Inhibitor MJN110 Reduces Neuronal Hyperexcitability, Restores Dendritic Arborization Complexity, and Regulates Reward-Related Behavior in Presence of HIV-1 Tat. Front. Neurol. 2021, 12, 651272. [Google Scholar] [CrossRef]
- Hermes, D.J.; Yadav-Samudrala, B.J.; Xu, C.; Paniccia, J.E.; Meeker, R.B.; Armstrong, M.L.; Reisdorph, N.; Cravatt, B.F.; Mackie, K.; Lichtman, A.H.; et al. GPR18 drives FAAH inhibition-induced neuroprotection against HIV-1 Tat-induced neurodegeneration. Exp. Neurol. 2021, 341, 113699. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.A.; Bauman, A.; Zhao, M.L.; Morgello, S.; Suh, H.S.; Lee, S.C. Cannabinoid receptor expression in HIV encephalitis and HIV-associated neuropathologic comorbidities. Neuropathol. Appl. Neurobiol. 2011, 37, 464–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swinton, M.K.; Sundermann, E.E.; Pedersen, L.; Nguyen, J.D.; Grelotti, D.J.; Taffe, M.A.; Iudicello, J.E.; Fields, J.A. Alterations in Brain Cannabinoid Receptor Levels Are Associated with HIV-Associated Neurocognitive Disorders in the ART Era: Implications for Therapeutic Strategies Targeting the Endocannabinoid System. Viruses 2021, 13, 1742. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Kim, W.K.; Chavarria, I.; Hillard, C.J.; Mackie, K.; Tolon, R.M.; Williams, K.; Romero, J. A glial endogenous cannabinoid system is upregulated in the brains of macaques with simian immunodeficiency virus-induced encephalitis. J. Neurosci. 2005, 25, 2530–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, J.; Narasimhan, M.; Guindon, J.; Benamar, K. Supraspinal interaction between HIV-1-gp120 and cannabinoid analgesic effectiveness. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Turchan-Cholewo, J.; Smart, E.J.; Geurin, T.; Chauhan, A.; Reid, R.; Xu, R.; Nath, A.; Knapp, P.E.; Hauser, K.F. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia 2008, 56, 1414–1427. [Google Scholar] [CrossRef] [Green Version]
- National Research Council. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Olah, S.; Fule, M.; Komlosi, G.; Varga, C.; Baldi, R.; Barzo, P.; Tamas, G. Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature 2009, 461, 1278–1281. [Google Scholar] [CrossRef]
- Petersen, C.C.; Crochet, S. Synaptic computation and sensory processing in neocortical layer 2/3. Neuron 2013, 78, 28–48. [Google Scholar] [CrossRef] [Green Version]
- Fitting, S.; Scoggins, K.L.; Xu, R.; Dever, S.M.; Knapp, P.E.; Dewey, W.L.; Hauser, K.F. Morphine efficacy is altered in conditional HIV-1 Tat transgenic mice. Eur. J. Pharmacol. 2012, 689, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Gouveia-Figueira, S.; Nording, M.L. Validation of a tandem mass spectrometry method using combined extraction of 37 oxylipins and 14 endocannabinoid-related compounds including prostamides from biological matrices. Prostaglandins Other Lipid Mediat. 2015, 121, 110–121. [Google Scholar] [CrossRef]
- Gaetani, S.; Cuomo, V.; Piomelli, D. Anandamide hydrolysis: A new target for anti-anxiety drugs? Trends Mol. Med. 2003, 9, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Farkas, I.; Kallo, I.; Deli, L.; Vida, B.; Hrabovszky, E.; Fekete, C.; Moenter, S.M.; Watanabe, M.; Liposits, Z. Retrograde endocannabinoid signaling reduces GABAergic synaptic transmission to gonadotropin-releasing hormone neurons. Endocrinology 2010, 151, 5818–5829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, K. Mechanisms of CB1 receptor signaling: Endocannabinoid modulation of synaptic strength. Int. J. Obes. 2006, 30 (Suppl. 1), S19–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, T.; Jiang, C.S.; Nakama, H.; Buchthal, S.; Chang, L. Lower brain glutamate is associated with cognitive deficits in HIV patients: A new mechanism for HIV-associated neurocognitive disorder. J. Magn. Reson. Imaging 2010, 32, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Ferrarese, C.; Aliprandi, A.; Tremolizzo, L.; Stanzani, L.; De Micheli, A.; Dolara, A.; Frattola, L. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 2001, 57, 671–675. [Google Scholar] [CrossRef]
- Schroecksnadel, K.; Zangerle, R.; Bellmann-Weiler, R.; Garimorth, K.; Weiss, G.; Fuchs, D. Indoleamine-2,3-dioxygenase and other interferon-gamma-mediated pathways in patients with human immunodeficiency virus infection. Curr. Drug Metab. 2007, 8, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.F. Tryptophan depletion and HIV infection: A metabolic link to pathogenesis. Lancet Infect. Dis. 2003, 3, 644–652. [Google Scholar] [CrossRef]
- Berger, J.R.; Arendt, G. HIV dementia: The role of the basal ganglia and dopaminergic systems. J. Psychopharmacol. 2000, 14, 214–221. [Google Scholar] [CrossRef]
- Nath, A.; Anderson, C.; Jones, M.; Maragos, W.; Booze, R.; Mactutus, C.; Bell, J.; Hauser, K.F.; Mattson, M. Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J. Psychopharmacol. 2000, 14, 222–227. [Google Scholar] [CrossRef]
- Gelman, B.B.; Spencer, J.A.; Holzer 3rd, C.E.; Soukup, V.M. Abnormal striatal dopaminergic synapses in National NeuroAIDS Tissue Consortium subjects with HIV encephalitis. J. Neuroimmune Pharmacol. 2006, 1, 410–420. [Google Scholar] [CrossRef]
- Wang, G.J.; Chang, L.; Volkow, N.D.; Telang, F.; Logan, J.; Ernst, T.; Fowler, J.S. Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain 2004, 127, 2452–2458. [Google Scholar] [CrossRef] [Green Version]
- Paganini-Hill, A.; Henderson, V.W. Estrogen deficiency and risk of Alzheimer’s disease in women. Am. J. Epidemiol. 1994, 140, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Pigott, T.A. Gender differences in the epidemiology and treatment of anxiety disorders. J. Clin. Psychiatry 1999, 60 (Suppl. 18), 4–15. [Google Scholar] [PubMed]
- Bristow, G.C.; Bostrom, J.A.; Haroutunian, V.; Sodhi, M.S. Sex differences in GABAergic gene expression occur in the anterior cingulate cortex in schizophrenia. Schizophr. Res. 2015, 167, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Zador, F.; Nagy-Grocz, G.; Kekesi, G.; Dvoracsko, S.; Szucs, E.; Tomboly, C.; Horvath, G.; Benyhe, S.; Vecsei, L. Kynurenines and the Endocannabinoid System in Schizophrenia: Common Points and Potential Interactions. Molecules 2019, 24, 3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorska, A.M.; Eugenin, E.A. The Glutamate System as a Crucial Regulator of CNS Toxicity and Survival of HIV Reservoirs. Front. Cell Infect. Microbiol. 2020, 10, 261. [Google Scholar] [CrossRef] [PubMed]
- Cirino, T.J.; Harden, S.W.; McLaughlin, J.P.; Frazier, C.J. Region-specific effects of HIV-1 Tat on intrinsic electrophysiological properties of pyramidal neurons in mouse prefrontal cortex and hippocampus. J. Neurophysiol. 2020, 123, 1332–1341. [Google Scholar] [CrossRef]
- Chevaleyre, V.; Takahashi, K.A.; Castillo, P.E. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 2006, 29, 37–76. [Google Scholar] [CrossRef]
- Wilson, R.I.; Kunos, G.; Nicoll, R.A. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 2001, 31, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Ledri, M.; Toth, B.; Marchionni, I.; Henstridge, C.M.; Dudok, B.; Kenesei, K.; Barna, L.; Szabo, S.I.; Renkecz, T.; et al. Multiple Forms of Endocannabinoid and Endovanilloid Signaling Regulate the Tonic Control of GABA Release. J. Neurosci. 2015, 35, 10039–10057. [Google Scholar] [CrossRef] [Green Version]
- Alamilla, J.; Gillespie, D.C. Maturation of calcium-dependent GABA, glycine, and glutamate release in the glycinergic MNTB-LSO pathway. PLoS ONE 2013, 8, e75688. [Google Scholar] [CrossRef]
- Zhuang, S.Y.; Bridges, D.; Grigorenko, E.; McCloud, S.; Boon, A.; Hampson, R.E.; Deadwyler, S.A. Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine-sensitive stores. Neuropharmacology 2005, 48, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, F.E.; Knop, T.; Urbanski, M.J.; Freiman, I.; Freiman, T.M.; Feuerstein, T.J.; Zentner, J.; Szabo, B. Exogenous and endogenous cannabinoids suppress inhibitory neurotransmission in the human neocortex. Neuropsychopharmacology 2012, 37, 1104–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellocchio, L.; Lafenetre, P.; Cannich, A.; Cota, D.; Puente, N.; Grandes, P.; Chaouloff, F.; Piazza, P.V.; Marsicano, G. Bimodal control of stimulated food intake by the endocannabinoid system. Nat. Neurosci. 2010, 13, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Shosaku, T.; Kano, M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr. Opin. Neurobiol. 2014, 29, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Bedse, G.; Bluett, R.J.; Patrick, T.A.; Romness, N.K.; Gaulden, A.D.; Kingsley, P.J.; Plath, N.; Marnett, L.J.; Patel, S. Therapeutic endocannabinoid augmentation for mood and anxiety disorders: Comparative profiling of FAAH, MAGL and dual inhibitors. Transl. Psychiatry 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Hlavacova, N.; Chmelova, M.; Danevova, V.; Csanova, A.; Jezova, D. Inhibition of fatty-acid amide hydrolyse (FAAH) exerts cognitive improvements in male but not female rats. Endocr. Regul. 2015, 49, 131–136. [Google Scholar] [CrossRef]
- Gorantla, S.; Makarov, E.; Roy, D.; Finke-Dwyer, J.; Murrin, L.C.; Gendelman, H.E.; Poluektova, L. Immunoregulation of a CB2 receptor agonist in a murine model of neuroAIDS. J. Neuroimmune Pharmacol. 2010, 5, 456–468. [Google Scholar] [CrossRef] [Green Version]
- Winsauer, P.J.; Molina, P.E.; Amedee, A.M.; Filipeanu, C.M.; McGoey, R.R.; Troxclair, D.A.; Walker, E.M.; Birke, L.L.; Stouwe, C.V.; Howard, J.M.; et al. Tolerance to chronic delta-9-tetrahydrocannabinol (Delta(9)-THC) in rhesus macaques infected with simian immunodeficiency virus. Exp. Clin. Psychopharmacol. 2011, 19, 154–172. [Google Scholar] [CrossRef] [Green Version]
- Benito, C.; Nunez, E.; Tolon, R.M.; Carrier, E.J.; Rabano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [Green Version]
- Duarte, E.A.C.; Benevides, M.L.; Martins, A.L.P.; Duarte, E.P.; Weller, A.B.S.; de Azevedo, L.O.C.; de Oliveira Thais, M.E.R.; Nunes, J.C. Female sex is strongly associated with cognitive impairment in HIV infection. Neurol. Sci. 2021, 42, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.H.; Neigh, G.N.; Sundermann, E.E.; Xu, Y.; Scully, E.P.; Maki, P.M. Sex Differences in Neurocognitive Function in Adults with HIV: Patterns, Predictors, and Mechanisms. Curr. Psychiatry Rep. 2019, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Sundermann, E.E.; Heaton, R.K.; Pasipanodya, E.; Moore, R.C.; Paolillo, E.W.; Rubin, L.H.; Ellis, R.; Moore, D.J.; Group, H. Sex differences in HIV-associated cognitive impairment. AIDS 2018, 32, 2719–2726. [Google Scholar] [CrossRef]
- Maki, P.M.; Martin-Thormeyer, E. HIV, cognition and women. Neuropsychol. Rev. 2009, 19, 204–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, P.M.; Rubin, L.H.; Springer, G.; Seaberg, E.C.; Sacktor, N.; Miller, E.N.; Valcour, V.; Young, M.A.; Becker, J.T.; Martin, E.M.; et al. Differences in Cognitive Function Between Women and Men With HIV. J. Acquir. Immune. Defic. Syndr. 2018, 79, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.H.; Sundermann, E.E.; Dastgheyb, R.; Buchholz, A.S.; Pasipanodya, E.; Heaton, R.K.; Grant, I.; Ellis, R.; Moore, D.J. Sex Differences in the Patterns and Predictors of Cognitive Function in HIV. Front. Neurol. 2020, 11, 551921. [Google Scholar] [CrossRef]
- DrDreyer, A.J.; Munsami, A.; Williams, T.; Andersen, L.S.; Nightingale, S.; Gouse, H.; Joska, J.; Thomas, K.G.F. Cognitive Differences between Men and Women with HIV: A Systematic Review and Meta-Analysis. Arch. Clin. Neuropsychol. 2022, 37, 479–496. [Google Scholar] [CrossRef]
- Failde-Garrido, J.M.; Alvarez, M.E.; Simon-Lopez, M.A. Neuropsychological impairment and gender differences in HIV-1 infection. Psychiatry Clin. Neurosci. 2008, 62, 494–502. [Google Scholar] [CrossRef]
- Hillard, C.J. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000, 61, 3–18. [Google Scholar] [CrossRef]
- Starowicz, K.; Nigam, S.; Di Marzo, V. Biochemistry and pharmacology of endovanilloids. Pharmacol. Ther. 2007, 114, 13–33. [Google Scholar] [CrossRef]
- Toth, A.; Blumberg, P.M.; Boczan, J. Anandamide and the vanilloid receptor (TRPV1). Vitam. Horm. 2009, 81, 389–419. [Google Scholar] [PubMed]
- Ross, R.A. Anandamide and vanilloid TRPV1 receptors. Br. J. Pharmacol. 2003, 140, 790–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Ruwe, D.; Saffari, R.; Kravchenko, M.; Zhang, W. Effects of TRPV1 Activation by Capsaicin and Endogenous N-Arachidonoyl Taurine on Synaptic Transmission in the Prefrontal Cortex. Front. Neurosci. 2020, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Martire, A.; Petrosino, S.; Popoli, P.; Di Marzo, V. Symptom-related changes of endocannabinoid and palmitoylethanolamide levels in brain areas of R6/2 mice, a transgenic model of Huntington’s disease. Neurochem. Int. 2008, 52, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Loria, F.; Petrosino, S.; Mestre, L.; Spagnolo, A.; Correa, F.; Hernangomez, M.; Guaza, C.; Di Marzo, V.; Docagne, F. Study of the regulation of the endocannabinoid system in a virus model of multiple sclerosis reveals a therapeutic effect of palmitoylethanolamide. Eur. J. Neurosci. 2008, 28, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, M.; McHugh, D.; Fernandez-Ruiz, J.; Bradshaw, H.; Walker, J.M. Short-term exposure to alcohol in rats affects brain levels of anandamide, other N-acylethanolamines and 2-arachidonoyl-glycerol. Neurosci. Lett. 2007, 421, 270–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schabitz, W.R.; Giuffrida, A.; Berger, C.; Aschoff, A.; Schwaninger, M.; Schwab, S.; Piomelli, D. Release of fatty acid amides in a patient with hemispheric stroke: A microdialysis study. Stroke 2002, 33, 2112–2114. [Google Scholar] [CrossRef] [Green Version]
- Borrelli, F.; Izzo, A.A. Role of acylethanolamides in the gastrointestinal tract with special reference to food intake and energy balance. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 33–49. [Google Scholar] [CrossRef]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti-inflammatory agent. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodriguez De Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef]
- Lo Verme, J.; Gaetani, S.; Fu, J.; Oveisi, F.; Burton, K.; Piomelli, D. Regulation of food intake by oleoylethanolamide. Cell Mol. Life Sci. 2005, 62, 708–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahern, G.P. Activation of TRPV1 by the satiety factor oleoylethanolamide. J. Biol. Chem. 2003, 278, 30429–30434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suardiaz, M.; Estivill-Torrus, G.; Goicoechea, C.; Bilbao, A.; Rodriguez de Fonseca, F. Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain 2007, 133, 99–110. [Google Scholar] [CrossRef]
- Wang, X.; Miyares, R.L.; Ahern, G.P. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J. Physiol. 2005, 564, 541–547. [Google Scholar] [CrossRef]
- Musella, A.; Fresegna, D.; Rizzo, F.R.; Gentile, A.; Bullitta, S.; De Vito, F.; Guadalupi, L.; Centonze, D.; Mandolesi, G. A novel crosstalk within the endocannabinoid system controls GABA transmission in the striatum. Sci. Rep. 2017, 7, 7363. [Google Scholar] [CrossRef] [Green Version]
- Sheskin, T.; Hanus, L.; Slager, J.; Vogel, Z.; Mechoulam, R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J. Med. Chem. 1997, 40, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Bisogno, T.; Petros, T.J.; Chang, S.Y.; Zavitsanos, P.A.; Zipkin, R.E.; Sivakumar, R.; Coop, A.; Maeda, D.Y.; De Petrocellis, L.; et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J. Biol. Chem. 2001, 276, 42639–42644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, N.; Ho, W.S. N-arachidonoyl glycine, an endogenous lipid that acts as a vasorelaxant via nitric oxide and large conductance calcium-activated potassium channels. Br. J. Pharmacol. 2010, 160, 594–603. [Google Scholar] [CrossRef] [Green Version]
- Grabiec, U.; Hohmann, T.; Ghadban, C.; Rothganger, C.; Wong, D.; Antonietti, A.; Groth, T.; Mackie, K.; Dehghani, F. Protective Effect of N-Arachidonoyl Glycine-GPR18 Signaling after Excitotoxical Lesion in Murine Organotypic Hippocampal Slice Cultures. Int. J. Mol. Sci. 2019, 20, 1266. [Google Scholar] [CrossRef] [Green Version]
- Roberto, M.; Cruz, M.; Bajo, M.; Siggins, G.R.; Parsons, L.H.; Schweitzer, P. The endocannabinoid system tonically regulates inhibitory transmission and depresses the effect of ethanol in central amygdala. Neuropsychopharmacology 2010, 35, 1962–1972. [Google Scholar] [CrossRef]
- Lutz, B.; Marsicano, G.; Maldonado, R.; Hillard, C.J. The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci. 2015, 16, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.J.; Fawcett-Patel, J.; Katzman, P.A.; Liu, S.J. Inhibitory neurotransmission drives endocannabinoid degradation to promote memory consolidation. Nat. Commun. 2020, 11, 6407. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipids | Sex Effect | Genotype Effect | Sex x Genotype | |||
|---|---|---|---|---|---|---|
| pg/mg | F1, 30 | p | F1, 30 | p | F1, 30 | p |
| 2-AG | 2.7 | 0.114 | <1.0 | 0.939 | 3.0 | 0.096 |
| 2-LG | <1.0 | 0.697 | 1.0 | 0.318 | 1.6 | 0.211 |
| AEA | 18.3 | <0.001 | <1.0 | 0.493 | 11.5 | 0.002 |
| DEA | 15.6 | <0.001 | <1.0 | 0.360 | 4.9 | 0.035 |
| DHEa | 18.4 | <0.001 | 1.0 | 0.325 | 7.0 | 0.013 |
| LEA | 25.6 | <0.001 | 1.3 | 0.261 | 4.7 | 0.038 |
| NAGly | 7.5 | 0.010 | <1.0 | 0.605 | 8.0 | 0.008 |
| OEA | 9.7 | 0.004 | <1.0 | 0.537 | 1.1 | 0.307 |
| PEA | 5.0 | 0.033 | <1.0 | 0.592 | 1.7 | 0.208 |
| POEA | 13.6 | <0.001 | <1.0 | 0.509 | 5.0 | 0.033 |
| SEA | 6.8 | 0.014 | 2.2 | 0.149 | <1.0 | 0.630 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.; Yadav-Samudrala, B.J.; Xu, C.; Nath, B.; Mistry, T.; Jiang, W.; Niphakis, M.J.; Cravatt, B.F.; Mukhopadhyay, S.; Lichtman, A.H.; et al. Inhibitory Neurotransmission Is Sex-Dependently Affected by Tat Expression in Transgenic Mice and Suppressed by the Fatty Acid Amide Hydrolase Enzyme Inhibitor PF3845 via Cannabinoid Type-1 Receptor Mechanisms. Cells 2022, 11, 857. https://doi.org/10.3390/cells11050857
Xu C, Yadav-Samudrala BJ, Xu C, Nath B, Mistry T, Jiang W, Niphakis MJ, Cravatt BF, Mukhopadhyay S, Lichtman AH, et al. Inhibitory Neurotransmission Is Sex-Dependently Affected by Tat Expression in Transgenic Mice and Suppressed by the Fatty Acid Amide Hydrolase Enzyme Inhibitor PF3845 via Cannabinoid Type-1 Receptor Mechanisms. Cells. 2022; 11(5):857. https://doi.org/10.3390/cells11050857
Chicago/Turabian StyleXu, Changqing, Barkha J. Yadav-Samudrala, Callie Xu, Bhupendra Nath, Twisha Mistry, Wei Jiang, Micah J. Niphakis, Benjamin F. Cravatt, Somnath Mukhopadhyay, Aron H. Lichtman, and et al. 2022. "Inhibitory Neurotransmission Is Sex-Dependently Affected by Tat Expression in Transgenic Mice and Suppressed by the Fatty Acid Amide Hydrolase Enzyme Inhibitor PF3845 via Cannabinoid Type-1 Receptor Mechanisms" Cells 11, no. 5: 857. https://doi.org/10.3390/cells11050857