
Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease



1
Institut National de la Santé et de la Recherche Médicale, Centre de Recherche des Cordeliers, Equipe-Immunopathologie et Immunointervention Thérapeutique, Sorbonne Université, Université de Paris, 75006 Paris, France
2
Service de Neurologie, Hôpitaux Universitaires de Strasbourg, 67000 Strasbourg, France
3
Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), INSERM-U964/CNRS-UMR7104/Université de Strasbourg, 67400 Illkirch, France
4
Fédération de Médecine Translationnelle de Strasbourg (FMTS), Université de Strasbourg, 67000 Strasbourg, France
5
CNRS and Strasbourg University, Unit Biotechnology and Cell Signaling/Strasbourg Drug Discovery and Development Institute (IMS), 67000 Strasbourg, France
6
University of Strasbourg Institute for Advanced Study (USIAS), 67000 Strasbourg, France
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(12), 3547; https://doi.org/10.3390/cells10123547
Submission received: 22 November 2021
/
Revised: 13 December 2021
/
Accepted: 13 December 2021
/
Published: 15 December 2021
(This article belongs to the Special Issue State-of-Art in Autophagy and Neurodegeneration)
Abstract
:Cellular quality control systems have gained much attention in recent decades. Among these, autophagy is a natural self-preservation mechanism that continuously eliminates toxic cellular components and acts as an anti-ageing process. It is vital for cell survival and to preserve homeostasis. Several cell-type-dependent canonical or non-canonical autophagy pathways have been reported showing varying degrees of selectivity with regard to the substrates targeted. Here, we provide an updated review of the autophagy machinery and discuss the role of various forms of autophagy in neurodegenerative diseases, with a particular focus on Parkinson’s disease. We describe recent findings that have led to the proposal of therapeutic strategies targeting autophagy to alter the course of Parkinson’s disease progression.
1. Introduction
Although some elements of Parkinson’s disease (PD) were described a very long time ago, the first clear medical description of this disease was published in 1817 by James Parkinson [1]. Since then, substantial efforts have been made to understand the underlying pathogenesis and pathological elements of this complex disease, in terms of neuropathological and anatomo-pathological changes [2,3,4,5]. PD is a multifactorial disease with heterogeneous causative factors, including genetic, environmental, molecular and cellular components. PD is characterized by a broad spectrum of motor and non-motor signs and symptoms. They include rest tremor, bradykinesia, postural instability/unsteady gait, rigidity, alongside psychiatric disorders, sleep disorders, dysautonomic disorders, pain, anosmia, and cognitive disorders. Motor signs result primarily from the loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and intracellular inclusions of aggregated and misfolded α-synuclein (α-syn), encapsulated or not in Lewy bodies (LB) and Lewy neurites (LN) in the neurons [3,6] (Figure 1; see Appendix A for definition).
The symptoms of PD develop gradually with age. They can start with a slight tremor in one hand and a feeling of stiffness in the body; bradykinesia is frequent. Recent studies confirm that more than 3% of the general population from 65 years of age are affected by PD. In 5%–10% of cases, however, symptoms of PD appear earlier; this is referred to as young-onset PD (YOPD). Men are 50% more likely to develop PD than women, but the risk for women appears to increase with age.
The root cause of PD remains largely unknown. Some cases of PD have been linked to genetic mutations, but clear hereditary causes of this disease are difficult to establish. Indeed, only 15% of patients with PD have a family history of the disease. Some genes have been associated with distinct, typical or rarer forms of the disease, which include juvenile or adult-onset, early or late, autosomal recessive, dominant or X-linked forms [4,7,8,9]. Causative risk factors associated with particular ethnic groups have also been identified. The genes most frequently linked to PD include GBA, LRRK2, PRKN, SNCA, ATP13A2, ATP10B, DJ-1, DNAJC6, FBXO7, HTRA2, MAPT, PINK1, PLA2G6, VPS35, and VPS13C [4,7,8,9,10,11,12,13]. The majority of these genes encode proteins that are linked either directly or indirectly to quality control mechanisms that are vital in maintaining cell homeostasis, vesicular transport pathways, autophagy processes, and the endo-lysosomal system. Other genetic alterations have also been associated with PD, including epigenetic changes, such as DNA methylation, chromatin remodeling, histone modifications, microRNAs and long non-coding RNAs [4,14].
2. Pathogenesis and Pathology
Clinicopathological studies reveal slow progression of PD from the ventrolateral region of the SNpc, with later spread to other brain regions [15]. Clinical symptoms of PD become detectable when the degeneration of the DA neurons progresses within the SNpc. LBs are observed at sites of neuronal damage (Figure 1). In normal physiology, the α-syn deposited in these structures performs central functions in endocytosis; vesicle trafficking; synthesis, storage and release of dopamine; Ca2+ homeostasis; microtubule dynamics; and other processes [16]. Thus, neuronal activity is completely dependent on α-syn, and also on mitochondrial homeostasis. Although α-syn is predominantly present in cytosolic eosinophilic LBs, it has also been detected in mitochondria, lysosomes, and other organelles in post-mortem PD brains. The presence of LBs in the peripheral, enteric, and central nervous systems (CNS) has been implicated in both motor and non-motor symptoms of PD [17,18]. Point mutations in the α-syn sequence or other pathological insults leads to the formation of oligomers, which may then group together as larger aggregates. These aggregates can alter numerous cellular and molecular pathways in neurons—in particular involving autophagy and proteasomal processes, such as mitochondrial function, vesicle trafficking, organelle, and protein degradation—all of which lead to neurodegeneration. Subsequently, as a result of the neurodegeneration, α-syn aggregates are deposited in the SN, where they activate microglia [19]. This uncontrollable activation can generate pro-inflammatory signals [20], which may lead to further neurodegeneration when a critical threshold is reached.
2.1. Neuropsychiatric Manifestations of PD
No specific test exists to diagnose PD. Consequently, diagnosis is based on medical history, a review of signs and symptoms, and a neurological and physical examination (Box 1). Motor signs of PD usually begin around 60 years of age [21], but YOPD is not rare, particularly in some hereditary forms [22]. Unilateral or asymmetric bradykinesia and/or rest tremor are the first symptoms of the disease [23]. Rest tremor is present in relaxed muscles and disappears during action and sleep. It may be increased by mental calculation. Bradykinesia, defined by slowness of movement and decreased amplitude or speed, leads to difficulties with repetitive movements, micrography, small-step gait, speech difficulties (hypophonia and dysarthria), which will emerge as the disease evolves. Rigidity may cause pain and contribute to postural deformity (thoracolumbar spinal flexion). Progression is slow with bilateral extension of akinesia, tremor, and hypertonia, followed by postural instability, freezing of gait, falls, and in some patients, camptocormia. Some non-motor signs (premotor) may occur several years before the first motor symptoms; these include depression, hyposmia, constipation, or rapid-eye-movement sleep disorders [24]. Anxiety and apathy may be present from the onset of PD, whereas severe dysautonomia (orthostatic hypotension, urinary dysfunction due to detrusor hyperactivity), sleep fragmentation, cognitive disorders (dysexecutive disorders), and hallucinations emerge later, and will contribute to loss of autonomy [24,25].
Box 1. Diagnosis of PD.
Parkinsonian syndrome, which may have numerous causes, is defined by the presence of bradykinesia and at least two features among rest tremor, plastic hypertonia, or both. Among parkinsonian syndromes, diagnosis of PD, based on the Movement Disorder Society international criteria [23], rests on
- -
- at least two supportive criteria (e.g., clear positive response to L-dopa, induced dyskinesia, rest tremor),
- -
- absence of absolute exclusion criteria (e.g., associated cerebellar signs, vertical ocular palsy, frontotemporal dementia or sensitive signs, no L-dopa response, isolated lower limb parkinsonism, treatment with dopamine receptor blockers), and
- -
- absence of red flags (e.g., rapid progression with, over the 5 first years, wheelchair use, bulbar dysfunction, severe dysautonomic dysfunction, recurrent falls during the first 3 years, inspiratory respiratory dysfunction, associated pyramidal signs or muscle contractures (neck, hands; feet), bilateral and symmetric evolution, absence of dopamine sensitivity and /or evolution, absence of non-motor signs).
2.2. Current Treatments for PD and Clinical Management
Symptomatic treatment is the only clinical option currently available [26], with therapies aiming to compensate for the dopaminergic deficit. Dopaminergic drugs (levodopa associated with dopa-decarboxylase inhibitor, dopaminergic agonists, or monoamine oxidase-type B inhibitors), used individually or in poly-therapy regimens, are very efficient during the early stages of the disease. However, management becomes more difficult as the years progress. Indeed, dopaminergic treatments, which improve motor signs, can have very disabling complications. A wearing-off phenomenon (end of dose failure) and dyskinesia occur after several years of levodopa treatment [27]. Impulse control disorders (pathological gambling or shopping, hyper sexuality; [28], hallucinations, or psychosis may also complicate dopaminergic treatments, and are more frequently encountered with dopamine agonists [29]. Other treatments, including catechol-O-methyltransferase inhibitors to treat motor fluctuations, or amantadine for dyskinesia [26] can be used later as the disease progresses. Second-line treatment (continuous subcutaneous infusion of apomorphine, continuous jejunal administration of duodopa gel, bilateral subthalamic nuclear stimulation) are proposed when motor fluctuations and dyskinesia become significant [30]. These treatments aim to achieve stable striatal dopaminergic stimulation but have no impact on disease progression. Furthermore, some axial symptoms (dysarthria, postural instability) are not dopa-sensitive, and clinical management of non-motor symptoms remains difficult [31,32].
Decades of investigations have led to the development of therapeutic strategies, which have undoubtedly improved quality of life for patients. However, slowing the disease’s progression still remains a challenge, and a persistent priority [33], and novel disease-modifying approaches are eagerly awaited [3,34]. Although the functions of the proteasome and of autophagy, including macroautophagy and chaperone-mediated autophagy (CMA), have long been known to contribute to α-syn clearance [35,36], dysregulation of these processes remains poorly understood in PD. Several gene mutations and alterations to proteins involved in PD are closely linked to autophagy, particularly mitophagy and autophagy-lysosomal pathways. In this review, we focus on the involvement of autophagy in PD, we comment on the major unanswered questions in the field and propose new directions for possible therapeutic interventions targeting autophagy pathways.
3. Autophagy
Autophagy is a major intracellular degradation system by which cytoplasmic materials are delivered to the lysosome for degradation. Based on the route by which content is delivered to lysosomes, several forms of autophagy have been defined. These different forms also have varying degrees of selectivity for targeted cargos (Table 1; Figure 2). The three main types of autophagy process are macroautophagy, CMA, and microautophagy/eMi. Whatever the delivery route, the main role of these processes is to degrade unwanted material that is defective, may be toxic, or has been produced in excess, and thus to maintain cell homeostasis.
3.1. The Autophagy Machinery
The mechanisms of autophagy have been thoroughly investigated and were reviewed in detail by numerous authors [61,62,63]. The general features of the three pathways are presented in Figure 2. Recent advances related to canonical and non-canonical autophagic processes—especially in mammalian systems—have added to our understanding of the mechanisms that may play important roles in neurodegenerative diseases such as PD. Nevertheless, many of the molecular discoveries upon which our current understanding of the regulation of autophagy is based emerged from analyses involving yeast. In cells, the three forms of autophagy coexist, and play vital roles in maintaining cellular homeostasis. However, the large majority of results available in this field relate to macroautophagy. This process has been divided into the following steps: nucleation, elongation, autophagosome formation, autophagosome-lysosome fusion, and degradation (Figure 2). Each step is finely genetically-regulated and plays its own specific role in maintaining the dynamic nature of the process. For example, a number of conserved autophagy-related proteins act in a hierarchical manner to mediate autophagosome formation. Upon upstream induction, the autophagy machinery comes into contact with the isolation membrane/phagophore. The early origins and definitive complex source [endoplasmic reticulum (ER), Golgi complex, endosomes, and mitochondria] of the nascent isolation membrane remains a matter of debate [64]. An ultrastructural study involving electron microscopy experiments confirmed that a specialized subdomain of the ER contributes to phagophore generation [65]. About 40 autophagy-related (ATG) proteins have been identified as involved in this dynamic process, they are hierarchically organized, starting from the initiation of the process and progressing through to the maturation of autophagosomes. These proteins work together in several functional complexes, notably (i) the Unc-51-like kinase 1 (ULK1)/ATG1 kinase complex; (ii) the class III phosphatidylinositol (PI) 3-kinase complex; (iii) the PI(3)P-binding ATG2-ATG18 complex; (iv) the two conjugation systems (ATG12 conjugation system and microtubule-associated protein 1A/1B-light chain 3 (MAP1LC3)/ATG8 conjugation system); and (v) the fusion machinery (Figure 2).
In fact, several so-called ATG proteins have alternative functions beyond autophagy [66]. Thus, for example, MAP1LC3 lipidation (a mechanism that has long been used to assess autophagic activity [67,68]) is also involved in non-autophagic cellular mechanisms such as phagocytosis, LAP, micropinocytosis, or viral infection. These processes are known as non-canonical autophagic processes [69]. In these non-canonical processes, the functions of which are still incompletely characterized [70,71] MAP1LC3 conjugates to single membranes (single membrane ATG8 conjugation, SMAC), and cytosolic constituents are not delivered to the lysosome [72].
3.2. Neuronal Autophagy Contributes to Neuronal Physiology
There is compelling evidence to support the idea that neuronal autophagy plays a decisive role in several aspects of neuron development and in preserving neuronal activity [73,74,75]. In post-mitotic cells like neurons, autophagy is especially important for survival and homeostasis, because these cells cannot eliminate accumulated toxic substances and damaged organelles during cell division. Autophagy, as well as the proteasomal system [76], is therefore one of the vital quality control mechanisms that ensure the longevity of neuronal cells. Presynaptic autophagy in the axon terminal is also essential for synaptic maintenance and plasticity [77].
Among nerve cells, only cortical neurons, Purkinje cells, and hypothalamic neurons can increase their autophagosome content upon a stimulus. The precise reasons for this niche mechanism are currently unknown [62,78]. One possible explanation is trivial and related to the fact that, like for some other cell types, measuring autophagy in neurons, especially in the brain, remains challenging [79,80]. Alternatively, because nerve cells are terminally differentiated—with a lower regenerative capacity than other cells—they are less autophagic. However, studies on brains from autophagy-deficient mice provided evidence that sequestosome-1 (SQSTM1)/p62 protein and polyubiquitinated proteins accumulate in most neuronal cells [81]. In contrast, SQSTM1 deficiency does not result in a complete lack of autophagy. It appears therefore that the autophagosome content depends on the type of cells and the type of stressor.
3.3. Autophagy and Neurodegenerative Diseases
As introduced above, to prevent neuronal and synaptic dysfunction, neurons have evolved mechanisms to remove toxic and defective components and organelles. These mechanisms are essential to maintain a high degree of neurotransmission and the integrity of the functional proteome in neurons. Autophagy is central to this protective system. Age-related functional loss of autophagy makes the neurons more vulnerable to stress and can lead to cell death [82]. Pathological disruption of autophagy pathways can also result in neurodegenerative disorders that may or may not be linked to ageing.
Compromised autophagy has been documented in many neurodegenerative diseases, including PD, Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) (for comprehensive reviews, see [63,83,84]). In investigating the mechanisms linking autophagy to these diseases, it has been observed, for example, that mice specifically deficient for Atg5 in neural cells develop progressive deficits in motor function while also accumulating cytoplasmic inclusion bodies in neurons [85]. Similarly, in mice deficient for Atg7, Atg5, or Ambra1, ubiquitin was found to accumulate in the CNS, and cytoplasmic inclusions were associated with motor dysfunctions, and neuronal tube defects in mouse embryos [86]. Mutation of genes linked to autophagic processes—for example, SQSTM1, optineurin/OPTN, E3 ubiquitin ligase PARKIN/PRKN, PINK1, TBK1—have also been implicated in many neurodegenerative diseases. In particular, defects in mitophagy that are also seen in organ-specific and systemic inflammatory diseases, have been documented in neurodegenerative diseases [87]. In addition to these genetic mutations, significant alterations to protein expression have been linked to neurodegenerative diseases. For example, abnormal expression of the protein glandular epithelial cell 1 (GABARAPL1/GEC1) has been associated with neurodegenerative diseases [88].
3.4. Autophagy and Parkinson’s Disease
Among the pathological hallmarks of PD are LBs that contain abnormally aggregated α-syn protein. Mutations or triplication of the gene encoding α-syn (SNCA) are rare, but are clearly involved in the initiation and progression of PD. Interestingly, any failure affecting one of the components of the degradative process, either directly or indirectly, impairs the other autophagy processes. The ubiquitin-proteasome system (UPS) is known to be the primary degradative pathway for monoubiquitinated α-syn, whereas the macroautophagy pathway degrades deubiquitinated α-syn [89,90]. In PD, therefore, both mitochondria and lysosomes play crucial roles (Figure 3).
3.4.1. Role of Mitophagy in PD
As an energy producing organelle, the mitochondrion is central to several neurodegenerative diseases, including PD [91,92,93,94,95]. Multiple investigations revealed that genetic mutations associated with PD (e.g., PRKN, PINK1, and others) are also closely linked to mitochondrial defects, including defects in mitophagy (Table 2) [96]. The type of damage caused to the mitochondria naturally depends on the type of α-syn (forming aggregates or not, produced from mutated or native forms of SNCA). Further studies confirmed that α-syn affects the interaction of the mitochondria-associated membrane with the ER. This interaction plays a pivotal role in regulating Ca2+ signaling and apoptosis. In addition, abnormal α-syn interferes with the peroxisome proliferator-activated receptor gamma coactivator 1-alpha, which plays a crucial role in mitochondrial biogenesis and in apoptosis. Mitochondrial dysfunction with the involvement of factors related to α-syn has been comprehensively discussed elsewhere [9,97,98].
Defects in the mitophagy pathway, especially PARK2 (PRKN mutations) and PARK6 (PINK1 mutations), have been proposed as a major cause of familial PD. In healthy conditions, PINK1, which localizes to the mitochondrion, is translocated into the mitochondrial inner membrane where it is degraded. Under certain unknown conditions, mitochondria become damaged and lose membrane potential (Figure 3). This leads to PINK1 activation and recruitment of PRKN, which helps to induce mitophagy while acting on other mitochondrial membrane proteins [OPTN and nuclear dot protein 52-kDa (NDP52)] [62,119,120,121,122,123,124]. PRKN mutations are the most frequent cause of autosomal recessive YOPD, followed by mutations in PINK1. Alongside its role in mitophagy, PRKN plays an essential role in lipid processing and the ubiquitination of the GTPase Rab7, which regulate lysosomal dynamics [125,126,127,128]. PRKN deficiency results in DA neuronal degeneration in mice, and embryonic fibroblasts derived from PINK1-deficient mice show lysosomal dysfunction [129]. In addition, mutations in PINK1 and PRKN lead to defects in the mitophagy process [62]. However, studies have yet to explain why PRKN is not recruited to mitochondria in DA neurons under depolarized conditions [130]. A consequence of mitophagy dysfunctions in neurons is uncontrolled stress (i.e., generation of reactive oxygen species), which causes neuronal cell death. In line with this effect, targeting mitophagy defects may be beneficial in PD. It has been shown, for example, that an inhibitor of the mitochondrial deubiquitinase USP30, which negatively regulates PRKN-mediated mitophagy, selectively increases mitophagic flux, thus it could be of interest for the development of novel therapeutic approaches [131,132].
In addition to the major effect of PINK1 and PRKN mutations, SNCA mutations have been studied in the context of mitophagy. α-syn interacts with the Miro proteins (outer mitochondrial membrane adapter proteins, useful in mitochondrial motility) and interferes with the Miro degradation process, which is an essential step in the mitophagy process [133]. Studies in mice and yeast harboring mutations in SCNA confirmed the role of α-syn in neuronal death, via mitochondrial dysfunction [134,135].
The transcription factor myocyte enhancer factor 2D (MEF2D) is another essential mitochondrial regulator (Table 2). It is a central factor in the transmission of extracellular signals and activation of genetic programs in response to a wide range of stimuli in several cell types, including neurons. MEF2D is a critical regulator of IL-10 gene expression, involved in negative control of the microglial inflammatory response, and preventing inflammation-mediated cytotoxicity [136]. Reduced MEF2D expression has been directly linked to reduced levels of nicotinamide adenine dinucleotide dehydrogenase 6 (NADH), a component of mitochondrial complex I. Post-mortem analysis of brain samples from PD patients revealed reduced levels of both MEF2D and NADH [137].
A number of other genetic mutations, including deficiencies in mitochondrial apoptosis-inducing factor (AIF) and mitochondrial transcription factor A (TFAM; [138] that perturb endo-lysosomal pathways, also affect mitochondrial physiology and function, leading for example to impaired mitophagy, dysfunctional oxidative phosphorylation, deregulated mitochondrial dynamics, altered mitogenesis, calcium imbalance, altered mitochondrial trafficking, and induction of oxidative stress (Table 2). PRKN-independent autophagy pathways are involved in the selective mitophagy process via receptor-mediated, lipid-mediated, and ubiquitin ligase-mediated pathways [97,139,140]. It is not currently known to what extent these pathways are linked to PD.
3.4.2. Role of Macroautophagy in PD
The involvement of macroautophagy and its defects have been extensively investigated in neurodegenerative diseases and synucleinopathy, with some studies also referring to PD [141,142,143,144]. Accumulating evidence indicates that several components of the macroautophagy pathway are involved in PD (Figure 3). Genetic analyses of PD patients have revealed abnormal expression levels for the genes encoding ATG5, ATG7, ATG12, and MAP1LC3B (Table 2). Two independent exome sequencing studies [145,146] also identified point mutations in the vacuolar protein sorting ortholog 35 (VPS35) gene, causing an autosomal dominant form of PD (PARK17) (Table 2). This monogenic subtype of PD is rare, and the precise role of the VPS35 mutations (especially VPS35 D620N) is not yet fully understood (discussed in [147]. Nevertheless, this pathogenic effect highlights the role of the retromer, a highly conserved membrane-associated protein complex in which VPS35 is an intrinsic component, in the α-syn degradation pathway [148,149]. Studies on DA neurons lacking the VPS35 gene show α-syn accumulation and toxicity, in particular loss of mitochondrial fusion and functions [150]. ATG9 trafficking defects (the ATG9 system is required for phagophore expansion) were also associated with mutant VPS35, leading to α-syn accumulation. Increasing VPS35 levels in PD mice rescued α-syn accumulation and induced neuroprotection, demonstrating that regulating VSP35 may be of interest to treat PD [147,151].
Although still a matter of debate, recent data suggest the involvement of more selective autophagy pathways particularly targeting α-syn, that is, synucleinphagy (Table 1) [58]. In Vitro and in vivo studies conducted on microglial cells clearly demonstrated that α-syn was degraded via macroautophagy. Toll-like receptor (TLR)-4 on the microglia recognizes α-syn and activates NF-κB signaling, which in turn alters SQSTM1 transcription. The protein produced, which is present at raised levels, selectively binds to the internalized α-syn leading to its colocalization with autophagosomes. Lack of TLR-4 and SQSTM1 alters autophagy-mediated α-syn degradation [58]. Thus, TLR-4 or SQSTM1 may serve as relevant markers in neurons where α-syn has accumulated.
3.4.3. Role of CMA in PD
In addition to mitophagy and lysosomal macroautophagy, which have relatively well-established roles in α-syn degradation, CMA also appears to be involved in this vital process. CMA is a central process that degrades proteins harboring a specific combination of five amino acids—the “KFERQ-motif” (Figure 2 and Figure 3). This motif allows the chaperone heat shock protein A8 (HSPA8)/cognate 70 kDa protein (HSC70) to bind the substrate protein and direct it to the lysosomal membrane, where it encounters lysosome-associated membrane protein type 2A (LAMP2A), which plays a decisive role. Binding of the complex promotes LAMP2A multimerization to form a membrane-bound higher-molecular–order complex. The substrate protein is then unfolded within this complex and translocated into the lysosome. A lysosome-resident HSPA8 isoform (Lys-HSPA8) contributes to the translocation of the substrate protein across the membrane towards the lysosomal lumen, where it is degraded by acidic hydrolases [36,152,153,154,155]. α-syn contains a KFERQ-like motif and its CMA-mediated degradation is significantly reduced for α-syn constructs lacking the KFERQ-like motif, and by LAMP2A knockdown [152,156].
Interestingly, another protein, leucine-rich repeat kinase 2 (LRRK2)—which is also linked to familial forms of PD (Table 2)—is degraded in lysosomes as part of CMA. In contrast, the most common pathogenic mutant form of LRRK2, G2019S, is poorly degraded by this pathway [154,157]. CMA activity can be modulated by the rate of assembly/disassembly of the translocation complex [40]. In this context, a key finding for PD was the discovery that lysosomal binding of both wild-type and several pathogenic mutant LRRK2 forms was increased in the presence of other CMA substrates. These substrates interfere with the organization of the CMA translocation complex as the enhanced binding inhibits assembly of the CMA translocation complex at the lysosomal membrane. In response to this inhibition, affected cells produced more LAMP2A. A similar feature has been observed in brains from PD patients with LRRK2 mutations. This mechanism leads to the accumulation of other CMA substrates, including α-syn, that remain bound longer than normal at the surface of the lysosomal membrane awaiting translocation [157]. Thus, in PD, the SCNA gene is not alone in contributing to the pathology, and genes involved in the clearance of aggregated α-syn may also be involved [154]. This finding is significant since both mutant α-syn and aggregated α-syn that have escaped autophagic degradation can hamper the CMA process in neurons, leading to neuronal cell death [158].
Several post-mortem studies have revealed that the levels of rate-limiting CMA components LAMP2A and HSPA8 are reduced in PD patients, especially in the SNpc [156,159,160]. Interestingly, the oxidized mitochondrial regulator MEF2D described above also binds to HSPA8 and is indirectly involved in CMA [161]. Studies on peripheral leukocytes from patients with sporadic PD revealed that LAMP2 transcript and protein levels are significantly reduced compared to the levels measured in healthy individuals [162]. Although in the same study, macroautophagy (measured by MAP1LC3II analysis) appeared to be induced, this conclusion would merit more complete independent confirmation, with the addition of measurement of autophagic flux (not performed in the initial study).
A few other autophagy-related genes and proteins involved in CMA have been associated with PD. For example, the peroxiredoxin-like redox sensor DJ-1 modulates the activity of SQSTM1 and the targeting of ubiquitin-conjugated proteins to macroautophagy under oxidative stress caused by tumor necrosis factor ligand superfamily, member 10 (TNFSF10/TRAIL) [163]. The implication of mutated DJ-1 (PARK7) in early-onset familial PD is well known (Table 2). DJ-1 is involved in a wide variety of cellular functions, including a role as an antioxidant, chaperone functions, transcriptional regulation, control of mitochondrial Ca2+ transients, among others. This protein regulates p53-induced mitochondrial dysfunction, stabilizes mitochondria-associated ER membranes, and interacts with anti-apoptotic proteins [164]. In addition to its effects on mitophagy and lysosomal macroautophagy, DJ-1 also modulates CMA through its interactions with LAMP2A and lysosomal HSPA8 [165]. In particular, it escorts wild-type α-syn for degradation by CMA. Lack of the DJ-1 gene inhibits CMA activity and α-syn degradation both in vitro and in vivo [165]. In contrast, DJ-1 is stabilized in cortical neurons by a CMA-mediated process that involves LAMP2A [166]. Interestingly, mutated α-syn proteins can escape from CMA-mediated degradation, which once again supports the CMA selective autophagy process.
In addition to impaired autophagy due to CMA-related genes in familial PD, alterations to CMA have also been implicated in sporadic PD, which accounts for the majority of PD cases. The etiology is more complicated to define in this setting and may be related to various factors—including environmental (e.g., pesticides) and cellular (e.g., oxidative stress; see above) stressors. The proposed involvement of CMA malfunction in PD pathogenesis is further supported by age-related changes to the LAMP2A protein itself, which lead to a gradual reduction in CMA and subsequent acceleration of the disease in older patients [154,156,157,167,168].
Recently, several notable data have highlighted various immune alterations underlying that PD is associated to autoimmune features and could be considered as an autoimmune disease [87]. The presence of serum autoantibodies reacting with LAMP2A has not yet been investigated in PD. These autoreactive antibodies have been described recently in the serum of autoimmune patients with lupus and closely related systemic autoimmune diseases [169].
3.4.4. Role of Lysosomes in PD
Regardless of the roles played by the different types of autophagy in PD (macroautophagy, CMA and even some forms of secretory autophagy [170]), lysosomes play a central role in α-syn degradation. Alterations to the lysosomal enzyme content (particularly hydrolases) influence the degradation process, leading to the accumulation of protein aggregates which cause neuronal damage [48]. Studies have reported decreased levels of lysosomal hydrolases such as α-mannosidase, β-mannosidase, and β-glucocerebrosidase (GBA) in the cerebrospinal fluid (CSF) from patients with PD. In contrast, in the same patients’ serum the activity of these hydrolases was not significantly altered [171]. The difference in levels of hydrolases in the CSF is now being used for diagnostic purposes [171]. In contrast, the level of α-syn in CSF from PD patients was found to be greatly diminished [172]. These reduced levels are probably due to α-syn accumulation in LBs [172]. Several genes associated with PD are also directly linked to lysosomal functions. As mentioned above, all the autophagy pathways at some point involve lysosomes, as the point to which the material is delivered for processing by various enzymes [48]. Among the enzymes implicated in these processes, cathepsin D is associated with α-syn degradation. Studies on Drosophila have shown that lack of cathepsin D leads to the accumulation of unprocessed substrates in lysosomes and late endosomes [10,148].
GBA, encoded by GBA1, is another lysosomal enzyme involved in α-syn degradation. Lack of GBA function or low levels of GBA in the human brain induce the accumulation of an oligomeric form of α-syn [173,174,175], which in turn interferes with the maturation of GBA [173], leading to a vicious cycle. Heterozygous mutations in ATP10B, which encodes ATP10B (Table 2), a late endo-lysosomal lipid flippase that translocates the lipids glucosylceramide and phosphatidylcholine towards the cytosolic membrane leaflet, have been implicated in PD [13]. These mutations alter the translocation functions of ATP10B, leading to an accumulation of glucosylceramide which can drive lysosomal dysfunction [13].
ATP13A2 (PARK9) (Table 2) is another lysosomal protein that is important in PD. It is responsible for cation transport in lysosome-like vesicles. Mutations in ATP13A2 have been observed in early-onset PD [12]. In Vitro studies confirmed that increased ATP13A2 protein levels reduce α-syn-induced toxicity [176,177]. Similarly, reduced levels of α-galactosidase A transcript and protein were observed in peripheral blood mononuclear cells from PD patients [178]. In addition, studies on the brain of PD patients revealed that DA neurons contained numerous lysosome-like vesicles and granules, suggesting that even in the final stages of the disease, these neurons undergo active apoptosis and engage in autophagy processes [179].
Human transmembrane protein 175 (TMEM175), one of the highly expressed genes that encode lysosome-bound K+ channels has also been linked to PD pathogenesis. Both in vitro and in vivo studies on neurons confirmed that TMEM175 deficiency provokes α-syn accumulation with defects in macroautophagy, lysosomal degradation, and mitochondrial respiration processes [117].
Another lysosomal protein with significant links to PD is the transcription factor EB (TFEB), which coordinates expression of lysosomal hydrolases, membrane proteins, and genes involved in autophagy. This master regulator of lysosome biogenesis is regulated by mammalian target of rapamycin (MTOR)C1 through phosphorylation of specific serine residues [180]. A study based on a rodent model of α-syn-induced toxicity confirmed that the autophagy-lysosomal pathway is impaired, with TFEB retained in the cytosol. Therefore, increasing autophagy-mediated degradation of SNCA via TFEB regulation could be a promising strategy for PD prevention and treatment [180,181,182,183]. Several direct and indirect TFEB agonists have been described as potent regulator in pre-clinical and clinical trials [183].
Taken together, these observations lead us to conclude that any drugs that increase the functional properties of lysosomes should have a potent effect, halting the progression of PD.
3.5. Is the Autophagy Machinery a Potential Target for Selective Intervention in PD?
The link between PD and autophagy dysfunction still remains largely unknown. Thus, there is insufficient information to answer the question of whether autophagy alteration occur in all patients with PD, and is not associated only to PD risk genes. In this context, it would be of great interest to examine the autophagic activity at the cellular level in patients with and without mutations in autophagy-linked genes. Despite the complexity of the autophagy defects in PD, this vital cellular system could represent a target of considerable interest for the treatment of the disease. Indeed, to date, although our understanding of the molecular bases of PD and its diagnosis [184,185] are improving, therapeutic aspects of PD remain below expectations, with a limited arsenal of efficient and specific drugs. Thus, current therapies are essentially symptomatic, aiming to partially maintain dopamine levels by limiting its degradation using monoamine oxidase B inhibitors, supplying dopamine precursors using levodopa [186] or dopamine agonists such as ropinirole, pramipexole, or rotigotine [187] (Figure 4). Today, numerous strategies, including regenerative medicine-based solutions—such as cell-based implants, fetal grafts, patient-specific tissue-engineered constructs—and some cutting-edge technological approaches involving the delivery of near-infrared light directly into the SN in the brain of PD patients, have been used or are being explored [188,189]. Biologics, such as antibodies to α-syn (prasinezumab, developed by Roche and Prothena) have been evaluated but unfortunately demonstrated limited success in a first phase II clinical trial. Other companies are also exploring the same line of possible intervention with monoclonal antibodies to α-syn [190]. However, this line of treatment remains uncertain; in February 2021, the failure of Biogen’s cinpanemab was announced—this monoclonal antibody has a mode of action similar to Roche’s prasinezumab. In parallel, novel pharmacological interventions are being evaluated in clinical trials. Some of these also target α-syn [191] whereas others target TNF, transcription factors, nuclear factor erythroid 2–related factor 2 (NRF2), and PPARγ, G protein-coupled receptors, glucocorticoid receptors, glucagon-like peptide 1 (GLP1), and inflammasome/NLR family pyrin domain-containing 3 (NLRP3) (see [187,192,193,194]). Microglial NLRP3 is a source of sustained neuroinflammation that can contribute to progressive DA neuron loss [195]. Molecules and biologics targeting B and T lymphocytes are also being investigated. In this context, compounds targeting autophagy remains an unexplored route for the development of innovative treatments for PD.
A number of proof-of-concept studies have been performed with compounds targeting autophagy in vitro in cells and in vivo, generally in genetically-deficient animal models. The molecules tested were designed to target mitophagy, macroautophagy, CMA, or lysosomes (Table 3 [196]). In the context of mitophagy, since oxidative stress is one of the principal causes of mitochondrial dysfunction, the agents assessed mainly protect against the production of, or neutralize, free radicals. In addition, agents targeting mitochondrial biogenesis, such as nuclear respiratory factor 1 and 2 (NRF1, NRF2), TFAM, and PGC1-α (described above) have been investigated (Table 3). This process could also be controlled by tuning a few of its crucial interactions. For example, the tumor suppressor protein p53 is a relevant target as it interacts with PRKN, inhibiting its translocation to the cytosol. Compared to healthy controls, significantly higher levels of p53 protein have been measured in the caudate nucleus in PD patients [197]. Experiments in PD models effectively showed that the inhibition of this interaction activates PRKN-dependent mitophagy and reduces the symptoms of PD [197]. Several other targets linked to the mitophagy pathway have been, or are currently being, explored in relation to PD [197,198,199]. Emerging promising molecules include selective inhibitor of the mitochondrial deubiquitinase, USP30 that negatively regulates PRKN-mediated mitophagy [132,200]. In addition, agents that target mitochondrial dysfunction rather than mitophagy per se (e.g., nimodipine or tetrahydroisoquinoline; Table 3) were found to have a protective effect against PD [201,202].
Independent studies on animal models have also demonstrated that enhancing macroautophagy by acting on TFEB or BECN1 can protect neurons against α-syn-induced toxicity [180,275]. Nitolinib, anbroxol, curcumin, spermidine, Torin1, 2-hydroxypropyl-β-cyclodextrin (2-HPβCD), or the well-known excipient trehalose are all representative of this pharmacological class (Table 3 and Table 4) [196,276]. An important molecule to mention here is the Tat-Beclin-1 peptide, a cell-permeable peptide consisting of BECN1 (residues 267–284) conjugated to the HIV-1 Tat protein. This peptide construct enhances autophagy initiation by interacting with the autophagy inhibitor Golgi-associated plant pathogenesis-related protein 1 (GAPR-1/GLIPR2). This interaction leads to BECN1 distribution throughout the cytosol while also increasing autophagosome formation in neurons. Analogues of this peptide are currently being explored in clinical trials.
Molecules that target CMA are also highly relevant. The decisive role of LAMP2A in α-syn degradation has been clearly demonstrated (see above). Cells and Drosophila overexpressing LAMP2A display a capacity to resist α-syn-induced neurotoxicity or neuronal degeneration and PD-related features [293,294]. These data along with the data presented above, undoubtedly indicate that CMA regulators LAMP2A and HSPA8 represent targets of choice for PD treatments [192,295]. Geranylgeranylacetone, a nontoxic acyclic isoprenoid compound that has been clinically applied as an antiulcer drug in Asian countries, and is a known inducer of HSPs acting via the activation of heat shock transcription factor-1, phorbol 12-myristate 13-acetate and others, could represent exploratory molecules to treat PD via modulation of the CMA pathway [296].
As indicated above, numerous molecules that target mitophagy or lysosomal autophagy pathways are under investigation in preclinical or clinical studies (Table 3 and Table 4). To the best of our knowledge, however, none of them have yet been approved and/or are already applied in the treatment of PD.
4. Awaiting Satisfactory Answers—Future Research
A significant number of new research lines suggest novel avenues of investigation centered on autophagy. In the specific context of PD, we reviewed above some of the key results indicating that targeting the mitophagy and CMA pathways could be a means to protect against α-syn-related toxicity (Appendix B provides a general significance statement). However, some limitations remain, related both to the development of efficient pharmacological molecules, to their administration, and to some theoretical considerations.
In fact, very few molecules are selective to one type of autophagy, or even autophagy as distinct from other cellular pathways, such as apoptosis [48,271,297,298,299,300,301] (Figure 4). Consequently, most molecules may display unwanted side-effects, especially when they are administered daily in the medium- or long-term to PD patients. Stability of a compound can also represent a limitation for its use. In general, the half-life in the body is around 10–25 min, especially in the liver. It should be mentioned, however, that in most organs and tissues, induction of autophagosome formation is very rapid. Therefore, activation of autophagy remains a rational target strategy when aggregated proteins accumulate. In addition, autophagosomes generally recycle quickly. This represents an advantage with regard to possible toxic events linked to the consequences of induced/activated autophagy. However, it can also be a limitation, in the sense that it will be necessary to extend the treatment periods. In fact, in the particular case of neurons, autophagosome biogenesis is extremely complex, and some heterogeneity has been described depending on the neuronal compartment considered. This aspect remains a matter of debate. Further in vivo information is also required regarding autophagosome formation and dynamics in developing versus mature systems [302].
Autophagy is a very dynamic compartmentalized process; it can be increased in certain organs and tissues but decreased in other organs in the same subject. This differential activation has been described in several models of chronic inflammation and autoimmune diseases, for example in murine models of Sjögren’s syndrome [303], chronic inflammatory demyelinating polyneuropathy [304] and chronic house dust mite-induced airway inflammation [305]. With regard to neurons, as indicated above, autophagy is even more complex, with specific stages of the pathway occurring in distinct subcellular compartments. As a consequence, treating an individual with a compound to induce or inhibit autophagy can have a range of individual effects. Due to this variety of effects, treating a subject with an inhibitor can restore abnormally weak levels of autophagic activity in another tissue, as illustrated by the effect of the CMA-modulator peptide P140 [303,304,305]. P140 that targets CMA and probably indirectly macroautophagy, has shown correcting effect on altered CMA activity, but display no effect on the basal, well balanced and vital autophagy process. This therapeutic peptide is currently evaluated in phase III clinical trials for lupus.
Another critical question that arises is the timing of treatment with regard to the course of the disease. How early should we intervene to see efficacy? This aspect has not really been solved and raises the general question of the benefit-risk of such treatments. The functionality of autophagy that declines with age also represents an aspect that must be taken into consideration in any autophagy-based treatment of PD.
5. General Conclusions
Although the considerations described in this review highlight some gaps in our understanding and appreciation of the potential of autophagy modulators to treat PD, several molecules hold promise for future specific treatment. An important aspect to underline here is that these molecules act on a cellular mechanism and not on the final damage induced. They could therefore be included in early treatment, or even as part of preventive strategies, to avoid or halt disease development.
Interestingly, in addition to the molecules described above in the context of PD, others that target autophagy have shown beneficial effects against neurodegenerative diseases [306,307,308,309], and could possibly be considered for review of their indications. These include, for example, neferine, Lu AE58054/idalopirdine, SB-742457, latrepirdine, MCI-186/Edaravone, SAGE217, GSK621, AICAR, Propofol, A769662, RSVA314, RSVA405, AUTEN-67, cystatin C, MSL, Digoxin, FTY720, carbamazepine, rilmenidine, clonidine, verapamil, SMER28, BRD5631, and AUTEN-67, among others. However, their selectivity, efficacy, and safety must be demonstrated in the context of PD. Extensive work is being undertaken to discover potent new chemical compounds for PD treatment [308,310]. Novel targets that are closely linked to autophagy pathways could also prove to be relevant in PD, for example, protein-O-linked N-acetyl-β-D-glucosaminidase (O-GlcNAcase) [311,312].
From a technical point of view, it is worth recalling here that it is highly recommended to study the efficacy of these novel strategies in several independent models, both in vitro and in vivo if we hope to achieve reproducible results and, in the context of autophagy, to monitor several relevant biomarkers [313,314,315], as well as measuring the autophagic flux [79,80].
PD affects 1–2 individuals per 1000 in the general population at any time. Its prevalence increases with age. In industrialized countries, it is estimated that the disease affects 0.6%–0.8% of 65–69-year-old individuals and 2.6%–3.5% of 85–89-year-olds. Currently, no specific test exists to diagnose PD, and it cannot be cured. Medications (dopaminergic drugs) as well as surgical treatment only act on symptoms. The ultimate goal of ongoing investigations is to develop, ideally non-invasive, therapies that could retune the cellular degradation pathways responsible for clearing abnormally folded or aggregated proteins that are toxic for neurons. Targeting autophagy without altering other vital cellular pathways is a challenge that may be achievable in PD and other neurodegenerative diseases if safe and selective molecules can be appropriately applied and delivered.
Author Contributions
All authors contributed equally. All authors have read and agreed to the published version of the manuscript.
Funding
This work received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to Béatrice Lannes for providing the images shown in Figure 1. We address our sincere apologies to authors whose work we could not cite due to space limitations. This research was funded in part by the French National Centre for Scientific Research (CNRS), Région Grand-Est, the University of Strasbourg Institute for Advanced Study (USIAS) and the Interdisciplinary Thematic Institute 2021–2028 program of the University of Strasbourg, CNRS and Inserm (ANR-10-IDEX-0002 and ANR-20-SFRI-0012) in the context of the Strasbourg drug discovery and development Institute (IMS). SRB is grateful to Jagadeesh Bayry (INSERM, Centre de Recherche des Cordeliers, Sorbonne Universités, Université de Paris, France and IIT Palakkad, Palakkad, Kerala, India) for providing a post-doctoral position. S.M. also acknowledges the support of the TRANSAUTOPHAGY COST Action (CA15138), the French-language autophagy club (CFATG), and the European Regional Development Fund of the European Union in the context of the INTERREG V Upper Rhine program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
α-syn, α-synuclein; Aβ, amyloid beta; AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; AMPK, AMP-activated protein kinase; APP, amyloid precursor protein; ATG, autophagy-related; AV, autophagic vacuole; BECLIN1/BECN1, B-cell lymphoma 2 interacting myosin/moesin-like coiled-coil protein 1; CMA, chaperone-mediated autophagy; CNS, central nervous system; DA, dopaminergic; eMI, endosomal microautophagy; ER, endoplasmic reticulum; HD, Huntington’s disease; LAMP2A, lysosomal-associated membrane protein 2A; LAP, LC3-associated phagocytosis; LB, Lewy body; LN, Lewy neurite; LRRK2/dardarin, leucine-rich repeat kinase; MAP1LC3, microtubule-associated protein 1A/1B-light chain 3; MTOR, mammalian target of rapamycin; PD, Parkinson’s disease; PE, phosphatidylethanolamine; PI3K, phosphatidyl inositol 3-kinase; PINK1/PARK6, PTEN-induced putative kinase 1; PRKN, PARKIN; SMAC, single membrane ATG8 conjugation; SN, substantia nigra; SNpc, SN pars compacta; SQSTM1/p62, sequestosome 1; TLR, Toll-like receptor; ULK1, Unc-51-like kinase 1; UPS, ubiquitin-proteasome system; YOPD, young-onset PD.
Appendix A
Alzheimer’s disease (AD)
The most common cause of dementia, a general term for loss of memory and other cognitive abilities serious enough to interfere with daily life. AD accounts for 60–80% of dementia cases.
Amyotrophic lateral sclerosis (ALS)
ALS (also known as motor neuron disease or Lou Gehrig’s disease) generally starts with muscle twitching and weakness in one limb, or slurred speech. It can affect control of the muscles needed to move, speak, eat, and breathe, and can be fatal.
Autophagosomes
A double membrane-bound vesicle, which encloses cellular constituents and fuses with lysosomes to form phagolysosomes where the engulfed material is digested or degraded and either released into the extracellular matrix via exocytosis, or released intracellularly to undergo further processing.
Autophagy
A vital, finely-regulated and evolutionarily-conserved intracellular pathway that continuously degrades, recycles, and clears unnecessary or dysfunctional cellular components. Autophagy is crucial for cellular adaptation to the environment and to maintain cell homeostasis, especially under stress conditions.
Chaperone-mediated autophagy (CMA)
Selective autophagy pathway in which proteins containing a KFERQ-like sequence are captured by the HSAP8 chaperone and translocated into lysosomes following interaction with LAMP2A, which acts as a rate-limiting factor in this process.
Endocytosis
A vesicle-mediated process by which cells engulf membrane and extracellular materials. Several endocytic pathways—phagocytosis, pinocytosis, and receptor-mediated endocytosis—use distinct mechanisms to internalize material. Clathrin-mediated endocytosis (CME) is the major endocytic pathway in mammalian cells.
Endosomal-lysosomal system
A set of intracellular membrane-delimited compartments that dynamically interconvert from early endosomes, recycling endosomes, late endosomes, and the lysosome.
GABARAP
A member of the ATG8 family that also includes MAP1LC3. It interacts with the GABAA receptor and tubulin, and promotes tubulin polymerization.
Huntington’s disease (HD)
A progressive brain disorder that causes uncontrolled movements, emotional problems, and loss of thinking ability (cognition).
Isolation membrane/phagophore
Small vesicular sac derived from the ER, mitochondria, vacuoles, and other unidentified sources, which sequesters and folds away a portion of the cytoplasm at the very first step of macroautophagy.
LC3-associated phagocytosis (LAP)
LAP combines the molecular machineries of phagocytosis and autophagy, resulting in conjugation of the autophagic marker MAP1LC3 to phosphatidylethanolamine (PE) on the phagosomal membrane, generating “LAPosomes” that undergo facilitated fusion with lysosomes.
Lewy bodies (LB)
Abnormal aggregates of α-syn and ubiquitin found in the cytoplasm of neurons from patients with PD. LB are initially present in dopaminergic neurons (substantia nigra), but as PD progresses they may also be found in cortical neurons. It is not fully understood how LBs contribute to neuronal cell death.
Lewy neurites (LN)
Aggregates of abnormal α-syn localized in axons. Like LBs, LNs may be found initially in the bulb, pons, and mesencephalon, but may extend later to the cortex.
Lysosome
An organelle located in the cytoplasm of eukaryotic cells, containing acid hydrolases, which can break down virtually any kind of biomolecule, including proteins, nucleic acids, carbohydrates, lipids, and cellular debris.
Macroautophagy
Finely-regulated process during which the cell forms a phagophore, a double membrane compartment sequestering its contents, which matures into an autophagosome.
Microglia
A type of small macrophage-like glial cell in the CNS
Mitophagy
A key process that selectively disrupts damaged mitochondria by autolysosomal degradation, preventing excessive production of reactive oxygen species, and activation of cell death.
PARKIN (PRKN)
A protein encoded by the human PARK2 gene, the precise functions of which are unknown. PARKIN is a component of a multiprotein E3 ubiquitin ligase complex, present in the ubiquitin-proteasome system (UPS) that targets proteins for degradation.
Phagocytosis
An endocytic process by which certain cells known as phagocytes (e.g., macrophages) internalize large particles (>0.5 µm) including bacteria, other microorganisms, foreign particles, and aged red blood cells to form a phagosome.
Proteasome
Protein complexes containing proteases, which degrade unneeded or damaged proteins. They are found in cells from all eukaryotes and archaea, and in some bacteria. Each human cell contains about 30,000 proteasomes.
Rab proteins
During the autophagy process, several vesicular structures are formed, transported and degraded by the fusion with lysosomes. The proteins that helps in the vesicular trafficking are called Rab protiens. Rab proteins are small GTPases, which belongs to the Ras-like GTPase, play a critical role in regulating autophagy.
SEQUESTOSOME 1(SQSTM1)
SQSTM1, also known as the ubiquitin-binding protein p62, acts as a major cargo receptor during the sequestration process. It acts both in a MAP1LC3-dependent and -independent manner.
α-SYNUCLEIN (α-syn)
A 140-amino acid residue soluble protein mostly found in the presynaptic neurons. The gene SNCA encodes α-synuclein, and mutations of this gene cause familial PD.
Ubiquitin-proteasome system (UPS)
A major proteolytic system that controls protein degradation and regulates many cellular processes in eukaryotic cells, such as DNA repair, stress responses, and cell proliferation. The UPS consists of specific enzymes that modify protein substrates using ubiquitin, and 26S proteasomes responsible for proteolysis of ubiquitin-tagged substrates.
Appendix B
Significance Statement
Autophagy plays an important role in the onset and development of neurodegeneration linked to the accumulation of misfolded proteins or dysfunctional mitochondria. Dysfunction of canonical and non-canonical autophagy processes are involved in neurodegenerative diseases, especially Parkinson’s disease (PD). Both genetic (ATG genes) and cellular autophagy defects have been described in young-onset and classical PD. Defects in the autophagy-lysosomal pathway in dopaminergic neurons may result in the excessive accumulation of damaged proteins in the cytoplasm, ultimately resulting in cell loss and the neuropsychiatric manifestations characteristic of PD. The rational introduction of specific agents to modulate mitophagy and other forms of lysosome-dependent autophagy processes, such as chaperone-mediated autophagy, could represent promising avenues for the treatment of PD.
References
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Charvin, D.; Medori, R.; Hauser, R.A.; Rascol, O. Therapeutic strategies for Parkinson disease: Beyond dopaminergic drugs. Nat. Rev. Drug Discov. 2018, 17, 804–822. [Google Scholar] [CrossRef]
- Del Rey, N.L.; Quiroga-Varela, A.; Garbayo, E.; Carballo-Carbajal, I.; Fernandez-Santiago, R.; Monje, M.H.G.; Trigo-Damas, I.; Blanco-Prieto, M.J.; Blesa, J. Advances in Parkinson’s disease: 200 years later. Front. Neuroanat. 2018, 12, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef]
- Beilina, A.; Cookson, M.R. Genes associated with Parkinson’s disease: Regulation of autophagy and beyond. J. Neurochem. 2016, 139, 91–107. [Google Scholar] [CrossRef]
- Gonzalez-Casacuberta, I.; Juarez-Flores, D.L.; Moren, C.; Garrabou, G. Bioenergetics and autophagic imbalance in patients-derived cell models of Parkinson disease supports systemic dysfunction in neurodegeneration. Front. Neurosci. 2019, 13, 894. [Google Scholar] [CrossRef]
- Van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.-P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Smolders, S.; Van den Haute, C.; Heeman, B.; van Veen, S.; Crosiers, D.; Beletchi, I.; Verstraeten, A.; Gossye, H.; Gelders, G.; et al. Mutated ATP10B increases Parkinson’s disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol. 2020, 139, 1001–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Lobato, B.L.; Vidal, A.F.; Ribeiro-dos-Santos, Â. Regulatory miRNA–mRNA Networks in Parkinson’s Disease. Cells 2021, 10, 1410. [Google Scholar] [CrossRef]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Yavich, L.; Tanila, H.; Vepsalainen, S.; Jakala, P. Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [Green Version]
- Todorova, A.; Jenner, P.; Ray Chaudhuri, K. Non-motor Parkinson’s: Integral to motor Parkinson’s, yet often neglected. Pract. Neurol. 2014, 14, 310–322. [Google Scholar] [CrossRef]
- Lin, K.J.; Lin, K.L.; Chen, S.D.; Liou, C.W.; Chuang, Y.C.; Lin, H.Y.; Lin, T.K. The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction in Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [Green Version]
- Pieri, L.; Madiona, K.; Melki, R. Structural and functional properties of prefibrillar α-synuclein oligomers. Sci. Rep. 2016, 6, 24526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.F. Non-motor symptoms in Parkinson’s disease. Park. Relat. Disord. 2016, 22, S119–S122. [Google Scholar] [CrossRef]
- Fox, S.H.; Katzenschlager, R.; Lim, S.Y.; Barton, B.; de Bie, R.M.A.; Seppi, K.; Coelho, M.; Sampaio, C.; Movement Disorder Society Evidence-Based Medicine, C. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov. Disord. 2018, 33, 1248–1266. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Lang, A.E. Levodopa-related motor complications-phenomenology. Mov. Disord. 2008, 23, S509–S514. [Google Scholar] [CrossRef]
- Weintraub, D.; Claassen, D.O. Chapter 22—Impulse control and related disorders in Parkinson’s disease. In International Review of Neurobiology; Chaudhuri, K.R., Titova, N., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 133, pp. 679–717. [Google Scholar]
- Garcia-Ruiz, P.J.; Martinez Castrillo, J.C.; Alonso-Canovas, A.; Herranz Barcenas, A.; Vela, L.; Sanchez Alonso, P.; Mata, M.; Olmedilla Gonzalez, N.; Mahillo Fernandez, I. Impulse control disorder in patients with Parkinson’s disease under dopamine agonist therapy: A multicentre study. J. Neurol. Neurosurg. Psychiatry 2014, 85, 840–844. [Google Scholar] [CrossRef]
- Dijk, J.M.; Espay, A.J.; Katzenschlager, R.; de Bie, R.M.A. The choice between advanced therapies for Parkinson’s disease patients: Why, what, and when? J. Park. Dis. 2020, 10, S65–S73. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Seppi, K.; Ray Chaudhuri, K.; Coelho, M.; Fox, S.H.; Katzenschlager, R.; Perez Lloret, S.; Weintraub, D.; Sampaio, C.; the collaborators of the Parkinson’s Disease Update on Non-Motor Symptoms Study Group on behalf of the Movement Disorders Society Evidence-Based Medicine Committee. Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov. Disord. 2019, 34, 180–198. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Bezard, E.; Brotchie, J.; Calon, F.; Collingridge, G.L.; Ferger, B.; Hengerer, B.; Hirsch, E.; Jenner, P.; Le Novere, N.; et al. Novel pharmacological targets for the treatment of Parkinson’s disease. Nat. Rev. Drug Discov. 2006, 5, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Elkouzi, A.; Vedam-Mai, V.; Eisinger, R.S.; Okun, M.S. Emerging therapies in Parkinson disease—Repurposed drugs and new approaches. Nat. Rev. Neurol. 2019, 15, 204–223. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [Green Version]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1981, 36, 219–234. [Google Scholar] [CrossRef]
- Landstrom, A.S. A role for crinophagy in pancreatic islet B-cells. Minireview based on a doctoral thesis. Ups. J. Med. Sci. 1987, 92, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, Y.; Kikuchi, H.; Aizawa, S.; Furuta, A.; Hatanaka, Y.; Konya, C.; Uchida, K.; Wada, K.; Kabuta, T. Direct uptake and degradation of DNA by lysosomes. Autophagy 2013, 9, 1167–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chino, H.; Mizushima, N. ER-Phagy: Quality control and turnover of endoplasmic reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef]
- Mancias, J.D.; Vaites, L.P.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Zhao, H.; Tang, M.; Liu, M.; Chen, L. Glycophagy: An emerging target in pathology. Clin. Chim. Acta 2018, 484, 298–303. [Google Scholar] [CrossRef]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging roles of lipophagy in health and disease. Front. Cell Dev. Biol. 2019, 7, 185. [Google Scholar] [CrossRef]
- Otomo, T.; Yoshimori, T. Lysophagy: A method for monitoring lysosomal rupture followed by autophagy-dependent recovery. In Lysosomes; Springer: Berlin/Heidelberg, Germany, 2017; Volume 1594, pp. 141–149. [Google Scholar]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijaljica, D.; Devenish, R.J. Nucleophagy at a glance. J. Cell Sci. 2013, 126, 4325–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, M.E.; Tavernarakis, N. Nucleophagy: From homeostasis to disease. Cell Death Differ. 2019, 26, 630–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.H.; Kim, Y.S.; Jo, D.S.; Choe, S.K.; Jo, E.K. Pexophagy: Molecular mechanisms and implications for health and diseases. Mol. Cells 2018, 41, 55–64. [Google Scholar]
- Tanaka, K.; Ichihara, A. Half-life of proteasomes (multiprotease complexes) in rat liver. Biochem. Biophys. Res. Commun. 1989, 159, 1309–1315. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Palmer, A.; Rivett, A.J.; Knecht, E. Degradation of proteasomes by lysosomes in rat liver. Eur. J. Biochem. 1995, 227, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Eat or be eaten: The autophagic plight of inactive 26S proteasomes. Autophagy 2015, 11, 1927–1928. [Google Scholar] [CrossRef] [Green Version]
- Cebollero, E.; Reggiori, F.; Kraft, C. Reticulophagy and ribophagy: Regulated degradation of protein production factories. Int. J. Cell Biol. 2012, 2012, 182834. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Furuta, A.; Kikuchi, H.; Aizawa, S.; Hatanaka, Y.; Konya, C.; Uchida, K.; Yoshimura, A.; Tamai, Y.; Wada, K.; et al. Discovery of a novel type of autophagy targeting RNA. Autophagy 2013, 9, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell. Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Grasso, D.; Ropolo, A.; Lo Re, A.; Boggio, V.; Molejon, M.I.; Iovanna, J.L.; Gonzalez, C.D.; Urrutia, R.; Vaccaro, M.I. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J. Biol. Chem. 2011, 286, 8308–8324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.; Menzies, F.M.; Narayanan, U.; Renna, M. Mammalian macroautophagy at a glance. J. Cell Sci. 2009, 122, 1707–1711. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biazik, J.; Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Noda, N.N.; Ohsumi, Y.; Inagaki, F. ATG systems from the protein structural point of view. Chem. Rev. 2009, 109, 1587–1598. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Durgan, J.; Lystad, A.H.; Sloan, K.; Carlsson, S.R.; Wilson, M.I.; Marcassa, E.; Ulferts, R.; Webster, J.; Lopez-Clavijo, A.F.; Wakelam, M.J.; et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol. Cell 2021, 81, 2031–2040.e8. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Leidal, A.M.; Debnath, J.; Hansen, M. Beyond autophagy: The expanding roles of ATG8 proteins. Trends Biochem. Sci. 2021, 46, 673–686. [Google Scholar] [CrossRef]
- Fletcher, K.; Ulferts, R.; Jacquin, E.; Veith, T.; Gammoh, N.; Arasteh, J.M.; Mayer, U.; Carding, S.R.; Wileman, T.; Beale, R.; et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 2018, 37, e97840. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy in neurons: It is not all about food. Trends Mol. Med. 2006, 12, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in neurons. Annu. Rev. Cell. Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Quality control in neurons: Mitophagy and other selective autophagy mechanisms. J. Mol. Biol. 2020, 432, 240–260. [Google Scholar] [CrossRef] [PubMed]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Azarnia Tehran, D.; Kuijpers, M.; Haucke, V. Presynaptic endocytic factors in autophagy and neurodegeneration. Curr. Opin. Neurobiol. 2018, 48, 153–159. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Bonam, S.R.; Bayry, J.; Tschan, M.P.; Muller, S. Progress and challenges in the use of MAP1LC3 as a legitimate marker for measuring dynamic autophagy in vivo. Cells 2020, 9, 1321. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) (1). Autophagy 2021, 17, 1–382. [Google Scholar] [PubMed]
- Yue, Z.; Friedman, L.; Komatsu, M.; Tanaka, K. The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1793, 1496–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavoe, A.K.H.; Gopal, P.P.; Gubas, A.; Tooze, S.A.; Holzbaur, E.L.F. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. eLife 2019, 8, e44219. [Google Scholar] [CrossRef]
- Kim, S.; Nam, Y.; Kim, C.; Lee, H.; Hong, S.; Kim, H.S.; Shin, S.J.; Park, Y.H.; Mai, H.N.; Oh, S.M.; et al. Neuroprotective and anti-inflammatory effects of low-moderate dose ionizing radiation in models of Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 3678. [Google Scholar] [CrossRef]
- Scheiblich, H.; Trombly, M.; Ramirez, A.; Heneka, M.T. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 2020, 41, 300–312. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.; Iqbal, H.; Singh, K.P.; Joshi, S.K. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: Current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonam, S.R.; Muller, S. Parkinson’s disease is an autoimmune disease: A reappraisal. Autoimmun. Rev. 2020, 19, 102684. [Google Scholar] [CrossRef]
- El Haddad, S.; Serrano, A.; Moal, F.; Normand, T.; Robin, C.; Charpentier, S.; Valery, A.; Brulé-Morabito, F.; Auzou, P.; Mollet, L.; et al. Disturbed expression of autophagy genes in blood of Parkinson’s disease patients. Gene 2020, 738, 144454. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Shani, V.; Bisharat, S.; Engelender, S. Alpha-synuclein ubiquitination and novel therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 630–637. [Google Scholar] [CrossRef]
- Popova, B.; Kleinknecht, A.; Braus, G.H. Posttranslational modifications and clearing of α-synuclein aggregates in yeast. Biomolecules 2015, 5, 617–634. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria-lysosome crosstalk: From physiology to neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Qi, H.; Tang, Y.; Shen, H.M. Post-translational modifications of key machinery in the control of mitophagy. Trends Biochem. Sci. 2020, 45, 58–75. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s disease: From pathogenesis to treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [Green Version]
- Dernie, F. Mitophagy in Parkinson’s disease: From pathogenesis to treatment target. Neurochem. Int. 2020, 138, 104756. [Google Scholar] [CrossRef]
- Leiva-Rodríguez, T.; Romeo-Guitart, D.; Marmolejo-Martínez-Artesero, S.; Herrando-Grabulosa, M.; Bosch, A.; Forés, J.; Casas, C. ATG5 overexpression is neuroprotective and attenuates cytoskeletal and vesicle-trafficking alterations in axotomized motoneurons. Cell Death Dis. 2018, 9, 626. [Google Scholar] [CrossRef]
- Hu, Z.-Y.; Chen, B.; Zhang, J.-P.; Ma, Y.-Y. Up-regulation of autophagy-related gene 5 (ATG5) protects dopaminergic neurons in a zebrafish model of Parkinson’s disease. J. Biol. Chem. 2017, 292, 18062–18074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Chen, D.; Pang, S.; Feng, X.; Huang, W.; Hawley, R.G.; Yan, B. Genetic analysis of the ATG7 gene promoter in sporadic Parkinson’s disease. Neurosci. Lett. 2013, 534, 193–198. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Korvatska, O.; Strand, N.S.; Berndt, J.D.; Strovas, T.; Chen, D.H.; Leverenz, J.B.; Kiianitsa, K.; Mata, I.F.; Karakoc, E.; Greenup, J.L.; et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum. Mol. Genet. 2013, 22, 3259–3268. [Google Scholar] [CrossRef] [Green Version]
- Rujano, M.A.; Cannata Serio, M.; Panasyuk, G.; Peanne, R.; Reunert, J.; Rymen, D.; Hauser, V.; Park, J.H.; Freisinger, P.; Souche, E.; et al. Mutations in the X-linked ATP6AP2 cause a glycosylation disorder with autophagic defects. J. Exp. Med. 2017, 214, 3707–3729. [Google Scholar] [CrossRef]
- Plotegher, N.; Duchen, M.R. Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front. Cell Dev. Biol. 2017, 5, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.K.; Cookson, M.R. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy 2011, 7, 531–532. [Google Scholar] [CrossRef] [Green Version]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H. Glucocerebrosidase and Parkinson disease: Recent advances. Mol. Cell. Neurosci. 2015, 66, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Westbroek, W.; Sidransky, E. The complicated relationship between gaucher disease and parkinsonism: Insights from a rare disease. Neuron 2017, 93, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaai7795. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Mira, M.T.; Alcais, A.; Nguyen, V.T.; Moraes, M.O.; Di Flumeri, C.; Vu, H.T.; Mai, C.P.; Nguyen, T.H.; Nguyen, N.B.; Pham, X.K.; et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 2004, 427, 636–640. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Deng, H.X.; Wong, Y.C.; Siddique, T.; Krainc, D. The Parkinson’s disease-linked protein TMEM230 is required for Rab8a-mediated secretory vesicle trafficking and retromer trafficking. Hum. Mol. Genet. 2017, 26, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Trajkovic, K.; Tsunemi, T.; Krainc, D. Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J. Neurosci. 2016, 36, 2425–2437. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple roles of the small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Belanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of mitochondrial function impairs lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [Green Version]
- Sterky, F.H.; Lee, S.; Wibom, R.; Olson, L.; Larsson, N.G. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12937–12942. [Google Scholar] [CrossRef] [Green Version]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Luo, H.; Krigman, J.; Zhang, R.; Yang, M.; Sun, N. Pharmacological inhibition of USP30 activates tissue-specific mitophagy. Acta Physiol. 2021, 232, e13666. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ren, Y.; Gui, C.; Zhao, M.; Wu, X.; Mao, K.; Li, W.; Zou, F. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T alpha-synuclein model of Parkinson’s disease. Cell Death Dis. 2018, 9, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Gao, L.; Lu, F.; Wang, B.; Gao, F.; Zhu, G.; Cai, Z.; Lai, J.; Yang, Q. Transcription factor myocyte enhancer factor 2D regulates interleukin-10 production in microglia to protect neuronal cells from inflammation-induced death. J. Neuroinflamm. 2015, 12, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, H.; Yang, Q.; Shepherd, K.; Smith, Y.; Miller, G.; Testa, C.; Mao, Z. Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Investig. 2011, 121, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltri, C.; Mazzeo, C.; Nunez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blazquez, A.; Martin, M.A.; Falcon-Perez, J.M.; et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Villa, E.; Marchetti, S.; Ricci, J.E. No Parkin zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef]
- Winslow, A.R.; Rubinsztein, D.C. The Parkinson disease protein alpha-synuclein inhibits autophagy. Autophagy 2011, 7, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Arotcarena, M.-L.; Teil, M.; Dehay, B. Autophagy in synucleinopathy: The overwhelmed and defective machinery. Cells 2019, 8, 565. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Rubinsztein, D.C. Autophagy in neuronal development and plasticity. Trends Neurosci. 2020, 43, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Eleuteri, S.; Albanese, A. VPS35-based approach: A potential innovative treatment in Parkinson’s disease. Front. Neurol. 2019, 10, 1272. [Google Scholar] [CrossRef]
- Miura, E.; Hasegawa, T.; Konno, M.; Suzuki, M.; Sugeno, N.; Fujikake, N.; Geisler, S.; Tabuchi, M.; Oshima, R.; Kikuchi, A.; et al. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 2014, 71, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Seaman, M.N.J.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Liu, W.; Hu, J.X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.C. VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.A.; Soto-Avellaneda, A.; Yong Jin, H.; Stojkovska, I.; Lai, N.K.; Albright, J.E.; Webb, A.R.; Oe, E.; Valarde, J.P.; Oxford, A.E.; et al. Enhanced hyaluronan signaling and autophagy dysfunction by VPS35 D620N. Neuroscience 2020, 441, 33–45. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front. Endocrinol. 2018, 9, 778. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Gao, L.; She, H.; Li, W.; Zeng, J.; Zhu, J.; Jones, D.P.; Mao, Z.; Gao, G.; Yang, Q. Oxidation of survival factor MEF2D in neuronal death and Parkinson’s disease. Antioxid. Redox Signal. 2014, 20, 2936–2948. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Wang, X.; Feng, X.; Zhang, A.; Li, J.; Gu, K.; Huang, J.; Pang, S.; Dong, H.; Gao, H.; et al. Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic Parkinson’s disease. Brain Res. 2011, 1394, 105–111. [Google Scholar] [CrossRef]
- Lim, H.; Lim, Y.-M.; Kim, K.H.; Jeon, Y.E.; Park, K.; Kim, J.; Hwang, H.-Y.; Lee, D.J.; Pagire, H.; Kwon, H.J.; et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef]
- Ottolini, D.; Calì, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum–mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar] [CrossRef]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits alpha-synuclein aggregation by regulating chaperone-mediated autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef] [Green Version]
- Brekk, O.R.; Makridakis, M.; Mavroeidi, P.; Vlahou, A.; Xilouri, M.; Stefanis, L. Impairment of chaperone-mediated autophagy affects neuronal homeostasis through altered expression of DJ-1 and CRMP-2 proteins. Mol. Cell. Neurosci. 2019, 95, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in age-associated neurodegeneration. Cells 2018, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, M.; Bonam, S.R.; Schall, N.; Bendorius, M.; Korganow, A.-S.; Lumbroso, C.; Muller, S. Implication of a lysosomal antigen in the pathogenesis of lupus erythematosus. J. Autoimmun. 2021, 120, 102633. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hunt, C.P.; Thacker, E.A.; Toste, C.M.; Boularand, S.; Deprets, S.; Dubois, L.; Sanders, L.H. Mitochondrial DNA damage as a potential biomarker of LRRK2 kinase activity in LRRK2 Parkinson’s disease. Sci. Rep. 2020, 10, 17293. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Pierguidi, L.; Persichetti, E.; Parnetti, L.; Sbaragli, M.; Tassi, C.; Orlacchio, A.; Calabresi, P.; Beccari, T.; Rossi, A. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov. Disord. 2007, 22, 1481–1484. [Google Scholar] [CrossRef]
- Tokuda, T.; Salem, S.A.; Allsop, D.; Mizuno, T.; Nakagawa, M.; Qureshi, M.M.; Locascio, J.J.; Schlossmacher, M.G.; El-Agnaf, O.M. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 349, 162–166. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef]
- Murphy, K.; Gysbers, A.; Abbott, S.; Tayebi, N.; Kim, W.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.E.; Cottle, L.; Gysbers, A.M.; Cooper, A.A.; Halliday, G.M. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol. Commun. 2013, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Huang, J.; Feng, X.; Zhang, A.; Li, J.; Pang, S.; Gu, K.; Dong, H.; Zhang, J.; Gao, H.; et al. Decreased expression of lysosomal alpha-galactosiase a gene in sporadic Parkinson’s disease. Neurochem. Res. 2011, 36, 1939–1944. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.-X.; Wang, S.-F.; Tan, Y.; Song, J.-X.; Zhu, Z.; Wang, Z.-Y.; Wu, M.-Y.; Cai, C.-Z.; Huang, Z.-J.; Tan, J.-Q.; et al. Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal death in oxidative stress-induced Parkinson’s disease models. Cell Death Dis. 2020, 11, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Dai, Y.; Liu, S.; Fan, Y.; Ding, Z.; Li, D. TFEB biology and agonists at a glance. Cells 2021, 10, 333. [Google Scholar] [CrossRef]
- Orrù, V.; Steri, M.; Sidore, C.; Marongiu, M.; Serra, V.; Olla, S.; Sole, G.; Lai, S.; Dei, M.; Mulas, A.; et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat. Genet. 2020, 52, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I. RT-QuIC for detection of prodromal α-synucleinopathies. Lancet Neurol. 2021, 20, 165–166. [Google Scholar] [CrossRef]
- Curtis, L.; Lees, A.J.; Stern, G.M.; Marmot, M.G. Effect of L-dopa on course of Parkinson’s disease. Lancet 1984, 2, 211–212. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.; Grealish, S.; Henchcliffe, C. The future of stem cell therapies for Parkinson disease. Nat. Rev. Neurosci. 2020, 21, 103–115. [Google Scholar] [CrossRef]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Dawson, V.L.; Dawson, T.M. Promising disease-modifying therapies for Parkinson’s disease. Sci. Transl. Med. 2019, 11, eaba1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujols, J.; Peña-Díaz, S.; Pallarès, I.; Ventura, S. Chemical chaperones as novel drugs for Parkinson’s disease. Trends Mol. Med. 2020, 26, 408–421. [Google Scholar] [CrossRef]
- Robinson, E.J.; Aguiar, S.; Smidt, M.P.; van der Heide, L.P. MCL1 as a therapeutic target in Parkinson’s disease? Trends Mol. Med. 2019, 25, 1056–1065. [Google Scholar] [CrossRef]
- Harris, J.P.; Burrell, J.C.; Struzyna, L.A.; Chen, H.I.; Serruya, M.D.; Wolf, J.A.; Duda, J.E.; Cullen, D.K. Emerging regenerative medicine and tissue engineering strategies for Parkinson’s disease. NPJ Parkinson’s Dis. 2020, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Marin-Aguilar, F.; Castejon-Vega, B.; Alcocer-Gomez, E.; Lendines-Cordero, D.; Cooper, M.A.; de la Cruz, P.; Andujar-Pulido, E.; Perez-Alegre, M.; Muntane, J.; Perez-Pulido, A.J.; et al. NLRP3 inflammasome inhibition by MCC950 in aged mice improves health via enhanced autophagy and PPARalpha activity. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1457–1464. [Google Scholar] [CrossRef]
- Panicker, N.; Sarkar, S.; Harischandra, D.S.; Neal, M.; Kam, T.-I.; Jin, H.; Saminathan, H.; Langley, M.; Charli, A.; Samidurai, M.; et al. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 2019, 216, 1411–1430. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.; Shin, C.; Lee, S. Intersection between redox homeostasis and autophagy: Valuable insights into neurodegeneration. Antioxidants 2021, 10, 694. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef]
- Koentjoro, B.; Park, J.-S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef]
- Yi, W.; MacDougall, E.J.; Tang, M.Y.; Krahn, A.I.; Gan-Or, Z.; Trempe, J.-F.; Fon, E.A. The landscape of Parkin variants reveals pathogenic mechanisms and therapeutic targets in Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 2811–2825. [Google Scholar] [CrossRef]
- Kluge, A.F.; Lagu, B.R.; Maiti, P.; Jaleel, M.; Webb, M.; Malhotra, J.; Mallat, A.; Srinivas, P.A.; Thompson, J.E. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 2018, 28, 2655–2659. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef]
- Banerjee, R.; Raju, A.; Ngima Nthenge-Ngumbau, D.; Singh, R.; Jaisankar, P.; Mohanakumar, K.P.; Biswas, S.C. Tetrahydroisoquinoline molecule of Indian ayurveda medicine: Therapeutic potential in Parkinson’s disease. Park. Relat. Disord. 2018, 46, e85. [Google Scholar] [CrossRef]
- Suresh, S.N.; Manjithaya, R. A small molecule autophagy inducer exerts cytoprotection against α-synuclein toxicity. Eur. J. Pharmacol. 2019, 862, 172635. [Google Scholar] [CrossRef]
- Kovács, T.; Billes, V.; Komlós, M.; Hotzi, B.; Manzéger, A.; Tarnóci, A.; Papp, D.; Szikszai, F.; Szinyákovics, J.; Rácz, Á.; et al. The small molecule AUTEN-99 (autophagy enhancer-99) prevents the progression of neurodegenerative symptoms. Sci. Rep. 2017, 7, 42014. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Jiang, C.; Chen, W.; Zhang, G.; Luo, D.; Cao, Y.; Wu, J.; Ding, Y.; Liu, B. Baicalein induces apoptosis and autophagy via endoplasmic reticulum stress in hepatocellular carcinoma cells. Biomed. Res. Int. 2014, 2014, 732516. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Ma, X.; Zhao, X.; Zhang, S. Baicalein induces apoptosis and autophagy of breast cancer cells via inhibiting PI3K/AKT pathway in vivo and vitro. Drug Des. Dev. Ther. 2018, 12, 3961–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Liu, X.; Di, C.; Wang, Z.; Mi, X.; Liu, Y.; Zhao, Q.; Mao, A.; Chen, W.; Gan, L.; et al. MitoQ regulates autophagy by inducing a pseudo-mitochondrial membrane potential. Autophagy 2017, 13, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, K.C.; Huang, H.J.; Wang, Y.T.; Lin, A.M. Baicalein attenuates alpha-synuclein aggregation, inflammasome activation and autophagy in the MPP(+)-treated nigrostriatal dopaminergic system in vivo. J. Ethnopharmacol. 2016, 194, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Uversky, V.N.; Huang, M.; Kang, H.; Xu, F.; Liu, X.; Lian, L.; Liang, Q.; Jiang, H.; Liu, A.; et al. Baicalein inhibits alpha-synuclein oligomer formation and prevents progression of alpha-synuclein accumulation in a rotenone mouse model of Parkinson’s disease. Biochim. Biophys. Acta 2016, 1862, 1883–1890. [Google Scholar] [CrossRef]
- Li, Y.; Lin, S.; Xu, C.; Zhang, P.; Mei, X. Triggering of autophagy by baicalein in response to apoptosis after spinal cord injury: Possible involvement of the PI3K activation. Biol. Pharm. Bull. 2018, 41, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, S.N.; Chavalmane, A.K.; Dj, V.; Yarreiphang, H.; Rai, S.; Paul, A.; Clement, J.P.; Alladi, P.A.; Manjithaya, R. A novel autophagy modulator 6-Bio ameliorates SNCA/α-synuclein toxicity. Autophagy 2017, 13, 1221–1234. [Google Scholar] [CrossRef]
- Deng, Y.N.; Shi, J.; Liu, J.; Qu, Q.M. Celastrol protects human neuroblastoma SH-SY5Y cells from rotenone-induced injury through induction of autophagy. Neurochem. Int. 2013, 63, 1–9. [Google Scholar] [CrossRef]
- Spinelli, K.J.; Osterberg, V.R.; Meshul, C.K.; Soumyanath, A.; Unni, V.K. Curcumin treatment improves motor behavior in α-synuclein transgenic mice. PLoS ONE 2015, 10, e0128510. [Google Scholar]
- Jiang, T.-F.; Zhang, Y.-J.; Zhou, H.-Y.; Wang, H.-M.; Tian, L.-P.; Liu, J.; Ding, J.-Q.; Chen, S.-D. Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef]
- Song, J.-X.; Sun, Y.-R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.-H.; Xu, Z.; Wang, M.-Z.; Liu, L.-F.; Huang, Y.-Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Moon, J.H.; Kim, S.W.; Jeong, J.K.; Nazim, U.M.D.; Lee, Y.J.; Seol, J.W.; Park, S.Y. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget 2015, 6, 9701–9717. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.H.; Guan, M.S.; Koh, C.; Ouyang, X.; Yu, F.; Tan, E.K.; O’Neill, S.P.; Zhang, X.; Chung, J.; Lim, K.L. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J. Neurosci. 2012, 32, 14311–14317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpatrick, K.; Zeng, Y.; Hancock, T.; Segatori, L. Genetic and chemical activation of TFEB mediates clearance of aggregated α-synuclein. PLoS ONE 2015, 10, e0120819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, R.L.; Wustman, B.A.; Huertas, P.; Powe, A.C.; Pine, C.W.; Khanna, R.; Schlossmacher, M.G.; Ringe, D.; Petsko, G.A. Structure of acid β-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 2007, 3, 101–107. [Google Scholar] [CrossRef]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.-Y.; Whitworth, A.J.; Schapira, A.H.V. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar]
- Lu, J.-H.; Tan, J.-Q.; Durairajan, S.S.K.; Liu, L.-F.; Zhang, Z.-H.; Ma, L.; Shen, H.-M.; Chan, H.Y.E.; Li, M. Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. Autophagy 2012, 8, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Mak, S.; Zuo, X.; Li, H.; Wang, Y.; Han, Y. Neuroprotection against MPP(+)-induced cytotoxicity through the activation of PI3-K/Akt/GSK3beta/MEF2D signaling pathway by rhynchophylline, the major tetracyclic oxindole alkaloid isolated from uncaria rhynchophylla. Front. Pharmacol. 2018, 9, 768. [Google Scholar] [CrossRef]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Graziani, I.; Piccirillo, S.; Rotilio, G.; Ciriolo, M.R. Carcinoma cells activate AMP-activated protein kinase-dependent autophagy as survival response to kaempferol-mediated energetic impairment. Autophagy 2010, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; Graziani, I.; De Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging 2012, 33, 767–785. [Google Scholar] [CrossRef]
- Myöhänen, T.; Hannula, M.; Van Elzen, R.; Gerard, M.; Van Der Veken, P.; García-Horsman, J.; Baekelandt, V.; Männistö, P.; Lambeir, A. A prolyl oligopeptidase inhibitor, KYP-2047, reduces α-synuclein protein levels and aggregates in cellular and animal models of Parkinson’s disease. Br. J. Pharmacol. 2012, 166, 1097–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savolainen, M.H.; Richie, C.T.; Harvey, B.K.; Männistö, P.T.; Maguire-Zeiss, K.A.; Myöhänen, T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014, 68, 1–15. [Google Scholar] [CrossRef]
- Kilpeläinen, T.P.; Hellinen, L.; Vrijdag, J.; Yan, X.; Svarcbahs, R.; Vellonen, K.-S.; Lambeir, A.-M.; Huttunen, H.; Urtti, A.; Wallen, E.A.A.; et al. The effect of prolyl oligopeptidase inhibitors on alpha-synuclein aggregation and autophagy cannot be predicted by their inhibitory efficacy. Biomed. Pharmacother. 2020, 128, 110253. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.W.; Ju, S.; Lachenmayer, M.L.; Liken, J.; Stock, A.; Kim, S.H.; Delgado, L.M.; Alfaro, I.E.; Bernales, S.; Verdile, G.; et al. Latrepirdine stimulates autophagy and reduces accumulation of α-synuclein in cells and in mouse brain. Mol. Psychiatry 2013, 18, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; De-Paula, V.J.R.; Diniz, B.S.O. Neuroprotective effects of lithium: Implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem. Neurosci. 2014, 5, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Motoi, Y.; Shimada, K.; Ishiguro, K.; Hattori, N. Lithium and autophagy. ACS Chem. Neurosci. 2014, 5, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Xiong, N.; Liu, L.; Huang, J.; Han, C.; Zhang, G.; Li, J.; Xu, X.; Lin, Z.; Wang, T. Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement. BMC Neurosci. 2015, 16, 82. [Google Scholar] [CrossRef] [Green Version]
- Lazzara, C.A.; Kim, Y.-H. Potential application of lithium in Parkinson’s and other neurodegenerative diseases. Front. Neurosci. 2015, 9, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavillard, L.E.; Cañadas-Lozano, D.; Alcocer-Gómez, E.; Marín-Aguilar, F.; Pereira, S.; Robertson, A.A.B.; Muntané, J.; Ryffel, B.; Cooper, M.A.; Quiles, J.L.; et al. NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget 2017, 8, 99740–99756. [Google Scholar] [CrossRef] [Green Version]
- Saber, S.; El-Kader, E.M.A. Novel complementary coloprotective effects of metformin and MCC950 by modulating HSP90/NLRP3 interaction and inducing autophagy in rats. Inflammopharmacology 2021, 29, 237–251. [Google Scholar] [CrossRef]
- Corcoran, S.E.; Reena, H.; Matthew, A.C. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharmacol. Rev. 2021, 73, 968–1000. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulovic, M.; Jovanovic, M.; Xilouri, M.; Stefanis, L.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Paunovic, V.; Ardah, M.T.; El-Agnaf, O.M.; Kostic, V.; et al. The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol. Dis. 2014, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Perez-Revuelta, B.I.; Hettich, M.M.; Ciociaro, A.; Rotermund, C.; Kahle, P.J.; Krauss, S.; Di Monte, D.A. Metformin lowers Ser-129 phosphorylated alpha-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. 2014, 5, e1209. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic-Tucovic, M.; Harhaji-Trajkovic, L.; Dulovic, M.; Tovilovic-Kovacevic, G.; Zogovic, N.; Jeremic, M.; Mandic, M.; Kostic, V.; Trajkovic, V.; Markovic, I. AMP-activated protein kinase inhibits MPP+-induced oxidative stress and apoptotic death of SH-SY5Y cells through sequential stimulation of Akt and autophagy. Eur. J. Pharmacol. 2019, 863, 172677. [Google Scholar] [CrossRef]
- Patil, S.P.; Jain, P.D.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.-G.; Wong, V.K.-W.; Xu, S.-W.; Chan, W.-K.; Ng, C.-I.; Liu, L.; Law, B.Y.-K. Onjisaponin B derived from radix polygalae enhances autophagy and accelerates the degradation of mutant α-synuclein and huntingtin in pc-12 cells. Int. J. Mol. Sci. 2013, 14, 22618–22641. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhai, Y.; Yuan, J.; Hu, Y. New insights into Paeoniaceae used as medicinal plants in China. Sci. Rep. 2019, 9, 18469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Cao, Y.B.; Hu, L.F.; Yang, Y.P.; Li, J.; Wang, F.; Liu, C.F. ASICs mediate the modulatory effect by paeoniflorin on alpha-synuclein autophagic degradation. Brain Res. 2011, 1396, 77–87. [Google Scholar] [CrossRef]
- Yang, L.-Y.; Greig, N.H.; Tweedie, D.; Jung, Y.J.; Chiang, Y.-H.; Hoffer, B.J.; Miller, J.P.; Chang, K.-H.; Wang, J.-Y. The p53 inactivators pifithrin-μ and pifithrin-α mitigate TBI-induced neuronal damage through regulation of oxidative stress, neuroinflammation, autophagy and mitophagy. Exp. Neurol. 2020, 324, 113135. [Google Scholar] [CrossRef]
- Duan, W.; Zhu, X.; Ladenheim, B.; Yu, Q.-S.; Guo, Z.; Oyler, J.; Cutler, R.G.; Cadet, J.L.; Greig, N.H.; Mattson, M.P. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol. 2002, 52, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-J.; Nam, Y.; Lee, J.W.; Nguyen, P.-K.T.; Yoo, J.E.; Tran, T.-V.; Jeong, J.H.; Jang, C.-G.; Oh, Y.J.; Youdim, M.B.H.; et al. N-Methyl, N-propynyl-2-phenylethylamine (MPPE), a selegiline analog, attenuates MPTP-induced dopaminergic toxicity with guaranteed behavioral safety: Involvement of inhibitions of mitochondrial oxidative burdens and p53 gene-elicited pro-apoptotic change. Mol. Neurobiol. 2016, 53, 6251–6269. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [Green Version]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2, ra36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Tain, L.S.; Mortiboys, H.; Tao, R.N.; Ziviani, E.; Bandmann, O.; Whitworth, A.J. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 2009, 12, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. mTOR: On target for novel therapeutic strategies in the nervous system. Trends Mol. Med. 2013, 19, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Wey, M.C.; Fernandez, E.; Hart, M.J.; Gelfond, J.; Bokov, A.F.; Rani, S.; Strong, R. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol. Aging Age Relat. Dis. 2015, 5, 28743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-z.; Chen, X.-p.; Zhao, K.; Bai, L.-m.; Zhang, H.; Zhou, X.-p. Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced Parkinsonism in mice: Possible mediation through enhanced autophagy. Int. J. Neurosci. 2012, 123, 73–79. [Google Scholar] [CrossRef]
- Price, P.A.; Parkes, J.D.; Marsden, C.D. Sodium valproate in the treatment of levodopa-induced dyskinesia. J. Neurol. Neurosurg. Psychiatry 1978, 41, 702–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutt, J.G.; Williams, A.C.; Calne, D.B. Effect of sodium valproate on parkinsonism and L-DOPA induced dyskinesia. Brain Res. Bull. 1980, 5, 589–593. [Google Scholar] [CrossRef]
- Ristic, A.J.; Vojvodic, N.; Jankovic, S.; Sindelic, A.; Sokic, D. The frequency of reversible parkinsonism and cognitive decline associated with valproate treatment: A study of 364 patients with different types of epilepsy. Epilepsia 2006, 47, 2183–2185. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.; Factor, S.A. Valproic acid-induced parkinsonism: Levodopa responsiveness with dyskinesia. Park. Relat. Disord. 2013, 19, 758–760. [Google Scholar] [CrossRef]
- Büttner, S.; Broeskamp, F.; Sommer, C.; Markaki, M.; Habernig, L.; Alavian-Ghavanini, A.; Carmona-Gutierrez, D.; Eisenberg, T.; Michael, E.; Kroemer, G.; et al. Spermidine protects against α-synuclein neurotoxicity. Cell Cycle 2014, 13, 3903–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiki, S.; Sasazawa, Y.; Fujimaki, M.; Kamagata, K.; Kaga, N.; Taka, H.; Li, Y.; Souma, S.; Hatano, T.; Imamichi, Y.; et al. A metabolic profile of polyamines in parkinson disease: A promising biomarker. Ann. Neurol. 2019, 86, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox Signal. 2009, 11, 2095–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano-López, I.; de Marañon, A.M.; Iannantuoni, F.; López-Domènech, S.; Abad-Jiménez, Z.; Díaz, P.; Solá, E.; Apostolova, N.; Rocha, M.; Víctor, V.M. The mitochondrial antioxidant SS-31 modulates oxidative stress, endoplasmic reticulum stress, and autophagy in type 2 diabetes. J. Clin. Med. 2019, 8, 1322. [Google Scholar] [CrossRef] [Green Version]
- Muller, S. Excipients: Not so inert? when the excipient plays the role of an active substance, as exemplified by systemic lupus. Swiss Med. Wkly. 2018, 148, w14631. [Google Scholar] [CrossRef] [Green Version]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L.; Higgins, C.B.; Crowley, J.R.; Kraft, T.E.; Chi, M.; Newberry, E.P.; Chen, Z.; Finck, B.N.; et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci. Signal. 2016, 9, ra21. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Rusilowicz-Jones, E.V.; Barone, F.G.; Lopes, F.M.; Stephen, E.; Mortiboys, H.; Urbé, S.; Clague, M.J. Benchmarking a highly selective USP30 inhibitor for enhancement of mitophagy and pexophagy. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.N.; Chavalmane, A.K.; Pillai, M.; Ammanathan, V.; Vidyadhara, D.J.; Yarreiphang, H.; Rai, S.; Paul, A.; Clement, J.P.; Alladi, P.A.; et al. Modulation of autophagy by a small molecule inverse agonist of ERRα Is neuroprotective. Front. Mol. Neurosci. 2018, 11, 109. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef]
- Javed, H.; Nagoor Meeran, M.F.; Azimullah, S.; Adem, A.; Sadek, B.; Ojha, S.K. Plant extracts and phytochemicals targeting alpha-synuclein aggregation in Parkinson’s disease models. Front. Pharmacol. 2018, 9, 1555. [Google Scholar]
- Vucicevic, L.; Misirkic-Marjanovic, M.; Harhaji-Trajkovic, L.; Maric, N.; Trajkovic, V. Mechanisms and therapeutic significance of autophagy modulation by antipsychotic drugs. Cell Stress 2018, 2, 282–291. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy activator drugs: A new opportunity in neuroprotection from misfolded protein toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [Green Version]
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Melone, M.A.B. The autophagy signaling pathway: A potential multifunctional therapeutic target of curcumin in neurological and neuromuscular diseases. Nutrients 2019, 11, 1881. [Google Scholar] [CrossRef] [Green Version]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Daly, L.; Bezard, E.; Schapira, A.H.V. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann. Neurol. 2016, 80, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Albani, D.; Polito, L.; Batelli, S.; De Mauro, S.; Fracasso, C.; Martelli, G.; Colombo, L.; Manzoni, C.; Salmona, M.; Caccia, S.; et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J. Neurochem. 2009, 110, 1445–1456. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, Á.; Cordero, M.D.; Salviati, L.; Artuch, R.; Pineda, M.; Briones, P.; Gómez Izquierdo, L.; Cotán, D.; Navas, P.; Sánchez-Alcázar, J.A. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy 2009, 5, 19–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Z.; Chen, Y.Z.; Su, M.; Zheng, H.F.; Yang, Y.P.; Chen, J.; Liu, C.F. dl-3-n-Butylphthalide prevents oxidative damage and reduces mitochondrial dysfunction in an MPP(+)-induced cellular model of Parkinson’s disease. Neurosci. Lett. 2010, 475, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ye, M.; Xu, W.; Yu, M.; Liu, X.; Chen, Y. DL-3-n-butylphthalide therapy for Parkinson’s disease: A randomized controlled trial. Exp. Ther. Med. 2019, 17, 3800–3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Ma, F.; Huang, L.; Zhang, Y.; Peng, Y.; Xing, C.; Feng, Y.; Wang, X.; Peng, Y. Dl-3-n-Butylphthalide (NBP): A promising therapeutic agent for ischemic stroke. CNS Neurol. Disord. Drug Targets 2018, 17, 338–347. [Google Scholar] [CrossRef]
- Xiong, N.; Huang, J.; Chen, C.; Zhao, Y.; Zhang, Z.; Jia, M.; Zhang, Z.; Hou, L.; Yang, H.; Cao, X.; et al. Dl-3-n-butylphthalide, a natural antioxidant, protects dopamine neurons in rotenone models for Parkinson’s disease. Neurobiol. Aging 2012, 33, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
- Tian, A.; Ma, X.; Li, H.; Zhang, R. Dl-3n-butylphthalide improves spatial learning and memory in rats with vascular dementia by reducing autophagy via regulation of the mTOR signaling pathway. Exp. Ther. Med. 2020, 19, 1940–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Zhang, Y.; Peng, T. Abstract 20228: Nicotinamide riboside improves autophagic flux and prevents doxorubicin-induced cardiac injury. Circulation 2017, 136, A20228. [Google Scholar]
- Maiese, K. New insights for nicotinamide: Metabolic disease, autophagy, and mTOR. Front. Biosci. 2020, 25, 1925–1973. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.-H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Abraham, N.; Gao, G.; Yang, Q. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Issa, A.-R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Chérif-Zahar, B.; Birman, S. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 2018, 14, 1898–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.R.; Ruff, M.; Muller, S. HSPA8/HSC70 in immune disorders: A molecular rheostat that adjusts chaperone-mediated autophagy substrates. Cells 2019, 8, 849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renna, M.; Jimenez-Sanchez, M.; Sarkar, S.; Rubinsztein, D.C. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J. Biol. Chem. 2010, 285, 11061–11067. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [Green Version]
- Gros, F.; Muller, S. Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br. J. Pharmacol. 2014, 171, 4337–4359. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardones, P.; Rubinsztein, D.C.; Hetz, C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 2016, 9, fs2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.E.; Colon-Ramos, D.A. The journey of the synaptic autophagosome: A cell biological perspective. Neuron 2020, 105, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Wang, F.; Schall, N.; Muller, S. Rescue of autophagy and lysosome defects in salivary glands of MRL/lpr mice by a therapeutic phosphopeptide. J. Autoimmun. 2018, 90, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Brun, S.; Schall, N.; Bonam, S.R.; Bigaut, K.; Mensah-Nyagan, A.-G.; de Sèze, J.; Muller, S. An autophagy-targeting peptide to treat chronic inflammatory demyelinating polyneuropathies. J. Autoimmun. 2018, 92, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, F.; Schall, N.; Petit-Demoulière, N.; Frossard, N.; Muller, S. An autophagy modulator peptide prevents lung function decrease and corrects established inflammation in murine models of airway allergy. Cells 2021, 10, 2468. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, R.; Poornima, P.; Priya, L.B.; Huang, C.Y.; Padma, V.V. Neferine prevents autophagy induced by hypoxia through activation of Akt/mTOR pathway and Nrf2 in muscle cells. Biomed. Pharmacother. 2016, 83, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.K.; Wu, A.G.; Wang, J.R.; Liu, L.; Law, B.Y. Neferine attenuates the protein level and toxicity of mutant huntingtin in PC-12 cells via induction of autophagy. Molecules 2015, 20, 3496–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, P.K.; Fahrner, A.; Vats, S.; Seranova, E.; Sharma, V.; Chipara, M.; Desai, P.; Torresi, J.; Rosenstock, T.; Kumar, D.; et al. Chemical screening approaches enabling drug discovery of autophagy modulators for biomedical applications in human diseases. Front. Cell Dev. Biol. 2019, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Suresh, S.N.; Chakravorty, A.; Giridharan, M.; Garimella, L.; Manjithaya, R. Pharmacological tools to modulate autophagy in neurodegenerative diseases. J. Mol. Biol. 2020, 432, 2822–2842. [Google Scholar] [CrossRef]
- Hatstat, A.K.; Ahrendt, H.D.; Foster, M.W.; Mayne, L.; Moseley, M.A.; Englander, S.W.; McCafferty, D.G. Characterization of small-molecule-induced changes in Parkinson’s-related trafficking via the nedd4 ubiquitin signaling cascade. Cell Chem. Biol. 2021, 28, 14.e19–25.e19. [Google Scholar] [CrossRef]
- Zhu, Y.; Shan, X.; Safarpour, F.; Erro Go, N.; Li, N.; Shan, A.; Huang, M.C.; Deen, M.; Holicek, V.; Ashmus, R.; et al. Pharmacological inhibition of O-GlcNAcase enhances autophagy in brain through an mTOR-independent pathway. ACS Chem. Neurosci. 2018, 9, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.; Xu, M.; Davey, A.K.; Danon, J.J.; Mellick, G.D.; Kassiou, M.; Rudrawar, S. O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease. ACS Chem. Neurosci. 2019, 10, 2209–2221. [Google Scholar] [CrossRef]
- Whitworth, A.J.; Wes, P.D.; Pallanck, L.J. Drosophila models pioneer a new approach to drug discovery for Parkinson’s disease. Drug Discov. Today 2006, 11, 119–126. [Google Scholar] [CrossRef]
- Chen-Plotkin, A.S.; Albin, R.; Alcalay, R.; Babcock, D.; Bajaj, V.; Bowman, D.; Buko, A.; Cedarbaum, J.; Chelsky, D.; Cookson, M.R.; et al. Finding useful biomarkers for Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaam6003. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.M.C.; Bannerman, D.M. Mouse models of neurodegeneration: Know your question, know your mouse. Sci. Transl. Med. 2019, 11, eaaq1818. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Neuropathological findings in Parkinson’s disease. (A,B) Post-mortem mesencephalon and pons from a control patient (A) and from a patient with PD (B): SN appeared paler in B due to dopaminergic denervation. (C), SN, H&E staining (×250). (D): H&E staining (×250) of LB in a cortical neuron. The black arrow shows a LB. Abbreviations not described in the text: H&E, hematoxylin and eosin.
Figure 1.
Neuropathological findings in Parkinson’s disease. (A,B) Post-mortem mesencephalon and pons from a control patient (A) and from a patient with PD (B): SN appeared paler in B due to dopaminergic denervation. (C), SN, H&E staining (×250). (D): H&E staining (×250) of LB in a cortical neuron. The black arrow shows a LB. Abbreviations not described in the text: H&E, hematoxylin and eosin.

Figure 2.
The three main types of autophagy. Macroautophagy: schematic representation of the sequential steps: initiation/nucleation, phagophore formation, phagophore elongation, closure (autophagosome), fusion, and degradation. Under nutrient/energy-rich conditions (insulin growth factors, amino acids) MTOR inhibits autophagy initiation by regulating the ULK1 complex. In contrast, AMPK activates autophagy in response to stress or starvation. CMA: process that degrades substrates containing a KFERQ-motif (or CMA-targeting recognition motif). The chaperone HSPA8 forms a complex with other co-chaperones (HSP90, HSP40, HSP70-interacting protein/HIP and HSP70-HSP90 organizing protein/HOP) that not only acts as a catalyst allowing unfolded proteins to refold, but also mediates delivery of proteins to the lysosome via the LAMP2A receptor at the lysosomal membrane. Microautophagy: in this process, the cytoplasmic content destined for autophagy is directly engulfed into the lysosome following formation of an invagination. Abbreviations not described in the text: DFCP1, double FYVE domain-containing protein 1; RB1CC1/FIP200, FAK-family interacting protein of 200 kDa; RER, rough ER; UVRAG, UV resistance-associated gene; WIPI, WD repeat domain phosphoinositide-interacting.
Figure 2.
The three main types of autophagy. Macroautophagy: schematic representation of the sequential steps: initiation/nucleation, phagophore formation, phagophore elongation, closure (autophagosome), fusion, and degradation. Under nutrient/energy-rich conditions (insulin growth factors, amino acids) MTOR inhibits autophagy initiation by regulating the ULK1 complex. In contrast, AMPK activates autophagy in response to stress or starvation. CMA: process that degrades substrates containing a KFERQ-motif (or CMA-targeting recognition motif). The chaperone HSPA8 forms a complex with other co-chaperones (HSP90, HSP40, HSP70-interacting protein/HIP and HSP70-HSP90 organizing protein/HOP) that not only acts as a catalyst allowing unfolded proteins to refold, but also mediates delivery of proteins to the lysosome via the LAMP2A receptor at the lysosomal membrane. Microautophagy: in this process, the cytoplasmic content destined for autophagy is directly engulfed into the lysosome following formation of an invagination. Abbreviations not described in the text: DFCP1, double FYVE domain-containing protein 1; RB1CC1/FIP200, FAK-family interacting protein of 200 kDa; RER, rough ER; UVRAG, UV resistance-associated gene; WIPI, WD repeat domain phosphoinositide-interacting.

Figure 3.
Autophagy impairment in PD. Impaired forms of autophagy have been observed in PD. Genetic mutations of α-syn are linked to impairment of the autophagy process. Many factors, such as genetic factors, defective mitochondrial trafficking, oxidative stress, dysfunctional ATP cycle, deregulated mitochondrial dynamics, and altered mitogenesis perturb healthy mitochondria. Damaged/dysfunctional mitochondria allow PINK1 to recruit PRKN, which in turn activates other essential proteins, such as OPTN and ubiquitin, Rab7 and others thus to initiate a quality control process, i.e., mitophagy. The function of the Rab7 is regulated by the TBC1D15/17 (belong to the TBC family with Rab-GAP functions), which is also regulate the shaping and target functions of isolation membrane by cross-linking with Fis1 and MAP1LC3B. The sequential steps of mitophagy are: formation of the phagophore, maturation into the mitoautophagosome, and fusion of the mitoautophagosome with the lysosome. Conventional autophagy also plays an essential role in (both naïve and aggregated) α-syn degradation. α-syn selectively binds to the pathogen-recognition receptor, TLR-4, which activates the downstream signaling pathway following NF-κB activation, to stimulate SQSTM1/p62 production. The SQSTM1 produced binds to the internalized α-syn and initiates the autophagy process. Dysregulation of the autophagy process leads to the accumulation of α-syn alongside SQSTM1. Apart from mitophagy and macroautophagy, CMA also selectively degrades α-syn, which contains a KFERQ-like motif. Selective CMA inhibition or altered CMA functioning affect α-syn degradation. Abbreviations not described in the text: Fis1, Mitochondrial fission 1 protein; GAP, GTPase-activating proteins; IKK, IκB kinase; MyD88, myeloid differentiation protein 88; Rab, Ras superfamily of small G proteins; TBC, Tre-2/Bub2/Cdc16; TIRAP, Toll-interleukin 1 receptor adaptor protein.
Figure 3.
Autophagy impairment in PD. Impaired forms of autophagy have been observed in PD. Genetic mutations of α-syn are linked to impairment of the autophagy process. Many factors, such as genetic factors, defective mitochondrial trafficking, oxidative stress, dysfunctional ATP cycle, deregulated mitochondrial dynamics, and altered mitogenesis perturb healthy mitochondria. Damaged/dysfunctional mitochondria allow PINK1 to recruit PRKN, which in turn activates other essential proteins, such as OPTN and ubiquitin, Rab7 and others thus to initiate a quality control process, i.e., mitophagy. The function of the Rab7 is regulated by the TBC1D15/17 (belong to the TBC family with Rab-GAP functions), which is also regulate the shaping and target functions of isolation membrane by cross-linking with Fis1 and MAP1LC3B. The sequential steps of mitophagy are: formation of the phagophore, maturation into the mitoautophagosome, and fusion of the mitoautophagosome with the lysosome. Conventional autophagy also plays an essential role in (both naïve and aggregated) α-syn degradation. α-syn selectively binds to the pathogen-recognition receptor, TLR-4, which activates the downstream signaling pathway following NF-κB activation, to stimulate SQSTM1/p62 production. The SQSTM1 produced binds to the internalized α-syn and initiates the autophagy process. Dysregulation of the autophagy process leads to the accumulation of α-syn alongside SQSTM1. Apart from mitophagy and macroautophagy, CMA also selectively degrades α-syn, which contains a KFERQ-like motif. Selective CMA inhibition or altered CMA functioning affect α-syn degradation. Abbreviations not described in the text: Fis1, Mitochondrial fission 1 protein; GAP, GTPase-activating proteins; IKK, IκB kinase; MyD88, myeloid differentiation protein 88; Rab, Ras superfamily of small G proteins; TBC, Tre-2/Bub2/Cdc16; TIRAP, Toll-interleukin 1 receptor adaptor protein.

Figure 4.
The macroautophagy pathway and potential drug targets for PD treatment. The diagram includes current small therapeutics and drug candidates (small molecules and peptides) that act at different stages of the macroautophagy pathways and might be beneficial in PD.
Figure 4.
The macroautophagy pathway and potential drug targets for PD treatment. The diagram includes current small therapeutics and drug candidates (small molecules and peptides) that act at different stages of the macroautophagy pathways and might be beneficial in PD.


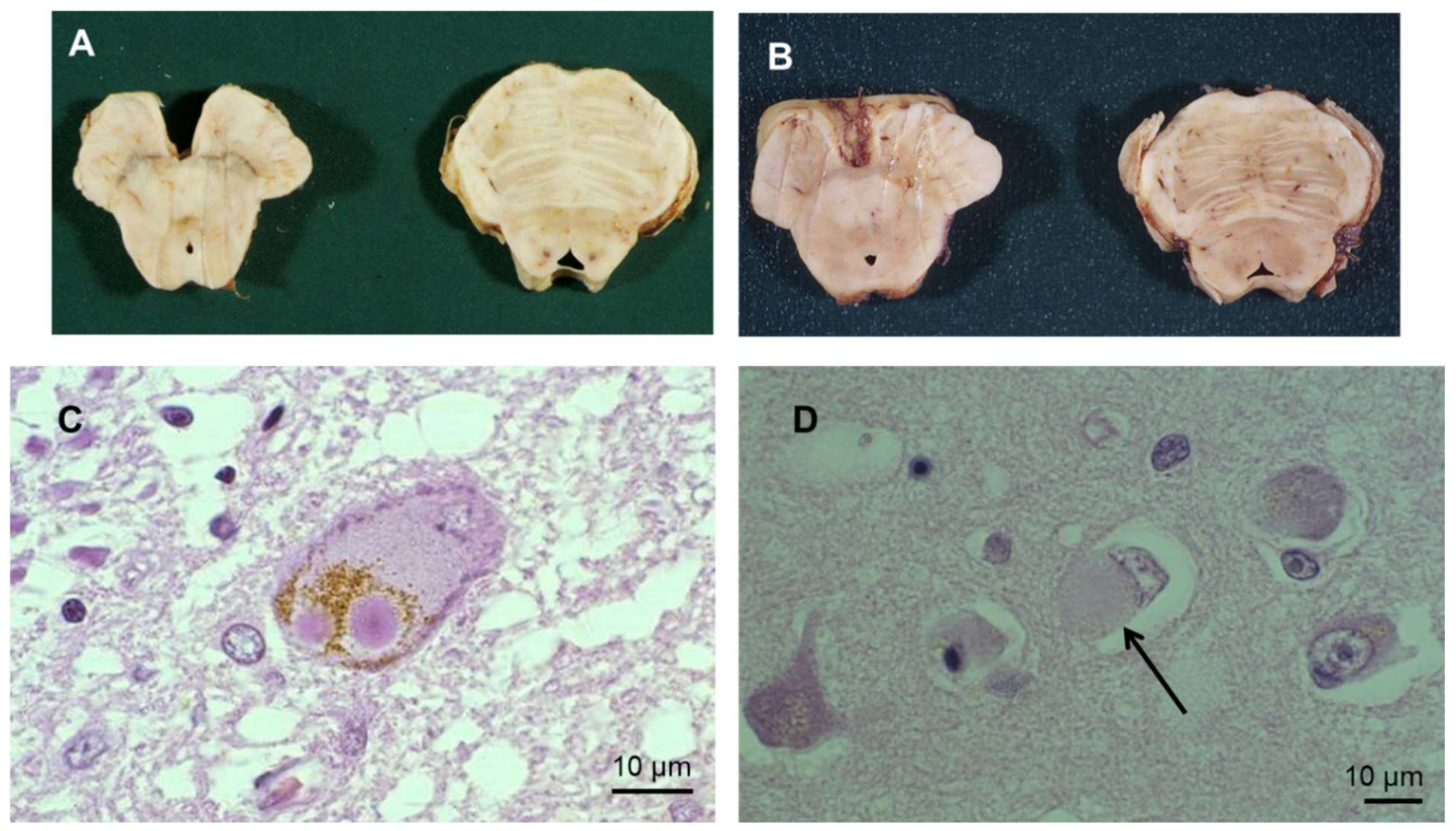{kind=link}
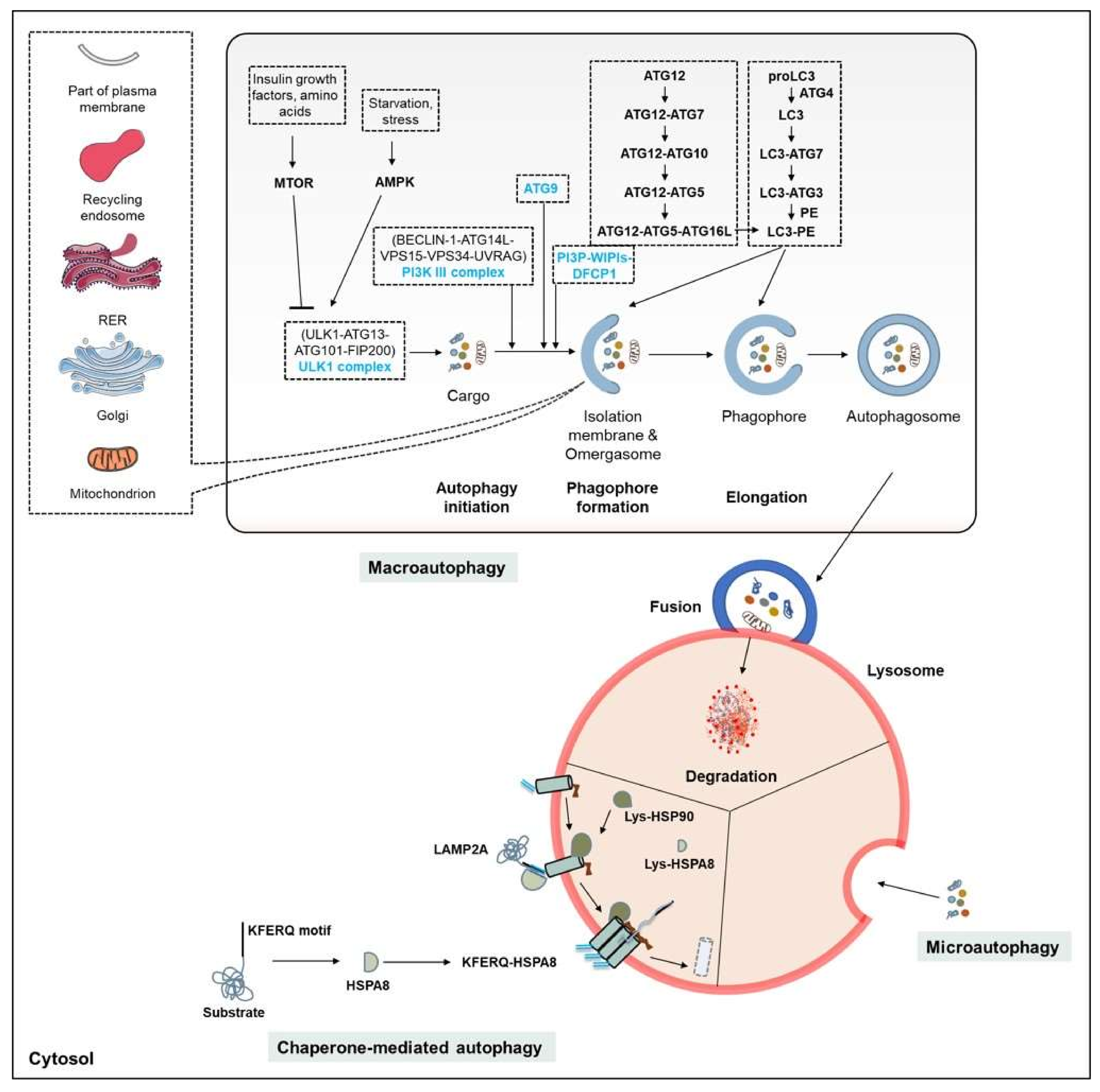{kind=link}
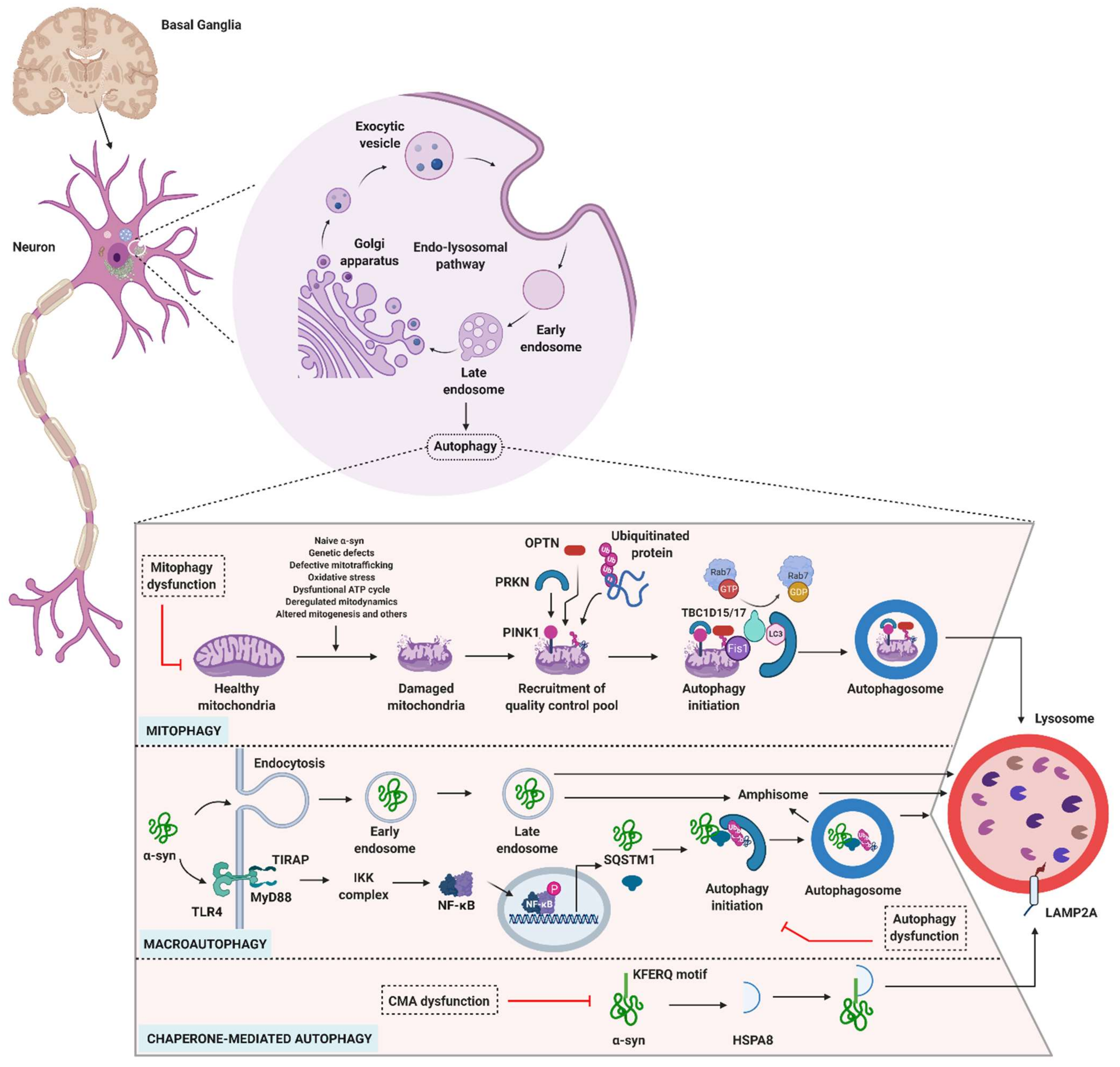{kind=link}
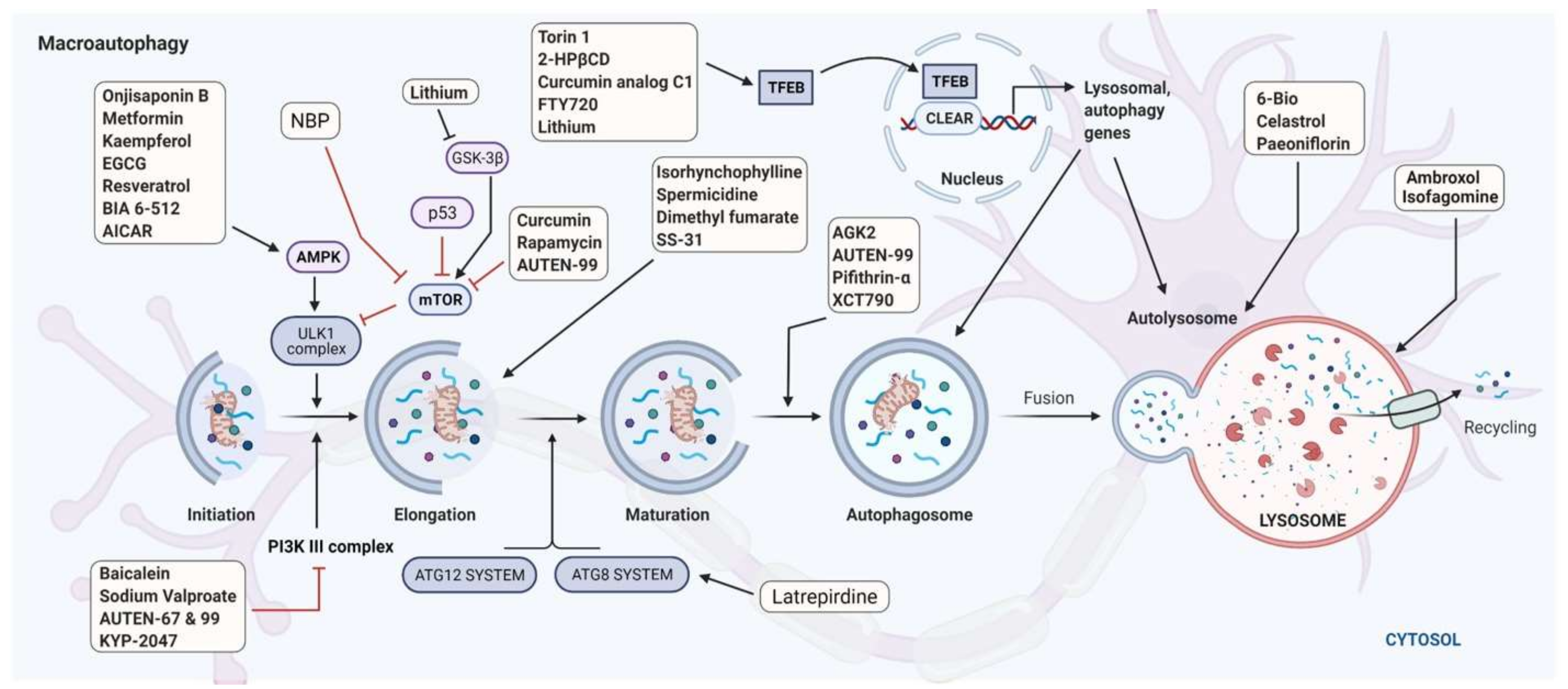{kind=link}
Table 1.
Types of selective autophagy.
| Processes | Organelles and Substrates | Selective Markers | Functions |
|---|---|---|---|
| Aggrephagy | Protein aggregates | MAP1LC3, Alfy |
|
| Crinophagy | Secretory vesicles or granules | HOPS |
|
| CMA | Proteins that contain KFERQ-motif | LAMP2A, HSPA8 |
|
| DNautophagy | DNA | LAMP2C |
|
| ER-phagy (also referred to as reticulophagy) | Endoplasmic reticulum | Syntaxin 17, DFCP1 |
|
| Ferritinophagy | Ferritin | NCOA4 |
|
| Glycophagy | Glycogen | STBD1/Genethonin-1 |
|
| LAP | Bacteria, living and dead cells | Lipidated LC3 |
|
| Lipophagy | Lipid droplets | LIPA |
|
| Lysophagy | Lysosomes | LAMP2A | |
| Mitophagy | Mitochondria | PINK1, RKN |
|
| Nucleophagy | Nucleus | ATG39 * |
|
| Pexophagy | Peroxisomes | ATG36 |
|
| Proteaphagy | Proteasomes | Proteasome (depends on the size of the proteasome) |
|
| Ribophagy | Ribosomes | Ubp3, Bre5, Rsp5 |
|
| RNautophagy | RNA | LAMP2C |
|
| Synucleinphagy | α-syn | SQSTM1 |
|
| Xenophagy | Invading microbes | CALCOCO2/NDP52 |
|
| Zymophagy | Pancreatic zymogen granules | VMP1 |
|
* Yeast-specific receptor; no specific receptor has been identified in mammals. Abbreviations not described in the text: Alfy, PI3P-binding Autophagy-linked FYVE domain protein; ATP, adenosine-triphosphate; Bre5, Ubp3-associated cofactor; CALCOCO2, calcium-binding and coiled-coil domain 2; DFCP1, double FYVE domain-containing protein; HOPS, homotypic fusion and vacuole protein sorting; LAMP2C, lysosomal-associated membrane protein 2C; LIPA, lysosomal acid lipase A; NCOA4, nuclear receptor coactivator 4; Rsp5, E3 ubiquitin ligase; STBD1, Starch-binding domain-containing protein 1; Ubp3, ubiquitin-specific protease 3; VMP1, vacuole membrane protein 1.
Table 2.
Genetic mutations linking autophagy components to PD *.
| Genes | Functions | Link to PD |
|---|---|---|
| ATG5 | ATG5-dependent autophagy protects various cells, including neurons, from apoptosis [99]. |
|
| ATG7 | ATG7 is a vital part of the ATG8 and ATG12 conjugating systems of autophagy, and plays an essential role in neuronal development [101]. |
|
| ATG12 | In conjunction with other ATGs, it forms an ATG12 system, which is essential for autophagosome formation. |
|
| ATP6AP2 | Vacuolar ATPase localized on the lysosomal membrane that regulates pH. | |
| ATP10B | P4-type ATPase present on the late endo/lysosomal compartment. In addition to its lipid flippase function (transfer of lipids from the exoplasmic to the cytoplasmic membrane leaflet), it also transports cell cycle regulator proteins from the ER to endosomes and lysosomes [9,13]. |
|
| ATP13A2/PARK9 | P5-type ATPase localized on vesicular structures, particularly lysosomes; functions as a cation transporter [10]. |
|
| DJ-1/PARK7 | Functions as an antioxidant and chaperone. Through its diversified functions, it maintains mitochondrial homeostasis [106]. |
|
| DNAJC13/PARK21 | Retromer-mediated endosomal protein sorting [106]. |
|
| FBOX7/PARK15 | In association with PINK1 and PRKN, regulates mitophagy [109]. |
|
| GBA1 | GBA is a lysosomal enzyme, which degrades cell membrane glycolipids, i.e., glycosylceramide [48]. |
|
| HSPA8 | Acts as a substrate carrier protein in CMA. |
|
| LRRK2 | LRRK2 is known for sorting various vesicles, based on its interactions with Rab5b, Rab7, Rab7L1, Hsc70, and others [10]. |
|
| MAP1LC3 | MAP1LC3B and its subfamily members are crucial to autophagosome formation, elongation and maturation [88]. |
|
| PRKN/PARK2 | Gene that encodes PRKN (E3 ligase), which plays an essential role in mitophagy in parallel with PINK1. |
|
| PINK1/PARK6 | Member of the serine-threonine kinases class, plays a crucial role in mitochondria quality control by inducing mitophagy. |
|
| TMEM175 | Gene that encodes endo/lysosomal K+ channel [117]. |
|
| TMEM230 | Gene that encodes transmembrane protein involved in retromer and secretory vesicle trafficking functions. |
|
| VPS13C/PARK23 | Mitophagy development. |
|
| VPS35/PARK17 | As a part of the retromer complex, it performs protein trafficking functions [106]. |
|
Table 3.
Therapeutic autophagy-targeting molecules with potential in PD *.
| Candidate | Mechanism of Action | Comments |
|---|---|---|
| AGK2 | Enhances autolysosome formation |
|
| AUTEN-99 (autophagy enhancer-99) | Inhibits the phosphatase activity of MTMR14 (Jumpy), a negative regulator of autophagy | |
| Baicalein | Induces PI3K-mediated autophagy |
|
| 6-Bio | Enhances autolysosome formation in DA neurons |
|
| Celastrol | Enhances autolysosome formation |
|
| Curcumin | Inhibits MTOR/p70S6K |
|
| Curcumin analogue C1 | MTOR-independent TFEB enhancer. Selectively binds to the N-terminus of TFEB and promotes its nuclear translocation | |
| Dimethyl fumarate | Modulator of SQSTM1-dependent autophagy |
|
| Epigallocatechin gallate (EGCG) | Activates AMPK | |
| 2-HPβCD | Activates TFEB |
|
| Isofagomine | GCase |
|
| (Iso)rhynchophylline | Activation of BECN-mediated autophagy |
|
| Kaempferol | AMPK/MTOR-mediated pathway | |
| KYP-2047 | Prolyl endopeptidase inhibitor III | |
| Latrepirdine (Dimebon; dimebolin) | Enhances ATG8-dependent autophagy | |
| Lithium | Reduces IP3 production by inhibiting inositol monophosphatase |
|
| MCC950 | Potent, selective inhibitor of NLRP3 with IC50 of 7.5 nM in BMDMs | |
| Metformin | Antihyperglycemic agent of the biguanide class. Acts via both AMPK-dependent and AMPK-independent mechanisms |
|
| Onjisaponin B | Triggers autophagy via the AMPK/MTOR-mediated signaling pathway |
|
| Paeoniflorin | Enhances autolysosome formation | |
| Pifithrin (PFT)-α | Specific inhibitor of p53 transcription activity. Displays potent p53-independent activity in cells. |
|
| Rapamycin and its derivatives (CCI-779, RAD001 and AP23573) | Inhibitors of MTOR | |
| Sodium valproate | Synergistic to lithium-induced autophagy |
|
| Spermidine | Autophagy inducer that maintains cellular and neuronal homeostasis. May achieve effects via BECN1 and TFEB-mediated pathways |
|
| SS-31 (D-Arg-2′6′-dimethylTyr-Lys-Phe-NH2) | BECN1-mediated autophagy | |
| Torin1 | MTOR-dependent TFEB enhancer | |
| Trehalose | Induces autophagy via lysosome-mediated TFEB activation | |
| MF-094 | Selective USP30 inhibitor | |
| XCT790 | Modulates autophagosome formation by estrogen-related receptor α-dependent manner |
|
* Further information about drug candidates or drug-like molecules that have potential in PD via their effects on autophagy processes can be found elsewhere [103,271,272,273,274]. Abbreviations not used in the text: BMDMs, bone-marrow-derived macrophages; GCase, β-glucocerebrosidase; GSK-3β, glycogen synthase kinase-3 beta; IP3, inositol-1,4,5-trisphosphate; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MTMRs, myotubularin-related phosphatases; SS, Szeto–Schiller, USP, ubiquitin specific peptidase.
Table 4.
Therapeutic autophagy-targeting molecules under clinical evaluation for use in PD *.
| Drugs | Characteristics and Mechanism of Action | Stage of Development/Sponsors | Identifiers |
|---|---|---|---|
| Ambroxol | Well-known expectorant and mucolytic agent. In addition to its diverse functions, also acts as a potent chaperone for GCase [277]. In Vitro and in vivo studies in wild-type and transgenic models indicate that its therapeutic effects in the context of PD involve modulating the autophagy process [221,278,279]. These effects are mediated by the TFEB pathway and through increased exocytosis [280]. A recent clinical study in PD patients with and without GBA1 mutations provided positive preliminary data for large cohort studies [281]. | Phase II/University College, London, UK (NCT02941822), Lawson Health Research Institute (NCT02914366) | NCT02941822; NCT02914366 |
| BIA 6-512 (trans-resveratrol) | An analogue of a natural compound, resveratrol, which has a wide pharmacological action, especially in PD [282,283]. Potential antioxidant; it also initiates Sirtuin 1 (SIRT1)-dependent AMPK activation [103,283]. | Phase I/Bial—Portela C S.A. | NCT03095092 |
| Coenzyme Q10 (CoQ) | Free radical scavenger used as a supplement in PD. Reduced CoQ in cells triggers mitophagy [284]. | Phase III/Weill Medical College of Cornell University (NCT00740714), Technische Universität Dresden, Germany (NCT00180037) | NCT00740714; NCT00180037 |
| DL-3-n-butylphthalide (NBP) | An active component isolated from Apium graveolens (Apiaceae) [285]. Its antioxidant function, and capacity to correct mitochondrial dysfunction is widely documented [286]. Approved by the Chinese FDA for the treatment of ischemic stroke [287]. Various preclinical models of PD have confirmed its efficacy in alleviating neuronal toxicity via autophagic mechanisms; indirectly inhibits MTOR-mediated autophagy [286,288,289]. | Phase II/First Affiliated Hospital of Bengbu Medical College (Bengbu, China) | ChiCTR1800018892 |
| MitoQ (mitoquinone) | A conjugate of lipophilic triphenylphosphonium cation and coenzyme Q10. Powerful antioxidant. Studies with MitoQ on leukemia cells (HL-60) and hepatocellular carcinoma cells (HepG2) confirmed the induction of autophagy following activation of AMPK and inhibition of MTOR [207]. | Phase II/Antipodean Pharmaceuticals, Inc. | NCT00329056 |
| Nicotinamide riboside | Used as a supplement. Nicotinamide (in various forms) induces autophagy by acting on SIRT1-dependent and MTOR-mediated mechanisms [290,291]. | ND/Haukeland University Hospital, Norway | NCT03816020 |
| Nilotinib | An Abelson kinase/tyrosine kinase inhibitor. Approved by the US FDA (2007) for the treatment of chronic myelogenous leukemia. Preclinical studies on transgenic mice models (A53T mutation of human α-syn) have shown that it increases α-syn clearance via BECN1-mediated autophagy [292]. Reduces cell death and improves motor function. Under evaluation for treatment of other neurodegenerative diseases (AD, NCT02947893; HD, NCT03764215). | Phase II/Georgetown University, USA | NCT02954978 |
* Updated: June 2021. Abbreviations not used in the text: FDA, Food and Drug Administration; GCase, β-glucocerebrosidase; ND, not disclosed.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bonam, S.R.; Tranchant, C.; Muller, S. Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease. Cells 2021, 10, 3547. https://doi.org/10.3390/cells10123547
AMA Style
Bonam SR, Tranchant C, Muller S. Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease. Cells. 2021; 10(12):3547. https://doi.org/10.3390/cells10123547
Chicago/Turabian StyleBonam, Srinivasa Reddy, Christine Tranchant, and Sylviane Muller. 2021. "Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease" Cells 10, no. 12: 3547. https://doi.org/10.3390/cells10123547
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.