
Treatment of Experimental Autoimmune Encephalomyelitis with an Inhibitor of Phosphodiesterase-8 (PDE8)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
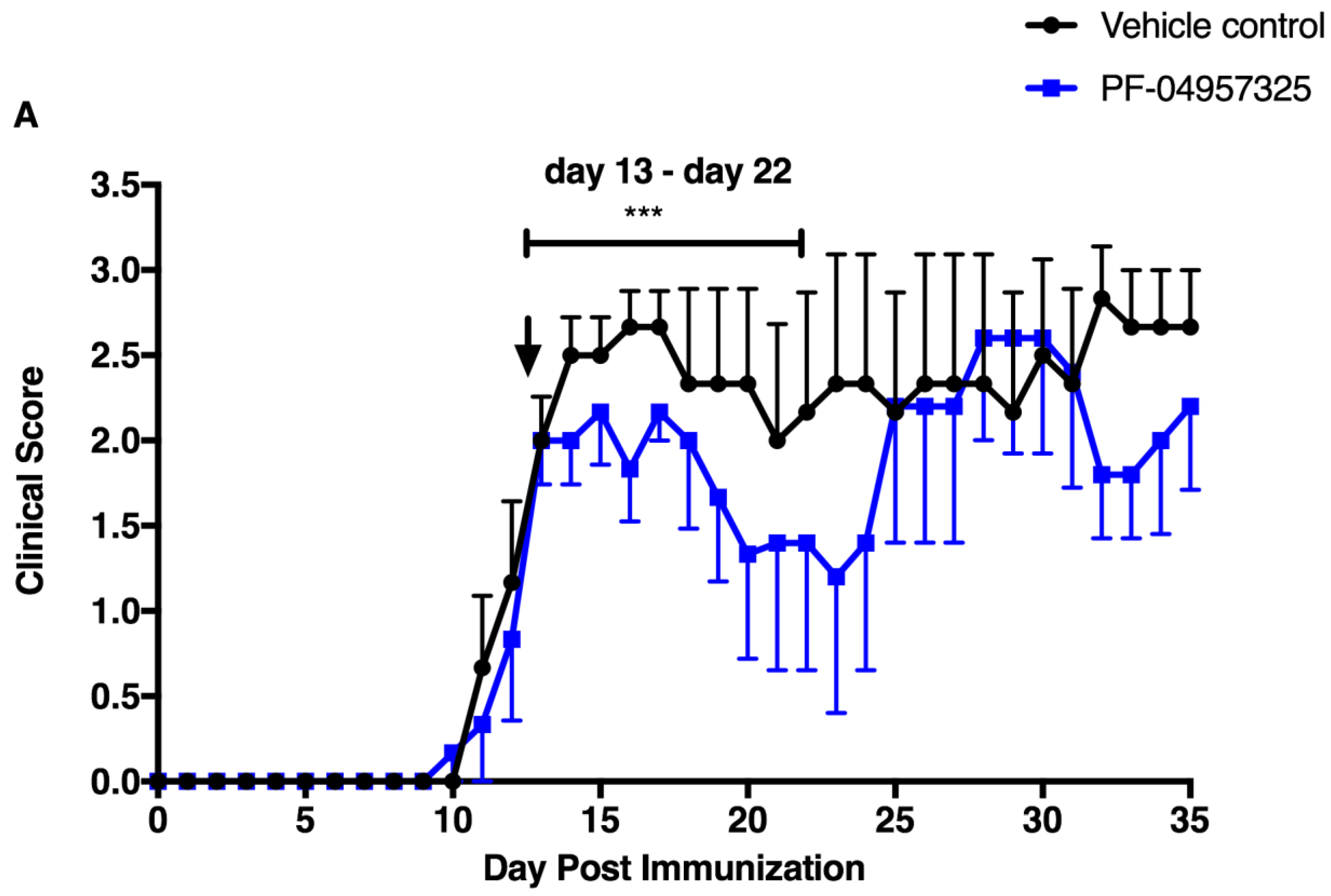
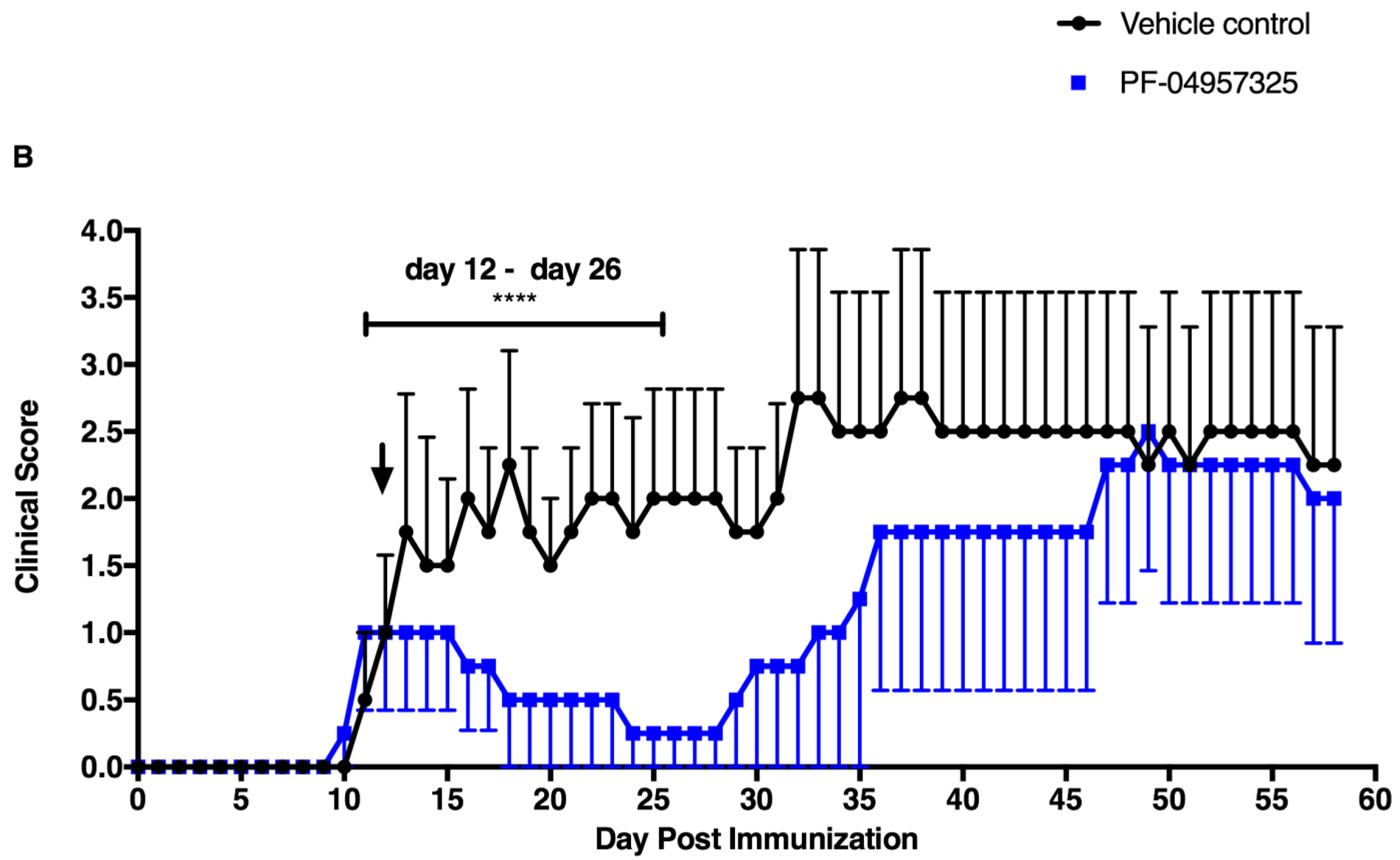
3.1. PF-04957325 Ameliorates Active EAE in a Therapeutic Fashion
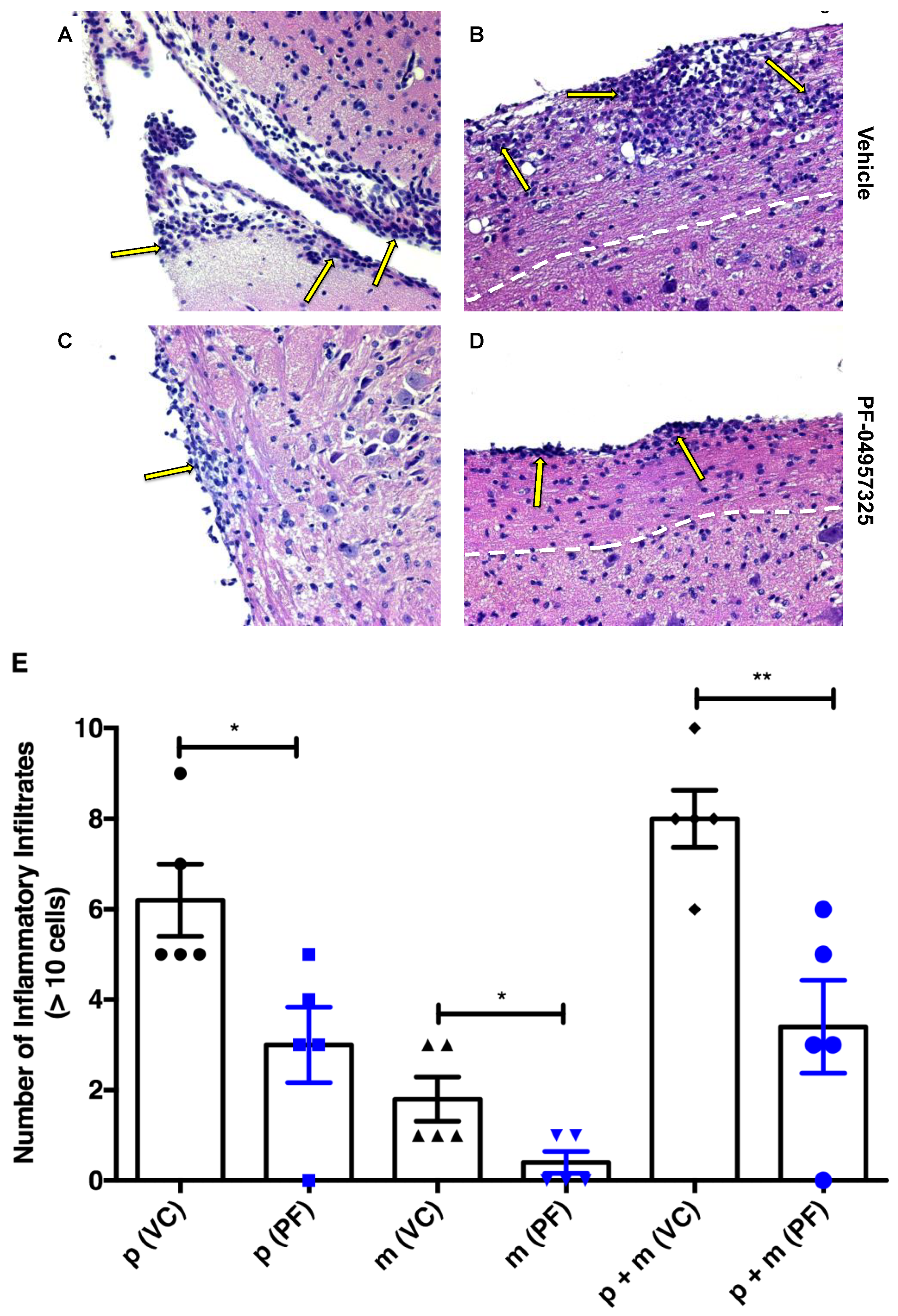
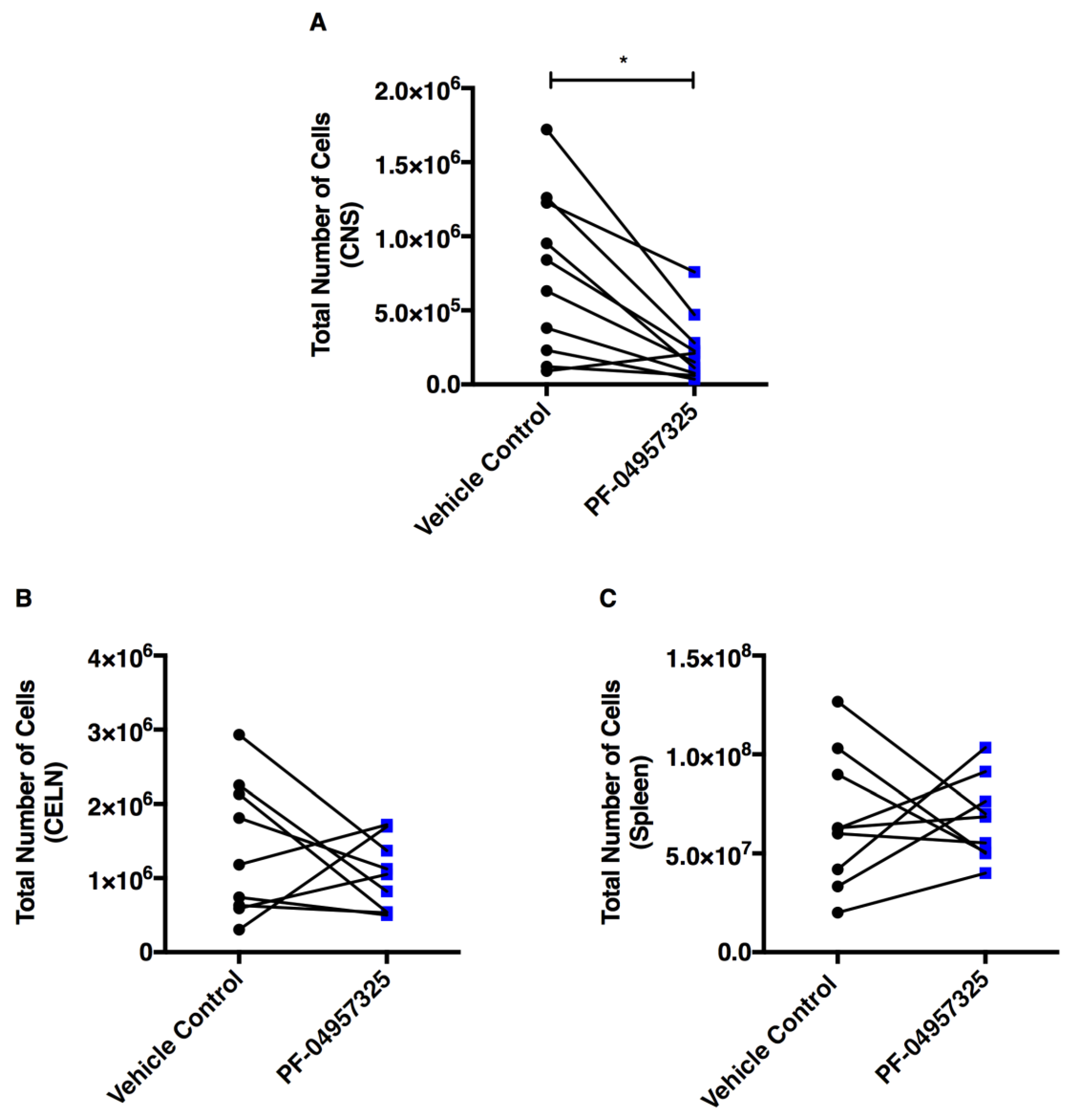
3.2. Treatment with PF-04957325 Suppresses Inflammatory Lesion Formation in the Spinal Cord and Brain, but Not in the Periphery
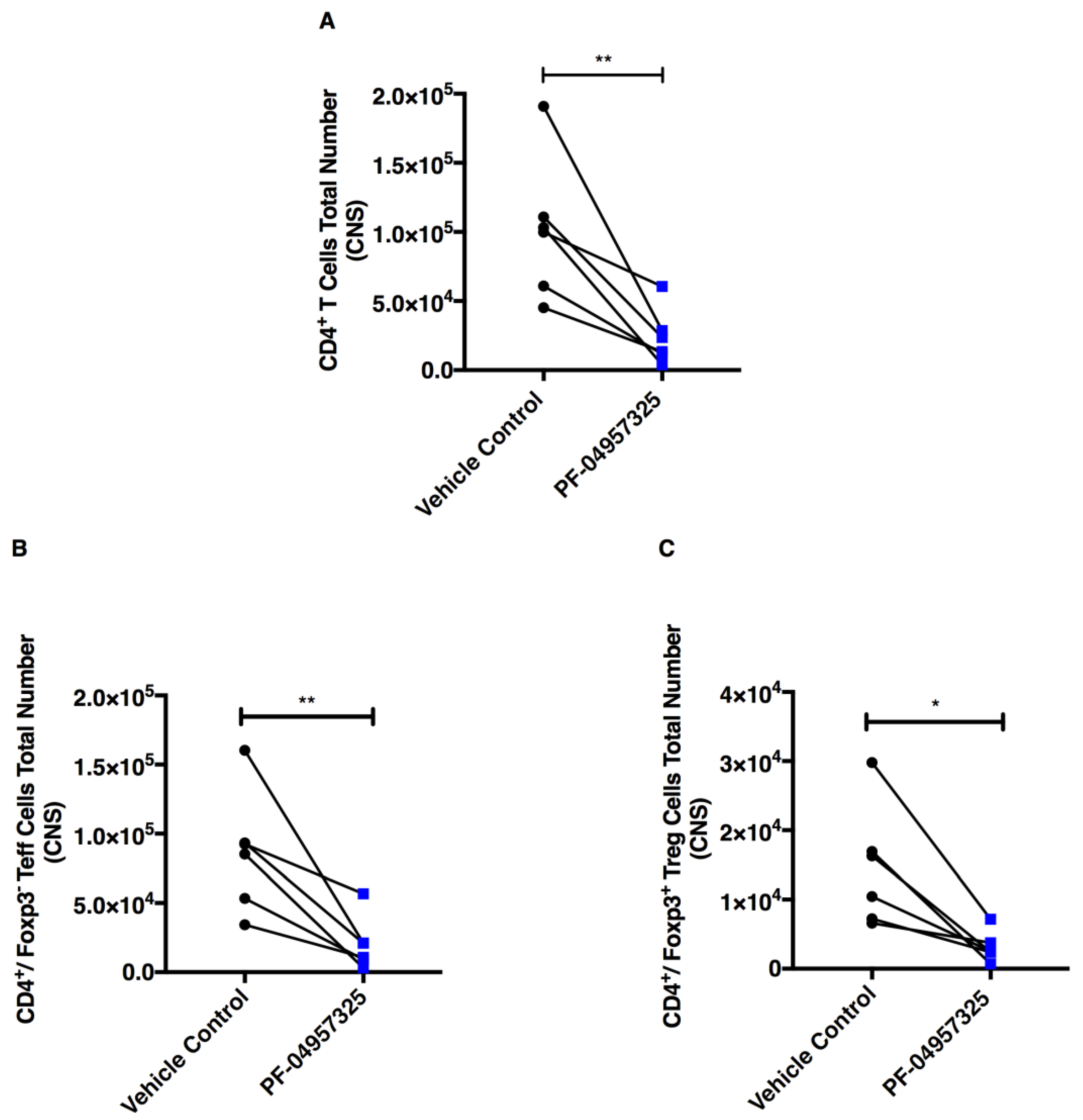
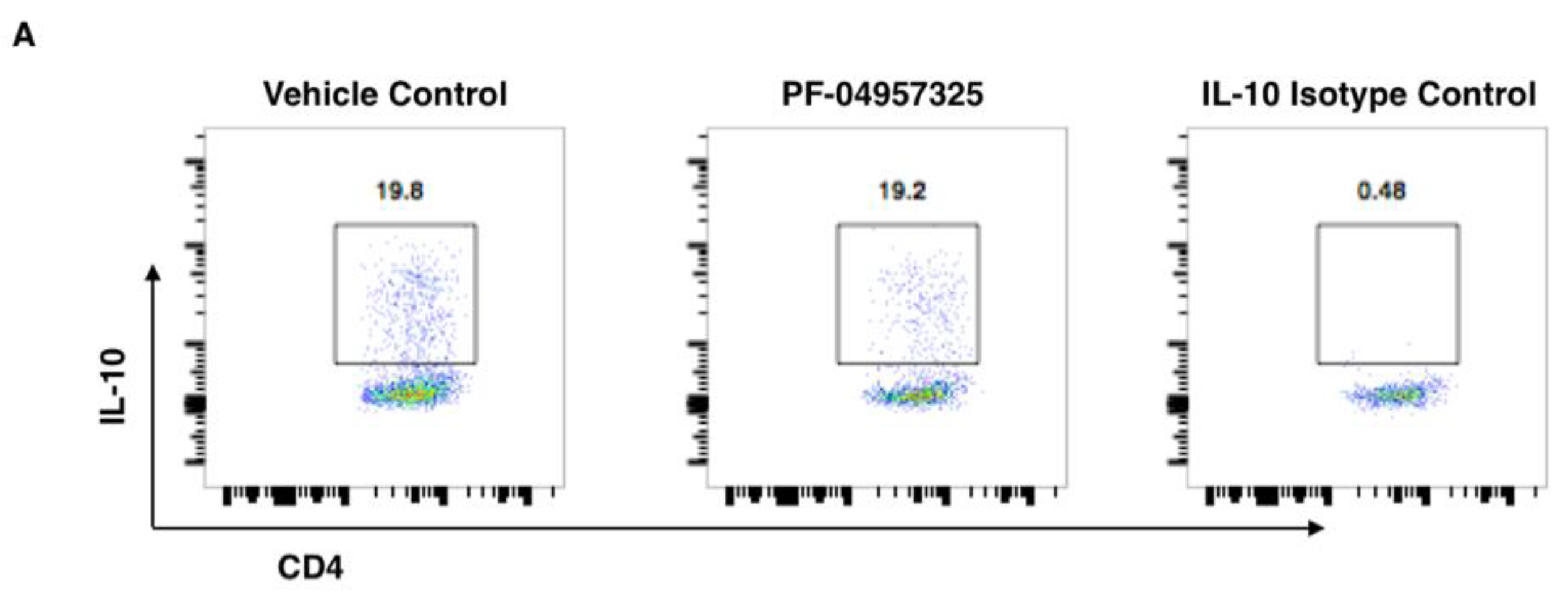
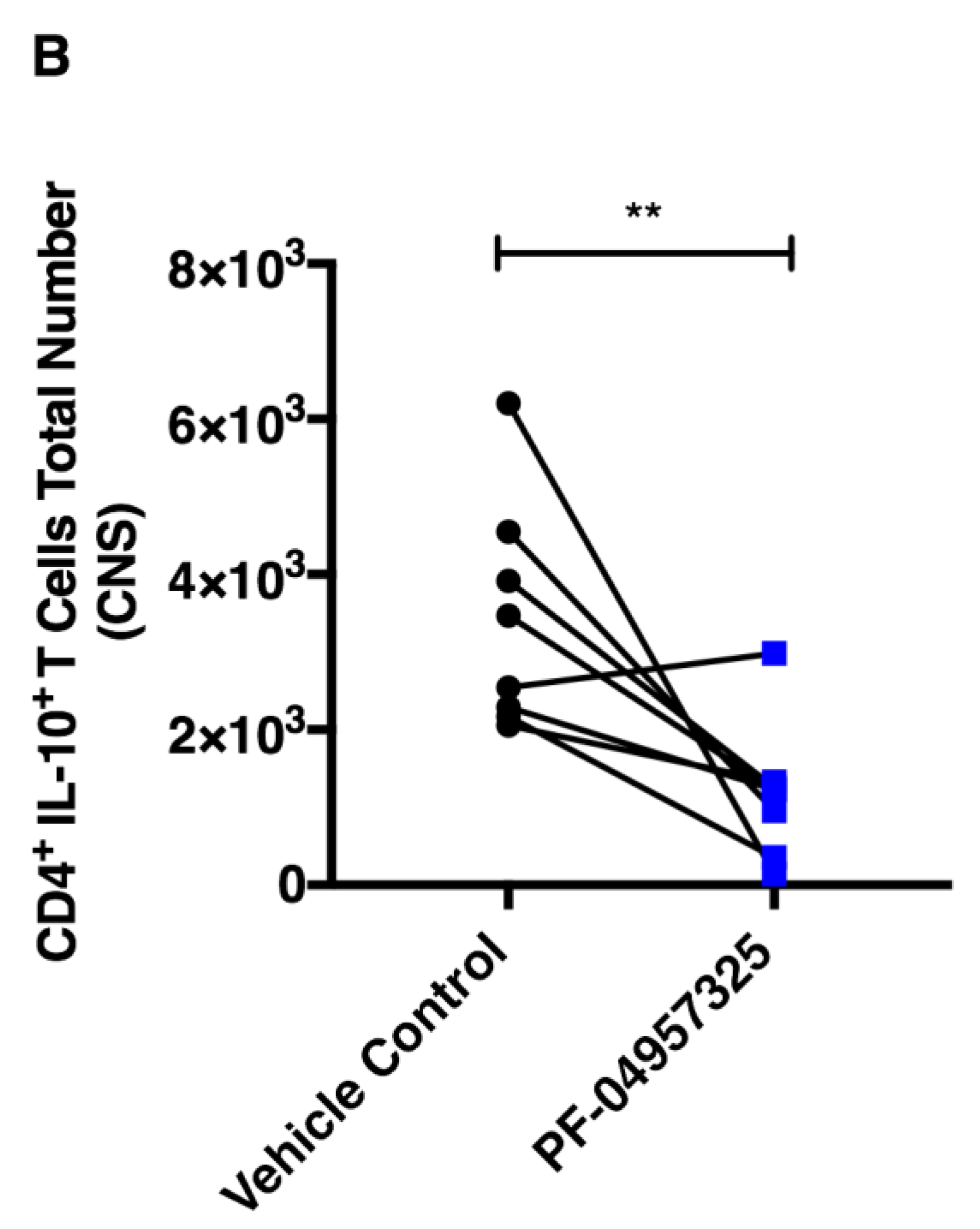
3.3. Treatment with PF-04957325 Suppresses CD4+ Teff Cell and Treg Cell Accumulation in the Spinal Cord
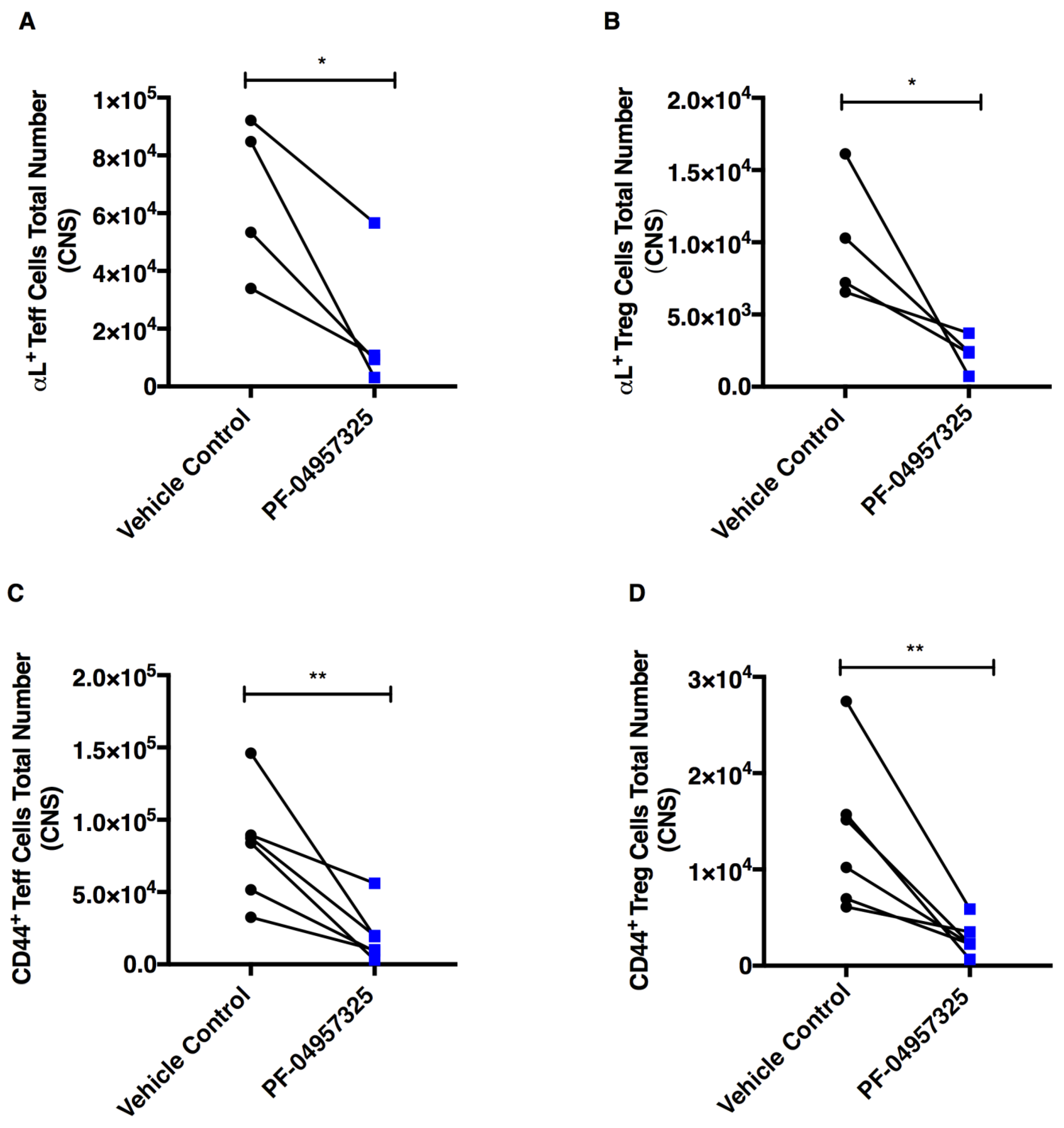
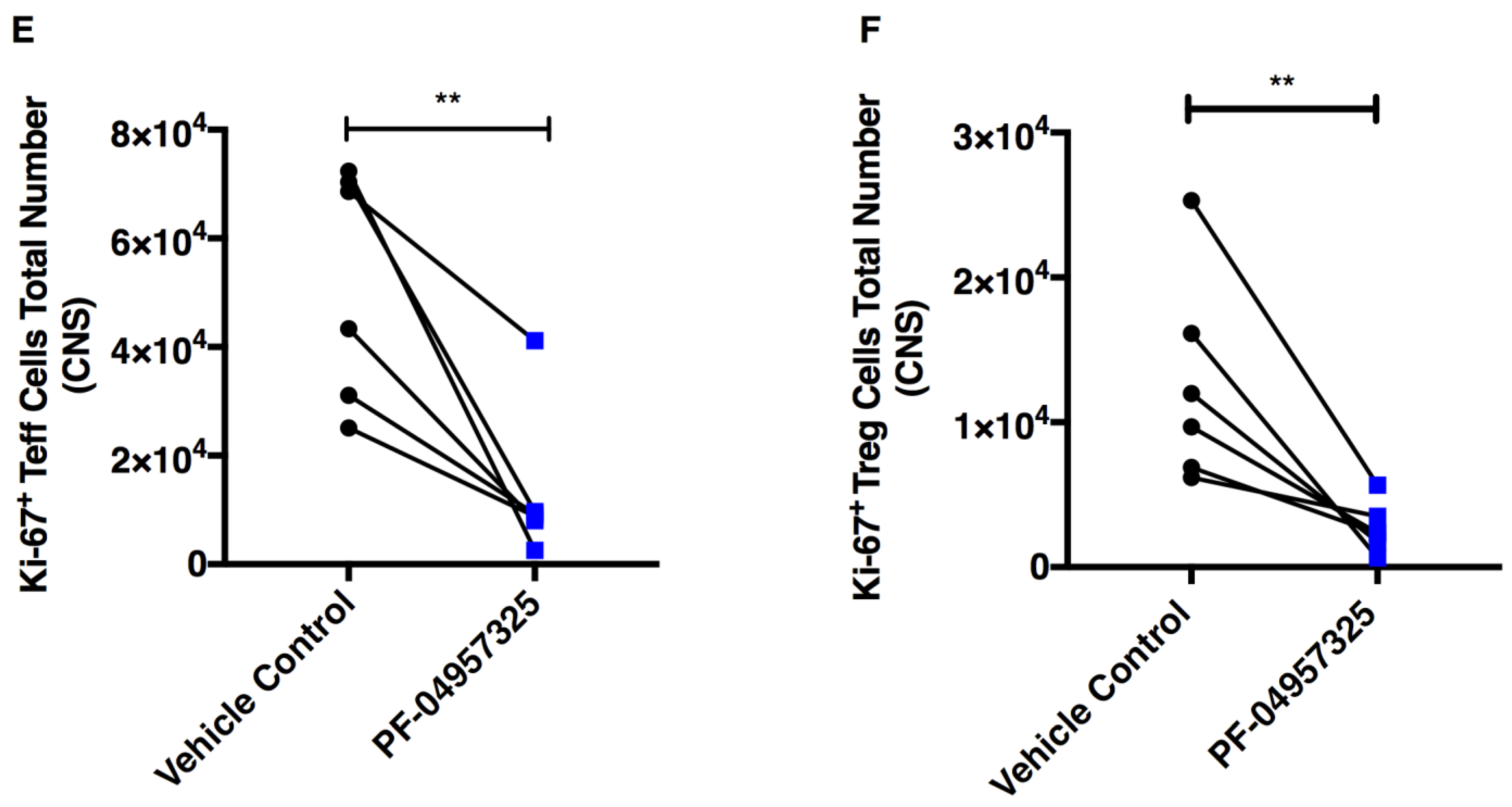
3.4. Treatment with PF-04957325 Suppresses the Accumulation of Activated and Proliferating Teff and Treg Cell Subpopulations in the Spinal Cord
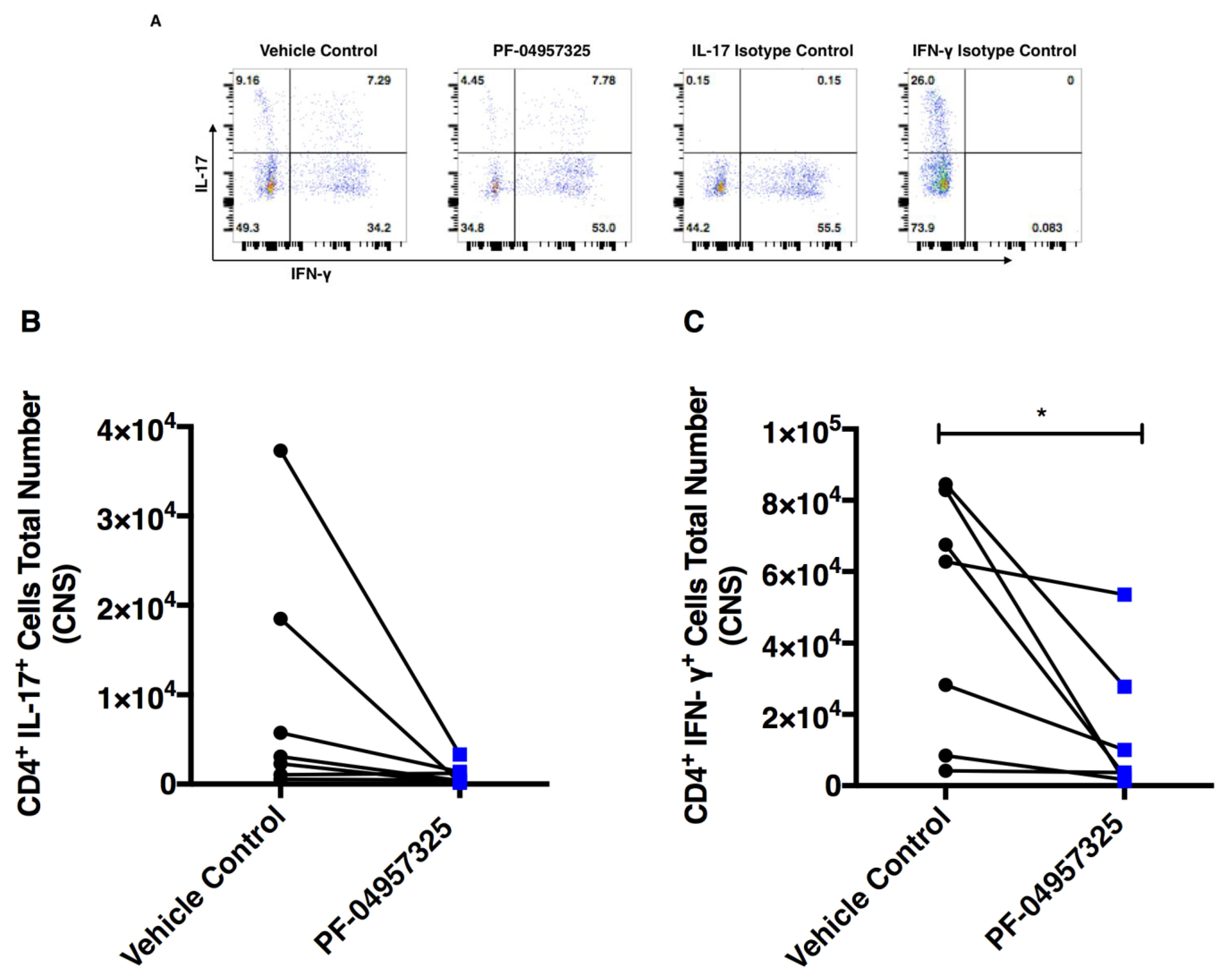
3.5. Treatment with PF-04957325 Suppresses the Accumulation of Pro-Inflammatory T Cell Subsets in the Spinal Cord
3.6. Treatment with PF-04957325 Does Not Affect Pro-Inflammatory Cytokine Production in the Spleen and CELN
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourne, H.R.; Weinstein, Y.; Melmon, K.L.; Lichtenstein, L.M.; Henney, C.S.; Shearer, G.M. Modulation of Inflammation and Immunity by Cyclic AMP. Science 1974, 184, 19–28. [Google Scholar] [CrossRef]
- Wang, T.; Sheppard, J.R.; Foker, J.E. Rise and Fall of Cyclic AMP Required for Onset of Lymphocyte DNA Synthesis. Science 1978, 201, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S. Compartmentalized signalling: Spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 2009, 276, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Epstein, P.M. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem. J. 2006, 393, 21–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzelmann, A.; Morcillo, E.J.; Lungarella, G.; Adnot, S.; Sanjar, S.; Beume, R.; Schudt, C.; Tenor, H. The preclinical pharmacology of roflumilast—A selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm. Pharm. Ther. 2010, 23, 235–256. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, O. Spondyloarthropathies: Apremilast: Welcome advance in treatment of psoriatic arthritis. Nat. Rev. Rheumatol. 2014, 10, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Callender, V.D.; Alexis, A.F.; Stein Gold, L.F.; Lebwohl, M.G.; Paller, A.S.; Desai, S.R.; Tan, H.; Ports, W.C.; Zielinski, M.A.; Tallman, A.M. Efficacy and Safety of Crisaborole Ointment, 2%, for the Treatment of Mild-to-Moderate Atopic Dermatitis Across Racial and Ethnic Groups. Am. J. Clin. Derm. 2019, 20, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paes, D.; Schepers, M.; Rombaut, B.; van den Hove, D.; Vanmierlo, T.; Prickaerts, J. The Molecular Biology of Phosphodiesterase 4 Enzymes as Pharmacological Targets: An Interplay of Isoforms, Conformational States, and Inhibitors. Pharm. Rev. 2021, 73, 1016–1049. [Google Scholar] [CrossRef]
- Schepers, M.; Tiane, A.; Paes, D.; Sanchez, S.; Rombaut, B.; Piccart, E.; Rutten, B.P.F.; Brone, B.; Hellings, N.; Prickaerts, J.; et al. Targeting Phosphodiesterases-Towards a Tailor-Made Approach in Multiple Sclerosis Treatment. Front. Immunol. 2019, 10, 1727. [Google Scholar] [CrossRef]
- Kelly, M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. 2018, 42, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.B.; Smith, K.H.; Koziol-White, C.J.; Li, F.; Kazarian, A.G.; Corpuz, M.L.; Shumyatcher, M.; Ehlert, F.J.; Himes, B.E.; Panettieri, R.A., Jr.; et al. PDE8 is Expressed in Human Airway Smooth Muscle and Selectively Regulates cAMP Signaling by beta2AR-AC6. Am. J. Respir. Cell Mol. Biol. 2018, 8, 530–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basole, C.P.; Nguyen, R.K.; Lamothe, K.; Vang, A.; Clark, R.; Baillie, G.S.; Epstein, P.M.; Brocke, S. PDE8 controls CD4(+) T cell motility through the PDE8A-Raf-1 kinase signaling complex. Cell. Signal. 2017, 40, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vang, A.G.; Basole, C.; Dong, H.; Nguyen, R.K.; Housley, W.; Guernsey, L.; Adami, A.J.; Thrall, R.S.; Clark, R.B.; Epstein, P.M.; et al. Differential Expression and Function of PDE8 and PDE4 in Effector T cells: Implications for PDE8 as a Drug Target in Inflammation. Front. Pharmacol. 2016, 7, 259. [Google Scholar] [CrossRef] [Green Version]
- Shimizu-Albergine, M.; Van Yserloo, B.; Golkowski, M.G.; Ong, S.E.; Beavo, J.A.; Bornfeldt, K.E. SCAP/SREBP pathway is required for the full steroidogenic response to cyclic AMP. Proc. Natl. Acad. Sci. USA 2016, 113, E5685–E5693. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Claffey, K.P.; Brocke, S.; Epstein, P.M. Inhibition of breast cancer cell migration by activation of cAMP signaling. Breast Cancer Res. Treat. 2015, 152, 17–28. [Google Scholar] [CrossRef]
- Maurice, D.H. PDE8A runs interference to limit PKA inhibition of Raf-1. Proc. Natl. Acad. Sci. USA 2013, 110, 6248–6249. [Google Scholar] [CrossRef] [Green Version]
- Tsai, L.C.; Shimizu-Albergine, M.; Beavo, J.A. The high affinity cAMP-specific phosphodiesterase 8B (PDE8B) controls steroidogenesis in the mouse adrenal gland. Mol. Pharmacol. 2010, 79, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.M.; Day, J.P.; Huston, E.; Zimmermann, B.; Hampel, K.; Christian, F.; Romano, D.; Terhzaz, S.; Lee, L.C.; Willis, M.J.; et al. Phosphodiesterase-8A binds to and regulates Raf-1 kinase. Proc. Natl. Acad. Sci. USA 2013, 110, E1533–E1542. [Google Scholar] [CrossRef] [Green Version]
- DeNinno, M.P.; Wright, S.W.; Visser, M.S.; Etienne, J.B.; Moore, D.E.; Olson, T.V.; Rocke, B.N.; Andrews, M.P.; Zarbo, C.; Millham, M.L.; et al. 1,5-Substituted nipecotic amides: Selective PDE8 inhibitors displaying diastereomer-dependent microsomal stability. Bioorganic Med. Chem. Lett. 2011, 21, 3095–3098. [Google Scholar] [CrossRef]
- Vang, A.G.; Ben-Sasson, S.Z.; Dong, H.; Kream, B.; DeNinno, M.P.; Claffey, M.M.; Housley, W.; Clark, R.B.; Epstein, P.M.; Brocke, S. PDE8 regulates rapid Teff cell adhesion and proliferation independent of ICER. PLoS ONE 2010, 5, e12011. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Zitt, C.; Auriga, C.; Hatzelmann, A.; Epstein, P.M. Inhibition of PDE3, PDE4 and PDE7 potentiates glucocorticoid-induced apoptosis and overcomes glucocorticoid resistance in CEM T leukemic cells. Biochem. Pharmacol. 2010, 79, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Osmanova, V.; Epstein, P.M.; Brocke, S. Phosphodiesterase 8 (PDE8) regulates chemotaxis of activated lymphocytes. Biochem. Biophys. Res. Commun. 2006, 345, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Glavas, N.A.; Ostenson, C.; Schaefer, J.B.; Vasta, V.; Beavo, J.A. T cell activation up-regulates cyclic nucleotide phosphodiesterases 8A1 and 7A3. Proc. Natl. Acad. Sci. USA 2001, 98, 6319–6324. [Google Scholar] [CrossRef] [Green Version]
- Soderling, S.H.; Bayuga, S.J.; Beavo, J.A. Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc. Natl. Acad. Sci. USA 1998, 95, 8991–8996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, D.A.; Smith, J.F.; Pillar, J.S.; St Denis, S.H.; Cheng, J.B. Isolation and characterization of PDE8A, a novel human cAMP-specific phosphodiesterase. Biochem. Biophys. Res. Commun. 1998, 246, 570–577. [Google Scholar] [CrossRef]
- Wang, H.; Yan, Z.; Yang, S.; Cai, J.; Robinson, H.; Ke, H. Kinetic and structural studies of phosphodiesterase-8A and implication on the inhibitor selectivity. Biochemistry 2008, 47, 12760–12768. [Google Scholar] [CrossRef] [Green Version]
- Demirbas, D.; Wyman, A.R.; Shimizu-Albergine, M.; Cakici, O.; Beavo, J.A.; Hoffman, C.S. A yeast-based chemical screen identifies a PDE inhibitor that elevates steroidogenesis in mouse Leydig cells via PDE8 and PDE4 inhibition. PLoS ONE 2013, 8, e71279. [Google Scholar] [CrossRef]
- Vang, A.G.; Housley, W.; Dong, H.; Basole, C.; Ben-Sasson, S.Z.; Kream, B.E.; Epstein, P.M.; Clark, R.B.; Brocke, S. Regulatory T-cells and cAMP suppress effector T-cells independently of PKA-CREM/ICER: A potential role for Epac. Biochem. J. 2013, 456, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Blanco, F.C.; Soria, M.A.; Klepp, L.I.; Bigi, F. ERAP1 and PDE8A Are Downregulated in Cattle Protected against Bovine Tuberculosis. J. Mol. Microbiol. Biotechnol. 2017, 27, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Chimienti, F.; Cavarec, L.; Vincent, L.; Salvetat, N.; Arango, V.; Underwood, M.D.; Mann, J.J.; Pujol, J.F.; Weissmann, D. Brain region-specific alterations of RNA editing in PDE8A mRNA in suicide decedents. Transl. Psychiatry 2019, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.J.; O’Rourke, K.S.; Olorenshaw, I.; Hawkins, G.A.; Maas, S.; Laxminarayana, D. Altered editing in cyclic nucleotide phosphodiesterase 8A1 gene transcripts of systemic lupus erythematosus T lymphocytes. Immunology 2008, 125, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.E.; Wong, Q.; Levine, D.M.; McHugh, C.; Laurie, C.; Doheny, K.; Lam, M.Y.; Baer, A.N.; Challacombe, S.; Lanfranchi, H.; et al. Genome-Wide Association Analysis Reveals Genetic Heterogeneity of Sjogren’s Syndrome According to Ancestry. Arthritis Rheumatol. 2017, 69, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamanuma, M.; Yuasa, K.; Sasaki, T.; Sakurai, N.; Kotera, J.; Omori, K. Comparison of enzymatic characterization and gene organization of cyclic nucleotide phosphodiesterase 8 family in humans. Cell. Signal. 2003, 15, 565–574. [Google Scholar] [CrossRef]
- Preller, V.; Gerber, A.; Wrenger, S.; Togni, M.; Marguet, D.; Tadje, J.; Lendeckel, U.; Rocken, C.; Faust, J.; Neubert, K.; et al. TGF-beta1-mediated control of central nervous system inflammation and autoimmunity through the inhibitory receptor CD26. J. Immunol. 2007, 178, 4632–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamvil, S.S.; Steinman, L. The T lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol. 1990, 8, 579–621. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L.; Zamvil, S.S. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005, 26, 565–571. [Google Scholar] [CrossRef]
- Steinman, L. Blocking adhesion molecules as therapy for multiple sclerosis: Natalizumab. Nat. Rev. Drug Discov. 2005, 4, 510–518. [Google Scholar] [CrossRef]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4βl integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef]
- Brocke, S.; Piercy, C.; Steinman, L.; Weissman, I.L.; Veromaa, T. Antibodies to CD44 and integrin alpha4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc. Natl. Acad. Sci. USA 1999, 96, 6896–6901. [Google Scholar] [CrossRef] [Green Version]
- Bielekova, B.; Richert, N.; Howard, T.; Packer, A.N.; Blevins, G.; Ohayon, J.; McFarland, H.F.; Sturzebecher, C.S.; Martin, R. Treatment with the phosphodiesterase type-4 inhibitor rolipram fails to inhibit blood-brain barrier disruption in multiple sclerosis. Mult. Scler. J. 2009, 15, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- DeNinno, M.P. Future directions in phosphodiesterase drug discovery. Bioorganic Med. Chem. Lett. 2012, 22, 6794–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biton, A.; Ansorge, S.; Bank, U.; Tager, M.; Reinhold, D.; Brocke, S. Divergent actions by inhibitors of DP IV and APN family enzymes on CD4+ Teff cell motility and functions. Immunobiology 2011, 216, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Fleming, K.K.; Bovaird, J.A.; Mosier, M.C.; Emerson, M.R.; LeVine, S.M.; Marquis, J.G. Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 170, 71–84. [Google Scholar] [CrossRef]
- Chakravarti, S.; Sabatos, C.A.; Xiao, S.; Illes, Z.; Cha, E.K.; Sobel, R.A.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. Tim-2 regulates T helper type 2 responses and autoimmunity. J. Exp. Med. 2005, 202, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Nichols, F.C.; Housley, W.J.; O’Conor, C.A.; Manning, T.; Wu, S.; Clark, R.B. Unique lipids from a common human bacterium represent a new class of Toll-like receptor 2 ligands capable of enhancing autoimmunity. Am. J. Pathol. 2009, 175, 2430–2438. [Google Scholar] [CrossRef] [Green Version]
- Epstein, P.M.; Hachisu, R. Cyclic nucleotide phosphodiesterase in normal and leukemic human lymphocytes and lymphoblasts. Adv. Cycl. Nucleotide Protein Phosphorylation Res. 1984, 16, 303–324. [Google Scholar]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Tenor, H.; Hatzelmann, A.; Beume, R.; Lahu, G.; Zech, K.; Bethke, T.D. Pharmacology, clinical efficacy, and tolerability of phosphodiesterase-4 inhibitors: Impact of human pharmacokinetics. Handb. Exp. Pharmacol. 2011, 204, 85–119. [Google Scholar]
- Bielekova, B.; Lincoln, A.; McFarland, H.; Martin, R. Therapeutic potential of phosphodiesterase-4 and -3 inhibitors in Th1-mediated autoimmune diseases. J. Immunol. 2000, 164, 1117–1124. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A.; Gil, C. cAMP-specific phosphodiesterase inhibitors: Promising drugs for inflammatory and neurological diseases. Expert Opin. Ther. Patents 2014, 24, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. The discovery of natalizumab, a potent therapeutic for multiple sclerosis. J. Cell Biol. 2012, 199, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorgo, E.; Moltu, K.; Tasken, K. Phosphodiesterases as targets for modulating T-cell responses. Handb. Exp. Pharmacol. 2011, 204, 345–363. [Google Scholar] [CrossRef]
- Warnke, C.; Menge, T.; Hartung, H.P.; Racke, M.K.; Cravens, P.D.; Bennett, J.L.; Frohman, E.M.; Greenberg, B.M.; Zamvil, S.S.; Gold, R.; et al. Natalizumab and progressive multifocal leukoencephalopathy: What are the causal factors and can it be avoided? Arch. Neurol. 2010, 67, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloka, S.; Metz, L.M.; Hader, W.; Starreveld, Y.; Yong, V.W. Reduction of microglial activity in a model of multiple sclerosis by dipyridamole. J. Neuroinflamm. 2013, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.J.; Coffey, C.S.; Conwit, R.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Naismith, R.T.; et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 846–855. [Google Scholar] [CrossRef]
- Goodman, A.D.; Gyang, T.; Smith, A.D., 3rd. Ibudilast for the treatment of multiple sclerosis. Expert Opin. Investig. Drugs 2016, 25, 1231–1237. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basole, C.P.; Nguyen, R.K.; Lamothe, K.; Billis, P.; Fujiwara, M.; Vang, A.G.; Clark, R.B.; Epstein, P.M.; Brocke, S. Treatment of Experimental Autoimmune Encephalomyelitis with an Inhibitor of Phosphodiesterase-8 (PDE8). Cells 2022, 11, 660. https://doi.org/10.3390/cells11040660
Basole CP, Nguyen RK, Lamothe K, Billis P, Fujiwara M, Vang AG, Clark RB, Epstein PM, Brocke S. Treatment of Experimental Autoimmune Encephalomyelitis with an Inhibitor of Phosphodiesterase-8 (PDE8). Cells. 2022; 11(4):660. https://doi.org/10.3390/cells11040660
Chicago/Turabian StyleBasole, Chaitali P., Rebecca K. Nguyen, Katie Lamothe, Puja Billis, Mai Fujiwara, Amanda G. Vang, Robert B. Clark, Paul M. Epstein, and Stefan Brocke. 2022. "Treatment of Experimental Autoimmune Encephalomyelitis with an Inhibitor of Phosphodiesterase-8 (PDE8)" Cells 11, no. 4: 660. https://doi.org/10.3390/cells11040660