
Advances in Allogeneic Cancer Cell Therapy and Future Perspectives on “Off-the-Shelf” T Cell Therapy Using iPSC Technology and Gene Editing

 ,
,
Abstract
:1. Introduction
2. Adoptive T Cell Therapy and Tumor Microenvironment
3. T Cell Exhaustion
4. Use of iPSC Technology to Reinvigorate Immunity
5. Rejuvenation of CTLs
6. Anti-Tumor Effect of iPSC-Derived CTLs Proven In Vivo
7. Rejuvenated CTLs for Solid Cancers
8. Rejuvenated CTLs as an Unlimited Source for “Off-the-Shelf” Therapy
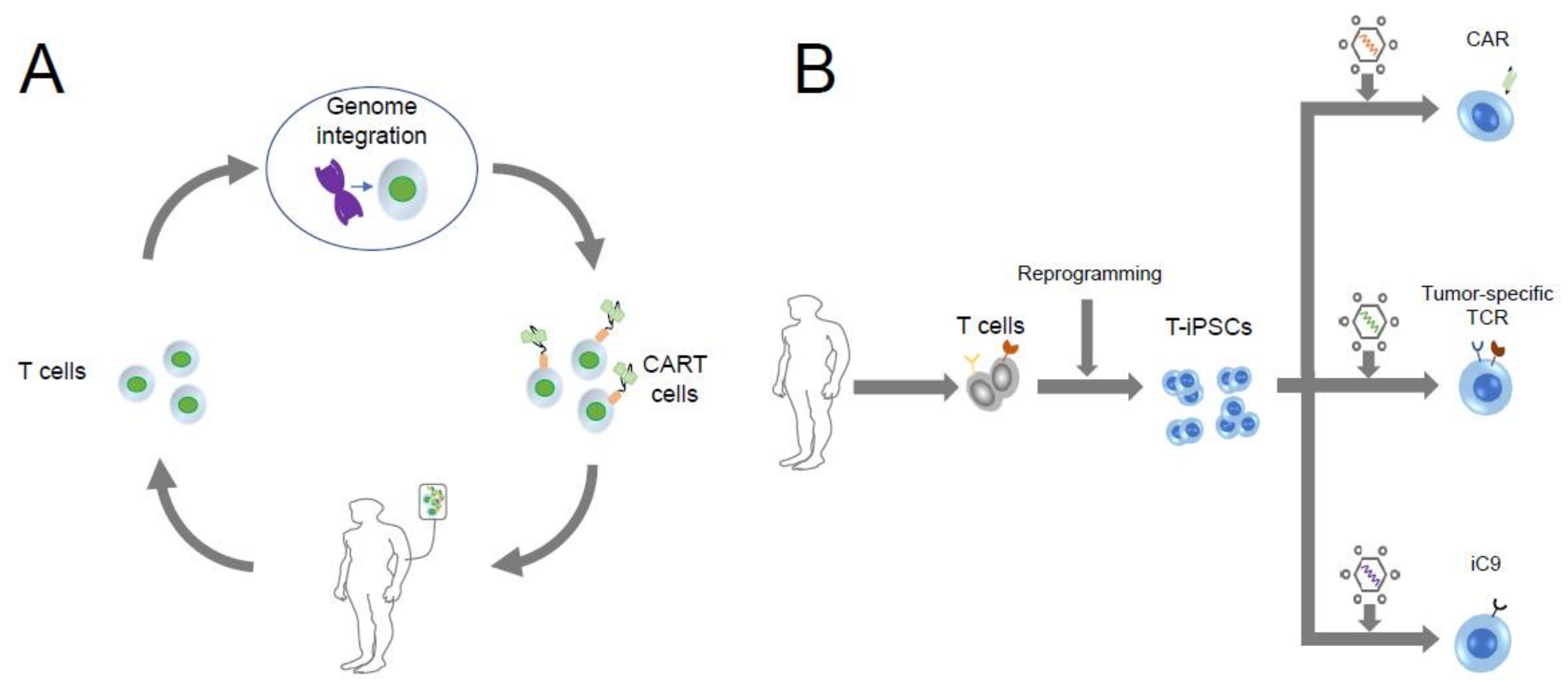
9. iPSC Technology Exerting Its “Off-the-Shelf” Advantage in CART Therapy
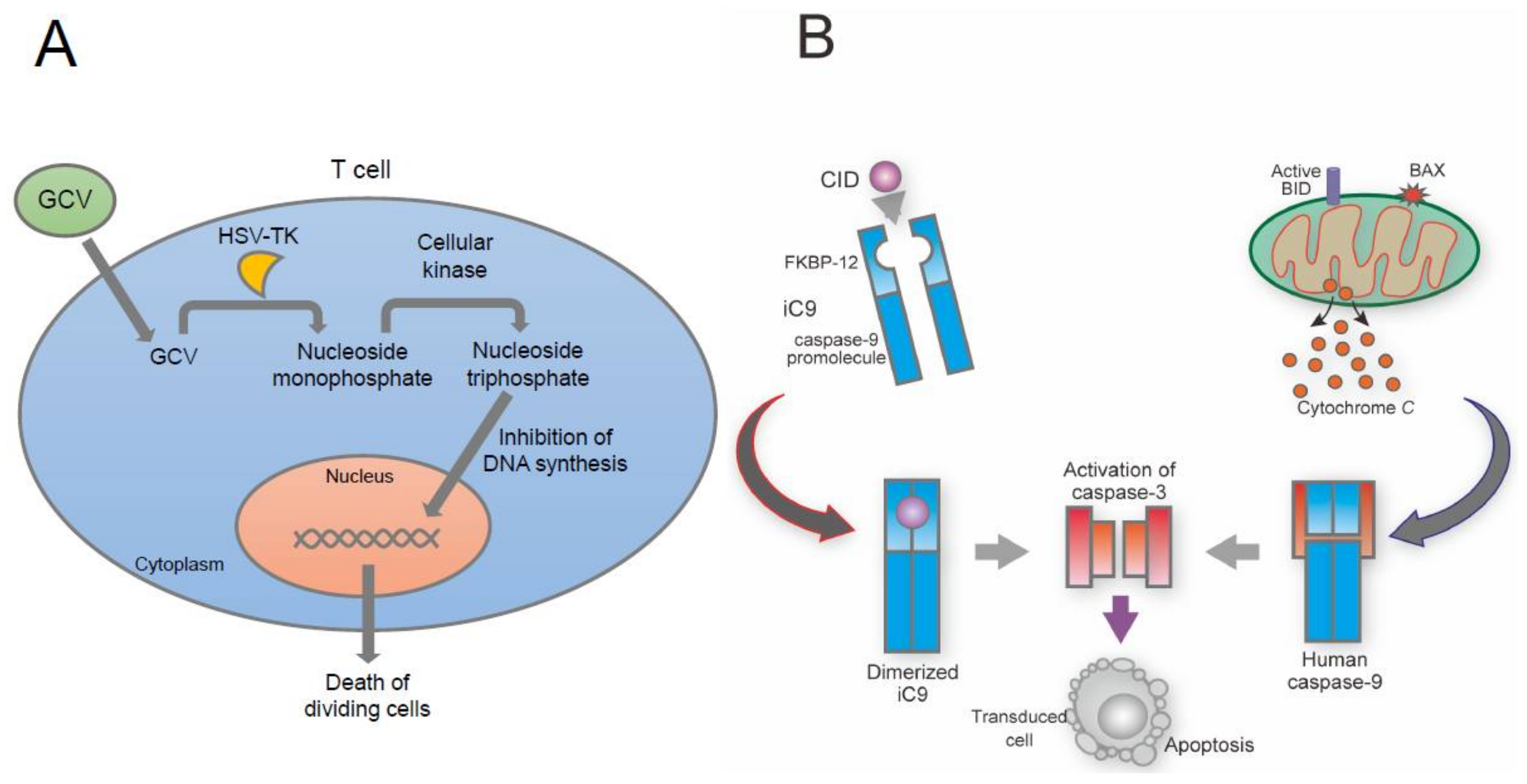
10. Suicide Gene-Based Safeguard System
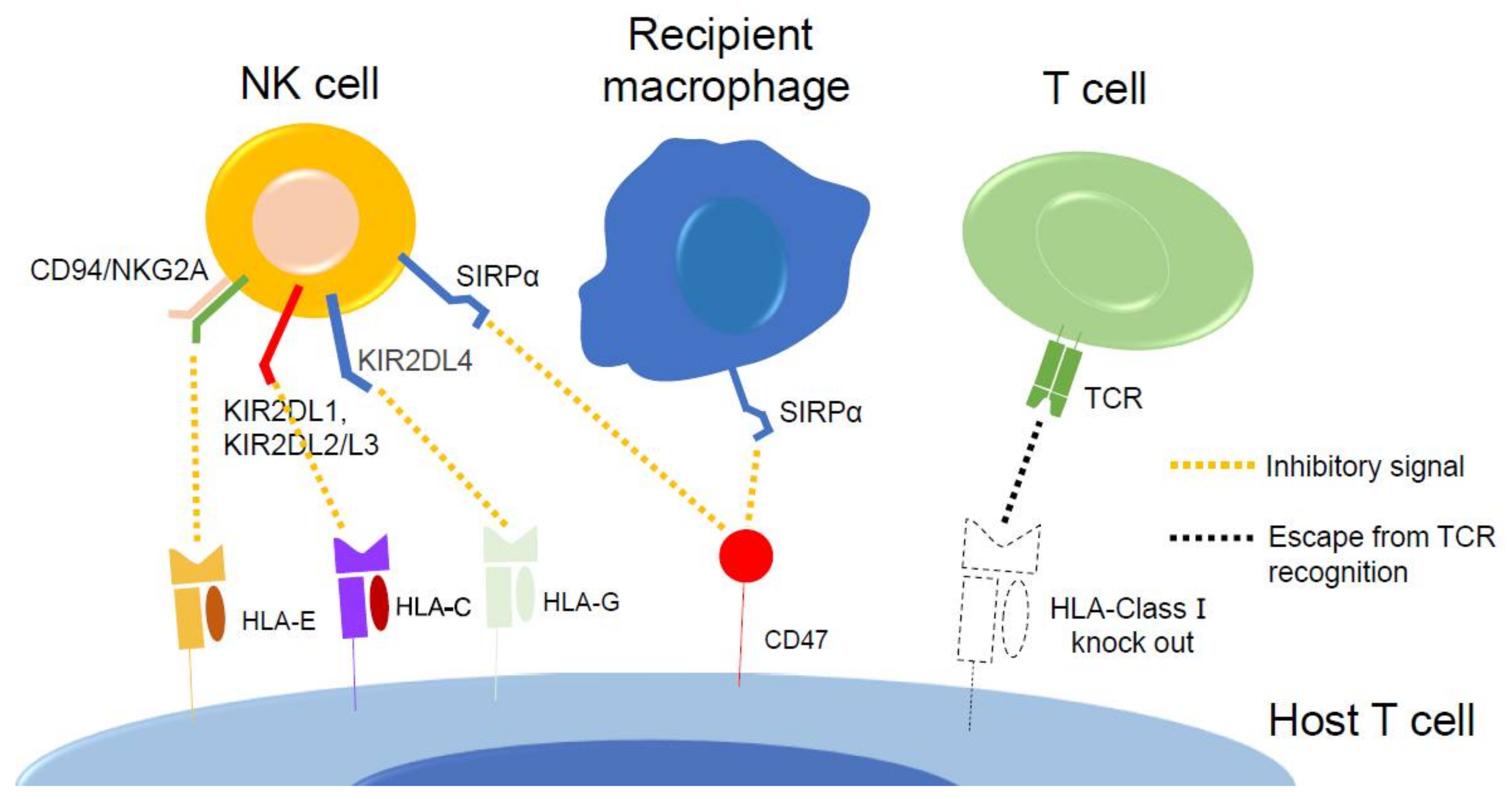
11. Gene Edited iPSC-Derived T Cell Therapy
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, E.D.; Lochte, H.L., Jr.; Lu, W.C.; Ferrebee, J.W. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N. Engl. J. Med. 1957, 257, 491–496. [Google Scholar] [CrossRef]
- Appelbaum, F.R. Hematopoietic-cell transplantation at 50. N. Engl. J. Med. 2007, 357, 1472–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; McGuirk, J.P. Allogeneic Stem Cell Transplantation: A Historical and Scientific Overview. Cancer Res. 2016, 76, 6445–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, E.D.; Buckner, C.D.; Banaji, M.; Clift, R.A.; Fefer, A.; Flournoy, N.; Goodell, B.W.; Hickman, R.O.; Lerner, K.G.; Neiman, P.E.; et al. One hundred patients with acute leukemia treated by chemotherapy, total body irradiation, and allogeneic marrow transplantation. Blood 1977, 49, 511–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, E.D.; Buckner, C.D.; Clift, R.A.; Fefer, A.; Johnson, F.L.; Neiman, P.E.; Sale, G.E.; Sanders, J.E.; Singer, J.W.; Shulman, H.; et al. Marrow transplantation for acute nonlymphoblastic leukemia in first remission. N. Engl. J. Med. 1979, 301, 597–599. [Google Scholar] [CrossRef]
- Thomas, E.D.; Buckner, C.D.; Rudolph, R.H.; Fefer, A.; Storb, R.; Neiman, P.E.; Bryant, J.I.; Chard, R.L.; Clift, R.A.; Epstein, R.B.; et al. Allogeneic marrow grafting for hematologic malignancy using HL-A matched donor-recipient sibling pairs. Blood 1971, 38, 267–287. [Google Scholar] [CrossRef]
- Hansen, J.A.; Clift, R.A.; Thomas, E.D.; Buckner, C.D.; Storb, R.; Giblett, E.R. Transplantation of marrow from an unrelated donor to a patient with acute leukemia. N. Engl. J. Med. 1980, 303, 565–567. [Google Scholar] [CrossRef]
- Tsoi, M.S.; Storb, R.; Dobbs, S.; Thomas, E.D. Specific suppressor cells in graft-host tolerance of HLA-identical marrow transplantation. Nature 1981, 292, 355–357. [Google Scholar] [CrossRef]
- Thomas, E.D. A history of haemopoietic cell transplantation. Br. J. Haematol. 1999, 105, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Billingham, R.E.; Brent, L. Quantitative studies on tissue transplantation immunity IV. Induction of tolerance in newborn mice and studies on the phenomenon of runt disease. Philos. Trans. R. Soc. B 1959, 242, 477. [Google Scholar] [CrossRef]
- Zeiser, R.; Blazar, B.R. Acute Graft-versus-Host Disease-Biologic Process, Prevention, and Therapy. N. Engl. J. Med. 2017, 377, 2167–2179. [Google Scholar] [CrossRef]
- Glucksberg, H.; Storb, R.; Fefer, A.; Buckner, C.D.; Neiman, P.E.; Clift, R.A.; Lerner, K.G.; Thomas, E.D. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 1974, 18, 295–304. [Google Scholar] [CrossRef]
- Deeg, H.J.; Storb, R.; Weiden, P.L.; Raff, R.F.; Sale, G.E.; Atkinson, K.; Graham, T.C.; Thomas, E.D. Cyclosporin A and methotrexate in canine marrow transplantation: Engraftment, graft-versus-host disease, and induction of intolerance. Transplantation 1982, 34, 30–35. [Google Scholar] [CrossRef]
- Storb, R.; Deeg, H.J.; Farewell, V.; Doney, K.; Appelbaum, F.; Beatty, P.; Bensinger, W.; Buckner, C.D.; Clift, R.; Hansen, J.; et al. Marrow transplantation for severe aplastic anemia: Methotrexate alone compared with a combination of methotrexate and cyclosporine for prevention of acute graft-versus-host disease. Blood 1986, 68, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Storb, R.; Deeg, H.J.; Whitehead, J.; Appelbaum, F.; Beatty, P.; Bensinger, W.; Buckner, C.D.; Clift, R.; Doney, K.; Farewell, V.; et al. Methotrexate and cyclosporine compared with cyclosporine alone for prophylaxis of acute graft versus host disease after marrow transplantation for leukemia. N. Engl. J. Med. 1986, 314, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Storb, R.; Raff, R.F.; Appelbaum, F.R.; Deeg, H.J.; Fitzsimmons, W.; Graham, T.C.; Pepe, M.; Pettinger, M.; Sale, G.; van der Jagt, R.; et al. FK-506 and methotrexate prevent graft-versus-host disease in dogs given 9.2 Gy total body irradiation and marrow grafts from unrelated dog leukocyte antigen-nonidentical donors. Transplantation 1993, 56, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Storb, R.; Gluckman, E.; Thomas, E.D.; Buckner, C.D.; Clift, R.A.; Fefer, A.; Glucksberg, H.; Graham, T.C.; Johnson, F.L.; Lerner, K.G.; et al. Treatment of established human graft-versus-host disease by antithymocyte globulin. Blood 1974, 44, 56–75. [Google Scholar] [CrossRef]
- Granot, N.; Storb, R. History of hematopoietic cell transplantation: Challenges and progress. Haematologica 2020, 105, 2716–2729. [Google Scholar] [CrossRef] [PubMed]
- Riesner, K.; Shi, Y.; Jacobi, A.; Kräter, M.; Kalupa, M.; McGearey, A.; Mertlitz, S.; Cordes, S.; Schrezenmeier, J.F.; Mengwasser, J.; et al. Initiation of acute graft-versus-host disease by angiogenesis. Blood 2017, 129, 2021–2032. [Google Scholar] [CrossRef] [Green Version]
- Loiseau, P.; Busson, M.; Balere, M.L.; Dormoy, A.; Bignon, J.D.; Gagne, K.; Gebuhrer, L.; Dubois, V.; Jollet, I.; Bois, M.; et al. HLA Association with hematopoietic stem cell transplantation outcome: The number of mismatches at HLA-A, -B, -C, -DRB1, or -DQB1 is strongly associated with overall survival. Biol. Blood Marrow Transplant. 2007, 13, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Flowers, M.E.; Inamoto, Y.; Carpenter, P.A.; Lee, S.J.; Kiem, H.P.; Petersdorf, E.W.; Pereira, S.E.; Nash, R.A.; Mielcarek, M.; Fero, M.L.; et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood 2011, 117, 3214–3219. [Google Scholar] [CrossRef] [PubMed]
- Jagasia, M.; Arora, M.; Flowers, M.E.; Chao, N.J.; McCarthy, P.L.; Cutler, C.S.; Urbano-Ispizua, A.; Pavletic, S.Z.; Haagenson, M.D.; Zhang, M.J.; et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood 2012, 119, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flowers, M.E.; Martin, P.J. How we treat chronic graft-versus-host disease. Blood 2015, 125, 606–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.J.; Counts, G.W., Jr.; Appelbaum, F.R.; Lee, S.J.; Sanders, J.E.; Deeg, H.J.; Flowers, M.E.; Syrjala, K.L.; Hansen, J.A.; Storb, R.F.; et al. Life expectancy in patients surviving more than 5 years after hematopoietic cell transplantation. J. Clin. Oncol. 2010, 28, 1011–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingard, J.R.; Majhail, N.S.; Brazauskas, R.; Wang, Z.; Sobocinski, K.A.; Jacobsohn, D.; Sorror, M.L.; Horowitz, M.M.; Bolwell, B.; Rizzo, J.D.; et al. Long-term survival and late deaths after allogeneic hematopoietic cell transplantation. J. Clin. Oncol. 2011, 29, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.P.; Hill, G.R.; Blazar, B.R. Chronic graft-versus-host disease: Biological insights from preclinical and clinical studies. Blood 2017, 129, 13–21. [Google Scholar] [CrossRef]
- Lee, S.J.; Vogelsang, G.; Flowers, M.E. Chronic graft-versus-host disease. Biol. Blood Marrow Transplant. 2003, 9, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Wolff, D.; Fatobene, G.; Rocha, V.; Kröger, N.; Flowers, M.E. Steroid-refractory chronic graft-versus-host disease: Treatment options and patient management. Bone Marrow Transplant. 2021, 56, 2079–2087. [Google Scholar] [CrossRef]
- Tzannou, I.; Papadopoulou, A.; Naik, S.; Leung, K.; Martinez, C.A.; Ramos, C.A.; Carrum, G.; Sasa, G.; Lulla, P.; Watanabe, A.; et al. Off-the-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections After Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2017, 35, 3547–3557. [Google Scholar] [CrossRef]
- Reisner, Y.; Kapoor, N.; Kirkpatrick, D.; Pollack, M.S.; Dupont, B.; Good, R.A.; O’Reilly, R.J. Transplantation for acute leukaemia with HLA-A and B nonidentical parental marrow cells fractionated with soybean agglutinin and sheep red blood cells. Lancet 1981, 2, 327–331. [Google Scholar] [CrossRef]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N. Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef]
- Kolb, H.J.; Mittermüller, J.; Clemm, C.; Holler, E.; Ledderose, G.; Brehm, G.; Heim, M.; Wilmanns, W. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood 1990, 76, 2462–2465. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, W.M.; Plougastel, B.F. Immune functions encoded by the natural killer gene complex. Nat. Rev. Immunol. 2003, 3, 304–316. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood 1999, 94, 333–339. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Schattenberg, A.; Cornelissen, J.J.; Thomas, L.L.; Verdonck, L.F. Donor leukocyte infusions are effective in relapsed multiple myeloma after allogeneic bone marrow transplantation. Blood 1997, 90, 4206–4211. [Google Scholar] [CrossRef]
- Kröger, N.; Badbaran, A.; Lioznov, M.; Schwarz, S.; Zeschke, S.; Hildebrand, Y.; Ayuk, F.; Atanackovic, D.; Schilling, G.; Zabelina, T.; et al. Post-transplant immunotherapy with donor-lymphocyte infusion and novel agents to upgrade partial into complete and molecular remission in allografted patients with multiple myeloma. Exp. Hematol. 2009, 37, 791–798. [Google Scholar] [CrossRef]
- Kolb, H.J.; Schattenberg, A.; Goldman, J.M.; Hertenstein, B.; Jacobsen, N.; Arcese, W.; Ljungman, P.; Ferrant, A.; Verdonck, L.; Niederwieser, D.; et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 1995, 86, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Donato, M.L.; Siegel, D.S.; Vesole, D.H.; McKiernan, P.; Nyirenda, T.; Pecora, A.L.; Baker, M.; Goldberg, S.L.; Mato, A.; Goy, A.; et al. The graft-versus-myeloma effect: Chronic graft-versus-host disease but not acute graft-versus-host disease prolongs survival in patients with multiple myeloma receiving allogeneic transplantation. Biol. Blood Marrow Transplant. 2014, 20, 1211–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hromas, R.; Cornetta, K.; Srour, E.; Blanke, C.; Broun, E.R. Donor leukocyte infusion as therapy of life-threatening adenoviral infections after T-cell-depleted bone marrow transplantation. Blood 1994, 84, 1689–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi, Y.; Kami, M.; Oki, Y.; Kazuyama, Y.; Kawabata, M.; Miyakoshi, S.; Morinaga, S.; Suzuki, R.; Mori, S.; Muto, Y. Donor lymphocyte infusion for treatment of life-threatening respiratory syncytial virus infection following bone marrow transplantation. Bone Marrow Transplant. 2000, 26, 573–576. [Google Scholar] [CrossRef]
- Greenberg, P.D. Adoptive T cell therapy of tumors: Mechanisms operative in the recognition and elimination of tumor cells. Adv. Immunol. 1991, 49, 281–355. [Google Scholar] [CrossRef]
- Cheever, M.A.; Chen, W. Therapy with cultured T cells: Principles revisited. Immunol. Rev. 1997, 157, 177–194. [Google Scholar] [CrossRef]
- Barry, M.; Bleackley, R.C. Cytotoxic T lymphocytes: All roads lead to death. Nat. Rev. Immunol. 2002, 2, 401–409. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Leen, A.M.; Heslop, H.E.; Brenner, M.K. Antiviral T-cell therapy. Immunol. Rev. 2014, 258, 12–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkart, P.A. Mechanism of lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 1985, 3, 31–58. [Google Scholar] [CrossRef]
- Froelich, C.J.; Orth, K.; Turbov, J.; Seth, P.; Gottlieb, R.; Babior, B.; Shah, G.M.; Bleackley, R.C.; Dixit, V.M.; Hanna, W. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J. Biol. Chem. 1996, 271, 29073–29079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkoski, M.J.; Hobman, M.; Heibein, J.A.; Tomaselli, K.; Li, F.; Seth, P.; Froelich, C.J.; Bleackley, R.C. Entry and trafficking of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood 1998, 92, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Eiz-Vesper, B.; Maecker-Kolhoff, B.; Blasczyk, R. Adoptive T-cell immunotherapy from third-party donors: Characterization of donors and set up of a T-cell donor registry. Front. Immunol. 2012, 3, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, J.N.; Doubrovina, E.; Sauter, C.; Jaroscak, J.J.; Perales, M.A.; Doubrovin, M.; Prockop, S.E.; Koehne, G.; O’Reilly, R.J. Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood 2010, 116, 5045–5049. [Google Scholar] [CrossRef] [PubMed]
- Leen, A.M.; Bollard, C.M.; Mendizabal, A.M.; Shpall, E.J.; Szabolcs, P.; Antin, J.H.; Kapoor, N.; Pai, S.Y.; Rowley, S.D.; Kebriaei, P.; et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood 2013, 121, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Gerdemann, U.; Katari, U.L.; Papadopoulou, A.; Keirnan, J.M.; Craddock, J.A.; Liu, H.; Martinez, C.A.; Kennedy-Nasser, A.; Leung, K.S.; Gottschalk, S.M.; et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol. Ther. 2013, 21, 2113–2121. [Google Scholar] [CrossRef] [Green Version]
- Alt, F.W.; Baltimore, D. Joining of immunoglobulin heavy chain gene segments: Implications from a chromosome with evidence of three D-JH fusions. Proc. Natl. Acad. Sci. USA 1982, 79, 4118–4122. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.K.; Sixbey, J.W.; Gan, Y.; Srivastava, D.K.; Bowman, L.C.; Krance, R.A.; Brenner, M.K.; et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood 1998, 92, 1549–1555. [Google Scholar] [CrossRef]
- Straathof, K.C.; Bollard, C.M.; Popat, U.; Huls, M.H.; Lopez, T.; Morriss, M.C.; Gresik, M.V.; Gee, A.P.; Russell, H.V.; Brenner, M.K.; et al. Treatment of nasopharyngeal carcinoma with Epstein-Barr virus--specific T lymphocytes. Blood 2005, 105, 1898–1904. [Google Scholar] [CrossRef] [Green Version]
- Comoli, P.; Pedrazzoli, P.; Maccario, R.; Basso, S.; Carminati, O.; Labirio, M.; Schiavo, R.; Secondino, S.; Frasson, C.; Perotti, C.; et al. Cell therapy of stage IV nasopharyngeal carcinoma with autologous Epstein-Barr virus-targeted cytotoxic T lymphocytes. J. Clin. Oncol. 2005, 23, 8942–8949. [Google Scholar] [CrossRef]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Aqui, N.; Badros, A.; Cotte, J.; Chrisley, L.; Veloso, E.; Zheng, Z.; Westphal, S.; Mair, R.; et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat. Med. 2005, 11, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Leen, A.M.; Rooney, C.M.; Foster, A.E. Improving T cell therapy for cancer. Annu. Rev. Immunol. 2007, 25, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Kinoshita, S.; Furukawa, Y.; Ando, J.; Nakauchi, H.; Brenner, M.K. Chapter 3—Improving the safety of iPSC-derived T cell therapy. In Molecular Players in iPSC Technology; Birbrair, A., Ed.; Academic Press: Cambridge, MA, USA, 2022; Volume 12, pp. 95–115. [Google Scholar]
- Strand, S.; Hofmann, W.J.; Hug, H.; Müller, M.; Otto, G.; Strand, D.; Mariani, S.M.; Stremmel, W.; Krammer, P.H.; Galle, P.R. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells--a mechanism of immune evasion? Nat. Med. 1996, 2, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Hahne, M.; Rimoldi, D.; Schröter, M.; Romero, P.; Schreier, M.; French, L.E.; Schneider, P.; Bornand, T.; Fontana, A.; Lienard, D.; et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: Implications for tumor immune escape. Science 1996, 274, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.W.; O’Connell, J.; O’Sullivan, G.C.; Brady, C.; Roche, D.; Collins, J.K.; Shanahan, F. The Fas counterattack in vivo: Apoptotic depletion of tumor-infiltrating lymphocytes associated with Fas ligand expression by human esophageal carcinoma. J. Immunol. 1998, 160, 5669–5675. [Google Scholar]
- Okada, K.; Komuta, K.; Hashimoto, S.; Matsuzaki, S.; Kanematsu, T.; Koji, T. Frequency of apoptosis of tumor-infiltrating lymphocytes induced by fas counterattack in human colorectal carcinoma and its correlation with prognosis. Clin. Cancer Res. 2000, 6, 3560–3564. [Google Scholar] [PubMed]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Peng, Y.; Laouar, Y.; Li, M.O.; Green, E.A.; Flavell, R.A. TGF-beta regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc. Natl. Acad. Sci. USA 2004, 101, 4572–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, S.; Chattopadhyay, S.; Chakraborty, N.G. Manipulation of regulatory T cells and antigen-specific cytotoxic T lymphocyte-based tumour immunotherapy. Immunology 2015, 144, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Scudellari, M. How iPS cells changed the world. Nature 2016, 534, 310–312. [Google Scholar] [CrossRef]
- Kimbrel, E.A.; Lanza, R. Current status of pluripotent stem cells: Moving the first therapies to the clinic. Nat. Rev. Drug Discov. 2015, 14, 681–692. [Google Scholar] [CrossRef]
- Hayashi, R.; Ishikawa, Y.; Ito, M.; Kageyama, T.; Takashiba, K.; Fujioka, T.; Tsujikawa, M.; Miyoshi, H.; Yamato, M.; Nakamura, Y.; et al. Generation of corneal epithelial cells from induced pluripotent stem cells derived from human dermal fibroblast and corneal limbal epithelium. PLoS ONE 2012, 7, e45435. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Nishimura, T.; Yamazaki, S.; Yamaguchi, T.; Kawana-Tachikawa, A.; Hayama, T.; Nakauchi, Y.; Ando, J.; Ota, Y.; Takahashi, S.; et al. A Safeguard System for Induced Pluripotent Stem Cell-Derived Rejuvenated T Cell Therapy. Stem Cell Rep. 2015, 5, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, M.; Ando, J.; Yamazaki, S.; Ishii, M.; Sakiyama, Y.; Harada, S.; Honda, T.; Yamaguchi, T.; Nojima, M.; Ohshima, K.; et al. Long-term eradication of extranodal natural killer/T-cell lymphoma, nasal type, by induced pluripotent stem cell-derived Epstein-Barr virus-specific rejuvenated T cells in vivo. Haematologica 2020, 105, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Gottschalk, S.; Edwards, O.L.; Sili, U.; Huls, M.H.; Goltsova, T.; Davis, A.R.; Heslop, H.E.; Rooney, C.M. Generating CTLs against the subdominant Epstein-Barr virus LMP1 antigen for the adoptive immunotherapy of EBV-associated malignancies. Blood 2003, 101, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Bollard, C.M.; Gottschalk, S.; Leen, A.M.; Weiss, H.; Straathof, K.C.; Carrum, G.; Khalil, M.; Wu, M.F.; Huls, M.H.; Chang, C.C.; et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood 2007, 110, 2838–2845. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Rooney, C.M.; Heslop, H.E. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat. Rev. Clin. Oncol. 2012, 9, 510–519. [Google Scholar] [CrossRef]
- Honda, T.; Ando, M.; Ando, J.; Ishii, M.; Sakiyama, Y.; Ohara, K.; Toyota, T.; Ohtaka, M.; Masuda, A.; Terao, Y.; et al. Sustainable Tumor-Suppressive Effect of iPSC-Derived Rejuvenated T Cells Targeting Cervical Cancers. Mol. Ther. 2020, 28, 2394–2405. [Google Scholar] [CrossRef]
- Kashima, S.; Maeda, T.; Masuda, K.; Nagano, S.; Inoue, T.; Takeda, M.; Kono, Y.; Kobayashi, T.; Saito, S.; Higuchi, T.; et al. Cytotoxic T Lymphocytes Regenerated from iPS Cells Have Therapeutic Efficacy in a Patient-Derived Xenograft Solid Tumor Model. iScience 2020, 23, 100998. [Google Scholar] [CrossRef]
- Ishii, M.; Ando, J.; Yamazaki, S.; Toyota, T.; Ohara, K.; Furukawa, Y.; Suehara, Y.; Nakanishi, M.; Nakashima, K.; Ohshima, K.; et al. iPSC-Derived Neoantigen-Specific CTL Therapy for Ewing Sarcoma. Cancer Immunol. Res. 2021, 9, 1175–1186. [Google Scholar] [CrossRef]
- Ando, M.; Nakauchi, H. ‘Off-the-shelf’ immunotherapy with iPSC-derived rejuvenated cytotoxic T lymphocytes. Exp. Hematol. 2017, 47, 2–12. [Google Scholar] [CrossRef]
- Lee, S.J.; Klein, J.; Haagenson, M.; Baxter-Lowe, L.A.; Confer, D.L.; Eapen, M.; Fernandez-Vina, M.; Flomenberg, N.; Horowitz, M.; Hurley, C.K.; et al. High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood 2007, 110, 4576–4583. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018, 15, 31–46. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 2013, 13, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.X.; Westwood, J.A.; Moeller, M.; Duong, C.P.; Wei, W.Z.; Malaterre, J.; Trapani, J.A.; Neeson, P.; Smyth, M.J.; Kershaw, M.H.; et al. Tumor ablation by gene-modified T cells in the absence of autoimmunity. Cancer Res. 2010, 70, 9591–9598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Themeli, M.; Rivière, I.; Sadelain, M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell 2015, 16, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, S.; Ando, M.; Ando, J.; Ishii, M.; Yamaguchi, T.; Yamazaki, S.; Toyota, T.; Ohara, K.; Ohtaka, M.; Nakanishi, M.; et al. Dual-antigen targeted iPSC-derived chimeric antigen receptor-T cell therapy for refractory lymphoma. Mol. Ther. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Restifo, N.P. Reassessing target antigens for adoptive T-cell therapy. Nat. Biotechnol. 2013, 31, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Bonini, C.; Ferrari, G.; Verzeletti, S.; Servida, P.; Zappone, E.; Ruggieri, L.; Ponzoni, M.; Rossini, S.; Mavilio, F.; Traversari, C.; et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 1997, 276, 1719–1724. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Gallo-Stampino, C.; Bordignon, C. Modulation of GvHD by suicide-gene transduced donor T lymphocytes: Clinical applications in mismatched transplantation. Cytotherapy 2005, 7, 144–149. [Google Scholar] [CrossRef]
- Berger, C.; Flowers, M.E.; Warren, E.H.; Riddell, S.R. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 2006, 107, 2294–2302. [Google Scholar] [CrossRef] [Green Version]
- Brenner, M.K.; Gottschalk, S.; Leen, A.M.; Vera, J.F. Is cancer gene therapy an empty suit? Lancet Oncol. 2013, 14, e447–e456. [Google Scholar] [CrossRef] [Green Version]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.C.; Pulè, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef]
- Clackson, T.; Yang, W.; Rozamus, L.W.; Hatada, M.; Amara, J.F.; Rollins, C.T.; Stevenson, L.F.; Magari, S.R.; Wood, S.A.; Courage, N.L.; et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA 1998, 95, 10437–10442. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Hoyos, V.; Yagyu, S.; Tao, W.; Ramos, C.A.; Dotti, G.; Brenner, M.K.; Bouchier-Hayes, L. Bortezomib sensitizes non-small cell lung cancer to mesenchymal stromal cell-delivered inducible caspase-9-mediated cytotoxicity. Cancer Gene Ther. 2014, 21, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Di Stasi, A.; Tey, S.K.; Krance, R.A.; Martinez, C.; Leung, K.S.; Durett, A.G.; Wu, M.F.; Liu, H.; Leen, A.M.; et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood 2014, 123, 3895–3905. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Dotti, G.; Krance, R.A.; Martinez, C.A.; Naik, S.; Kamble, R.T.; Durett, A.G.; Dakhova, O.; Savoldo, B.; Di Stasi, A.; et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015, 125, 4103–4113. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Brenner, M.K. Improving the safety of T-Cell therapies using an inducible caspase-9 gene. Exp. Hematol. 2016, 44, 1013–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Hong, S.G.; Winkler, T.; Spencer, D.M.; Jares, A.; Ichwan, B.; Nicolae, A.; Guo, V.; Larochelle, A.; Dunbar, C.E. Development of an inducible caspase-9 safety switch for pluripotent stem cell-based therapies. Mol. Ther. Methods Clin. Dev. 2014, 1, 14053. [Google Scholar] [CrossRef] [PubMed]
- Barese, C.N.; Felizardo, T.C.; Sellers, S.E.; Keyvanfar, K.; Di Stasi, A.; Metzger, M.E.; Krouse, A.E.; Donahue, R.E.; Spencer, D.M.; Dunbar, C.E. Regulated apoptosis of genetically modified hematopoietic stem and progenitor cells via an inducible caspase-9 suicide gene in rhesus macaques. Stem Cells 2015, 33, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Yagyu, S.; Hoyos, V.; Del Bufalo, F.; Brenner, M.K. An Inducible Caspase-9 Suicide Gene to Improve the Safety of Therapy Using Human Induced Pluripotent Stem Cells. Mol. Ther. 2015, 23, 1475–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, G.; Kawabata, S.; Ando, M.; Nishiyama, Y.; Sugai, K.; Ozaki, M.; Iida, T.; Ookubo, T.; Kojima, K.; Kashiwagi, R.; et al. Fail-Safe System against Potential Tumorigenicity after Transplantation of iPSC Derivatives. Stem Cell Rep. 2017, 8, 673–684. [Google Scholar] [CrossRef]
- Martin, R.M.; Fowler, J.L.; Cromer, M.K.; Lesch, B.J.; Ponce, E.; Uchida, N.; Nishimura, T.; Porteus, M.H.; Loh, K.M. Improving the safety of human pluripotent stem cell therapies using genome-edited orthogonal safeguards. Nat. Commun. 2020, 11, 2713. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Ikeda, K.; Uchida, N.; Nishimura, T.; White, J.; Martin, R.M.; Nakauchi, H.; Sebastiano, V.; Weinberg, K.I.; Porteus, M.H. Efficient scarless genome editing in human pluripotent stem cells. Nat. Methods 2018, 15, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 2019, 24, 566–578.e567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzue, K.; Reinherz, E.L.; Koyasu, S. Critical role of NK but not NKT cells in acute rejection of parental bone marrow cells in F1 hybrid mice. Eur. J. Immunol. 2001, 31, 3147–3152. [Google Scholar] [CrossRef]
- Masuda, K.; Kawamoto, H. Possible NK cell-mediated immune responses against iPSC-derived cells in allogeneic transplantation settings. Inflamm. Regen. 2021, 41, 2. [Google Scholar] [CrossRef] [PubMed]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.A.; Clegg, D.O.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, M.; Duan, S.; Franco, P.J.; Kenty, J.H.; Hedrick, P.; Xia, Y.; Allen, A.; Ferreira, L.M.R.; Strominger, J.L.; et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10441–10446. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.; Zhou, Y.; Ni, X.; Tong, X.; Xu, X.; Dong, Z.; Sun, R.; Tian, Z.; Wei, H. Natural Killer Cells Promote Fetal Development through the Secretion of Growth-Promoting Factors. Immunity 2017, 47, 1100–1113.e1106. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.X.; Jiang, F.; Zhu, Y.J.; Wang, L.; Li, K.; Li, Y.; Wang, X.H.; Li, L.S.; Yao, Y.Q. Enhanced Immunological Tolerance by HLA-G1 from Neural Progenitor Cells (NPCs) Derived from Human Embryonic Stem Cells (hESCs). Cell Physiol. Biochem. 2017, 44, 1435–1444. [Google Scholar] [CrossRef]
- Zhao, L.; Teklemariam, T.; Hantash, B.M. Heterelogous expression of mutated HLA-G decreases immunogenicity of human embryonic stem cells and their epidermal derivatives. Stem Cell Res 2014, 13, 342–354. [Google Scholar] [CrossRef] [Green Version]
- Cudkowicz, G.; Stimpfling, J.H. Deficient growth of c57bl marrow cells transplanted in f1 hybrid mice. association with the histocompatibility-2 locus. Immunology 1964, 7, 291–306. [Google Scholar] [PubMed]
- Raulet, D.H. Bone Marrow Cell Rejection, MHC, NK Cells, and Missing Self Recognition: Ain’t That Peculiar (with Apologies to Marvin Gaye). J. Immunol. 2015, 195, 2923–2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, W.J.; Kumar, V.; Bennett, M. Rejection of bone marrow allografts by mice with severe combined immune deficiency (SCID). Evidence that natural killer cells can mediate the specificity of marrow graft rejection. J. Exp. Med. 1987, 165, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Iriguchi, S.; Waseda, M.; Ueda, N.; Ueda, T.; Xu, H.; Minagawa, A.; Ishikawa, A.; Yano, H.; Ishi, T.; et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat. Biomed. Eng. 2021, 5, 429–440. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Company | Products | CAR | Allogeneic Cell Source | Strategy to Evade GVHD or Immune Rejection | Target Gene | Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| Allogene Therapeutics | ALLO-501 (in USA) | CD19 | T cell | TALEN | TRAC and CD52 | NCT03939026 |
| ALLO-715 | BCMA | NCT04093596 | ||||
| ALLO-316 | CD70 | NCT04696731 | ||||
| Adicet Bio, Inc. | ADI-001 | CD20 | γδT cell | NCT04735471 | ||
| Caribou Biosciences, Inc. | CB010A | CD19 | T cell | CRISPR/Cas9 | TRAC, PD1 (KO) | NCT04637763 |
| Cellectis | UCART-123 | CD123 | T cell | TALEN | TRAC (KO) | NCT03190278 (AML) NCT03203369 (BPDCN) NCT04150497 NCT04142619 |
| UCART-22 | CD22 | TRAC and CD52 (KO) | ||||
| UCART-CS1 | CS1 | TRAC and CS1 (KO) | ||||
| Celyad | CYAD-211 | BCMA | T cell | shRNA | CD3ζ | NCT03692429 |
| CYAD-101 | NKG2DL | Overexpression of TIM | - | NCT03692429 | ||
| CRISPR Therapeutics AG | CTX110 | CD19 | T cell | CRISPR/Cas9 | TRAC and B2M (KO) | NCT04035434 |
| CTX120 | BCMA | NCT04244656 | ||||
| CTX130 | CD70 | NCT04502446 | ||||
| Fate Therapeutics | FT-819 | CD19 | iPSC-derived T cell | CRISPR/Cas9 | TRAC | NCT04629729 |
| Great Ormond Street Hospital for Children | PBLTT52CAR19 | CD19 | T cell | CRISPR/Cas9 | CD52 and TRAC (KO) | NCT04557436 |
| Kurr therapeutics | KUR-502 | CD19 | NKT | NCT03774654 | ||
| Precision Biosciences | PBCAR0191 | CD19 | T cell | meganuclease mRNA shRNA | B2M (shRNA), HLA-E | NCT03666000 |
| PBCAR19B | CD19 | NCT04649112 | ||||
| PBCAR20A | CD20 | NCT04030195 | ||||
| PBCAR269A | BCMA | NCT04171843 | ||||
| Tessa Therapeutics Pte Ltd. | CD30.CAR-EBVST cells | CD30 | EBVST | NCT04288726 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furukawa, Y.; Hamano, Y.; Shirane, S.; Kinoshita, S.; Azusawa, Y.; Ando, J.; Nakauchi, H.; Ando, M. Advances in Allogeneic Cancer Cell Therapy and Future Perspectives on “Off-the-Shelf” T Cell Therapy Using iPSC Technology and Gene Editing. Cells 2022, 11, 269. https://doi.org/10.3390/cells11020269
Furukawa Y, Hamano Y, Shirane S, Kinoshita S, Azusawa Y, Ando J, Nakauchi H, Ando M. Advances in Allogeneic Cancer Cell Therapy and Future Perspectives on “Off-the-Shelf” T Cell Therapy Using iPSC Technology and Gene Editing. Cells. 2022; 11(2):269. https://doi.org/10.3390/cells11020269
Chicago/Turabian StyleFurukawa, Yoshiki, Yasuharu Hamano, Shuichi Shirane, Shintaro Kinoshita, Yoko Azusawa, Jun Ando, Hiromitsu Nakauchi, and Miki Ando. 2022. "Advances in Allogeneic Cancer Cell Therapy and Future Perspectives on “Off-the-Shelf” T Cell Therapy Using iPSC Technology and Gene Editing" Cells 11, no. 2: 269. https://doi.org/10.3390/cells11020269